
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green synthesis of silver nanoparticles using seed extract of Alpinia katsumadai, and their antioxidant, cytotoxicity, and antibacterial activities†
Yangqing He *a,
Fenfei Weia,
Zhanying Mab,
Hao Zhangc,
Qian Yanga,
Binghua Yaoa,
Zhengrui Huanga,
Jie Lia,
Cun Zenga and
Qian Zhang*a
*a,
Fenfei Weia,
Zhanying Mab,
Hao Zhangc,
Qian Yanga,
Binghua Yaoa,
Zhengrui Huanga,
Jie Lia,
Cun Zenga and
Qian Zhang*a
aDepartment of Applied Chemistry, Xi'an University of Technology, No. 5 South Jinhua Road, Xi'an, Shaanxi 710048, China. E-mail: yqhe6@xaut.edu.cn; qzh@xaut.edu.cn
bDepartment of Chemistry, Xianyang Normal University, Wenling Road, Xianyang, Shaanxi 712000, China
cDepartment of Oncology, The First Affiliated Hospital of Xi'an Jiaotong University, No. 227 West Yanta Road, Xi'an, Shaanxi 710061, China
First published on 15th August 2017
Abstract
The green synthesis of nanoparticles using plants and their extracts is considered as a representative approach in material syntheses for environmental benignity. Numerous metal nanoparticles with potential bioactivities have been prepared based on plant extracts. In this paper, we report an eco-friendly technique for the preparation of silver nanoparticles (AgNPs) using seed extract of Alpinia katsumadai, and the evaluation of their antioxidant, antibacterial, and cytotoxicity activities. UV-visible spectroscopy, FETEM, EDX, SAED, XRD, DLS and FT-IR analysis were used to characterize the morphology and crystalline phase of AgNPs. The FETEM and DLS analysis showed AgNPs were quasi-spherical in shape, with an average diameter of 12.6 nm for the size distribution. The SAED spectrum of the AgNPs indicated their crystalline nature, which was further confirmed by the XRD studies. The FT-IR spectra revealed that phytochemicals from Alpinia katsumadai seed extract act as a capping and reducing agent for AgNP formation. The AgNPs showed distinctive free radical scavenging, potent antibacterial activity against different types of bacteria, and cytotoxicity activities. The results confirmed that Alpinia katsumadai is a potential bio-resource/biomaterial for synthesizing AgNPs with applications in antibacterial and antioxidant agents.
1. Introduction
In the last two decades, nanoparticle research has become one of the most important areas in modern materials science research. It has attracted considerable interest in the fields of electronics, biology, and medicine, due to the distinctive properties of nanoparticles, such as optical, antimicrobial, cancer therapeutic, and catalytic properties.1,2 Currently, metal nanoparticles with their unparalleled characteristics are extensively studied owing to their potential applicability in diverse areas such as packaging, coatings, biological tagging, and pharmaceutical applications.3,4 The non-destructive determination of single-stranded DNA using metal nanoparticles opens up the prospect of medical diagnostic applications.5 Furthermore, for their unusual physical and chemical properties, noble metal (gold, silver, platinum and palladium) nanoparticles have been widely used in the electronic, industrial, and biomedical fields.6 Among all the noble metal nanoparticles, silver nanoparticles (AgNPs) have attracted intensive research interest owing to their unique properties, such as catalytic and good conductivity, but specifically their wide range of promising bioactivities. These include antifungal, anti-oxidant, antibacterial, anti-inflammatory and anticancer effects.7–14 To date, silver and its salts have been used as potent antimicrobial agents in curative and preventative health care, with low toxicity.15 Currently, silver nanoparticles are widely employed as an antimicrobial agent for the treatment of infections for open wounds and chronic ulcers.8,16 Many effective approaches have been devoted to the synthesis of silver nanoparticles, including physical and chemical techniques, such as electrochemical, photochemical, and chemical reductions of silver ions,17 heat evaporation,18 and so on. However, conventional chemical and physical methods raise environmental and biological risks on account of the poisonous reducing agents used during the synthesis procedures. To fulfil the growing demand for environmentally friendly methodology to produce metal nanoparticles, biological or green-chemistry synthesis has received much attention because of its economy in the use of time and the minimal use of external hazardous reducing/stabilizer chemicals compared to physical and chemical synthesis. For this purpose, the utilization of eco-friendly benign reducing agents and solvents, as well as renewable and non-toxic materials, is one of the important issues that should be evaluated for a green synthesis strategy. Previously, various green synthetic approaches to silver nanoparticles using microorganisms such as bacteria,19 yeast,20 and fungi21 have been reported. However, there are several drawbacks, including multiple purification steps and the elaborate process of maintaining the microbial culture.Now, using plant extracts as reducing and stabilizing agents for the synthesis of metallic nanoparticles is considered to be an eco-friendly and rapid strategy. It can significantly eliminate the above problems and make the nanoparticles more biocompatible and non-toxic.22,23 Numerous investigations focused on the synthesis of silver nanoparticles with potential anticancer and antibacterial activities have been reported, such as using different plant extracts of Sesbania grandiflora,9 Rubus glaucus Benth,11 Alpinia calcarate,12 and Artocarpus heterophyllus.24
Alpinia katsumadai Hayata is a perennial herb native to India and Ceylon. It is now widely cultivated in China and Southeast Asia. The A. katsumadai seed is widely used as a condiment and flavoring material in the preparation of various desserts, spicy fish, and other foods in China. The crude extract from the A. katsumadai seed has been used for the treatment of indigestion, as an antiemetic, and for stomach complaints, and is claimed to improve spleen function. Previous photochemical investigations of A. katsumadai have resulted in the isolation of a number of compounds, including flavonoids, diarylheptanoids, and chalcones.25–27 Several active biomolecules isolated from this plant have been proved to possess antiemetic,28 antiviral,29,30 anti-inflammatory,31 and anticancer activities.32
To our knowledge, there have been no reports of silver nanoparticle synthesis using A. katsumadai seed aqueous extract (AKSE) as a reducing agent without the addition of external reducing and capping agents. We consider that using A. katsumadai extract is an effective method for the synthesis of silver nanoparticles with potential biological activities. Therefore, the present study focused on the green synthesis of silver nanoparticles using AKSE (Scheme 1) and the evaluation of their antibacterial and antioxidant activities and cytotoxicity against human gastric carcinoma SGC-7901 cell lines. The AgNPs have been extensively characterized by UV-Vis spectral analysis, FETEM, SAED, EDX, XRD, and FT-IR.
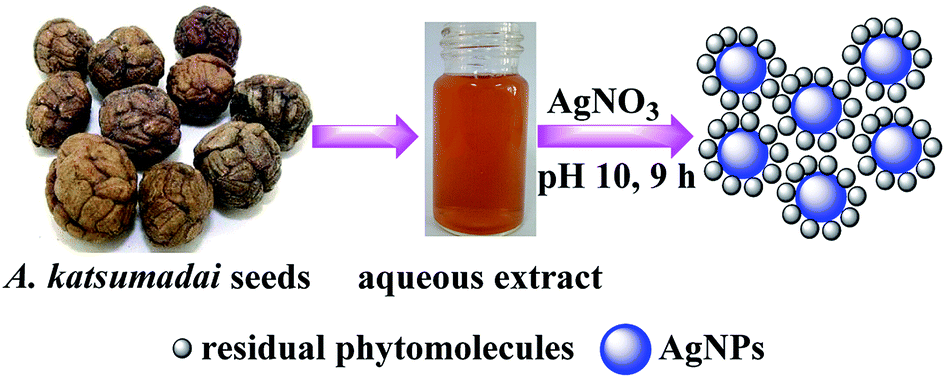 | ||
| Scheme 1 Schematic of the green synthesis of AgNPs using Alpinia katsumadai seed extracts as the bioreductant. | ||
2. Experimental section
2.1. Materials
All the reagents in this study were analytical grade, without further purification. Silver nitrate (AgNO3) was purchased from Tianjin Kemiou Chemical Reagents Co. 2,2-Diphenyl-1-picrylhydrazyl (DPPH) and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromine (MTT) were supplied by Sigma-Aldrich (Shanghai, China); 2,4,6-tri(2-pyridyl)-s-triazine (TPTZ) and other reagents were purchased from Aladdin Reagents Co. Dry seeds of A. katsumadai were collected from Nanning, Guangxi Province, China. Human gastric carcinoma SGC-7901 and healthy transformed human gastric epithelial GES-1 cell lines were purchased from the cell repository of the Chinese Academy of Sciences (Shanghai, China). They were isolated from the lymph nodes of patients with adenocarcinoma of the stomach and from the gastric glands of healthy subjects. Informed consents from patients and healthy subjects were obtained prior to any human subject experimentation.2.2. Preparation and HPLC analysis of the extract
The dried and chopped seeds of A. katsumadai (25 g) were sonicated for 50 min with 300 mL of deionized water in a 500 mL Erlenmeyer flask at 30 °C. The solution was then filtered using no. 1 Whatman filter paper and stored at 4 °C for further studies. 1 mL of the filtrate was further filtered through 0.22 μm filters, prior to HPLC analysis. The extract (20 μL) was analyzed by analytical HPLC on a Shimadzu LC-20A instrument with a Shimadzu SPD-M20A UV/Vis photodiode detector. The samples were loaded on a Shim-park VP-ODS column (5 μm; 4.6 × 150 mm) and eluted with 40% MeOH in H2O + 0.1% CH3COOH for 30 min, with subsequent washing of the column with 100% MeOH at a flow rate 0.5 mL min−1. Rutin was used as the standard sample and its λmax at 255 nm was selected as the detection wavelength.2.3. Synthesis of silver nanoparticles
The synthesis of silver nanoparticles was achieved by mixing 15 mL of A. katsumadai seed extract with 150 mL of 10.0 mM AgNO3 and stirring at 200 rpm for 90 min in the dark at room temperature. The reaction mixture was adjusted to basic pH (pH 10.0), which was determined as the optimum pH by initially adjusting the reaction mixtures (5 mL of plant extract with 15 mL of 1 mM AgNO3 solution) from pH 4 to pH 12 using 0.1–1.0 M HCl and NaOH solutions. Subsequent experiments were carried out at the same pH. The effect of the Ag+ ion concentration on the formation of AgNPs was evaluated by changing the concentration of AgNO3 to 0.10, 1.0, 3.0, 5.0 and 10 mM. Thereafter, the AgNPs were obtained by repeated centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min, and then, to remove the water-soluble biomolecules, the pellet was dispersed in deionized water, repeated thrice. The purified pellets were then dried on Petri plates at 60 °C under vacuum for 24 h. Then the dried AgNPs were used for further study.
000 rpm for 20 min, and then, to remove the water-soluble biomolecules, the pellet was dispersed in deionized water, repeated thrice. The purified pellets were then dried on Petri plates at 60 °C under vacuum for 24 h. Then the dried AgNPs were used for further study.
2.4. Characterization of silver nanoparticles
The reduction of Ag+ ions was monitored periodically by using a UV-Vis spectrophotometer (UV-2102, China) and the UV-Vis spectra of the reaction solutions (1:5 diluted) were measured in the range of 280–760 nm. The morphology of the AgNPs was characterized using field emission TEM (FETEM) images with a Tecnai G2F30 S-Twin equipped with an EDX attachment at 300 kV. The crystalline nature of the AgNPs was examined by Selected Area Electron Diffraction (SAED). The crystalline phase analysis of the synthesized nanoparticles was measured by a Shimadzu XRD-7000S X-ray diffraction (Japan) at 10° min−1 scanning speed in different ranges of two-theta angles (2–80°), with a tube current of 30 mA and a voltage of 40 kV. The particle size was evaluated by dynamic light scattering (DLS) on a DelsaNano C instrument supplied by Beckman Coulter. FT-IR spectra were recorded for the A. katsumadai seed extract and for the AgNPs, using a Shimadzu FT-IR 8900 (Japan) at a resolution of 4.0 cm−1, with KBr as the reference.
2.5. In vitro antioxidant assays
| DPPH scavenging effect (%) = [(A0 − At)/A0] × 100 |
2.6. In vitro anticancer activity of silver nanoparticles
| Loss of viability (%) = [1 − (At − Ab)/(Ac − Ab)] × 100% |
2.7. Antimicrobial activity of silver nanoparticles
Growth inhibition studies against both Gram positive (S. aureus) and Gram negative (P. aeruginosa, E. coli) of silver nanoparticles were carried out in Luria–Bertani (LB) broth media (Becton-Dickinson and Co., USA). The experiments were conducted according to the following procedure, with some modifications.37 In order to maintain the initial bacterial concentration within the same range, in each sterile 100 mL Erlenmeyer flask, 50 mL of LB broth media and silver nanoparticles ranging from 5 to 40 μg mL−1 were mixed with 1 mL of freshly prepared bacterial suspension. Then, the flasks were incubated at 150 rpm at 37 °C in a rotary shaker. A flask containing only the broth media and bacteria was used as the control. The growth of the bacteria was monitored at 600 nm every hour for 9 h using a UV-Vis spectrophotometer (UV-2102, China).3. Results and discussion
3.1. Synthesis of silver nanoparticles
Silver nanoparticles were initially prepared by the reduction of Ag+ into Ag0 with 5 mL of A. katsumadai seed extract, at its original pH value of 4, with 15 mL of 1 mM AgNO3 solution at 25 °C. The colorless solution of AgNO3 turned yellow to dark brown with time, which indicated that the silver nanoparticles were formed. As an efficient technique, the reaction kinetics were monitored in the visible range of 400–500 nm by UV-Vis absorption spectroscopy, in which silver nanoparticles are known to exhibit a surface plasmon resonance (SPR) band due to the free electron excitation.38 The SPR of AgNPs so prepared was observed at 417 nm and slightly shifted to 436 nm during the reaction (ESI Fig. S1a†). This shift may be attributed to the aggregation of smaller particles according to Mie theory.39 However, the absorption increased steadily as a function of reaction time after 8 days, indicating the slow synthesis kinetics of AgNPs.As reported previously, A. katsumadai is a rich source of flavonoids and chalcones. The hydroxyl groups in these compounds, as powerful reduction and stabilizing functional groups, could participate in the synthesis of AgNPs. However, in the acidic pH 4 and pH 6 solutions, the competing biosorption process and the hydroxyl protonation might be the key issue that makes the formation of AgNPs slow.41 On the contrary, at higher pH values (10.0 and 12.0) of the reaction medium, the greater degree of ionization could generate strong complexing ligands for the silver ions,1 so that more complex compounds of silver ions are formed, which would have an effect on the capping ability of biomolecules in AKSE and promote the reduction of silver ions. A possible mechanism was proposed using chrysin as an example, which was isolated from A. katsumadai as shown in Fig. 1.
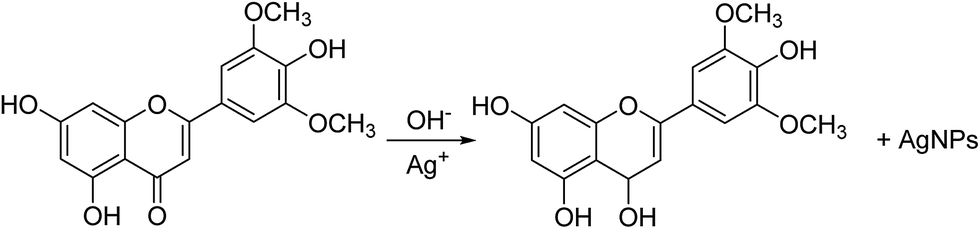 | ||
| Fig. 1 Mechanism of the synthesis of Ag nanoparticles. | ||
In addition, the effect of AKSE quantity on the formation of the AgNPs was also investigated. Different dosages of seed extract, varying from 0.4, 0.8, 1.2, 1.6 to 2.0 mL, were incubated with 15 mL of 10 mM AgNO3 solutions at room temperature at pH 10 (ESI Fig. S1d†). One hour after mixing the AKSE and AgNO3 solutions, visible color changes were noted, from reddish yellow to deep red, with increasing AKSE dosage in each reaction solution. Interestingly, the absorbance values correlated with the AKSE quantity in the UV-Vis spectrum, reflecting a higher amount of AgNPs prepared due to the increased amount of reducing biomolecules available at a higher dosage. Similar results were reported by Sathishkumar37 and Dubey.42 It was noticeable that the slight red shift of the maximum absorption peak from 421 nm to 428 nm when the AKSE quantity was increased from 0.4 mL to 2.0 mL suggested an increasing size of the AgNPs. In order to confirm the key roles AKSE played during the synthesis of AgNPs, a series of experiments at various pHs was carried out without AKSE and the absorbance values for each of them in 2 h were recorded, as shown in ESI Fig. S2.† None of them show the SPR band in the visible range of 400–500 nm, suggesting that no AgNPs were formed without AKSE. Finally, we used 150 mL of 10 mM AgNO3 and 15 mL of aqueous AKSE in a further study to control the reaction kinetics and obtain more products with smaller particle sizes.
3.2. Characterization of AgNPs
The DLS and FETEM were used to evaluate the particle size and surface morphology of AgNPs as prepared. It was obvious that the synthesized AgNPs were nearly quasi-spherical in shape, and well dispersed and scattered in nature (Fig. 2a and b). A narrow distribution of the particle size with an average diameter of ∼12.6 nm was recorded by DLS (Fig. 2a). However, the few traces of AgNP clusters resulting from its aggregation might be attributed to the solvent evaporation during sample preparation. Furthermore, these contributions can also lead to particle size variation.24 It is worth noting that a faint thin layer of other material was clearly visualized around the AgNPs, which further confirms that the plant constituents, such as proteins and flavonoid compounds, capped the AgNPs and prevented their aggregation. This was definitively proven by the EDX and HPLC (ESI Fig. S3†) experiments.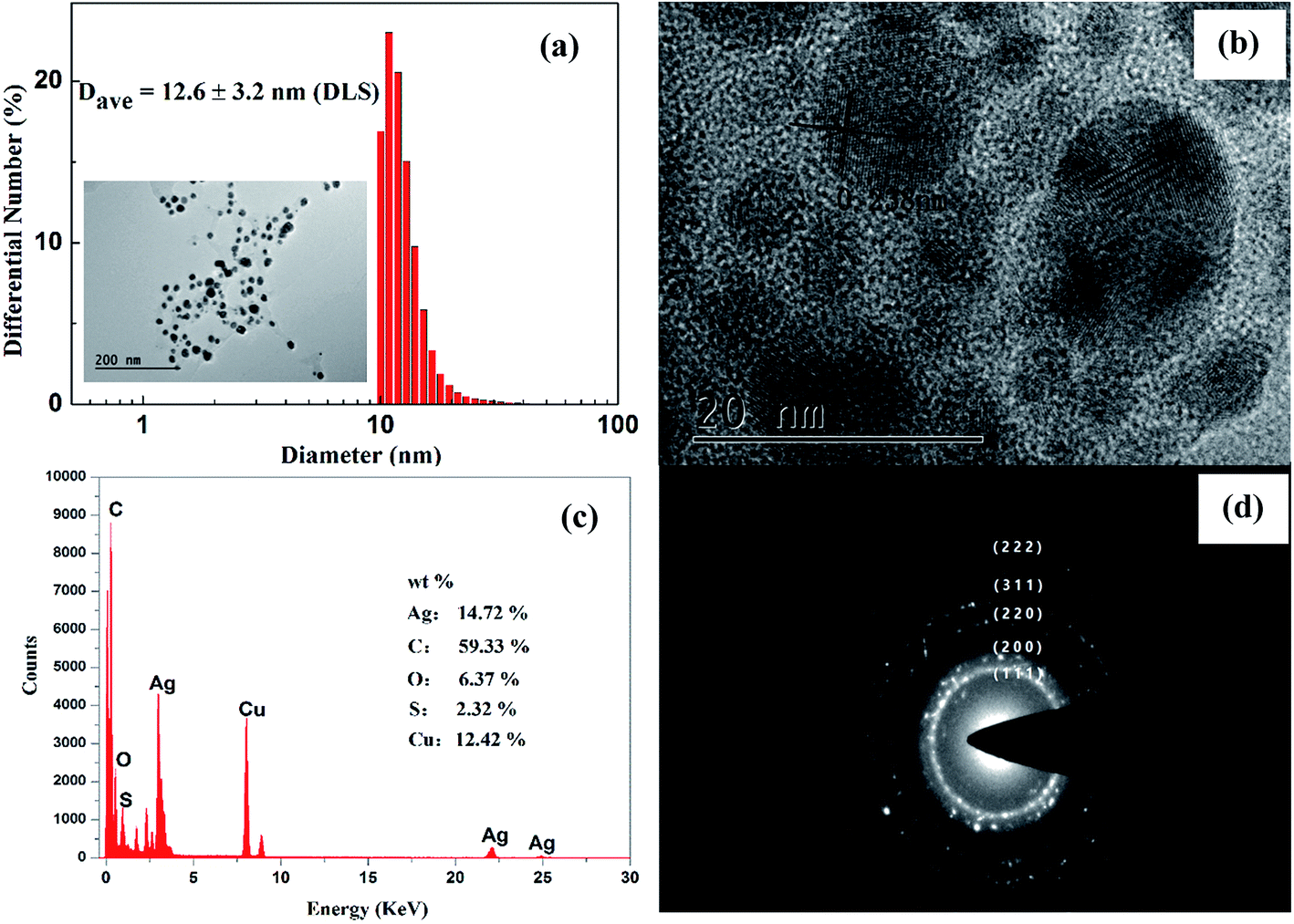 | ||
| Fig. 2 DLS diagram (a) and FETEM images of the synthesized silver nanoparticles with different magnification (a and b), EDX spectrum (c) and SAED pattern (d). | ||
The elemental constituents of the AgNPs were measured by subjecting the TEM grid to energy-dispersive X-ray spectroscopy (EDX) of FETEM (Fig. 2c). The presence of elemental silver in the nanoparticles was proved by a strong absorption peak located at 3 keV. The XRD diffraction peaks observed for carbon, oxygen, and sulfur suggested that the AgNPs were successfully capped by the extracellular organics from AKSE on the surface of the AgNPs, or in the vicinity of them. However, the strongest signal was for carbon, partially originating from the carbon-coated copper grid used in the TEM sample preparation and EDX analysis, which also accounted for the relatively weak signal of copper.
The five bright circular rings assigned to (111), (200), (220), (311) and (222) observed in the selected area electron diffraction (SAED) pattern (Fig. 2d) are the characteristic reflections of face centered cubic crystalline silver.23 Furthermore, the lattice fringe with a distance of 2.38 Å shown in the HRTEM image (Fig. 2b) further confirmed the crystalline nature of the AgNPs.
The crystalline nature of the synthesized AgNPs was further proved by X-ray diffraction. The XRD observed with 2θ degrees at 37.8°, 44.0°, 64.4° and 77.4° can be assigned to the (111), (200), (220), and (311) Bragg's reflections of the face-centered cubic crystalline structure of metallic nanoparticles, respectively (JCPDS no. 04-0783). Similar results were reported previously for silver nanoparticles.11,23 Combined with the TEM micrograph images, the resultant XRD spectrum clearly suggests that the AgNPs prepared by using AKSE were crystalline. These data led to the preliminary conclusion that the AgNPs were successfully prepared by using AKSE.
3.3. Possible functional groups involved in the synthesis of AgNPs
The functional groups which might be responsible for the reducing and efficient stabilizing of the synthesized AgNPs were studied by FT-IR analysis (ESI Fig. S4†). The spectra of AKSE and synthesized AgNPs revealed a shift in the peaks: 3371–3377 cm−1 (stretching vibration for the OH of phenol groups on the flavone rings), 2980–2978 cm−1 (asymmetric stretching of the C–H of alkanes), 1649–1653 cm−1 (characteristic of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of flavone derivatives), 1389–1385 cm−1 (C–O–H bending), 1045–1049 cm−1 (C–O stretching), and 888–881 cm−1 (C–H, flexural vibrations out of the page for the aromatic ring). Compared with AKSE, the AgNP spectrum features the characteristic stretching vibration of the CO or N–H bond at 2359 cm−1. But the signal at 2141 cm−1 of the stretching vibration of the CC bond variably present in plant proteins4 disappeared, which demonstrates that molecules in the AKSE that contain an abundance of CC groups, such as proteins or flavonoids, might play an important role in the reduction and stabilization of AgNPs. In addition, the signal at 2359 cm−1 in the spectrum of the AgNPs indicated the attachment of some CO or N–H groups on the surface of the AgNPs during the synthesis process. Thus, the above information reveals that Ag+ might be reduced to Ag0 by the proteins or flavonoids of AKSE, and consequently the synthesized AgNPs were capped and stabilized by functional groups comprising the free amine groups of the proteins or the carbonyl groups of the flavonoids. To confirm the existence of flavonoids, the AKSE was investigated by analytical HPLC (Shim-park VP-ODS column; 5 μm; 4.6 × 150 mm) experiments using rutin as the standard sample. One major peak with the characteristic retention time of rutin, at 22.5 min, is shown in ESI Fig. S3† and further confirmed the presence of rutin in AKSE; it partially supported the involvement of the above candidate biomolecules in the formation of AgNPs.
O stretching vibration of flavone derivatives), 1389–1385 cm−1 (C–O–H bending), 1045–1049 cm−1 (C–O stretching), and 888–881 cm−1 (C–H, flexural vibrations out of the page for the aromatic ring). Compared with AKSE, the AgNP spectrum features the characteristic stretching vibration of the CO or N–H bond at 2359 cm−1. But the signal at 2141 cm−1 of the stretching vibration of the CC bond variably present in plant proteins4 disappeared, which demonstrates that molecules in the AKSE that contain an abundance of CC groups, such as proteins or flavonoids, might play an important role in the reduction and stabilization of AgNPs. In addition, the signal at 2359 cm−1 in the spectrum of the AgNPs indicated the attachment of some CO or N–H groups on the surface of the AgNPs during the synthesis process. Thus, the above information reveals that Ag+ might be reduced to Ag0 by the proteins or flavonoids of AKSE, and consequently the synthesized AgNPs were capped and stabilized by functional groups comprising the free amine groups of the proteins or the carbonyl groups of the flavonoids. To confirm the existence of flavonoids, the AKSE was investigated by analytical HPLC (Shim-park VP-ODS column; 5 μm; 4.6 × 150 mm) experiments using rutin as the standard sample. One major peak with the characteristic retention time of rutin, at 22.5 min, is shown in ESI Fig. S3† and further confirmed the presence of rutin in AKSE; it partially supported the involvement of the above candidate biomolecules in the formation of AgNPs.
3.4. In vitro antioxidant activity
Reactive oxygen species (ROS) are the detrimental by-products of cellular respiration and have an important role in cell signaling but can also lead to cellular oxidative damage.43 ROS are highly active on account of their unpaired valence shell electrons, being free radicals besides being molecules and ions. Many diseases such as cancer, aging, cataracts, diabetes milieus, cardiovascular disease, and brain dysfunction have been reported to be in part a consequence of an excess of ROS in the body.44 Due to their radical scavenging power, antioxidants play a significant role in terminating free radicals before they attack cells and biological targets and in preventing various diseases.45 In order to accurately reflect the antioxidant properties of AKSE and the as-prepared AgNPs, three different assays were used, including DPPH, ferric reducing antioxidant power (FRAP) and hydroxyl radical scavenging, and each of them was performed at different concentrations from 100 to 500 μg mL−1.Fig. 3 shows the dose–response bar chart for the DPPH-scavenging activity of the AgNPs and AKSE. It is clearly observed that both AgNPs and AKSE are potential free radical scavengers with effective inhibition activity in a dose-dependent manner. However, as compared with AKSE, the inhibition of the DPPH radical scavenging activity of AgNPs increases rapidly with increasing concentration. Interestingly, the AgNPs exhibited a better scavenging activity for DPPH, with a maximum scavenging efficacy at 500 μg mL−1 of 89.9% within 30 min, higher than that of AKSE (85.0%), which might due to more oxidant being incorporated onto the surface of AgNPs because of their high surface area, as the silver quantity increased. But the values for both AgNPs and AKSE were lower than for standard ascorbic acid (96.3%).
 | ||
| Fig. 3 The DPPH radical scavenging activity of AgNPs, AKSE and ascorbic acid. | ||
A similar trend was found in the FRAP assay for AgNPs and AKSE. However, the reducing power was consistently lower for both the samples in comparison with their DPPH-scavenging activities at 400 and 500 μg mL−1 dosages. As shown in Fig. 4, the reducing powers of AgNPs and AKSE increased with increasing concentration, and reached peaks at 79.0% and 73.7% at 500 μg mL−1, respectively. By comparison, as the standard sample, ascorbic acid exhibited the highest reducing power, with a scavenging efficacy of 94.5% at 100 μg mL−1, which was similar to the previously reported data.46
 | ||
| Fig. 4 The reducing power assay of the AgNPs, AKSE and ascorbic acid. | ||
As described above, the inhibition of AgNPs for the oxidation of the hydroxyl radical, which is the most damaging of ROS in the body,44 was also evaluated. Most molecules in biological tissues, such as amino acid and proteins, can be destroyed by the hydroxyl radical.47 Therefore, it is necessary to scavenge the culprit of the hydroxyl radical to avert cell oxidative damage. In this study, Fenton reagents (Fe2+ + H2O2) were used to generate the hydroxyl radical. Five parallel experiments were carried out, including the control (no H2O2), bleaching (no AgNPs), and three different amounts of 500 μg mL−1 AgNPs, from 1 mL to 5 mL. Fig. 5 shows the efficiency of different concentrations of AgNPs for scavenging the hydroxyl radical. As shown in Fig. 5, the results clearly indicated that AgNPs possessed a dose-dependent scavenging activity for the hydroxyl radical, with a maximum scavenging efficacy of 92.3% within 60 min with 5 mL of AgNPs (equivalent to 100 μg mL−1). It was found that a quick color change from blue to light yellow occurred with the absorbance decreasing from 0.93 to 0.04 in 10 min. However, a significant increase of absorbance to 0.78 was observed as 1 mL AgNPs were added into the reaction mixture, suggesting that the oxidation reaction was effectively inhibited by scavenging of the hydroxyl radical. Accordingly, as the concentrations of AgNPs increased in the following tests, the remaining absorbance significantly increased to the absorbance at a 5 mL dose compared with that of the control indicated that the hydroxyl radical was almost completely scavenged by AgNPs.
 | ||
| Fig. 5 The hydroxyl radical scavenging activity of various concentrations of AgNPs. | ||
The positive antioxidant activity of the AgNPs in all three antioxidant assays might be attributed to two aspects; one may be associated with capping agents such as flavonoids described in the FT-IR analysis, and the other is obviously due to the contribution of silver nanoparticles on account of their large surface to volume ratios and crystallographic surface structure. As free radical scavengers, the efficiency of flavonoids depends mainly on the number of hydroxyl groups and the location of these groups on the molecule.48 The mechanism of action of biosynthesized AgNPs needs more sophisticated experimental proof.
3.5. In vitro cytotoxicity activity
An initial screening for the cytotoxic effect against human gastric carcinoma SGC-7901 of the AgNPs was determined by an MTT-assay. As shown in Fig. 6a, the synthesized nanoparticles showed potent cytotoxicity toward SGC-7901 cancer cells. Significant cytotoxic effects of 96% and 93% SGC-7901 cell death were observed at treatment concentrations of 22.5 and 15.0 μg mL−1 in 72 h, respectively. Moreover, 62% cell death was observed at 7.5 μg mL−1. Cytotoxicity against healthy transformed human gastric epithelial GES-1 cell lines was also investigated. Fig. 6b illustrates the results for normal cells treated with the same range of concentrations of particles as tested for SGC-7901 cells. Similar inhibition behavior from 54% to 91% in 72 h was observed for the GES-1 cell line with increasing AgNP concentrations from 7.5 to 22.5 μg mL−1. Unfortunately, no selectivity was demonstrated between cancer and normal cells, excepting a slight distinction at the concentration of 15.0 μg mL−1 with 71% inhibition for GES-1 cells. Thus, further scanning for other cancer cell lines is warranted. | ||
| Fig. 6 Cytotoxic effects of various concentrations of AgNPs on SGC-7901 (a) and GES-1 cell lines (b). | ||
It is noteworthy that, although cancer still remains one of the most devastating diseases in the world, its mortality has significantly decreased due to the rapid developments of diagnostic devices and a deeper understanding of cancer pathogenesis. Over the last decade, nanoparticles have become particularly interesting for cancer diagnosis and therapy on account of their potential to selectivity bind and target cancer cells.49 Many researchers have found that the cytotoxic mechanism of AgNPs can be attributed to the formation of ROS.9 As described above, ROS, such as hydrogen peroxide, can cause cell death by a direct or indirect interaction with intracellular macromolecules like DNA, lipids and proteins.2 Jeyaraj9 reported that AgNPs synthesized using Sesbania grandiflora leaf extract exhibited potential oxidative damage and apoptosis for human breast cancer cells (MCF-7) via the formation of ROS. Furthermore, it was identified that besides increasing intracellular ROS, AgNPs also can lead to cancer cell death through destroying the cell membrane integrity, and through DNA damage and apoptosis.9
3.6. Antibacterial activity
The antibacterial properties of synthesized AgNPs were assessed against both the Gram-positive bacterium Streptococcus aureus and the Gram-negative bacteria Escherichia coli and Pseudomonas aeruginosa. Optical densities of the three pathogens were measured for 9 h at regular intervals with different concentrations of AgNPs from 0 to 40 μg mL−1, which were plotted as a function of time as shown in Fig. 7. The AgNPs exhibited dose-dependent antibacterial activity against all three pathogens, with very fast killing kinetics in comparison with the control. The same minimum inhibition concentration (MIC) was observed for S. aureus and E. coli to be 20 μg mL−1 (Fig. 7a and b), whereas it was observed to be 40 μg mL−1 for P. aeruginosa (Fig. 7c). In all three cases, there was obviously no bacterial growth in 7 h with 40 μg mL−1 of AgNPs as the effective bactericides. While the antibacterial activity decreased significantly in the case of P. aeruginosa after 7 h of treatment, the percent inhibition for S. aureus and E. coli remained the same as at the beginning. In addition, the remarkable growth inhibition at 20 μg mL−1 for S. aureus and E. coli and at 40 μg mL−1 for P. aeruginosa reached 65%, 64%, and 63% after 9 h (Fig. 7d), respectively. Taking into account the previously synthesized AgNPs with potent bactericidal activity obtained by using the red alga Gracilaria dura,14 Artocarpus heterophyllus seed,24 Cinnamon zeylanicum bark,36 and so on, it is fair to say that synthesized AgNPs are a good antibacterial bio-resource with potential applications in biomedical and related areas. | ||
| Fig. 7 Batch growth profiles of (a) S. aureus, (b) E. coli and (c) P. aeruginosa at various concentrations of AgNPs for a time period of 9 hours. (d) Percent inhibition of pathogenic bacteria after the addition of different concentration of AgNPs to media over a time period of 9 hours. | ||
Although many possible mechanisms of action for the growth inhibition of AgNPs against bacterial have been proposed, the exact mechanism has not yet been elucidated. In general, it has been believed that the silver cations from AgNPs appear to attach to the negatively charged bacterial cell wall and rupture it, leading to protein denaturation and eventual cell death.50 The attachment of silver cations or nanoparticle onto the cell wall leads to an increase in envelope protein precursors, which results in the loss of the proton motive force and finally cell death. It was also reported that the outer and plasma membranes can be ruptured by AgNPs, leading to depletion of intracellular ATP.51 Another mechanism that has been proposed is that R–S–S–R bonds are formed through reactions related to silver and oxygen with sulfhydryl groups on the cell wall, therefore blocking respiration and causing cell death.52
4. Conclusions
In summary, this study demonstrated an ecofriendly, rapid, green approach to the synthesis of AgNPs by using AKSE, which supplies a simple, cost-effective and ecological method for the synthesis of AgNPs. Water-soluble secondary metabolites, most likely flavonoids or proteins, in AKSE were mainly responsible for the biosynthesis of AgNPs. The AgNPs so-prepared exhibited effective antioxidant, antibacterial and anti-cancer activities, suggesting AgNPs might be useful as a silver dressing for wounds or as an alternative material. Therefore, further investigations are needed to fully characterize the cytotoxicity and the mechanism of action associated with the antioxidant and antibacterial activity of their particles.Acknowledgements
This work was supported by the Scientific and Technological Project of Shaanxi Province in China (2015SF087); the Scientific Research Program Funded by Shaanxi Provincial Education Department (15JK1500); the Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shaanxi Province (302-253081601) and the Xi'an University of Technology Starting Grant awarded to Dr Yangqing He. The authors also thank Mr Ting Zhang and Xuyang Zhu for XRD measurements.Notes and references
- A. M. Abdel-Mohsen, R. Hrdina, L. Burgert, G. Krylová, R. M. Abdel-Rahman, A. Krejčová, M. Steinhart and L. Beneš, Carbohydr. Polym., 2012, 89, 411–422 CrossRef CAS PubMed.
- S. J. P. Jacob, J. S. Finub and A. Narayanan, Colloids Surf., B, 2012, 91, 212–214 CrossRef PubMed.
- J. Y. Chen, D. L. Wang, J. F. Xi, L. Au, A. Siekkinen, A. Warsen, Z. Y. Li, H. Zhang, Y. N. Xia and X. D. Li, Nano Lett., 2007, 7, 1318–1322 CrossRef CAS PubMed.
- V. Gopinath, D. MubarakAli, S. Priyadarshini, N. M. Priyadharsshini, N. Thajuddin and P. Velusamy, Colloids Surf., B, 2012, 96, 69–74 CrossRef CAS PubMed.
- C. Liu, X. Yang, H. Yuan, Z. Zhou and D. Xiao, Sensors, 2007, 7, 708–718 CrossRef CAS.
- P. Mohanpuria, N. K. Rana and S. K. Yadav, J. Nanopart. Res., 2008, 10, 507–517 CrossRef CAS.
- K. Chaloupka, Y. Malam and A. M. Seifalian, Trends Biotechnol., 2010, 28, 580–588 CrossRef CAS PubMed.
- C. Dipankar and S. Murugan, Colloids Surf., B, 2012, 98, 112–119 CrossRef CAS PubMed.
- M. Jeyaraj, G. Sathishkumar, G. Sivanandhan, D. MubarakAli, M. Rajesh, R. Arun, G. Kapildev, M. Manickavasagam, N. Thajuddin, K. Premkumar and A. Ganapathi, Colloids Surf., B, 2013, 106, 86–92 CrossRef CAS PubMed.
- B. Kumar, K. Smita, L. Cumbal and A. Debut, BioNanoScience, 2016, 6, 15–21 CrossRef.
- B. Kumar, K. Smita, R. Seqqat, K. Benalcazar, M. Grijalva and L. Cumbal, J. Photochem. Photobiol., B, 2016, 159, 8–13 CrossRef CAS PubMed.
- S. Pugazhendhi, E. Kirubha, P. K. Palanisamy and R. Gopalakrishnan, Appl. Surf. Sci., 2015, 357, 1801–1808 CrossRef CAS.
- A. Saxena, R. M. Tripathi, F. Zafar and P. Singh, Mater. Lett., 2012, 67, 91–94 CrossRef CAS.
- M. K. Shukla, R. S. Singh, C. R. K. Reddy and B. Jha, Bioresour. Technol., 2012, 107, 295–300 CrossRef CAS PubMed.
- D. H. Williams and B. Bardsley, Angew. Chem., Int. Ed., 1999, 38, 1172–1193 CrossRef.
- J. Chastre, Clin. Microbiol. Infect., 2008, 14, 3–14 CrossRef CAS PubMed.
- N. Ahmad, S. Sharma, M. K. Alam, V. N. Singh, S. F. Shamsi, B. R. Mehta and A. Fatma, Colloids Surf., B, 2010, 81, 81–86 CrossRef CAS PubMed.
- A. B. Smetana, K. J. Klabunde and C. M. Sorensen, J. Colloid Interface Sci., 2005, 284, 521–526 CrossRef CAS PubMed.
- D. Mandal, M. E. Bolander, D. Mukhopadhyay, G. Sarkar and P. Mukherjee, Appl. Microbiol. Biotechnol., 2006, 69, 485–492 CrossRef CAS PubMed.
- M. Kowshik, S. Ashtaputre, S. Kharrazi, W. Vogel, J. Urban, S. K. Kulkarni and K. M. Paknikar, Nanotechnology, 2003, 14, 95–100 CrossRef CAS.
- N. Samadi, D. Golkaran, A. Eslamifar, H. Jamalifar and M. R. Fazeli, J. Biomed. Nanotechnol., 2009, 5, 247–253 CrossRef CAS PubMed.
- D. Philip, C. Unni, S. A. Aromal and V. K. Vidhu, Spectrochim. Acta, Part A, 2011, 78, 899–904 CrossRef PubMed.
- V. K. Shukla, R. S. Yadav, P. Yadav and A. C. Pandey, J. Hazard. Mater., 2012, 213–214, 161–166 CrossRef CAS PubMed.
- U. B. Jagtap and V. A. Bapat, Ind. Crops Prod., 2013, 46, 132–137 CrossRef CAS.
- K. S. Nog and G. D. Brown, Phytochemistry, 1998, 47, 1117–1123 CrossRef.
- S. Z. Hua, X. B. Wang, J. G. Lou, J. S. Wang and L. Y. Kong, Tetrahedron Lett., 2008, 49, 5658–5661 CrossRef CAS.
- X. Xiao, X. Si, T. Xing and G. K. Li, Sep. Purif. Technol., 2011, 81, 265–269 CrossRef CAS.
- Y. Yang, K. Kinoshita, K. Koyama, K. Takahashi, K. Tai, Y. Nunoura and K. Watanabe, Nat. Prod. Res., 1999, 5, 20–24 CAS.
- H. H. Kim, H. J. Kwon, Y. B. Ryu, J. S. Chang, K. O. Cho, M. D. T. Hosmillo, M. C. Rho, S. J. Park and W. S. Lee, Res. Vet. Sci., 2010, 92, 320–323 CrossRef PubMed.
- H. J. Kwon, H. H. Kim, S. Y. Yoon, Y. B. Ryu, J. S. Chang, K. O. Cho, M. C. Rho, S. J. Park and W. S. Lee, Virol. J., 2010, 7, 307–315 CrossRef PubMed.
- M. Y. Lee, C. S. Seo, J. A. Lee, I. S. Shin, S. J. Kim, K. Ha and H. K. Shin, Inflammation, 2011, 35, 746–757 CrossRef PubMed.
- E. R. Hahm, S. Park and C. H. Yang, Nat. Prod. Res., 2003, 17, 431–436 CrossRef CAS PubMed.
- C. W. Choi, S. C. Kim, S. S. Hwang, B. K. Choi, H. J. Ahn, M. Y. Lee, S. H. Park and S. K. Kim, Plant Sci., 2002, 163, 1161–1168 CrossRef CAS.
- R. Pulido, L. Bravo and F. Saura-Calixto, J. Agric. Food Chem., 2008, 48, 3396–3402 CrossRef.
- Y. Wang, Q. Zhang, C. Xie, X. Zhao, J. F. Niu, J. Zhao, J. K. Oh, P. Li and P. F. Li, RSC Adv., 2015, 5, 95666–95673 RSC.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- M. Sathishkumar, K. Sneha, S. W. Won, C. W. Cho, S. Kim and Y. S. Yun, Colloids Surf., B, 2009, 73, 332–338 CrossRef CAS PubMed.
- M. Sastry, K. S. Mayya and K. Bandyopadhyay, Colloids Surf., A, 1997, 127, 221–228 CrossRef CAS.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- D. Andreescu, C. Eastman, K. Balantrapu and D. V. Goia, J. Mater. Res., 2007, 22, 2488–2496 CrossRef CAS.
- P. P. Gan, S. H. Ng, Y. Huang and S. F. Y. Li, Bioresour. Technol., 2012, 113, 132–135 CrossRef CAS PubMed.
- S. P. Dubey, M. Lahtinen and M. Sillanpää, Process Biochem., 2010, 45, 1065–1071 CrossRef CAS.
- U. Krengel and S. Törnroth-Horsefield, Science, 2015, 347, 125–126 CrossRef CAS PubMed.
- M. T. Zimmerman, C. A. Bayse, R. R. Ramoutar and J. L. Brumaghim, J. Inorg. Biochem., 2015, 14, 30–40 CrossRef PubMed.
- B. Kumar, K. Smita, R. Seqqat, K. Benalcazar, M. Grijalva and L. Cumbal, J. Photochem. Photobiol., B, 2016, 159, 8–13 CrossRef CAS PubMed.
- N. Basavegowda, A. Idhayadhulla and Y. R. Lee, Ind. Crops Prod., 2014, 52, 745–751 CrossRef CAS.
- W. G. Li, C. H. Ge, L. Yang, R. X. Wang, Y. M. Lu, Y. Gao, Z. H. Li, Y. H. Wu, X. F. Zheng, Z. Y. Wang and C. G. Zhang, Int. J. Biol. Macromol., 2016, 82, 97–103 CrossRef CAS PubMed.
- C. C. Denardin, G. E. Hirsch, R. F. D. Rocha, M. Vizzotto, A. T. Henriques, J. C. F. Moreira, F. T. C. R. Guma and T. Emanuelli, J. Food Drug Anal., 2015, 23, 387–398 CrossRef CAS.
- J. W. Tian, L. Ding, H. J. Xu, Z. Shen, H. X. Ju, L. Jia, L. Bao and J. S. Yu, J. Am. Chem. Soc., 2013, 135, 18850–18858 CrossRef CAS PubMed.
- Y. S. Lin, R. D. Vidic, J. E. Stout, C. A. MaCartney and V. L. Yu, Water Res., 1998, 32, 1997–2000 CrossRef CAS.
- C. N. Lok, C. M. Ho, R. Chen, Q. Y. He, W. Y. Yu, H. Z. Sun, K. S. P. Tam, J. F. Chiu and C. M. Che, J. Proteome Res., 2006, 5, 916–924 CrossRef CAS PubMed.
- V. S. Kumar, B. M. Nagaraja, V. Shashikala, A. H. Padmasri, S. S. Madhavendra, B. D. Raju and K. S. R. Rao, J. Mol. Catal. A: Chem., 2004, 223, 313–319 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05286c |
| This journal is © The Royal Society of Chemistry 2017 |