
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDrug and dye binding induced folding of the intrinsically disordered antimicrobial peptide CM15†
Ferenc Zsila *a,
Szilvia Bőszeb,
Kata Horvátib,
Imola Cs. Szigyártóa and
Tamás Beke-Somfaia
*a,
Szilvia Bőszeb,
Kata Horvátib,
Imola Cs. Szigyártóa and
Tamás Beke-Somfaia
aBiomolecular Self-Assembly Group, Institute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences, P.O. Box 286, H-1519, Budapest, Hungary. E-mail: zsila.ferenc@ttk.mta.hu
bMTA-ELTE Research Group of Peptide Chemistry, Hung. Acad. Sci., Eötvös Loránd University, Budapest 112, P.O. Box 32, H-1518 Budapest, Hungary
First published on 23rd August 2017
Abstract
The rapid increase of antimicrobial resistance against conventional antibiotics has resulted in a significant focus on the use of peptides as antimicrobial agents. Understanding the structure and function relationships of these compounds is thus highly important, however, their in vivo actions are a complex issue, including interactions with small molecule agents. Here we report the folding inducing capability of some pharmaceutical substances and synthetic dyes on the intrinsically disordered (ID) cationic antimicrobial peptide CM15 (KWKLFKKIGAVLKVL). By employing circular dichroism (CD) spectroscopy, it is shown that some therapeutic drugs (suramin, pamoic acid, cromolyn) and polysulfonated dyes (Congo red, trypan blue) trigger the disorder-to-order conformational transition of CM15. The cooperative binding of 2–4 acidic molecules per peptide chain provokes its folding in a concentration dependent manner. Secondary structure analysis indicated the sharp and moderate rise of the α-helical and β-sheet content, respectively. According to semi-empirical quantum chemical calculations, these organic molecules may induce folding by forming multiple salt-bridges with lysine residues from both N- and C-terminals as well as from the middle of the CM15 sequence. Due to the mutual neutralization of the positive and negative charges, the water solubility of the resulting complexes decreases which favours their aggregation as detected by dynamic light scattering measurements. Our findings suggest that small molecules can dramatically affect the structure of antimicrobial peptides, which may potentially alter, either enhancing or attenuating, their efficiency. It is proposed that CM15 or similar ID peptides could be useful for preliminary screening of folding inducer effect of anionic drugs and biomolecules. The data presented herein may stimulate further studies on the structural and functional impacts of related compounds on ID peptides.
Introduction
Cationic antimicrobial peptides (AMPs) are instrumental constituents of the innate immune system and ubiquitous in all living organisms.1 Their size typically ranges from ∼10 to 50 residues and most of them carry a net positive charge due to the preponderance of lysine and arginine residues. The electrostatic repulsion between these side chains renders the solution structure of the majority of cationic AMPs intrinsically disordered (ID) with no discernible secondary structure.2,3 However, upon membrane binding or in helix promoting organic solvents such as trifluoroethanol (TFE) many AMPs undergo a conformational change and often fold into a well ordered, mostly α-helical structure.4,5 The membrane associated conformational transition is a crucial step in mediating their biological activities against a broad spectrum of bacteria, fungi, parasites and even cancer cells.1 Besides the elimination and inactivation of the invading pathogens, these multifaceted substances also exhibit immunomodulatory activities.6 Importantly, disordered regions are characteristic not only to AMPs but are highly abundant in eukaryotic proteomes as well.7 For instance, about 60% of all proteins contain a disordered stretch of 15 or more amino acids.8 Due to their exceptional structural plasticity, intrinsically disordered proteins (IDPs) and ID protein regions (IDPRs) are engaged in a plethora of biological functions and thus in a variety of human diseases as well.9 Similarly to AMPs, their sequences are significantly enriched in charged (Arg, Lys, Glu, Asp) and other structure-breaking residues.7,10The natively unfolded CM15 is a synthetic hybrid peptide containing five cationic residues (Lys-Trp-Lys-Leu-Phe-Lys-Lys-Ile-Gly-Ala-Val-Leu-Lys-Val-Leu).5,11–13 It is composed of the first seven amino acids of the insect AMP, cecropin A, and residues 2–9 of the bee venom peptide melittin (cecropin A (1–7)-melittin (2–9)amide, CAS No. 157606-25-2). CM15 retains the bactericidal effect of cecropins but lacks the hemolytic activity of melittin. It is to be noted that cationic AMP families of human and animal origins such as cathelicidins and NK-lysins abundantly possess ID regions.14,15 Like CM15, NK-lysins are amphiphilic and besides the disorder promoting Lys residues they also contain some structure stabilizing aromatic side chains (Trp, Phe).14 In addition, several examples of experimentally verified ID protein regions are known consisting of both order- and disorder promoting residues (ESI Table S1†). From this point of view, CM15 can be considered as an, though less typical, ID model sequence.
Employing experimental as well as computational methods, our work demonstrates that some biologically active small molecules deeply affect the secondary structure of this peptide inducing disorder-to-order conformational transition.
Materials and methods
Materials
Congo red sodium salt, trypan blue sodium salt, cromolyn sodium salt, pamoic acid sodium salt and heparin sodium salt (from porcine intestinal mucosa) were obtained from Sigma-Aldrich and used as supplied. Suramin sodium salt was purchased from Calbiochem. All other chemicals were of analytical reagent grade.Preparation of working solutions
All materials were dissolved in pH 7.3 potassium phosphate buffer (10 mM) containing 50 mM Na2SO4. Due to its polydisperse nature, the molar concentration of heparin is expressed in the average repeating disaccharide unit (Mw ≈ 665).Circular dichroism (CD) spectroscopic measurements
CD spectra were measured on a JASCO J-715 spectropolarimeter at 25 ± 0.2 °C in a 0.1 cm path length rectangular quartz cuvette (Hellma, USA). Temperature control was provided by a Peltier thermostat. Peptide CD data were collected in continuous scanning mode between 185 and 260 nm at a rate of 50 nm min−1, with a step size of 0.1 nm, response time of 4 s, three accumulations, and 2 nm bandwidth. CD curves of peptide and ligand–peptide samples were corrected by the spectral contribution of the blank buffer solution. JASCO CD spectropolarimeters record CD data as ellipticity (Θ) in units of millidegrees (mdeg). Θ values were converted into molar circular dichroic absorption per residues (Δε/n, for CM15 n is 15). Δε = Θ/(32982cl), where ‘c’ is the molar concentration of the peptide (mol L−1), and ‘l’ is the optical path length expressed in cm.Estimation of the secondary structure content of CM15 from CD spectroscopic data
In order to compute the fractions of α-helices, β-sheets, turns and unordered content of ligand-free and ligand-loaded forms of CM15, the DichroWeb online server was used.16 Taking into consideration the disordered structure of CM15, reference data set 7 was applied against the experimental CD data between 190–240 nm. In contrast to other reference sets, it contains spectra of five denatured proteins as well.16 The reference set 6 could also be used but it requires quality spectral data measured down to 185 nm. Among the algorithms available on DichroWeb for fitting CD data to reference sets of proteins with known secondary structure, the CDSSTR method yielded the lowest (<0.03) and most consistent normalized root mean square deviations (NRMSD) and, thus, was adopted here.17 Before secondary structure analysis, the experimental CD spectra were smoothed with a convolution width of 17 using the Means-Movement method (JASCO Spectra Analysis software, version 1.53.00).Dynamic light scattering (DLS) analysis
DLS measurements were performed on a W130i instrument (Avid Nano Ltd., High Wycombe, UK) with diode laser (660 nm) and a photodiode detector at 20 °C. Low volume disposable cuvettes having a 1 cm path length were used (UVette, Eppendorf Austria GmbH). Different amount of drugs, dyes and heparin were added to 53 μL CM15 sample (30.5 μM) which then diluted to a final volume of 80 μL (20 μM CM15) with the same buffer used for the CD spectroscopic measurements. The first data acquirement was performed 120 s after addition of the ligands to the peptide solution to ensure the homogeneity of the sample. The time-dependent autocorrelation functions were measured for 10 seconds, repeated 10 times and the average distributions were reported. The average and the standard deviation values of the sizes corresponding to the peak of interest in each of these distributions represent the apparent hydrodynamic diameter and the experimental error for each sample, respectively. DLS data were processed with the iSize 3.0 software (Avid Nano Ltd., High Wycombe, UK).Peptide synthesis, purification and characterization
See the ESI.†Computational details
See the ESI.†Results and discussion
Chiroptical evaluation of the induced folding of CM15
Circular dichroism (CD) spectroscopy is an inherently sensitive, non-destructive tool for the conformational analysis of peptides and proteins.18,19 The far-UV CD curve recorded in buffer solution indicates that the structure of CM15 is intrinsically disordered (Fig. 1A). The spectrum is dominated by a sole negative band at around 199–200 nm which is typical of highly dynamic conformational ensembles.18 Furthermore, there is no shoulder in the 210–230 nm region which also refers to the lack of dominant secondary structural elements. In full concordance with this qualitative assessment, quantitative evaluation of the CD data shows the decisive contribution of the disordered state (Table 1). However, in TFE, which provides a strong dehydration force allowing the backbone to collapse into a self-H-bonded form, profound spectral changes occur (Fig. 1A). The negative band is replaced by an intense couplet consisting of a longer wavelength negative and a shorter wavelength positive peak that correspond to the π–π* exciton components of the peptide bonds. In addition, there is an unresolved minimum at about ∼222 nm allied to the n–π* transition of the amide chromophores. All of these spectral features complemented with the quantitative analysis refer to the α-helical folding of CM15 (Table 1).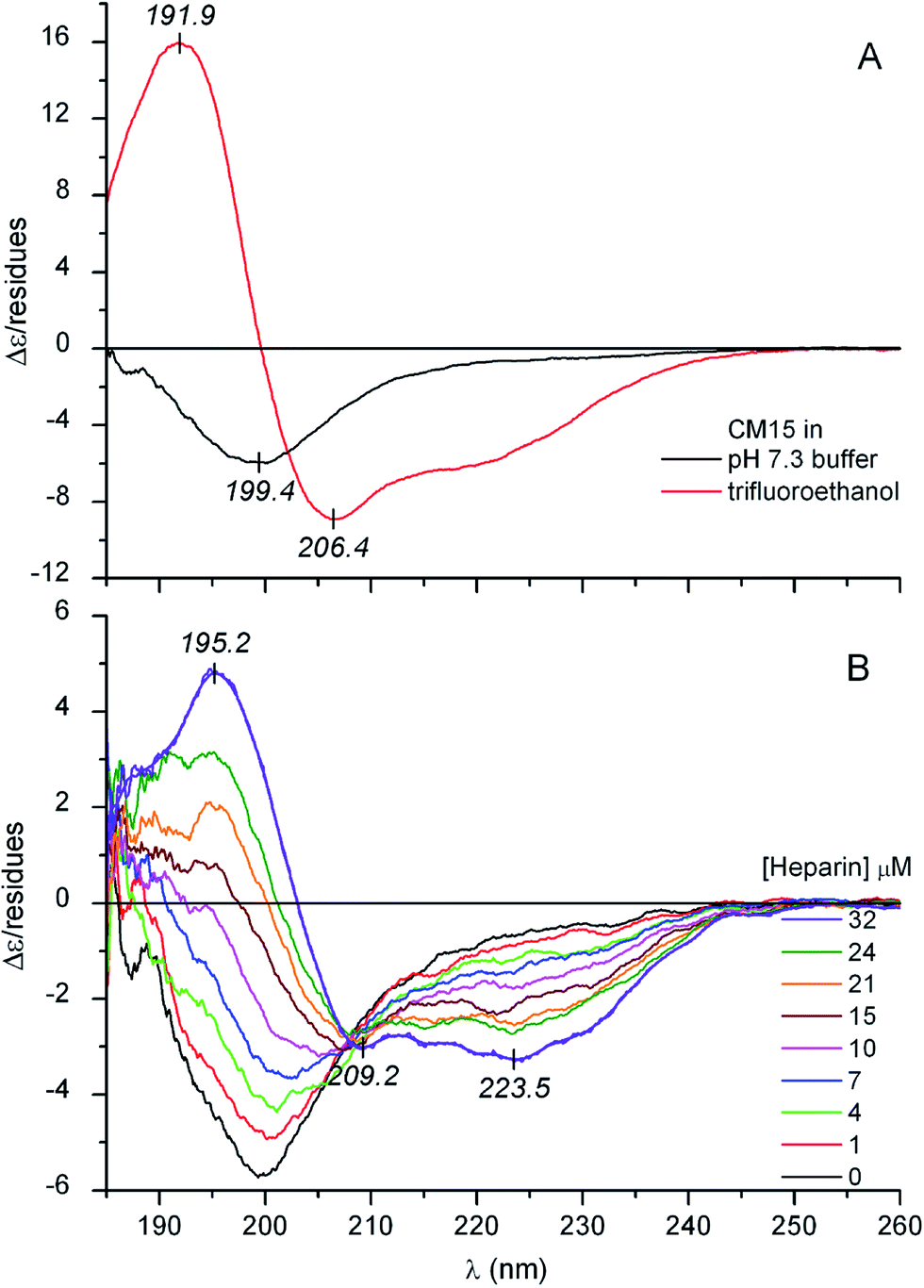 | ||
| Fig. 1 (Panel ‘A’) Far-UV CD spectrum of 30 μM CM15 in buffer and in trifluoroethanol. (Panel ‘B’) Far-UV CD spectra of 23 μM CM15 measured at increasing concentrations of heparin in the sample solution (10 mM potassium phosphate buffer at pH 7.3, 50 mM Na2SO4, 25 °C). | ||
| [CM15] (μM) | [Ligand] (μM) | α-Helix | β-Sheet | Turn | Unordered | NRMSD | |
|---|---|---|---|---|---|---|---|
| CM15 in TFE | 30 | — | 77 | 7 | 7 | 9 | 0.004 |
| CM15 in buffer | 23 | — | 4 | 13 | 9 | 74 | 0.017 |
| +Heparin | 23 (16) | 32 | 43 | 12 | 15 | 30 | 0.024 |
| CM15 in buffer | 40 | — | 4 | 14 | 9 | 73 | 0.012 |
| +Congo red | 39 (32) | 80 | 19 | 26 | 17 | 37 | 0.021 |
| CM15 in buffer | 39 | — | 5 | 13 | 10 | 72 | 0.017 |
| +Trypan blue | 39 (28) | 100 | 24 | 20 | 19 | 36 | 0.017 |
| CM15 in buffer | 39 | — | 4 | 12 | 9 | 74 | 0.016 |
| +Suramin | 39 (36) | 45 | 15 | 26 | 21 | 37 | 0.019 |
| CM15 in buffer | 27 | — | 5 | 13 | 9 | 72 | 0.018 |
| Pamoic acid | 27 (14) | 136 | 21 | 26 | 17 | 37 | 0.027 |
It is well documented that besides membrane binding and TFE, polyanionic biopolymers such as sulfated glycosaminoglycans (e.g., heparin, heparan sulfate) can also provoke the folding of AMPs.20–22 In line with these reports, our CD spectroscopic results verify that albeit in a less extent to that observed in TFE, the coil-to-helix transition of CM15 can be successfully induced by heparin (Fig. 1B and Table 1). For the first time, this communication demonstrates that similar structural rearrangement can also be prompted by pharmaceutical drugs and synthetic dyes possessing acidic functional groups (Scheme 1). Sequential addition of drugs (suramin, pamoic acid, cromolyn) as well as sulfonated dyes (Congo red, trypan blue) substantially altered the disorder-associated CD profile of CM15 (Fig. 2 and 3). The broad negative band is gradually converted into a polyphasic curve displaying two negative and a positive extremum above and below 200 nm, respectively. These spectral transformations occurred promptly after addition of the ligands and exhibited no time dependence. In relation to the α-helical pattern measured in TFE, the negative–positive couplets are less intense and the λmax values are red shifted (Fig. 2 and 3). Additionally, the spectral positions of the isodichroic (zero crossover) points are also at higher wavelengths than that displayed in Fig. 1A (202 nm) and they are shifted to longer wavelengths during the titrations (ESI Fig. S1†). The smallest shift was observed with heparin followed by trypan blue and Congo red. Of note, the classical coil ⇌ helix equilibrium generates a family of CD spectra with an isodichroic point near 203 nm.18,23 In contrast, the isodichroic points of coil ⇌ β-sheet transitions are at longer wavelengths ranging between 208 and 215 nm.24,25 Consideration of these results refers to the ligand binding induced disorder-to-order structural transition of CM15 which involves more than two conformational states. It seems that besides the α-helix, ligand molecules also affect the percentage of additional structural elements such as β-sheets and turns. That is supported by the estimation of the secondary content of ligand-loaded CM15 (Table 1). At the cost of the unstructured component, the α-helical fraction multiplied by a factor of 4–5 but the β-sheet and turn content rose significantly as well. Taking into account the CD changes in the function of the ligand/peptide molar ratio, suramin was the most effective folding inducer whilst trypan blue and Congo red increased the helix content to the greatest extent. In marked contrast to heparin which left the β-sheet fraction unaltered, all compounds raised its proportion (Table 1). Since the heparin chain lacks aromatic rings, this structural motif may be responsible for the sheet promoting activity of the small molecules.
 | ||
| Scheme 1 Chemical structures of folding inducer small molecules used in this study. | ||
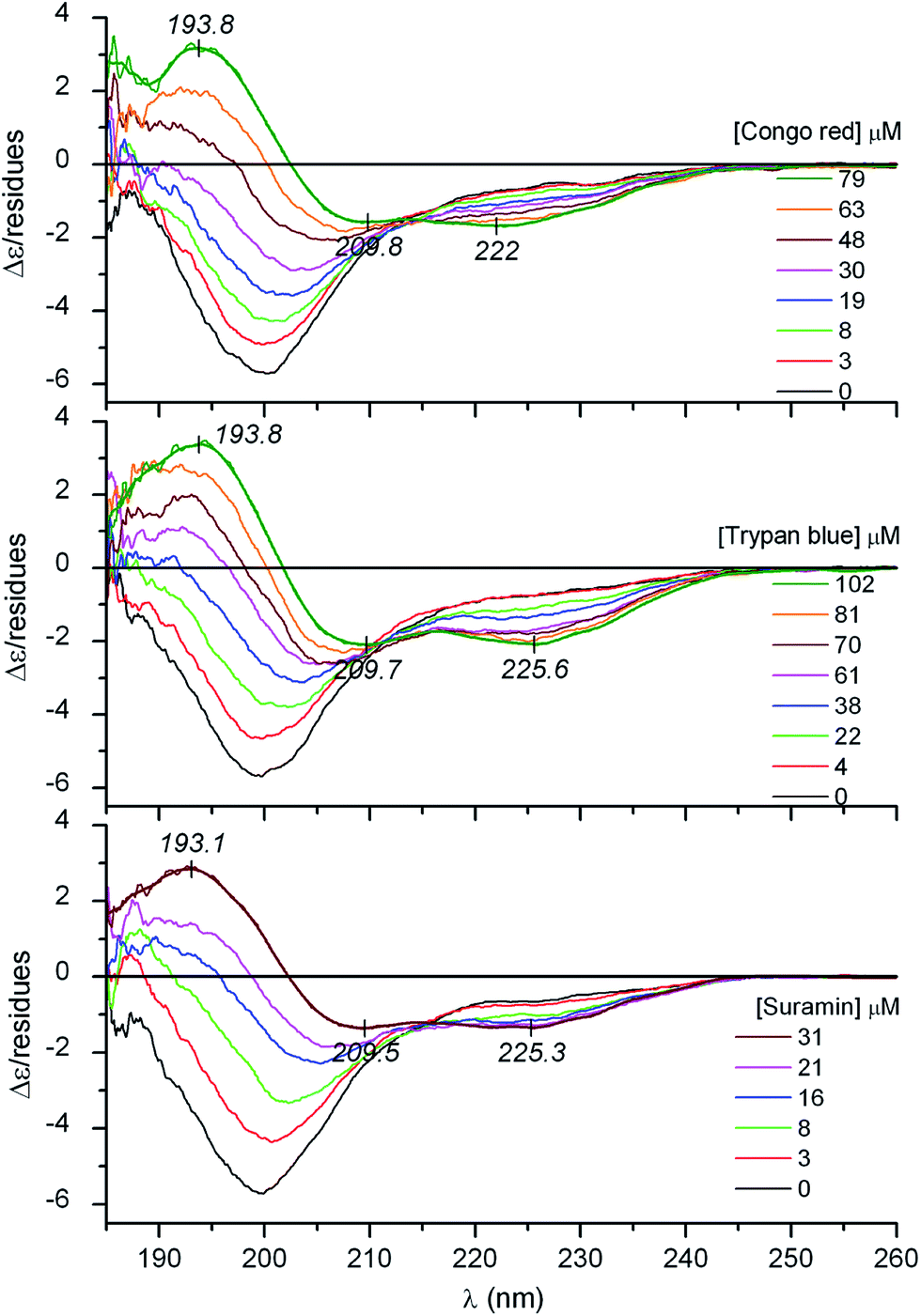 | ||
| Fig. 2 Far-UV CD spectroscopic changes of CM15 (39 μM) measured upon consecutive increase of the concentrations of sulfonated azo dyes and the antiparasitic drug suramin in the sample solution (10 mM potassium phosphate buffer at pH 7.3, 50 mM Na2SO4, 25 °C). Thin lines are smoothed versions of the CD curves obtained at the highest concentration of the ligands. | ||
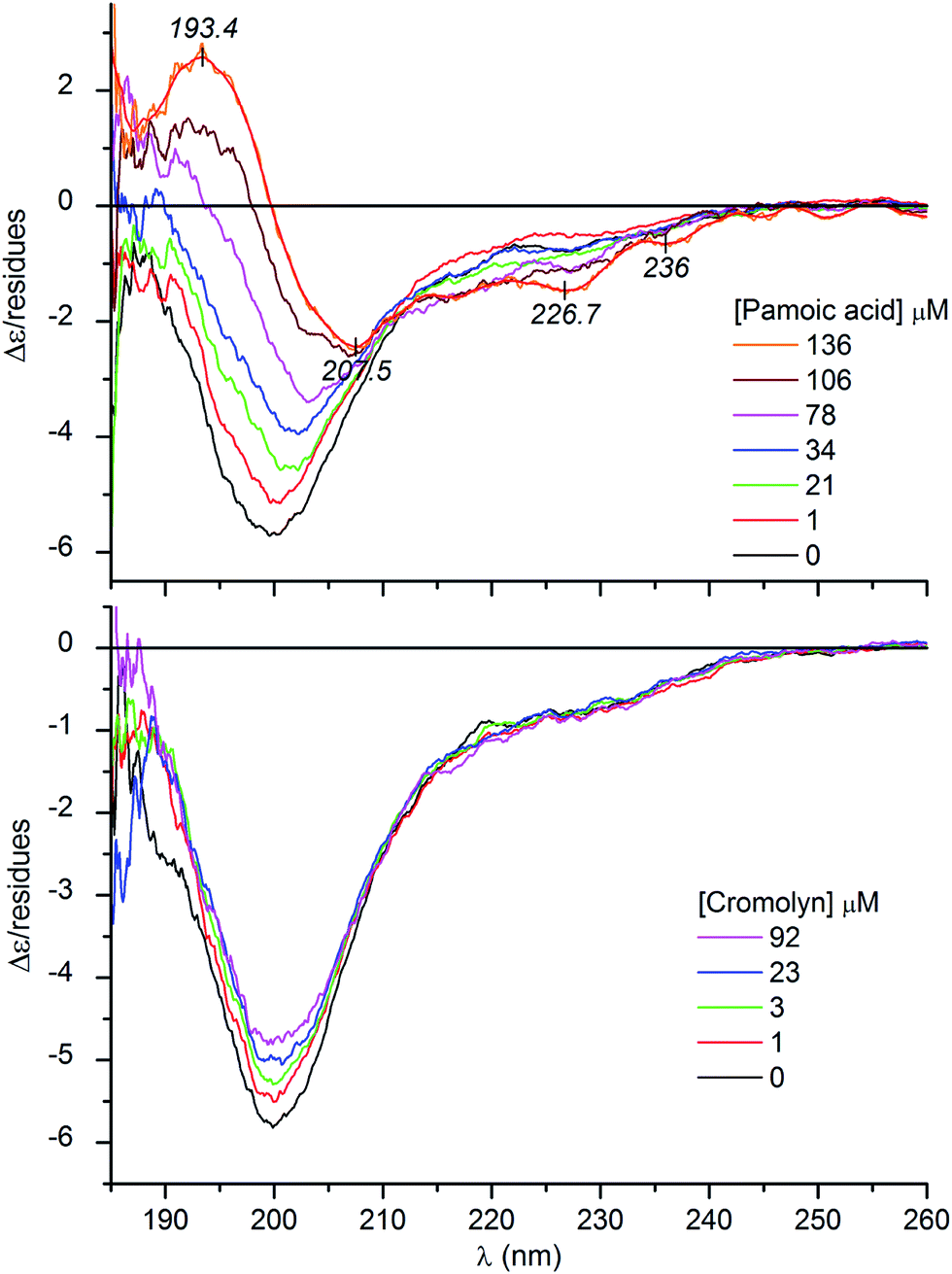 | ||
| Fig. 3 Representative CD curves of CM15 (27 and 30 μM) measured upon consecutive increase of the concentration of pamoic acid and the antiallergic drug cromolyn in the sample solution (10 mM potassium phosphate buffer at pH 7.3, 50 mM Na2SO4, 25 °C). Thin line (orange) is the smoothed version of the CD curve obtained at the highest concentration of pamoic acid. | ||
It is to be noted that at high pamoic acid concentrations, two negative CD peaks are displayed at 227 and 236 nm in the strong UV absorption band of the naphthyl units (Fig. 3). Most likely, these signals are not of peptide origin but allied to the asymmetrically perturbed π–π* transitions of pamoic acid. As such, these are induced (extrinsic) CD effects associated to the interaction between the chiral peptide and the achiral ligand.26 Encoded by its structure, pamoic acid has a pronounced tendency to conformational enantiomerism induced by asymmetric hosts.27,28 Interestingly, considered as a pharmacologically inactive constituent, pamoic acid has long been used in several drug formulations as pamoate salt. It has been disclosed, however, that it is a potent agonist of the human G-protein coupled receptor 35.29,30
CD spectroscopic changes were utilized to estimate the binding affinity and stoichiometry of the folding inducer agents. Upon successive addition of the ligands, the original CD curve gradually transformed including the appearance of a zero crossing point at shorter wavelengths which then shifted bathochromically during the titration (Fig. 1–3, ESI Fig. S1†). However, no further shift was observed at the end of the titrations at high ligand/peptide molar ratios. The measure of the shift in wavenumber was calculated as the arithmetic difference between the local maximum of the native peptide at 188 nm (53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 191 cm−1) and the actual position of the crossing point at increasing concentrations of the ligand. It is a better approach than using CD intensity changes since rising the concentration of UV-active ligands in the sample solution progressively deteriorates the signal-to-noise ratio resulting in less reliable data points for the estimation of the binding parameters. Non-linear regression analysis of the shift values plotted against the concentrations of the inducers gave rise to a sigmoidal curve in each case suggesting cooperative binding interactions (ESI Fig. S2†). The Hill coefficients (h) indicate the formation of multimeric complexes where 2–4 ligand molecules are accommodated to a peptide chain (Table 2). According to the apparent Kd values, suramin binds most tightly to CM15 followed by the azo dyes and pamoic acid. Importantly, the CM15 affinity of suramin and heparin is equally strong which could be due to the high degree of sulfation and thus the heparin-mimicking ability of this drug as reported previously in the literature.31–34 The development of suramin was based on earlier observations on the trypanocidal activity of related polysulfonated dyes such as trypan blue.35,36 As a highly promiscuous drug showing antiprotozoal, antihelminthic, antiviral and anticancer activities, suramin affects the function of diverse range of receptors, growth factors and enzymes.34,35 Interestingly, many of them possess ID regions raising the possibility that this drug may alter protein functions by inducing disorder-to-order conformational transitions (ESI Table S2†). Similarly to suramin, some protein targets of Congo red (ESI Table S3†), pamoic acid (ESI Table S4†) and even cromolyn (ESI Table S5†) contain ID sequences as well.
191 cm−1) and the actual position of the crossing point at increasing concentrations of the ligand. It is a better approach than using CD intensity changes since rising the concentration of UV-active ligands in the sample solution progressively deteriorates the signal-to-noise ratio resulting in less reliable data points for the estimation of the binding parameters. Non-linear regression analysis of the shift values plotted against the concentrations of the inducers gave rise to a sigmoidal curve in each case suggesting cooperative binding interactions (ESI Fig. S2†). The Hill coefficients (h) indicate the formation of multimeric complexes where 2–4 ligand molecules are accommodated to a peptide chain (Table 2). According to the apparent Kd values, suramin binds most tightly to CM15 followed by the azo dyes and pamoic acid. Importantly, the CM15 affinity of suramin and heparin is equally strong which could be due to the high degree of sulfation and thus the heparin-mimicking ability of this drug as reported previously in the literature.31–34 The development of suramin was based on earlier observations on the trypanocidal activity of related polysulfonated dyes such as trypan blue.35,36 As a highly promiscuous drug showing antiprotozoal, antihelminthic, antiviral and anticancer activities, suramin affects the function of diverse range of receptors, growth factors and enzymes.34,35 Interestingly, many of them possess ID regions raising the possibility that this drug may alter protein functions by inducing disorder-to-order conformational transitions (ESI Table S2†). Similarly to suramin, some protein targets of Congo red (ESI Table S3†), pamoic acid (ESI Table S4†) and even cromolyn (ESI Table S5†) contain ID sequences as well.
| Ligand | Kd (μM) | h | R2 |
|---|---|---|---|
| Heparin | 13.3 (±0.2) | 2.9 (±0.1) | 0.9974 |
| Suramin | 16.2 (±0.5) | 2.1 (±0.1) | 0.9968 |
| Pamoic acid | 85.8 (±3.6) | 2.8 (±0.3) | 0.9917 |
| Congo red | 42.2 (±0.4) | 4.0 (±0.1) | 0.9990 |
| Trypan blue | 49.6 (±0.7) | 2.8 (±0.1) | 0.9982 |
Distinctly from the other compounds, the antiallergic drug cromolyn failed to transform the monophasic CD profile. It diminished the amplitude of the negative peak but did not produce appreciable changes above 210 nm (Fig. 3). As seen during the initial phase of ligand–CM15 titrations, intensity decrease of the negative band is the prelude to further profound spectral modifications. Accordingly, this suggests that cromolyn triggers the transition of CM15 to a more ordered conformation. Presumably due to steric reasons (see below), this drug possesses a weaker folding activity and thus its structure ordering effect lags behind that of the pamoic acid and sulfonated compounds. It is to be noted that closely resembling CD spectroscopic changes were witnessed upon titration of the natively unfolded α-synuclein with DDT.37 In parallel with the increasing concentration of the pesticide, the single negative CD band lost intensity which was considered as the sign of a disordered-to-misfolded conformational shift of the protein.
In analogy with heparin–AMP complexes, coulombic interactions and intermolecular H-bonds between lysine residues of CM15 and anionic/hydroxylic groups of the ligands may be decisive in the folding by screening intra-chain electrostatic repulsions. In this regard, the sulfonate and carboxylate groups seem to be equipotent since pamoic acid showed similar helix promoting ability to that of trypan blue and Congo red (Table 1). The aromatic units connected by single bonds may serve as a semi-rigid framework holding the anionic groups in suitable positions for binding to the peptide. Moreover, upon accommodation of the ligands, the water molecules along the peptide chain are replaced by aromatic moieties. Therefore, the local dielectric constant between the positively charged residues declines which may also contribute to the stabilization of the adducts. The chemical constitution of cromolyn allows much larger conformational freedom than that of pamoic acid or the sulfonated inducers (Scheme 1) which may account by for its impaired folding promoting capability.
Ligand-induced aggregate formation monitored by dynamic light scattering (DLS) method
DLS measurements were conducted to detect the formation of molecular aggregates and to determine their sizes. The experiments were designed to simulate the conditions of the CD titrations employing similar peptide and ligand concentrations. No particle formation could be detected in the 20 μM sample solution of the native CM15. The same result was obtained after addition of the first aliquot of stock solutions setting 1:2 ligand:peptide molar ratio. However, upon increase this value to 1:1 and above, large aggregates, except for pamoic acid, appeared with a hydrodynamic radius of ∼1000 nm (ESI Table S6†). This finding can be explained by the mutual charge neutralization within the complexes composed from the cationic CM15 and its anionic partners. As a consequence, the resulting adducts become less hydrophilic and are prone to aqueous aggregation. According to the largest values of the polydispersity index, the particle size distribution is most heterogeneous at 2:1 ligand:CM15 ratio. Further rising of the ligand concentration decreases the broadness of the size distribution. Pamoic acid triggered less effectively the aggregation process that might be related to its lowest CM15 affinity (Table 2).
Computational studies
To provide a molecular level insight into the potential interactions manifesting in the ligand–CM15 complexes, semi-empirical calculations were performed employing the PM3MM method. For all the five ligands several relative conformations were considered and the most stable ones were analyzed in detail. The obtained lowest energy structures all show strong interactions with the lysine side chains. All small molecules form salt bridges, H-bonds or, in the case of pamoic acid, cation–π interaction with at least three lysine residues (ESI Table S7†). The disposition of the lysine side chains seems also important, as residues from the N-terminal, from the middle of the sequence, as well as from the C-terminal are involved. Accordingly, Congo red, cromolyn, pamoic acid form salt bridges with Lys1, Lys6 and Lys13. Note that for pamoic acid, Lys6 coordinates on one of the naphthyl groups forming a less common cation–π interaction. For all molecules studied here, except pamoic acid, intermolecular π–π stacking with either Trp2 or Phe5 is present stabilizing further the lowest energy structures. The most intermolecular salt bridges and H-bonds, 8 altogether, can be observed for the suramin–CM15 complex, with the participation of all lysines. The cationic amino and the anionic sulfonate groups of lysines and suramin form a salt bridge network (ESI Fig. S3†), which may give an extra stabilization to the complex, as supported by the highest affinity observed for this ligand (Table 2). In summary, the theoretical calculations indicate that all the ligands could span their negatively charged groups along the CM15 chain enabling formation of a salt-bridge with at least one lysine from the N-, Lys1 or Lys3, one lysine from the C-terminal, Lys13, as well as one from the middle of the sequence, i.e. either Lys6 or Lys7. Forming a favorable interaction simultaneously with a Lys from these three positions seems a prerequisite to induce folding of CM15. This is also supported by those optimized structures where the small organic molecule is located only on one half of CM15. For these instances, the region of CM15 which does not interact with the small molecule adopts a random conformation (ESI Fig. S4†).Overall, the first experimental evidence on the folding promoting activity of therapeutic drugs communicated here suggests a potential strategy for targeting ID sequences in peptides and proteins. The structural ordering effect of small molecules on disordered regions would prevent their interaction with natural peptide/protein partners thereby allowing the modulation of various activation and signaling pathways.38,39 On the other hand, the recognition of such property may help to better understand therapeutic actions and adverse effects of pharmaceutical agents. The identification of ID target sequences of approved drugs may expand their therapeutic repertoire beyond the conventional indications.
Conflicts of interest
There are no conflicts to declare.Abbreviations
| AMP | Antimicrobial peptide |
| CD | Circular dichroism |
| DLS | Dynamic light scattering |
| ID | Intrinsic disorder |
| TFE | Trifluoroethanol |
Acknowledgements
This work was funded by the Momentum programme of the Hungarian Academy of Sciences (LP2016-2), the National Competitiveness and Excellence Program, Hungary (NVKP_16-1-2016-0007), the BIONANO_GINOP-2.3.2-15-2016-00017b project and the Hungarian Research Fund (115431, 124077, 104275). The Marie Curie fellowship for T. Beke-Somfai (MSCA-IF BARREL 660030) and the János Bolyai Research Scholarship for Kata Horváti are greatly acknowledged. The authors also thank to Dr. Hedvig Medzihradszky-Schweiger for the amino acid analysis.References
- J. M. Ageitos, A. Sánchez-Pérez, P. Calo-Mata and T. G. Villa, Biochem. Pharmacol., 2017, 133, 117–138 CrossRef CAS PubMed.
- E. H. Mattar, H. A. Almehdar, H. A. Yacoub, V. N. Uversky and E. M. Redwan, Cytokine Growth Factor Rev., 2016, 28, 95–111 CrossRef CAS PubMed.
- H. A. Yacoub, O. A. Al-Maghrabi, E. S. Ahmed and V. N. Uversky, J. Biomol. Struct. Dyn., 2016, 1–21 Search PubMed.
- J. P. Wang, M. Mura, Y. H. Zhou, M. Pinna, A. V. Zvelindovsky, S. R. Dennison and D. A. Phoenix, Biochim. Biophys. Acta, 2014, 1838, 2870–2881 CrossRef CAS PubMed.
- M. Respondek, T. Madl, C. Gobl, R. Golser and K. Zangger, J. Am. Chem. Soc., 2007, 129, 5228–5234 CrossRef CAS PubMed.
- M. Hemshekhar, V. Anaparti and N. Mookherjee, Pharmaceuticals, 2016, 9, 40 CrossRef PubMed.
- J. Habchi, P. Tompa, S. Longhi and V. N. Uversky, Chem. Rev., 2014, 114, 6561–6588 CrossRef CAS PubMed.
- J. L. Ross, Biophys. J., 2016, 111, 909–916 CrossRef CAS PubMed.
- V. N. Uversky, V. Dave, L. M. Iakoucheva, P. Malaney, S. J. Metallo, R. R. Pathak and A. C. Joerger, Chem. Rev., 2014, 114, 6844–6879 CrossRef CAS PubMed.
- V. N. Uversky, J. R. Gillespie and A. L. Fink, Proteins, 2000, 41, 415–427 CrossRef CAS.
- S. Pistolesi, R. Pogni and J. B. Feix, Biophys. J., 2007, 93, 1651–1660 CrossRef CAS PubMed.
- D. E. Schlamadinger, Y. Wang, J. A. McCammon and J. E. Kim, J. Phys. Chem. B, 2012, 116, 10600–10608 CrossRef CAS PubMed.
- H. Sato and J. B. Feix, Biochim. Biophys. Acta, 2006, 1758, 1245–1256 CrossRef CAS PubMed.
- H. A. Yacoub, O. A. Al-Maghrabi, E. S. Ahmed and V. N. Uversky, J. Biomol. Struct. Dyn., 2017, 35, 836–856 CAS.
- H. A. Yacoub, A. M. Elazzazy, M. M. Mahmoud, M. N. Baeshen, O. A. Al-Maghrabi, S. Alkarim, E. S. Ahmed, H. A. Almehdar and V. N. Uversky, Dev. Comp. Immunol., 2016, 65, 8–24 CrossRef CAS PubMed.
- L. Whitmore and B. A. Wallace, Biopolymers, 2008, 89, 392–400 CrossRef CAS PubMed.
- N. Sreerama and R. W. Woody, Anal. Biochem., 2000, 287, 252–260 CrossRef CAS PubMed.
- R. W. Woody, in Instrumental Analysis of Intrinsically Disordered Proteins, John Wiley & Sons, Inc., 2010, pp. 303–321 Search PubMed.
- Z. S. Shi, R. W. Woody and N. R. Kallenbach, Adv. Protein Chem., 2002, 62, 163–240 CrossRef CAS PubMed.
- G. Klocek and J. Seelig, Biochemistry, 2008, 47, 2841–2849 CrossRef CAS PubMed.
- A. Tchoumi Neree, P. T. Nguyen, D. Chatenet, A. Fournier and S. Bourgault, FEBS Lett., 2014, 588, 4590–4596 CrossRef CAS PubMed.
- J. Yang, H. Tsutsumi, T. Furuta, M. Sakurai and H. Mihara, Org. Biomol. Chem., 2014, 12, 4673–4681 CAS.
- M. E. Holtzer and A. Holtzer, Biopolymers, 1995, 36, 365–379 CrossRef CAS PubMed.
- H. Maeda and K. Ooi, Biopolymers, 1981, 20, 1549–1563 CrossRef CAS.
- R. Mandel and G. D. Fasman, Biopolymers, 1975, 14, 1633–1649 CrossRef CAS PubMed.
- F. Zsila, J. Phys. Chem. B, 2016, 120, 10611–10613 CrossRef CAS PubMed.
- K. Kano, M. Tatsumi and S. Hashimoto, J. Org. Chem., 1991, 56, 6579–6585 CrossRef CAS.
- A. Tanaka, S. Fujiyoshi, K. Motomura, O. Hayashida, Y. Hisaeda and Y. Murakami, Tetrahedron, 1998, 54, 5187–5206 CrossRef CAS.
- L. Jenkins, J. Brea, N. J. Smith, B. D. Hudson, G. Reilly, N. J. Bryant, M. Castro, M. I. Loza and G. Milligan, Biochem. J., 2010, 432, 451–459 CrossRef CAS PubMed.
- P. Zhao, H. Sharir, A. Kapur, A. Cowan, E. B. Geller, M. W. Adler, H. H. Seltzman, P. H. Reggio, S. Heynen-Genel, M. Sauer, T. D. Chung, Y. Bai, W. Chen, M. G. Caron, L. S. Barak and M. E. Abood, Mol. Pharmacol., 2010, 78, 560–568 CrossRef CAS PubMed.
- M. Rusnati, G. Tulipano, C. Urbinati, E. Tanghetti, R. Giuliani, M. Giacca, M. Ciomei, A. Corallini and M. Presta, J. Biol. Chem., 1998, 273, 16027–16037 CrossRef CAS PubMed.
- V. K. Ganesh, S. K. Muthuvel, S. A. Smith, G. J. Kotwal and K. H. M. Murthy, Biochemistry, 2005, 44, 10757–10765 CrossRef CAS PubMed.
- D. Marchetti, J. Reiland, B. Erwin and M. Roy, Int. J. Cancer, 2003, 104, 167–174 CrossRef CAS PubMed.
- C. R. Middaugh, H. Mach, C. J. Burke, D. B. Volkin, J. M. Dabora, P. K. Tsai, M. W. Bruner, J. A. Ryan and K. E. Marfia, Biochemistry, 1992, 31, 9016–9024 CrossRef CAS PubMed.
- R. P. McGeary, A. J. Bennett, Q. B. Tran, K. L. Cosgrove and B. P. Ross, Mini-Rev. Med. Chem., 2008, 8, 1384–1394 CrossRef CAS PubMed.
- H. P. Morgan, I. W. McNae, M. W. Nowicki, W. Zhong, P. A. Michels, D. S. Auld, L. A. Fothergill-Gilmore and M. D. Walkinshaw, J. Biol. Chem., 2011, 286, 31232–31240 CrossRef CAS PubMed.
- B. A. Silva, O. Einarsdóttir, A. L. Fink and V. N. Uversky, Res. Rep. Biol., 2011, 2, 43–56 CrossRef CAS.
- Y. Wang, T. I. Cesena, Y. Ohnishi, R. Burger-Caplan, V. Lam, P. D. Kirchhoff, S. D. Larsen, M. J. Larsen, E. J. Nestler and G. Rudenko, ACS Chem. Neurosci., 2012, 3, 546–556 CrossRef CAS PubMed.
- V. N. Uversky, Expert Opin. Drug Discovery, 2012, 7, 475–488 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05290a |
| This journal is © The Royal Society of Chemistry 2017 |