DOI:
10.1039/C7RA05223E
(Paper)
RSC Adv., 2017,
7, 35648-35654
A global potential energy surface and dynamics study of the Au+ + H2 → H + Au+H reaction
Received
9th May 2017
, Accepted 8th July 2017
First published on 17th July 2017
Abstract
A global potential energy surface (PES) of the ground state of the Au+H2 system was constructed using a neural network method with permutation invariant polynomials. More than 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ab initio points were calculated using a multi-reference configuration interaction approach, and these were adopted into a fitting process; the root mean square error was 0.02 eV. The topography of the present PES was compared in detail with the PES reported by Dorta-Urra (J. Chem. Phys., 135, 2011, 091102). The potential well in the present PES is about 0.12 eV deeper than the previous PES. The dynamics calculations based on these two PESs were performed by the time-dependent quantum wave packet method with a second-order split operator. The reaction probability, integral cross section and differential cross section based on these two PESs were calculated in this work. The results obtained from these two PESs were compared with the experimental data. It was clear that the integral cross section calculated by using the present PES is closer to the experimental data than the one obtained from Dorta-Urra PES. In addition, the reaction mechanism of the title reaction changed from an indirect reaction to an abstraction reaction as the collision energy increased.
000 ab initio points were calculated using a multi-reference configuration interaction approach, and these were adopted into a fitting process; the root mean square error was 0.02 eV. The topography of the present PES was compared in detail with the PES reported by Dorta-Urra (J. Chem. Phys., 135, 2011, 091102). The potential well in the present PES is about 0.12 eV deeper than the previous PES. The dynamics calculations based on these two PESs were performed by the time-dependent quantum wave packet method with a second-order split operator. The reaction probability, integral cross section and differential cross section based on these two PESs were calculated in this work. The results obtained from these two PESs were compared with the experimental data. It was clear that the integral cross section calculated by using the present PES is closer to the experimental data than the one obtained from Dorta-Urra PES. In addition, the reaction mechanism of the title reaction changed from an indirect reaction to an abstraction reaction as the collision energy increased.
I. Introduction
Since gold and gold alloys are effective catalysts, reactions involving gold and its ions have been investigated extensively both theoretically and experimentally. Catalytic activity is closely related to the electronic structure and geometry of the cluster. Gold clusters are dominated by planar structures, which are caused by the strong hybridization of the atomic 5d and 6s orbitals. Häkkinen et al.1 found that the strong hybridization of the atomic 5d and 6s orbitals of the gold atom is intimately related to relativistic effects. In 1987, Haruta et al.2 found that a variety of gold catalysts exhibit high catalytic activity for the oxidation of carbon monoxide at low temperatures (about −70 °C). In 2004, Kimble et al.3 investigated, both theoretically and experimentally, the catalytic activity of atomic gold anions with respect to oxygen and the oxidation of CO. Using a fast-flow reactor mass spectrometer, they found that AuO− and AuO3− promote the oxidation of CO. In addition, according to density functional theory (DFT) calculations of structures and energetics, they confirmed the experimental findings and proposed mechanisms for the oxidation of carbon monoxide. In 2016, Zhang et al.4 used DFT and studied the AuVO3+ system which is a barrier-free catalyst for oxidation of CO. Additionally, the atomic structure and electronic structure of gold clusters has received much attention in previous theoretical investigations.5–11
Recently, the collision reaction of hydrogen and gold atoms or gold ions has received a great deal of attention.12–23 The electronic structure and the reaction of the AuH2 system were calculated by Chambaud et al. in 2005.12 The ground state potential energy surface (PES) was constructed by Zanchet et al.13 using multi-reference configuration interaction (MRCI)24,25 with the Gaussian-type basis set ECP60MDF for the Au atom14 and the correlation consistent polarized valence triple zeta basis set for the H atom.15 Some dynamic properties of the Au + H2 reaction have been calculated using a quantum wave packet and quasi-classical trajectories (QCT) approach based on the PES. Using guided ion beam tandem mass spectrometry, Li et al. studied the reactions of the Au ion with H2 and the reaction isotope, and the integral cross sections (ICS) of above reactions.16 In 2011, Dorta-Urra et al. constructed the PES of Au+H2 (Dorta-Urra PES) and studied the reactive collisions of Au+ with H2, D2, and HD using QCT.17 In 2015, Dorta-Urra et al. made a comparative study of the Au + H2, Au+ + H2, and Au− + H2 systems using the MRCI method to construct the PES and using QCT to study the reactive collision.18
With the aspect of dynamics, Yuan et al. investigated the Au + H2/HD reaction using a time-dependent wave packet at the state-to-state level of theory.19,20 For the Au+ + H2/Au− + H2 reaction, the QCT method is always employed to study reaction scattering.17,18 For the Au+ + H2 reaction, the theoretical calculations deviate largely from the experimental data.16 To further investigate the reaction mechanism of the Au+ + H2 system, it is necessary to construct a more accurate PES. Additionally, the reaction involves two hydrogen atoms, and there is a deep well on the reaction path. Therefore, it is meaningful to investigate the role of quantum effects on the reaction.
This paper is organized as follows: Section II gives the details of the ab initio potential calculation and the fitting method. Section III gives the detailed information of the dynamic calculations. Section IV concentrates on the topological features of the ground state PES. Section V analyzes the dynamic properties of the title reaction. The conclusions are given in Section VI.
II. Construction of the PES
Ab initio calculations
To map the AuH2+ PES accurately, a total of 40352 grid points were generated using the atom–diatom Jacobi coordinates (r, R, θ). The grid points for the Au+ + H2 region are defined by 0.6 ≤ rHH/a0 ≤ 25, 0 ≤ RAu+–HH/a0 ≤ 25 and 0 ≤ θ/deg ≤ 90. For the H + Au+H region, the grid points are defined by 1.0 ≤ RAu+H/a0 ≤ 25, 0 ≤ RAu+H–H/a0 ≤ 25 and 0 ≤ θ/deg ≤ 180. Considering that the topographical attributes of strong interactions are more complicated than the asymptotic region, most of the grid points are distributed in the strong interaction region, and a relatively small amount of grid points are distributed in the asymptotic region. In this work, all ab initio energies for the AuH2+ were obtained at the internally contracted MRCI level using the complete active space for the self-consistent field (CASSCF)26,27 wave functions as reference. In addition, the Davidson correction was used in the MRCI calculation to compensate for effects of the higher order correlation energy. The augmented correlation consistent polarized valence quadruple-zeta (AVQZ) basis set was employed for H atom. To account for the relativistic effects of the heavy element, the ECP-based correlation consistent basis set (WCVQZ-PP) was employed for the Au atom. 12 active orbitals (9a′ + 3a′′) and six states (11A′, 21A′, 31A′, 41A′, 11A′′ and 11A′′) of AuH2+ system were involved in the CASSCF calculations. The double many-body expansion (DMBE) strategy, which includes the two-body term and the three-body term, was used to construct the AuH2+ PES. For the two-body term, we calculated 99 ab initio energy points for H2 and 93 ab initio energy points for Au+H. All of the calculations were carried out by the Molpro 2012 software package.28
Fitting the PES
The analytical expression for the global surface of the Au+H2 system can be written as follows:| |
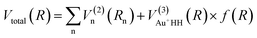 | (1) |
R is a collective variable of all internuclear distances, V(2)n (n = HH, Au+Ha, Au+Hb) is the diatomic potential, Rn is the bond length between the two atoms, and 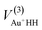 represents the three-body term. f(R) is a switch function, which is used to better describe the PES in the asymptotic area, and it can be presented as:
represents the three-body term. f(R) is a switch function, which is used to better describe the PES in the asymptotic area, and it can be presented as:| |
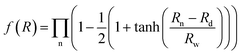 | (2) |
where n = HH, Au+Ha, and Au+Hb. Rd is the position of the switch, and Rw is the constant of the switch strength. We used a neural network (NN)29 method to fit the PES; the NN method was inspired by the central nervous system of animals. This fitting method has been applied to many PESs.30–33 A neuron is the basic unit for the NN. The neuron functions as a synapse, receiving input signals and emitting an output signal. We can express the output signal y as follows:| |
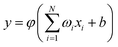 | (3) |
where xi (i = 1, …, N) represents the input signals, wi is the connection weight, b is a bias, and φ is a transfer function. To enforce the permutation symmetry in the NN representation of the AuH2+ PES, low-order permutation invariant polynomials (PIP)34–36 were applied during the fitting procedure. For the title reaction system, Au+, H and H are labelled as 1, 2 and 3, respectively. The input signals can be written as:
So far, many different NNs have been developed for different purposes, and the feed-forward NN, which is employed in this PES to fit the two-body and three-body term, is the common one. To obtain a higher computational efficiency and fitting precision, we used a series of tests to determine the structures of the NN. In the present work, two hidden layers were used in the fitting process, and each layer involved 6 neurons for the two-body terms of H2 and Au+H. The root mean square errors (RMSEs) of two-body terms are only 5.25 × 10−4 and 1.37 × 10−4 eV for H2 and Au+H molecules, respectively. For the three-body terms, there were 11 neurons in each hidden layer, and the overall RMSE of the present PES is 0.02 eV.
III. Dynamic method
The time-dependent quantum wave packet (TDWP) method has been widely applied to atom–diatom reactions and reactions involving polyatoms, such as O + H2, Li + HF, F + HD, and H + H2O.37–40 An abbreviated introduction of the TDWP approach is given below.
The Hamiltonian of the Au+ + H2 reaction in body-fixed (BF) reactant Jacobi coordinates for a given total angular momentum J can be written as
| |
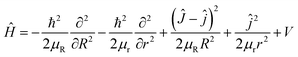 | (7) |
where
R is the distance from the H atom to the center of mass of the Au
+H molecule, and
r is the bond length of the Au
+H molecule.
μR and
μr correspond to the reduced masses that are respectively associated with the
R and
r coordinates.
J is the total angular momentum operators of the Au
+H
2 system, and
j is the rotational angular momentum operator of the reactant diatomic molecule.
V is the potential energy of the Au
+H
2 system. The reactant-coordinate-based (RCB) method
41 is used to extract the state-to-state
S-matrix. The reaction probability of title reaction can be expressed as
| |
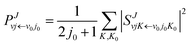 | (8) |
where
v0, and
j0 are the initial quantum numbers used for denoting the initial rovibrational state. The state-to-state ICS and differential cross sections (DCS) are calculated by
| |
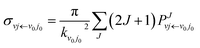 | (9) |
and
| |
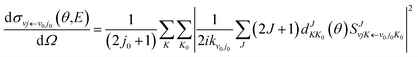 | (10) |
in which
θ is the scattering angle of the products. The Wigner function
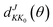
is used for calculating the reduced rotation matrix element. The numerical parameters used in the TDWP calculation are listed in
Table 1.
Table 1 Numerical parameters for reactant Jacobi coordinate propagation
| Grid/basis range and size |
R ∈ [0.01 a.u., 17.0 a.u.], NR = 215 |
| r ∈ [0.01 a.u., 17.0 a.u.], NR = 129 |
| jmin = 0 to jmax = 100, Nj = 51 over [0°, 180°] |
| Initial wavepacket |
R0 = 12.0 a.u. |
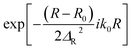 |
ΔR = 0.3 a.u. |
| k0 = (2E0μR)1/2 with E0 = 3.5 eV |
| Total propagation time |
30000 a.u. |
| Time step |
15 a.u. |
IV. Features of the Au+H2 PES
The dissociation energies and equilibrium positions of Au+H and H2 obtained from the new AuH2+ PES as well as the available experimental data and theoretical values are listed in Table 2.14,16,17,42 As shown in Table 2, the equilibrium positions and dissociation energies of two-body term are in good agreement with available theoretical and experimental values. Fig. 1 presents the three-dimensional ground-state PES obtained at four different approaching angles (45°, 90°, 135°, and 180°). As shown in Fig. 1, E = 0 corresponds to the dissociation limit of H–Au+–H. There are two valleys in each map, the left valley corresponds to the Au+ + H2 products and the right valley corresponds to H + Au+H reactant. The left valley is deeper than the right valley, which means that the Au+ + H2 → H + Au+H reaction is endothermic. It is clear that there are no barriers in the reactants and products valleys.
Table 2 The equilibrium bond lengths and dissociation energies for the ground electronic states of Au+H ion and H2 molecule
| Au+H |
This work |
Exp.16 |
Exp.14 |
Theo.17 |
| Re (Å) |
1.528 |
— |
— |
1.523 |
| De (eV) |
1.969 |
2.170 |
1.940 |
1.900 |
| H2 |
This work |
Exp.42 |
Theo. |
| Re (Å) |
0.741 |
0.742 |
0.741 |
| De (eV) |
4.741 |
4.742 |
4.74117 |
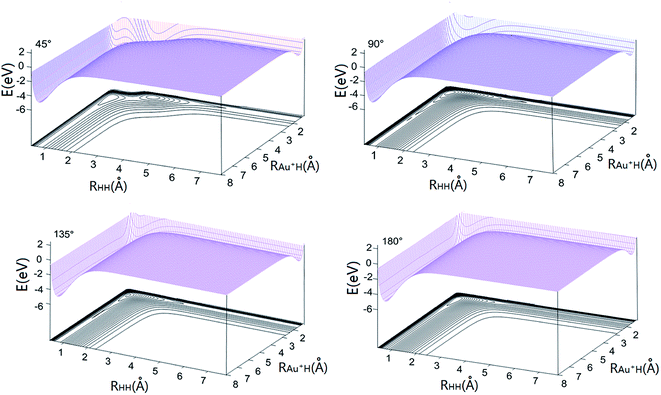 |
| | Fig. 1 Potential energy surface for Au+–H–H angles 45°, 90°, 135° and 180°. | |
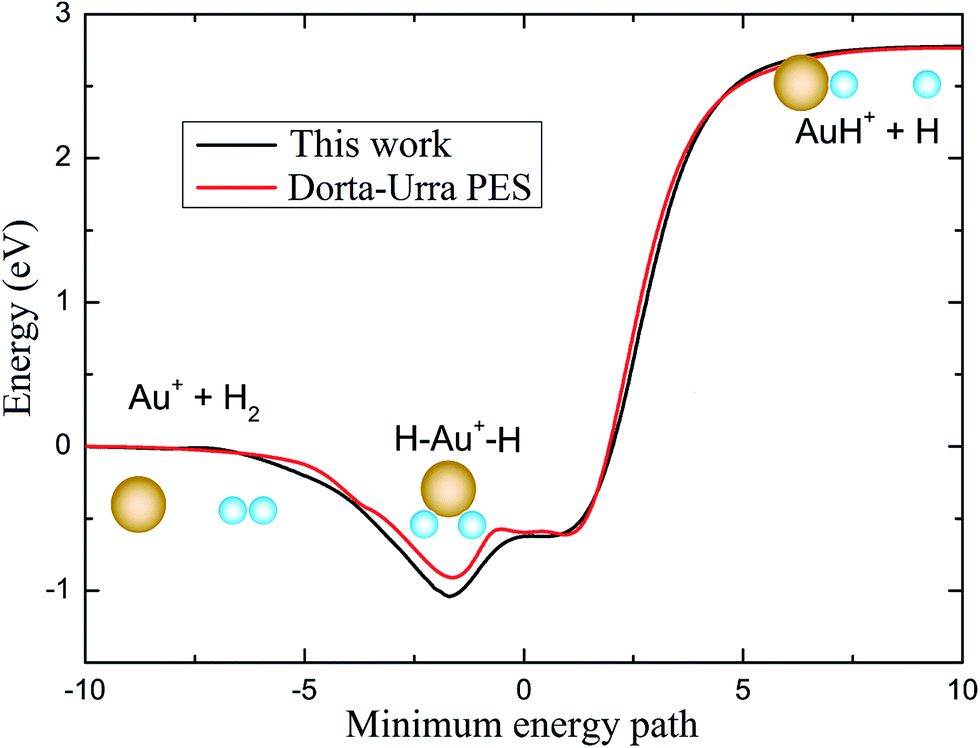
To have a clear picture and to compare with the PES reported by Dorta-Urra et al.,17 the minimum energy paths (MEPs) of the Au+ + H2 → H + Au+H reaction based on the present PES and the Dorta-Urra PES at four approaching angles (45°, 90°, 135°, and 180°) are displayed in the left and right panels of Fig. 2, respectively. As shown in Fig. 2, the endothermic energy of the title reaction is about 2.770 and 2.786 eV for the present PES and the Dorta-Urra PES, respectively. The potential wells located at the MEPs with 90°, 135° and 180° Au+–H–H angles are about 0.960, 0.583, 0.452 eV, respectively, for present PES, and the correspond potential wells are about 0.835, 0.501, 0.433 eV, respectively, for the Dorta-Urra PES. Moreover, there are two potential wells located at the MEPs with 45° Au+–H–H angle. For the present PES, the two wells are 0.707 and 0.552 eV, whereas the well depths are 0.604 and 0.588 eV for Dorta-Urra PES. The global minimum energy reaction paths of Au+ + H2 → H + Au+H reaction for these two PESs are shown in Fig. 3. Energies refer to the asymptotic valley of the Au+ + H2 reactants. In Fig. 3, the global minimum energy reaction paths obtained using our new AuH2+ PES is similar to the one obtained using the Dorta-Urra PES. In addition, the present PES is about 0.1 eV deeper than the Dorta-Urra PES, and this can lead to different dynamic results.
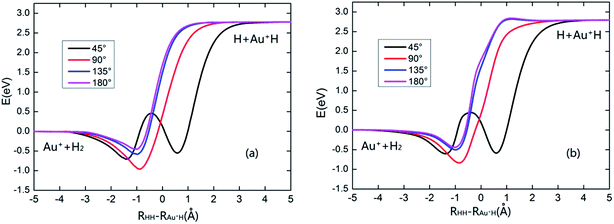 |
| | Fig. 2 The minimum energy paths of present PES and the Dorta-Urra PES at four different angles are collected in the left and right panels, respectively. | |
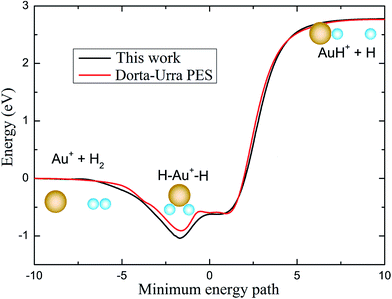 |
| | Fig. 3 Minimum reaction path of present PES and the Dorta-Urra PES. | |
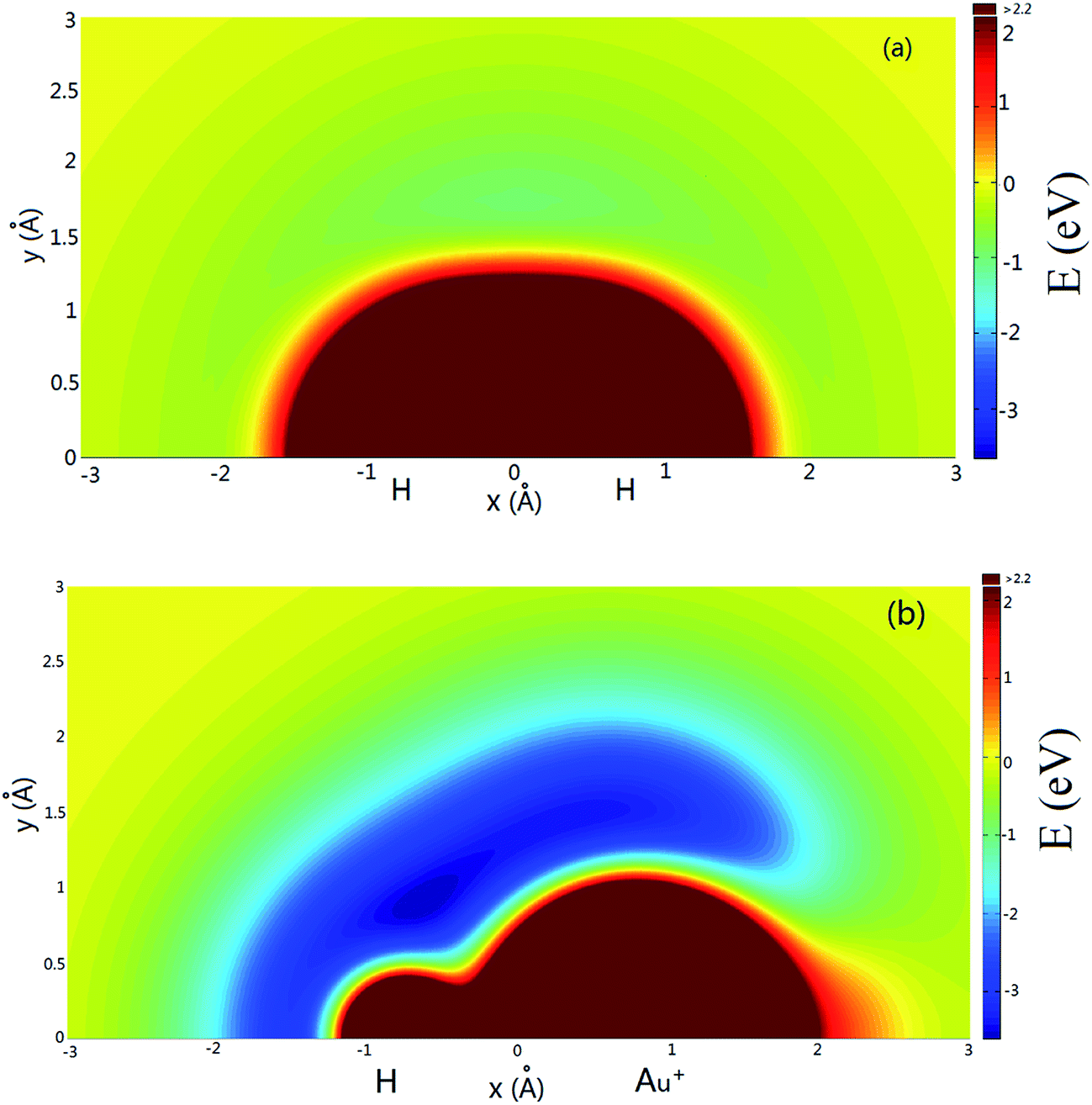
Fig. 4(a) shows the potential energy plot for the Au+ atom moving around the H2 diatom with a fixed bond length at an equilibrium distance of RHH = 0.741 Å. The zero energy is set as the energy in the configuration when the Au+ ion is far from the H2 molecule. There is a shallow well at x = 0.0 Å, y = 1.7 Å. The potential energy plot for a H atom moving around a AuH+ ion with a bond length fixed at the equilibrium distance is shown in Fig. 4(b), and the zero energy is set as the energy in the configuration when the H atom is far from the Au+H molecule. As shown in Fig. 4(b), there are two wells around the Au+H molecule that are located about x = −0.7 Å, y = 0.9 Å and x = 0.5 Å, y = 1.5 Å, respectively.
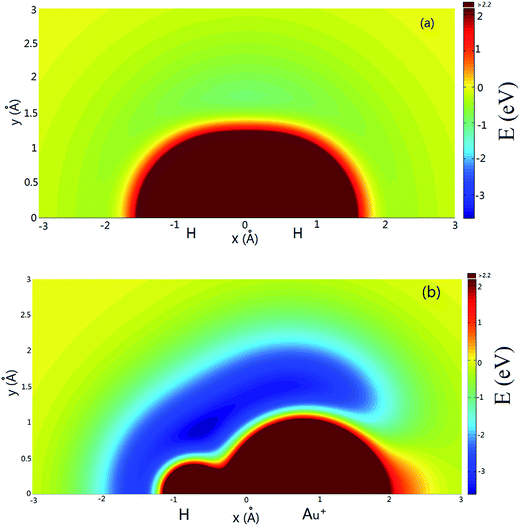 |
| | Fig. 4 (a) Color plot of the potential energy when the Au+ atom moves around the H2 diatom with the bond length fixed at RHH = 0.741 Å. (b) Color plot of the potential energy when the H atom moves around the Au+H ion with the bond length fixed at RAu+H = 1.528 Å. | |
V. Dynamical results
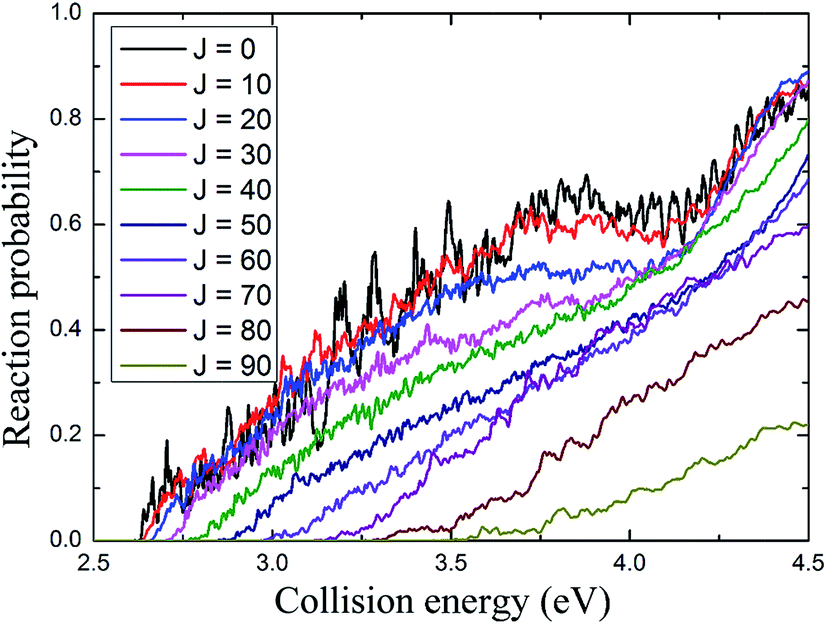
The total reaction probabilities for several selected values of angular momentum J are displayed in Fig. 5 and are shown as a function of collision energy. As shown in Fig. 5, the energy threshold for J = 0 is about 2.63 eV which is lower than the endothermic energy of reaction (about 2.8 eV) obtained from Fig. 3, which leaded by that the zero-point energy of H2 molecule is higher than that of the AuH+ ion. The energy threshold increases with the total angular momentum quantum J, because the centrifugal barrier increases with J (J + 1). In addition, the resonance peaks are less apparent as the collision energy and total angular momentum increase.
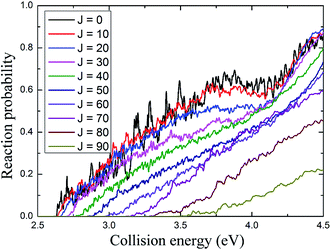 |
| | Fig. 5 The total reaction probability of several selected angular momentum J values depends on the collision energy. | |
As shown in Fig. 6, the profiles of the total reaction probabilities based on the present PES and the Dorta-Urra PES are very similar to each other. Both curves monotonically increase with the collision energy, and exhibit apparent resonances that attribute to the potential well along the reaction path. However, there are some differences between the two curves. The reaction probability obtained using the present PES has relatively small threshold energy, and the peaks seem to be sharper and more intensive, because the well in the present PES is deeper than the well in the Dorta-Urra PES.
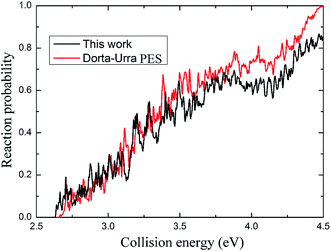 |
| | Fig. 6 The total reaction probabilities obtained based on present PES and the Dorta-Urra PES for the angular momentum J = 0. | |
The maximum total angular momentum quantum number in the TDWP calculations is 90. With total angular momentum up to J = 90, the ICS and DCS are convergent when the collision energy is below 3.5 eV. The ICSs of title reaction based on these two PESs are collected in Fig. 7 and are shown as a function of collision energy. For comparison, the experimental data, the experimental data and model data reported by Li et al.16 are also shown in Fig. 7. The ICSs obtained using the two PESs are generally very similar to each other. Both ICSs increase when the collision energy increases monotonously. In addition, the ICS obtained using our PES is larger than the one obtained using the Dorta-Urra PES over the whole collision energy range. Also, the ICSs based on our AuH2+ PES are closer to the experiment results than those based on the Dorta-Urra PES. The experimental ICSs are larger than theoretical results, and the experimental threshold energy, which is about 2.0 eV, is lower than the theoretical results. The threshold energy, which is about 2.7 eV, is similar for these two PESs. We speculate that the deviations between experiment and theoretical calculations may be attributed to the experimental error, which is as high as ±20%.
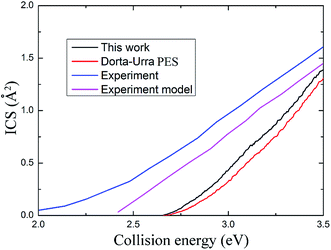 |
| | Fig. 7 The integral cross section based on present PES and the Dorta-Urra PES as well as experimental data. | |
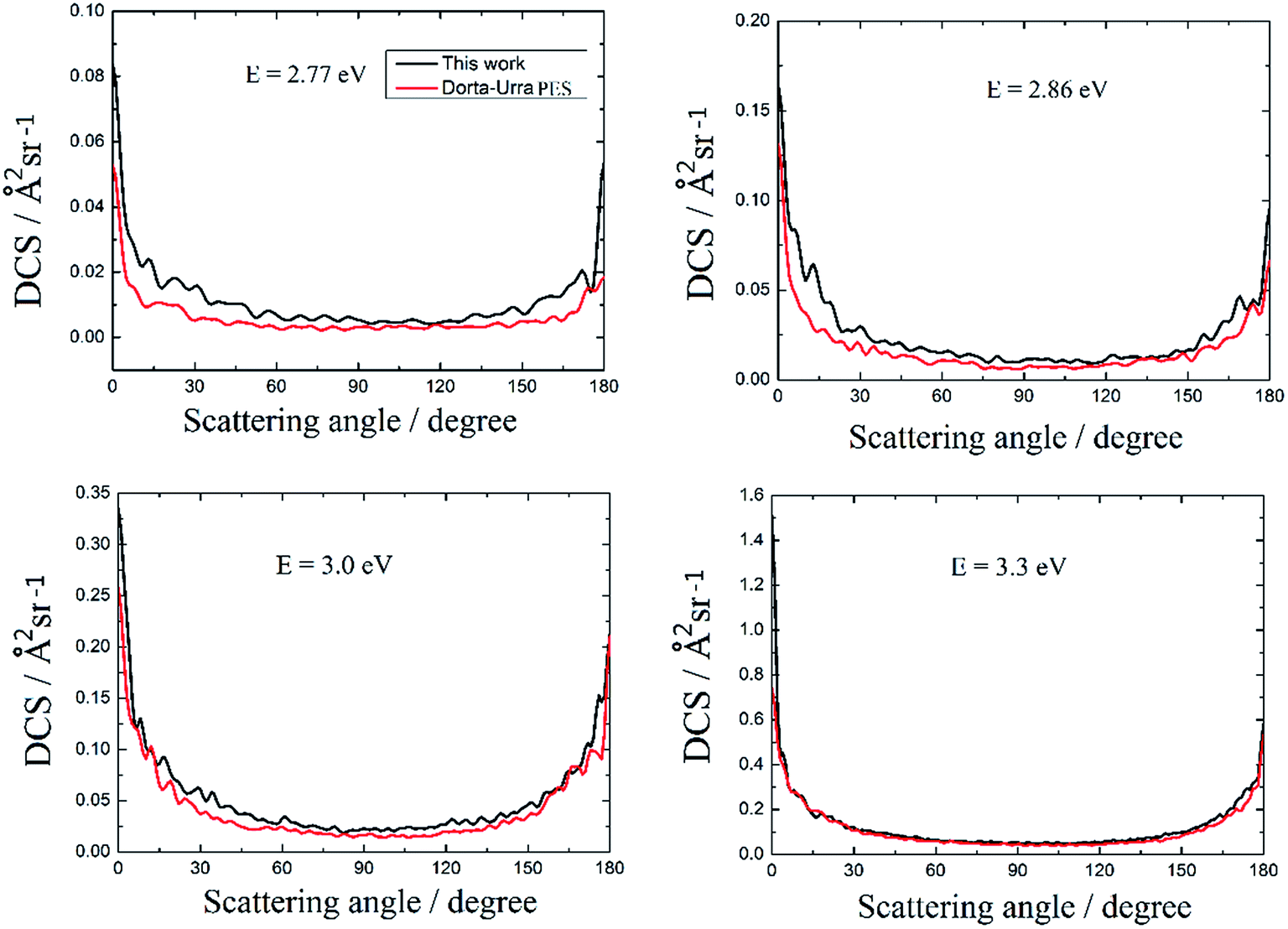
The DCSs of the title reaction based on these two PESs and the four selected collision energies are shown in Fig. 8. The overall shapes of the DCSs are rather similar. All of the peaks are located at two extreme angles (0° and 180°) for the four selected collision energies. The DCSs are symmetrical about π/2 except in the vicinity of the 0° and 180° poles, and this is attributed to the well, which supports long lifetime of the Au+H2 complex, located on the reaction path. Comparing the four panels in Fig. 8, it is noted that the forward scattering peak becomes much higher than the backward scattering peak as the collision energy increases. This implies that more and more products are formed via short-lived complexes with increasing collision energy, which means that the Au+ + H2 → H + Au+H reaction is gradually dominated by the abstraction mechanism with an increase in the collision energy.
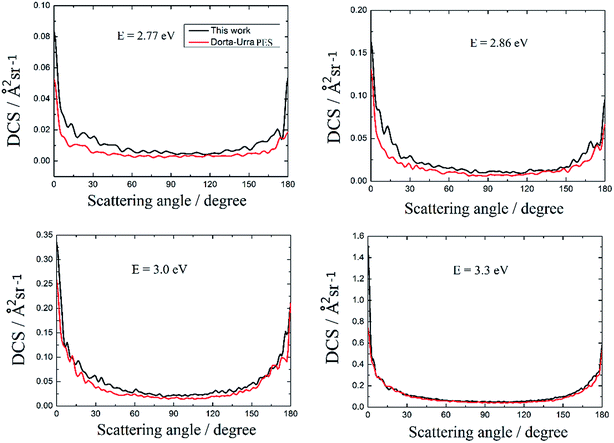 |
| | Fig. 8 The differential cross sections of the Au+ + H2 → H + Au+H reaction at four selected collision energies. | |
VI. Conclusion
In this study, a new AuH2+ PES has been constructed based on a mass of high-level energy points. The ab initio energies were calculated at the MRCI level with Davidson correction. The AVQZ and WCVQZ-PP basis sets were employed for the H and Au atoms, respectively. The two-body and the three-body terms were fitted via the NN method. The PIP was applied in the fitting process to solve the problem of the adaptation of the permutation symmetry. The attributes of the present PES are compared with the Dorta-Urra PES in detail. Both PESs have similar topography in general. In addition, the potential well on present PES is deeper than the one on the Dorta-Urra PES. Dynamics calculations based on these two PESs were performed using the time-dependent quantum wave packet method with a second-order split operator. The dynamics results indicate that the potential well supports the long lifetime complex. The dynamics properties obtained using these two PESs are compared with available theoretical and experimental values. All of the ICSs based on the theoretical calculations are lower than experimental results. However, the ICS obtained using the present PES are closer to the experimental data than that obtained using the Dorta-Urra PES.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 11374045), and the Program for New Century Excellent Talents in University (Grant No. NCET-12-0077).
References
- H. Häkkinen, M. Moseler and U. Landman, Phys. Rev. Lett., 2002, 89, 033401 CrossRef PubMed
 .
. - M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405–408 CrossRef .
- M. L. Kimble, A. W. Castleman, R. Mitrić, C. Burgel and V. Bonačić-Koutecký, J. Am. Chem. Soc., 2003, 126, 2526–2535 CrossRef PubMed .
- H. X. Zhang and X. L. Ding, Chem. Phys., 2016, 475, 69–76 CrossRef CAS .
- S. Bulusu and X. C. Zeng, J. Chem. Phys., 2006, 125, 154303 CrossRef PubMed .
- R. B. King, Z. F. Chen and P. V. R. Schleyer, Inorg. Chem., 2004, 43, 4564–4566 CrossRef CAS PubMed .
- A. Lechtken, C. Neiss, M. M. Kappes and D. Schooss, Phys. Chem. Chem. Phys., 2009, 11, 4344–4350 RSC .
- S. Gilb, P. Weis, F. Furche, R. Ahlrichs and M. M. Kappes, J. Chem. Phys., 2002, 116, 4094–4101 CrossRef CAS .
- H. Häkkinen, R. N. Barnett and U. Landman, J. Phys. Chem. B, 1999, 103, 8814–8816 CrossRef .
- H. Häkkinen, Chem. Soc. Rev., 2008, 39, 1847–1859 RSC .
- H. Auml and K. Landman, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 4, R2287–R2290 Search PubMed .
- M. Guitouguichemerre and G. Chambaud, J. Chem. Phys., 2005, 122, 204325 CrossRef CAS PubMed .
- A. Zanchet, O. Roncero, S. Omar, M. Paniagua and A. Aguado, J. Chem. Phys., 2010, 132, 034301 CrossRef PubMed .
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS .
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS .
- F. Li, C. S. Hinton, M. Citir, F. Liu and P. B. Armentrout, J. Phys. Chem., 2011, 134, 024310 CrossRef PubMed .
- A. Dortaurra, A. Zanchet, O. Roncero, A. Aguado and P. B. Armentrout, J. Chem. Phys., 2011, 135, 091102 CrossRef PubMed .
- A. Dortaurra, A. Zanchet, O. Roncero and A. Aguado, J. Chem. Phys., 2015, 142, 154301 CrossRef PubMed .
- J. C. Yuan, D. H. Cheng and M. D. Chen, RSC Adv., 2014, 4, 36189–36195 RSC .
- J. C. Yuan, D. H. Cheng, Z. G. Sun and M. D. Chen, Mol. Phys., 2014, 112, 2945–2953 CrossRef CAS .
- S. A. Varganov, R. M. Olson, M. S. Gordon, G. Mills and H. Metiu, J. Chem. Phys., 2004, 120, 5169–5175 CrossRef CAS PubMed .
- A. Zanchet, A. Dorta-Urra, O. Roncero, F. Flores, C. Tablero, M. Paniagua and A. Aguado, Phys. Chem. Chem. Phys., 2009, 11, 10122–10131 RSC .
- K. Balasubramanian and M. Z. Liao, J. Phys. Chem., 1989, 93, 89–94 CrossRef CAS .
- H. J. Werner and P. J. Knowles, J. Chem. Phys., 1988, 89, 5803–5814 CrossRef CAS .
- P. J. Knowles and H. J. Werner, Chem. Phys. Lett., 1988, 145, 514–522 CrossRef CAS .
- P. J. Knowles and H. J. Werner, Chem. Phys. Lett., 1985, 115, 259–267 CrossRef CAS .
- H. J. Werner and P. J. Knowles, J. Chem. Phys., 1985, 82, 5053–5063 CrossRef CAS .
- H. J. Werner, P. J. Knowles and G. Knizia, MOLPRO, version 2012.1, a package of ab initio programs Search PubMed .
- S. Manzhos and T. Carrington, J. Chem. Phys., 2006, 125, 084109 CrossRef PubMed .
- J. C. Yuan, D. He and M. D. Chen, Sci. Rep., 2014, 5, 14594 CrossRef PubMed .
- D. He, J. C. Yuan, H. X. Li and M. D. Chen, J. Chem. Phys., 2016, 145, 234312 CrossRef PubMed .
- D. He, J. C. Yuan, H. X. Li and M. D. Chen, Sci. Rep., 2016, 6, 25083 CrossRef CAS PubMed .
- J. C. Yuan, D. He and M. D. Chen, Sci. Rep., 2015, 5, 11732–11739 CrossRef PubMed .
- B. Jiang, J. Li and H. Guo, Int. Rev. Phys. Chem., 2016, 35, 479–506 CrossRef CAS .
- B. Jiang and H. Guo, J. Chem. Phys., 2014, 141, 034109 CrossRef PubMed .
- B. J. Braams and J. M. Bowman, Int. Rev. Phys. Chem., 2009, 28, 577–606 CrossRef CAS .
- S. Y. Lin and H. Guo, J. Chem. Phys., 2008, 129, 124311 CrossRef PubMed .
- W. T. Li, M. D. Chen and Z. G. Sun, Chin. J. Chem. Phys., 2015, 28, 415–425 CrossRef CAS .
- Z. G. Sun and D. H. Zhang, Int. J. Quantum Chem., 2015, 115, 689–699 CrossRef CAS .
- B. Zhao, Z. G. Sun and H. Guo, J. Chem. Phys., 2016, 144, 064104 CrossRef PubMed .
- Z. G. Sun, X. Lin, S. Y. Lee and D. H. Zhang, J. Phys. Chem. A, 2009, 113, 4145–4154 CrossRef CAS PubMed .
- W. Kolos and L. Wolniewicz, J. Chem. Phys., 1965, 43, 2429–2441 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ac and
Maodu Chen
ac and
Maodu Chen
 represents the three-body term. f(R) is a switch function, which is used to better describe the PES in the asymptotic area, and it can be presented as:
represents the three-body term. f(R) is a switch function, which is used to better describe the PES in the asymptotic area, and it can be presented as: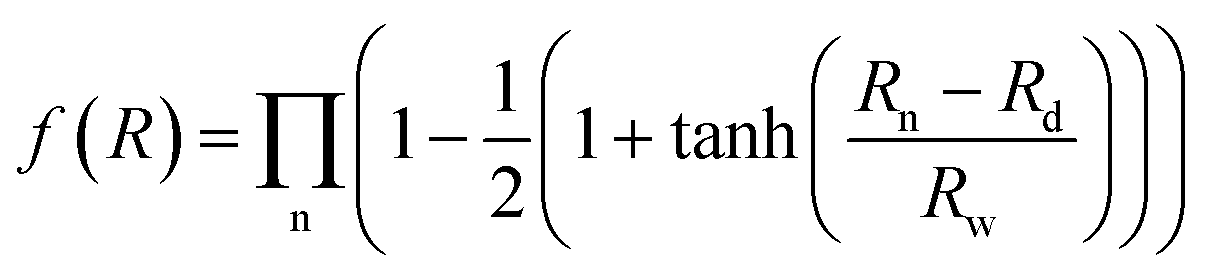
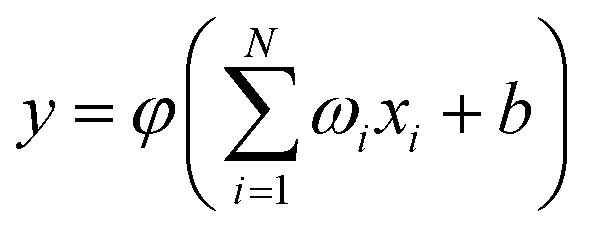

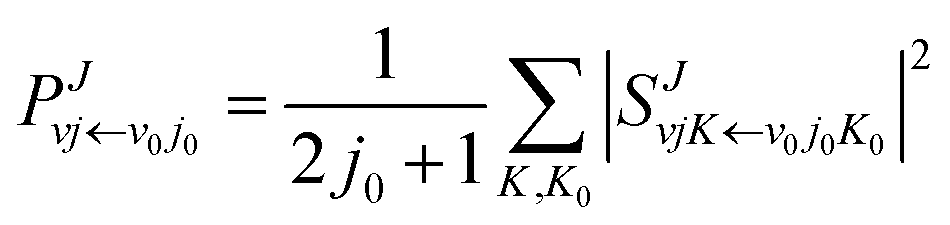

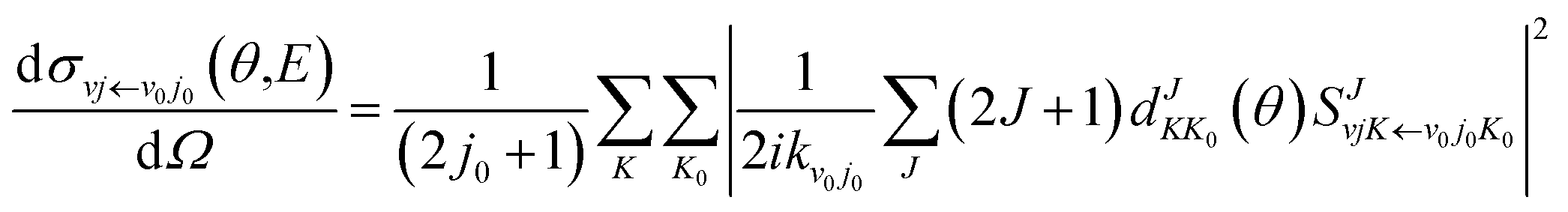
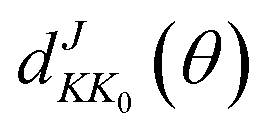 is used for calculating the reduced rotation matrix element. The numerical parameters used in the TDWP calculation are listed in Table 1.
is used for calculating the reduced rotation matrix element. The numerical parameters used in the TDWP calculation are listed in Table 1.