
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoselective acylation of 2-amino-8-quinolinol in the generation of C2-amides or C8-esters†
Yongseok Park‡
a,
Xiang Fei‡a,
Yue Yuana,
Sanha Leea,
Joonseong Hurb,
Sung Jean Parka,
Jae-Kyung Jung c and
Seung-Yong Seo*a
c and
Seung-Yong Seo*a
aCollege of Pharmacy and Gachon Institute of Pharmaceutical Sciences, Gachon University, Incheon 21936, South Korea
bCollege of Pharmacy, Seoul National University, Seoul 08826, South Korea
cCollege of Pharmacy, Chungbuk National University, Cheongju, Chungbuk 28644, South Korea
First published on 30th August 2017
Abstract
Two different ways to carry out the chemoselective acylation of 2-amino-8-quinolinol with unique features to generate C2-amides or C8-esters were developed. The coupling reaction with a variety of carboxylic acids using EDCI and DMAP provided C8-ester derivatives, whereas N-heteroaromatic acids were not introduced on the C8-hydroxy group, but rather on the C2-amino group under the same conditions. To obtain C2-amides selectively, the anionic nucleophile from 2-amino-8-quinolinol was treated with less reactive acyl imidazolides or esters.
Introduction
Ester and amide functionalities are important structural components that serve as key motifs in a variety of bioactive natural products and pharmaceuticals. Amide and ester for-mation are fundamental reactions in chemical synthesis.1 Typically, carbodiimides such as dicyclohexylcarbodiimide (DCC) and 1-ethyl-3-(3′-dimethylaminopropyl)-carbodiimide (EDCI) are applied as coupling reagents for esterification and amidation reactions. To increase the overall rate of the cou-pling reaction, DMAP (4-dimethylaminopyridine), 1-hydroxy-1H-benzotriazole (HOBt) and 1-hydroxy-7-azabenzotriazole (HOAt) are commonly used for carbodiimide-mediated reac-tions in either stoichiometric or catalytic fashions. Meanwhile, 1,1′-carbonyl diimidazole (CDI) is used because of the en-hanced stability relative to that of acid halides and viability in large-scale syntheses.2 In addition, numerous other methods have been developed for ester and amide bond formation.1cQuinolines are valuable synthons and precursors in synthetic organic chemistry, as they comprise important heterocyclic pharmacophores with a broad spectrum of biological and me-dicinal activities. In particular, 8-hydroxyquinoline (8-HQ) is a well-known metal ion chelator and represents an excellent scaffold with a wide spectrum of pharmacological applica-tions.3 Similarly, 2-aminoquinoline (2-AQ) exhibits various biological activities such as anthelmintic, antiprotozoal, anti-depressant, some effectiveness in the treatment of Alzheimer's disease.4 Toward that end, we paid attention to unique 2-amino-8-quinolinol (1), which contains important structural components of 8-HQ and 2-AQ.5 The synthesis of its derivatives may prove to be useful, owing to the utility of the parent compounds; however, the previous works for O- and N-acylation of 1 have hardly been reported to date.6 We sought the synthesis of its derivatives, especially the esterification on C8 and the amidation on C2 (Fig. 1).
 | ||
| Fig. 1 2-Amino-8-quinolinol (1) and its acyl derivatives. | ||
Recently, we reported an acyl derivative of 2-amino-8-quinolinol (1) called SG-HQ2. This compound is an anti-allergic inflammatory agent.7 Unfortunately, NMR spectro-scopic studies cast doubt on the originally proposed structure, which was regarded as the amide form. To prove that the ester form was obtained as the major product of the EDCI-mediated coupling reaction of 2-amino-8-quinolinol (1) and 3,4,5-tris(benzyloxy)benzoic acid, we reexamined the acylation and analyzed the structure of SG-HQ1 via NMR spectroscopy and X-ray crystallography.8 SG-HQ2 from the catalytic hydrogenation of SG-HG1 was determined to be the C8-ester instead of the amide as reported previously (Scheme 1).
 | ||
| Scheme 1 Synthesis of SG-HQ1 and SG-HQ2 and their revised structures.8 | ||
Results and discussion
Encouraged by the chemoselective O-acylation of 2-amino-8-quinolinol, our synthetic investigations regarding C2-amide derivatives began with initial studies of the 2 step synthesis comprised of the diacylation with excess of aromatic acid (up to 5 equiv.) and the hydrolysis of the ester moiety (Scheme 2). C2-amides of various aromatic acids were prepared, and the structures of 3b and 4b were confirmed by X-ray crystallog-raphy.8 Interestingly, the acylation with heteroaromatic acids such as 2-picolinic acid and pyrazinecarboxylic acid afforded only the C2-amides (4h and 4i), excluding C8-ester and diacyl compounds, albeit in low yield. Thus, the selective and efficient introduction of various carboxylic acids on 2-amino-8-quinolinol remains a challenge,9 although some C2 and C8-acyl derivatives were prepared successfully. Inspired by the unique characteristics of 2-amino-8-quinolinol, herein we present the optimization of the chemoselective O- or N-acylation.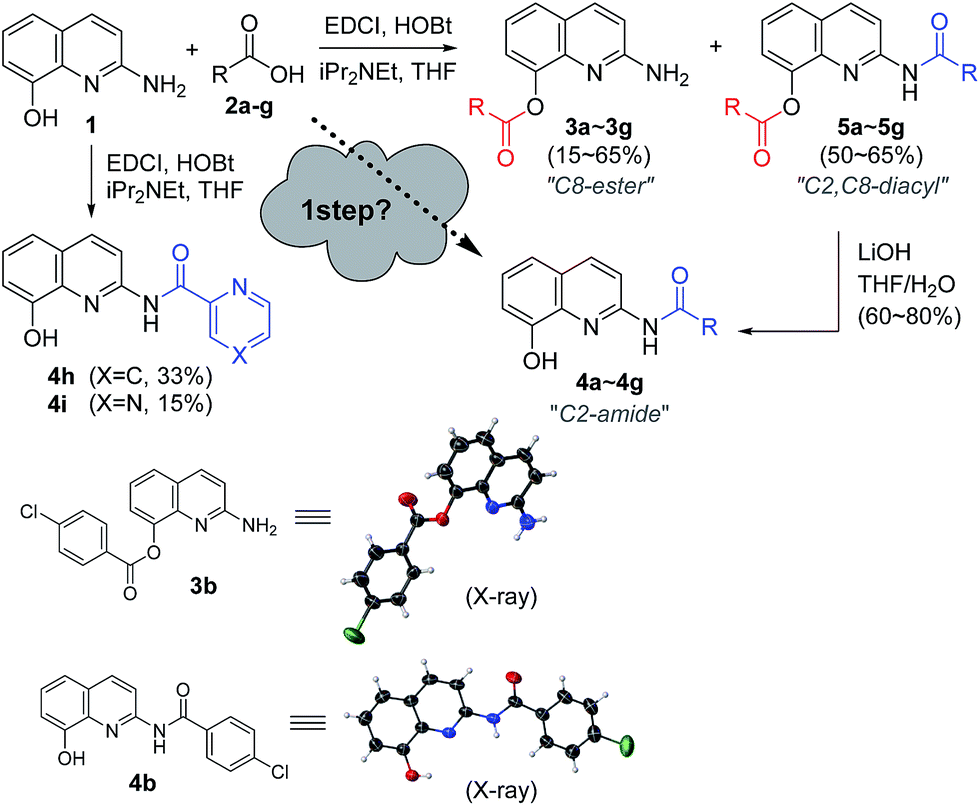 | ||
| Scheme 2 Initial synthesis of C8-ester and C2-amide derivatives of 2-amino-8-quinolinol. Aromatic acids including 4-methoxybenzoic (a), 4-chlorobenzoic (b), 4-(trifluoromethyl)benzoic (c), 4-tert-butylbenzoic (d), 3-methoxybenzoic (e), 3,4,5-tris(benzyloxy)benzoic (f), 2-furoic (g), picolinic (h), and pyrazinecarboxylic (i) acids. | ||
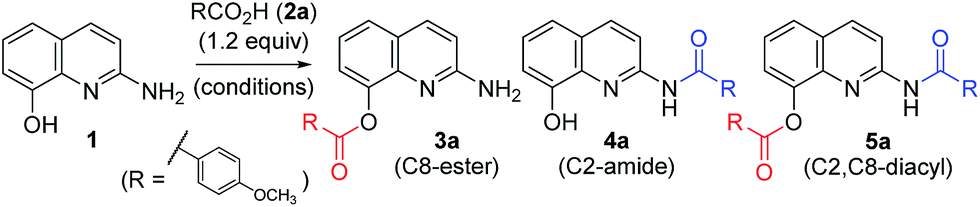
To investigate possible synthetic routes towards C2-amide and C8-ester derivatives of 2-amino-8-quinolinol, we first examined various acylation conditions with 4-methoxybenzoic acid (Table 1). THF was selected as the most favorable solvent in terms of solubility and purification. The use of carbodiimides provided the C8-ester 3a preferentially as the major product. DMAP as an additive increased the chemical yield and ester selectivity (entry 1 and 7). However, when HOBt was employed, the formation of diacyl derivative 5a increased with that of C8-ester (entry 3–5). Though the use of HOAt provided the C2-ester selectively, the isolated yield of 3a was not satisfactory (entry 6). Uranium (HATU) and phosphonium (PyBop) salts did not exhibit significant effects (entry 8 and 9). The use of 4-methoxybenzoyl chloride with iPr2NEt gave the C8-ester as the major product (entry 10). The yield decreased slightly when pyridine was used as a solvent (entry 11). Finally, we chose CDI for the amidation through the acyl imidazolide, leading to the formation of C2-amide 4a although the C8-ester formed as the major product. Higher temperature increased the formation of C2-amide slightly (entry 12–14). C2-amide 4a could be obtained in 75% yield with the ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :88:7, when the anionic nucleophile by the treatment of 2-amino-8-quinolinol with NaH reacted with the acyl imidazolide derived from 4-methoxybenzoic acid and CDI (entry 15).
:88:7, when the anionic nucleophile by the treatment of 2-amino-8-quinolinol with NaH reacted with the acyl imidazolide derived from 4-methoxybenzoic acid and CDI (entry 15).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Activator (equiv.) | Base | Time (h) | % yieldb | Ratioc (3a:4a:5a) |
|
| 3a | 4a | |||||
| a The reactions were carried out three times.b Isolated yield of 3a and 4a after column chromatography.c Ratio of 3a, 4a and 5a determined by HPLC analysis.d At reflux.e 4-Methoxybenzoyl chloride was used.f Reaction conditions: a THF solution of CDI and 4-methoxybenzoic acid was added to 2-amino-8-quinolinol and NaH (2 equiv.) in THF. | ||||||
| 1 | DCC (1.3), DMAP (0.5) | 24 | 54 | 0 | >99:0:<1 |
|
| 2 | EDCI (1.3) | iPr2NEt | 24 | 21 | 0 | >99:0:<1 |
| 3 | EDCI (1.3), HOBt (0.5) | iPr2NEt | 24 | 60 | 0 | 67:1:32 |
| 4d | EDCI (1.3), HOBt (0.5) | iPr2NEt | 24 | 65 | 4 | 73:5:22 |
| 5 | EDCI (1.3), HOBt (0.5) | Et3N | 24 | 58 | 1 | 79:1:20 |
| 6 | EDCI (1.3), HOAt (0.5) | iPr2NEt | 24 | 35 | 0 | 93:0:7 |
| 7 | EDCI (1.3), DMAP (0.5) | iPr2NEt | 3 | 92 | 0 | 99:0:1 |
| 8 | HATU (1.3) | iPr2NEt | 24 | 65 | 0 | 83:0:17 |
| 9 | PyBOP (1.3) | iPr2NEt | 4 | 50 | 1 | 58:1:41 |
| 10e | Et3N | 4 | 82 | 1 | 86:1:13 |
|
| 11e | Pyridine | 24 | 65 | 0 | 81:0:19 |
|
| 12 | CDI (1.3) | 24 | 30 | 9 | 70:22:8 |
|
| 13d | CDI (1.3) | 24 | 40 | 14 | 56:19:24 |
|
| 14d | CDI (1.0) | 24 | 36 | 19 | 44:23:33 |
|
| 15f | CDI (1.3) | NaH | 3 | 4 | 75 | 5:88:7 |
The optimized acylation conditions were further investigated using a cheap and readily available ester instead of acyl imidazolide 6 with other strong bases (Table 2).10 Methyl 4-methoxybenzoate 7 was used as an acyl donor. Even though the use of the methyl ester provided a lower yield than acyl imidazolide 6, the acylation resulted in the selective formation of C2-amide. When NaH and n-BuLi were used to generate the anionic intermediate of 2-amino-8-quinolinol, the excellent selectivity was observed (entries 2 and 3). The use of iPrMgCl as a base resulted in a significant loss of reactivity and selectivity (entry 4). The formation of C2-amide 4a was improved as the amount of t-BuOK was increased (entry 5–8). The anionic nucleophile of 2-amino-8-quinolinol could mostly be converted into C2-amide rather than C8-ester.
|
|||||
|---|---|---|---|---|---|
| Entry | Acyl donor | Base (equiv.) | Time (h) | % yielda | Ratiob |
| a Isolated yield of amide 3a.b Ratio (3a:4a:5a) determined by HPLC analysis. |
|||||
| 1 | 6 | NaH (2.0) | 4 | 75 | 5:88:7 |
| 2 | 7 | NaH (2.0) | 24 | 25 | 2:98:0 |
| 3 | 7 | n-BuLi (2.0) | 4 | 56 | 0:99:1 |
| 4 | 7 | iPrMgCl (2.0) | 24 | 8 | 0:86:14 |
| 5 | 7 | t-BuOK (1.0) | 24 | 11 | 4:93:3 |
| 6 | 7 | t-BuOK (2.0) | 2 | 63 | 3:96:0 |
| 7 | 7 | t-BuOK (2.5) | 1 | 72 | 1:93:7 |
| 8 | 7 | t-BuOK (3.0) | 0.5 | 85 | 1:96:3 |

Additive effect on reactivity and selectivity was evaluated in order to reveal the role of acyl imidazolide in C8-acylation. Some amines (such as pyridine, DMAP, and HOBt) and their HCl salts were treated in the reaction of 1 with the isolated acyl imidazolide 6 (Table 3). Without additives, C2-amide (4a) was obtained even though the reaction yield and ratio was slightly different with CDI-mediated acylation. However, the use of pyridine and DMAP at reflux provided C8-ester (3a) reversely. The use of their HCl salt (Pyr·HCl and DMAP·HCl) and HOBt provided selectively the C8-ester in good yield even at room temperature.
|
|||||
|---|---|---|---|---|---|
| Entry | Additive (equiv.) | Temp (°C) | % yieldb | Ratioc (3a:4a:5a) |
|
| 3a | 4a | ||||
| a Reaction conditions: 2-amino-8-quinolinol (1 equiv.), acyl imidazolide (1.2 equiv.), an additive (1.5 equiv.), THF (4 mL), at room temperature.b Isolated yield.c Ratio determined by HPLC analysis.d At reflux.e Added iPr2NEt (3 equiv.). | |||||
| 1d,e | — | 65 | 9 | 15 | 15:25:60 |
| 2d | Pyridine (1.5) | 65 | 67 | 0 | 80:0:20 |
| 3d | DMAP (1.5) | 65 | 63 | 0 | 73:0:27 |
| 4 | HOBt (1.5) | rt | 56 | 0 | 80:0:20 |
| 5 | Pyr·HCl (1.5) | rt | 78 | 0 | 87:0:13 |
| 6 | DMAP·HCl (1.5) | rt | 75 | 0 | 87:0:13 |
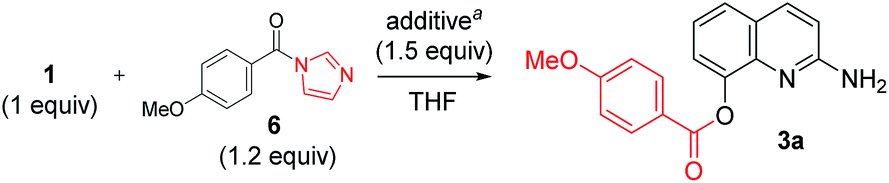
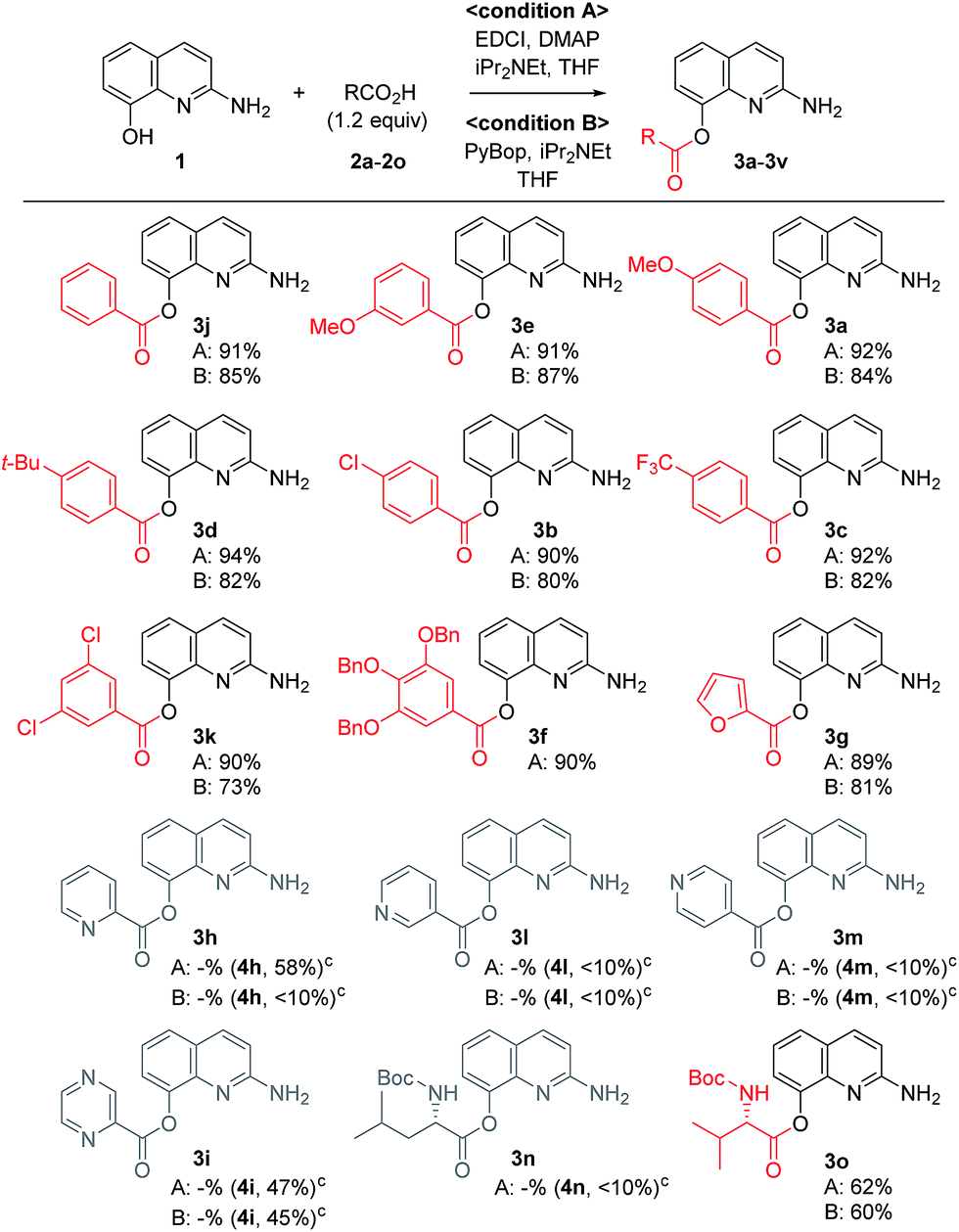
A variety of carboxylic acids were introduced on 2-amino-8-quinolinol under the optimized acylation conditions. Either condition A (EDCI, iPr2NEt and catalytic DMAP) or condition B (PyBop and iPr2NEt) was used for the synthesis of the C8-esters (Table 4). Various substituted-benzoic acids and furoic acid (i.e. 3a–3f, 3j and 3k) were introduced on the C8 position in good yields, regardless of electron-withdrawing and donating groups. However, N-heteroaromatic acids were not incorporated on the phenol group in the C8 position; C2-amides (4h–4i, 4l–4m) were isolated even in low yields instead. Interestingly, Boc-leucinyl C8-ester (3n) seems to be unstable during flash column chromatography like heteroaromatic acid, however Boc-valinyl C8-ester (3o) was obtained in moderate yield.
Next, we turned our attention to the amidation of 2-amino-8-quinolinol on the C2 position. The CDI-mediated amidation of some aromatic acids with electron-withdrawing substituents provided C2-amides as the major product in contrast to carbodiimide-mediated acylation (condition C, Table 5). Nevertheless, this general amidation was accompanied by the formation of the C8-ester species, which was difficult to separate. With the optimized reaction conditions in hand, the substrate scope was examined (condition D, Table 5). 4-Methoxybenzoyl derivatives (2a) was obtained in 75% yield, whereas the isolated yields of other substituted benzoic acids and heteroaromatic acids were improved up to 99% (4a vs. 4b–4m). Interestingly amides with N-protected amino acids (4n and 4o) were easily prepared from the corresponding acyl imidazolides.
| a Reaction conditions: a mixture of the carboxylic acid (1.2 equiv.) and CDI (1.3 equiv.) was added to a THF (4 mL) solution of 1 (0.20 mmol) and NaH (2 equiv.) at room temperature.b Isolated yield from flash column chromatography.c Isolated yield based on recovered starting material in parenthesis. |
|---|
 |
By using EDCI-mediated acylation, the bulky aliphatic acyl moieties such as piperidinylcarboxy, isovaleroyl, pivaloyl and 1-adamantylcarbonyl were introduced on the C8-hydroxy group successfully (3p–3s) as shown in Scheme 3. Similarly, sulfonyl chloride, benzyl chloroformate and Boc2O were used to prepare the sulfonate and carbonate derivatives (3t–3v) on the C8-position in good yield, respectively. Meanwhile, the C2-amides with bulky aliphatic amides bearing piperidinylcarboxy, isovaleroyl, pivaloyl and 1-adamantylcarbonyl (4p–4s), were easily prepared from the corresponding acyl imidazolides. Finally, a similar method was used to prepare two carbamates with Cbz and Boc on C2 position, in spite of the unsatisfactory result obtained with p-toluenesulfonamide (4t vs. 4u and 4v).
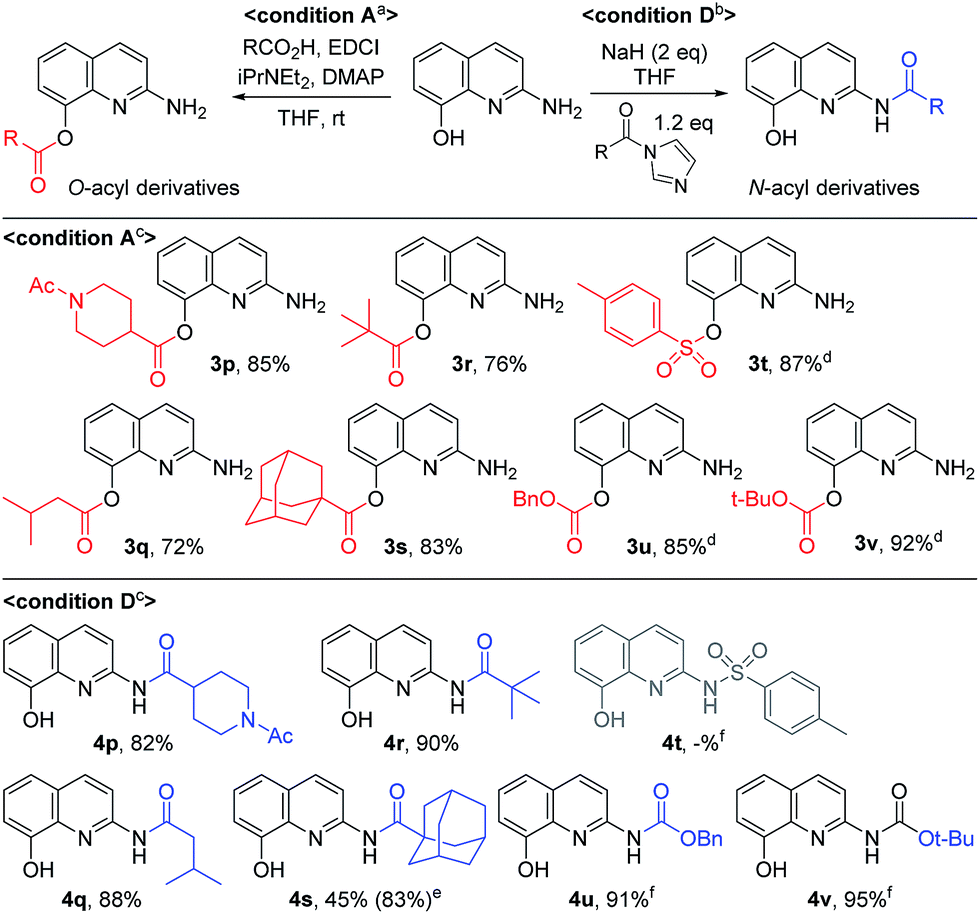 | ||
| Scheme 3 The introduction of aliphaticacyl, sulfonyl and alkoxycarbonyl groups. aReaction conditions: 1 (0.2 mmol), carboxylic acid (0.4 mmol), EDCI (2.5 equiv.), DMAP (10 mol%), iPr2NEt (3 equiv.), THF (1 mL). bReaction conditions: a mixture of the carboxylic acid (1.2 equiv.) and CDI (1.3 equiv.) was added to a THF (4 mL) solution of 1 (0.20 mmol) and NaH (2 equiv.). cIsolated yield by flash column chromatography. dp-toluenesulfonyl chloride, benzyl chloroformate and Boc2O were used for 3t, 3u, and 3v respectively. eIsolated yield based on recovered starting material in parenthesis. fp-Toluenesulfonyl chloride, benzyl chloroformate and Boc2O were used with imidazole for 4t, 4u, and 4v respectively, but 4t was not isolated. | ||
Additive effects besides imidazole are shown in Table 3 to shed light on the mechanism of the chemoselective synthesis of C8-ester and C2-amide derivatives. Thus, representative amines (such as pyridine, DMAP, and HOBt) and their HCl salts were treated with the imidazolide 6 to afford the C8-ester (Scheme 4). With reactive acyl donors, C8-ester was regarded as the kinetic product. On the contrary, two C8-esters (3a and 3k) were refluxed with imidazole to afford the corresponding amides (4a and 4k) in 40% and 82% yields, respectively. The observed conversion from the C8-ester to the C2-amide revealed that the C2-amide was the thermodynamically stable product. The conversion of 3k to 4k was superior to that of 3a, because of the electronic effect on substituents of benzene ring. This result is relevant to the lack of formation of C8-esters with N-heteroaromatic carbonyls (3h–3n) under most of conditions, as shown in Scheme 3. In order to determine the origin of the unique reactivity of 2-amino-8-quinolinol, 8-hydroxyquinoline (8) and 2-aminoquinoline (9) reacted with 4-methoxybenzoic acid under various coupling conditions. The yields with EDCI, PyBop, HBTU, and CDI were not satisfactory, but the acylation was successful with using the reactive acid chloride in good yields (up to 92%). As the weak reactivity of 8 and 9, it is considered to have a beneficial effect between the 2-amino and 8-hydroxy moieties for the introduction of an acyl group to 1.
 | ||
| Scheme 4 Mechanistic investigation. | ||
To investigate the acylation pattern of 1, we monitored the progress of C2-amide and C8-ester formation via CDI-mediated acylation by HPLC. Ester 3a formed faster than amide 4a (0–6 h). After 24 h, 4a was the major component, while starting material 1, 3a, and 5a were formed in similar amounts (almost 20%, Table 6).8
|
||||
|---|---|---|---|---|
| Time (h) | 1 (mmol) | 3a (mmol) | 4a (mmol) | 5a (mmol) |
| a Reaction conditions: a THF (4 mL) solution of 2-amino-8-quinolinol (1 equiv.), p-methoxybenzoic acid (1 equiv.), and CDI (1.3 equiv.) was stirred at reflux. | ||||
| 0 | 0.3000 | 0 | 0 | 0 |
| 1 | 0.2780 | 0.0201 | 0.0014 | 0.0005 |
| 3 | 0.2096 | 0.0593 | 0.0252 | 0.0060 |
| 6 | 0.1666 | 0.0697 | 0.0512 | 0.0125 |
| 12 | 0.0753 | 0.0628 | 0.1201 | 0.0418 |
| 24 | 0.0681 | 0.0656 | 0.1087 | 0.0576 |

Based on the above mechanistic investigation, a proposed reaction pathway is outlined in Scheme 5. Notably, Joussef et al. reported the chemoselectivity O-sulfonylation at C8.11 For the C8-ester formation, the nitrogen nucleophile at N1 induced from the lone pair of the amino at C2 attacks acyl donors such as acid chlorides preferentially; this step is followed by an intra- or intermolecular acyl transfer to the neighboring hydroxy at C8.12 Meanwhile, the formation of C2-amides involves the anions of 1, generated by strong bases such as NaH. We speculated that a sodium ion which interacts with the anion at C8 might coordinate to the nitrogen atom at N1, whereas the second anion at C2 might attack acyl donors such as acyl imidazolides and esters. As with the unstable C8-ester (i.e. 3k) could be converted into the C2-amide by imidazole, the reactivity of the amino at C2 with acyl imidazolides under neutral conditions remains to be elucidated.
 | ||
| Scheme 5 Proposed reaction pathways. | ||
Conclusions
In conclusion, we developed chemoselective acylation of 2-amino-8-quinolinol on amino group at C2 and hydroxy group at C8. The coupling reaction with a variety of carboxylic acids using EDCI and DMAP provided C8-ester derivatives in excellent yield. When the dianion of 2-amino-8-quinolinol reacts with less reactive acylimidazoles and esters, the corresponding C2-amides and carbamate derivatives could be prepared as the major product. Biological evaluations of the C2-amide and C8-ester derivatives of 2-amino-8-quinolinol are underway in our laboratory and will be reported in due course.Experimental
All the starting materials, solvents and reagents were obtained from commercial suppliers and were used without further purification. Flash column chromatography was performed using silica gel 60 (230–400 mesh, Merck) with the indicated solvents. Thin-layer chromatography was performed using 0.25 mm silica gel plates (Merck). 1H and 13C NMR spectra were recorded on a Bruker 600 MHz spectrometer as solutions in deuterated CDCl3, methanol-d4 or DMSO-d6. 1H NMR data were reported in the order of chemical shift, multiplicity (s, singlet; d, doublet; t, triplet; m, multiplet and/or multiple resonances), number of protons, and coupling constant (J) in hertz (Hz). High-resolution mass spectra (HRMS) were recorded on a JEOL JMS-700 (FAB and EI) and an Agilent 6530 Q-TOF LC/MS/MS system (ESI). For conversion measurements, the samples prepared for the HPLC were analyzed at 256 nm using an Agilent 1260 HPLC system equipped with a 6 mm × 50 mm Sunfire 5μ C18 column, in which the mobile phase was a gradient from water to acetonitrile for 30 min.General procedure of condition A (Table 4) for O-acylation at C8
To a solution of 2-amino-8-quinolinol (30 mg, 0.19 mmol) and a carboxylic acid (1.2 equiv.) in anhydrous tetrahydrofuran (4 mL) were added EDCI (1.3 equiv.), DMAP (0.5 equiv.) and N,N-diisopropylethylamine (3.0 equiv.) under N2 atmosphere with an ice bath. After stirring for 3 h at ambient temperature, the mixture was diluted with dichloromethane, washed by sat. NH4Cl, sat. NaHCO3, and brine, dried by MgSO4, filtered, and concentrated. The residue was purified by silica gel column chromatography to afford the desired product.General procedure of condition B (Table 4) for O-acylation at C8
To a solution of 2-amino-8-quinolinol (30 mg, 0.19 mmol) and a carboxylic acid (1.2 equiv.) in anhydrous tetrahydrofuran were added PyBop (1.3 equiv.) and N,N-diisopropylethylamine (3.0 equiv.) under N2 atmosphere with an ice bath and stirring for 4 h at ambient temperature. The reaction mixture was diluted with dichloromethane, washed by NH4Cl, saturated NaHCO3, and brine, dried by MgSO4, filtered, and concentrated. The residue was purified by flash column chromatography to the desired product.General procedure of condition C (Table 5) for N-acylation at C2
To a solution of a carboxylic acid (1.2 equiv.) in anhydrous tetrahydrofuran was added 1,1′-carbodiimidazole (1.3 equiv.). After stirring for 1 h under N2 atmosphere, 2-amino-8-quinolinol (30 mg, 0.19 mmol) was added to the reaction mixture. After stirring for 24 h at reflux, the mixture was diluted with tetrahydrofuran, washed by NH4Cl, saturated NaHCO3, and brine, dried by MgSO4, filtered, and concentrated. The residue was purified by flash column chromatography to the desired product.General procedure of condition D (Table 5) for N-acylation at C2
To a solution of carboxylic acid (1.2 equiv.) in anhydrous tetrahydrofuran (4 mL), 1,1′-carbonyldiimidazole (1.3 equiv.) was added under N2 atmosphere at ambient temperature. After stirred for 1 h. The mixture was diluted with dichloromethane, extracted by brine and dichloromethane, dried by MgSO4, filtered, and concentrated to obtain the acyl imidazolide intermediate. To a solution of 2-amino-8-quinolinol (30 mg, 0.19 mmol) and NaH (2.0 equiv.) in anhydrous tetrahydrofuran (4 mL) was added the acyl imidazolide intermediate under N2 atmosphere with an ice bath. After stirring for 2 h at ambient temperature, the reaction was quenched with sat. NH4Cl, washed by dichloromethane and dried by MgSO4, filtered, and concentrated. The residue was purified by silica gel column chromatography to afford the desired product.:3, Rf = 0.1). 1H-NMR (600 MHz, CDCl3) δ 8.28 (dd, 2H, J = 9.0 Hz), 7.86 (d, 1H, J = 9.0 Hz), 7.53 (dd, 1H, J = 8.4 and 1.2 Hz), 7.39 (dd, 1H, J = 7.8 and 1.2 Hz), 7.26 (t, 1H, J = 7.8 Hz), 6.99 (dd, 2H, J = 9.0 and 1.8 Hz), 6.66 (d, 1H, J = 9.0 Hz), 4.85 (bs, 2H), 3.89 (s, 3H); 13C-NMR (150 MHz, CDCl3) δ 165.3, 163.7, 157.0, 145.3, 140.7, 137.9, 132.5, 125.4, 125.0, 122.2, 122.1, 121.9, 113.7, 112.3, 55.5; HRMS (EI): m/z calcd for C17H14N2O3: 294.1004; found: 294.1006.:1, Rf = 0.25). 1H-NMR (600 MHz, DMSO-d6) δ 10.68 (s, 1H), 9.29 (bs, 1H), 8.34 (d, 1H, J = 9.0 Hz), 8.28 (d, 1H, J = 9.0 Hz), 8.10 (d, 2H, J = 8.4 Hz), 7.38 (d, 1H, J = 7.8 Hz), 7.35 (t, 1H, J = 7.8 Hz), 7.10 (t 3H, J = 7.5 Hz), 3.86 (s, 3H); 13C-NMR (150 MHz, DMSO-d6) δ 165.9, 162.8, 152.4, 150.7, 138.4, 137.2, 130.5, 127.0, 126.3, 126.2, 118.2, 116.4, 114.1, 111.9, 55.9; HRMS (EI): m/z calcd for C17H14N2O3: 294.1004; found: 294.1003.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2016R1D1A1A09917300), the Pioneer Research Center Program through the NRF funded by the Ministry of Science, ICT & Future Planning (2014M3C1A3001556), and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare (Grant number HI14C1135).Notes and references
- For selected reviews of esterification and amidation, see: (a) F. Albericio, Curr. Opin. Chem. Biol., 2004, 8, 211–221 CrossRef CAS PubMed; (b) S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS; (c) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS; (d) R. M. de Figueiredo, J. S. Suppo and J. M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed; (e) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- (a) R. Bright, D. J. Dale, P. J. Dunn, F. Hussain, Y. Kang, C. Mason, J. C. Mitchell and M. Snowden, Org. Process Res. Dev., 2004, 8, 1054–1058 CrossRef CAS; (b) R. Vaidyanathan, V. G. Kalthod, D. P. Ngo, J. M. Manley and S. P. Lapekas, J. Org. Chem., 2004, 69, 2565–2568 CrossRef CAS PubMed; (c) C. Larrivée-Aboussafy, B. P. Jones, K. E. Price, M. A. Hardink, R. W. McLaughlin, B. M. Lillie, J. M. Hawkins and R. Vaidyanathan, Org. Lett., 2010, 12, 324–327 CrossRef PubMed; (d) S. T. Heller, T. Fu and R. Sarpong, Org. Lett., 2012, 14, 1970–1973 CrossRef CAS PubMed.
- (a) J. P. Phillips, Chem. Rev., 1956, 56, 271–297 CrossRef CAS; (b) M. Albrecht, M. Fiege and O. Osetska, Coord. Chem. Rev., 2008, 252, 812–824 CrossRef CAS; (c) Y. N. Song, H. Xu, W. Chen, P. Zhan and X. Liu, Med. Chem. Commun., 2015, 6, 61–74 RSC.
- (a) Y. Cheng, T. C. Judd, M. D. Bartberger, J. Brown, K. Chen, R. T. Fremeau, D. Hickman, S. A. Hitchcock, B. Jordan, V. Li, P. Lopez, S. W. Louie, Y. Luo, K. Michelsen, T. Nixey, T. S. Powers, C. Rattan, E. A. Sickmier, D. J. St Jean, R. C. Wahl, P. H. Wen and S. Wood, J. Med. Chem., 2011, 54, 5836–5857 CrossRef CAS PubMed; (b) T. Tomioka, Y. Takahashi and T. Maejima, Org. Biomol. Chem., 2012, 10, 5113–5118 RSC; (c) M. A. Cinelli, H. Li, G. Chreifi, P. Martasek, L. J. Roman, T. L. Poulos and R. B. Silverman, J. Med. Chem., 2014, 57, 1513–1530 CrossRef CAS PubMed; (d) L. Wang, J. Ferguson and F. Zeng, Org. Biomol. Chem., 2015, 13, 11486–11491 RSC.
- (a) T. Storz, R. Marti, R. Meier, P. Nury, M. Roeder and K. Zhang, Org. Process Res. Dev., 2004, 8, 663–665 CrossRef CAS; (b) P. K. Nayak, N. Agarwal, F. Ali, M. P. Patankar, K. L. Narasimhan and N. Periasamy, J. Chem. Sci., 2010, 122, 847–855 CrossRef CAS; (c) S. M. Shiriskar, N. Agarwal, R. R. Pissurlenkar and B. Ahmad, Eur. Biophys. J., 2015, 44, 193–205 CrossRef CAS PubMed.
- 89% yield in N-acetylation. For details, see: (a) M. Albrecht, K. Witt, R. Frohlich and O. Kataeva, Tetrahedron, 2002, 58, 561–567 CrossRef CAS. 32% yield in 8-hydroxyquinoline-2-carbonyl amide. For details, see (b) C. Deraeve, C. Boldron, A. Maraval, H. Mazarguil, H. Gornitzka, L. Vendier, M. Pitie and B. Meunier, Chem.–Eur. J., 2008, 14, 682–696 CrossRef CAS PubMed. 36% yield in ethyl malonyl amide. For details, see (c) K. Serafin, P. Mazur, A. Bak, E. Laine, L. Tchertanov, J.-F. Mouscadet and J. Polanski, Bioorg. Med. Chem., 2011, 19, 5000–5005 CrossRef CAS PubMed.
- (a) I.-G. Je, H.-H. Kim, P.-H. Park, T. K. Kwon, S.-Y. Seo, T.-Y. Shin and S.-H. Kim, Exp. Biol. Med., 2015, 240, 631–638 CrossRef CAS PubMed; (b) S.-Y. Seo and W. Kang, J. Pharm. Biomed. Anal., 2016, 131, 103–106 CrossRef CAS PubMed.
- See ESI†, for details.
- For chemoselective acylation, see: (a) M. Movassaghi and M. A. Schmidt, Org. Lett., 2005, 7, 2453–2456 CrossRef CAS PubMed; (b) J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS PubMed; (c) S. D. Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef PubMed; (d) S. M. Mali, R. D. Bhaisare and H. N. Gopi, J. Org. Chem., 2013, 78, 5550–5555 CrossRef CAS PubMed.
- (a) Y.-J. Yoon, J. Park, B. Kim, H.-G. Lee, S.-B. Kang, G. Sung, J.-J. Kim and S.-G. Lee, Synthesis, 2012, 44, 42–50 CrossRef; (b) M. Choi, S.-W. Won, H. Jo, M. Viji, S.-Y. Seo, Y.-J. Lee, H.-S. Lee, H. Lee, J. T. Hong, Y.-S. Kwak and J.-K. Jung, Tetrahedron Lett., 2014, 55, 6582–6584 CrossRef CAS.
- (a) L. Everson Da Silva, P. Teixieira De Sousa and A. C. Joussef, Synth. Commun., 2009, 39, 1378–1388 CrossRef CAS; (b) M. M. Bader, T. Hamada and A. Kakuta, J. Am. Chem. Soc., 1992, 114, 6475–6479 CrossRef CAS.
- The pKa values of 2-amino-8-quinolinol (1) and its conjugate acid are 10.5 and 6.7, respectively, calculated using the ACD/Percepta 14 (ACD/Labs, Toronto, Canada). The chemical shift perturbation of the ring proton of N1 positon of 1 depending on pHs was monitored by NMR. The result also showed three plateaus exist around the calculated pKa values, which supported that the pKa values should be correct. For details, see ESI.†.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral and analytical data (PDF) and X-ray crystallographic analyses of SG-HQ1 (3f), 3b, and 4b (CIF). CCDC 1548392–1548394. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra05287a |
| ‡ Both authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2017 |