
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNiobium-doped titanium dioxide on a functionalized carbon supported palladium catalyst for enhanced ethanol electro-oxidation†
Weichuan Xua,
Litao Yana,
Huiqiang Wangb,
Steven Liawa and
Hongmei Luo *a
*a
aDepartment of Chemical and Materials Engineering, New Mexico State University, New Mexico 88003, USA. E-mail: hluo@nmsu.edu
bCollege of Mechanical and Electrical Engineering, Agricultural University of Hebei, Baoding 071001, China
First published on 11th July 2017
Abstract
Pd nanoparticles anchored on Nb-doped TiO2 with functionalized carbon support (denoted as Pd/Nb–TiO2–C) is synthesized through a controllable hydrolysis and impregnation method. The as-synthesized catalyst is characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM). Pd nanoparticles exhibit a uniform distribution with an average particle size of 3 nm. The electrochemical performance is tested by cyclic voltammetry (CV) and chronoamperometry (CA). Compared with Pd supported by functionalized carbon (Pd/C), Pd/Nb–TiO2–C demonstrates 15.7% higher metallic Pd content, 23% higher electrochemical active surface area, 75% higher current density in ethanol electro-oxidation, 5% higher durability, and better tolerance of carbonaceous species. The performance enhancement is attributed to the increased conductivity from Nb-doping and the synergistic effect between Pd and TiO2.
1. Introduction
The direct ethanol fuel cell (DEFC) is a clean energy generation device in terms of environmental benignity compared with fossil fuel based energy sources. As a type of direct alcohol fuel cell, DEFCs have advantages over other renewable fuel sources. Ethanol, with a relatively higher H/C ratio in its molecules, presents higher energy density (26.4 MJ kg−1) compared with methanol (19.7 MJ kg−1). Fewer health issues are recorded for ethanol in either electro-oxidation processes or daily contact.1 Large quantities of ethanol can be obtained from biomass fermentation,2 giving it incomparably wide availability. These factors ensure ethanol as a suitable fuel in clean renewable energy devices. Among all the catalysts for ethanol electro-oxidation, noble metals i.e. platinum (Pt),3–5 palladium (Pd),6,7 ruthenium (Ru),8,9 rhodium (Rh)9,10 and iridium (Ir)11,12 along with their combinations have been widely investigated because of their superior performance. Compared with Pt, Pd has higher abundance and shows much more active electrocatalytic ability in basic solutions due to its higher oxophilicity and relatively inert nature.13,14 Bi-metallic and tri-metallic catalyst usually have higher electrochemical activity and better stability over single noble metal because of their increased ability of breaking C–C bonds and its blocking effect of further oxidation of poisoning species.15To fully utilize and maintain their catalytic ability of precious metals, catalyst support with large surface area is required to uniformly disperse catalysts. Carbon black has been identified as a classic alcohol cell catalyst support due to its high electro-conductivity16 and large surface area.17 Typically, functionalized carbon is acid-treated carbon, where oxidative functional groups18 are introduced, and this support can further increase solubility and density of catalyst reaction sites.16 Carbon alone however suffers from fast degradation and low electrochemical stability, especially at high potential in alkaline media. Carbon oxidation reaction in high pH environment is shown below:16
| C + 6OH− → CO32− + 3H2O + 4e− | (1) |
Therefore, the search of a catalyst support with corrosion resistance in electrolyte, during alcohol electro-oxidation, is demanded. To tackle the corrosion problem, transition metal oxides such as TiO2,19–21 WO3,22–24 and V2O5 (ref. 25–27) offer high stability in both acidic and alkaline media. TiO2 stands out as a dominating candidate for electro-catalyst support due to their superiorities in many aspects. Corrosion resistant TiO2 can dramatically increase cycle numbers and catalyst efficiency in the long run. Also, TiO2-based material is noted for its contribution to strong metal-support interaction (SMSI) and three-phase junction,28 i.e. junction of carbon, TiO2 and Pd/Pt. Pt immobilized on high niobium content rutile titania and carbon nanotubes shows smaller particle size, narrower and more uniform distribution.5 Produced by either photoreduction method29 or hydrothermal synthesis,30 the three-phase junction effect on the electrochemical performance is beneficial owing to more catalyst exposure, less agglomeration of catalyst nanoparticles, and extended catalyst life span. However, TiO2 has relatively low surface area31 and low electro-conductivity.32 One way to increase its conductivity is utilizing sub-stoichiometric TiO2−x, but it can be oxidized to TiO2 in fuel cell operation condition.33,34 Another method to lower impedance is to dope with a donor type metal.35 Typically, Nb has one free electron in the outer shell when it is doped into TiO2 lattice.16,36 It has been reported that doping 10 at% Nb into TiO2 would increase its conductivity by ∼1600 fold.16 Besides, Nb-doping decreases the particle size of TiO2 and increases its surface area. However, increasing the doping amount by over 30% would damage the crystal structure of TiO2, thus reducing the stability of catalyst support.16 We herein report Nb-doped TiO2 on functionalized carbon supported Pd catalyst for electro-oxidation of ethanol in alkaline media. As compared with the functionalized carbon supported Pd catalyst, it shows higher ethanol electro-oxidation current density and higher durability, thanks from the increased conductivity from Nb-doping and the synergistic effect between Pd and TiO2.
2. Experimental
2.1 Material synthesis
2.2 Materials characterization
Phase and crystal structure of Pd/Nb–TiO2/C and Pd/C were analyzed with X-ray diffraction (XRD) on a MINIFlex II, Rigaku, using filtered Cu Kα radiation (λ = 1.54005 Å) at 30 kV and 15 mA. The morphology was investigated by transmission electron microscopy (TEM) on H-7650, Hitachi High-Technologies Corp. The TEM used an accelerating voltage at 80 kV with an integrated, side-mounted CCD digital image camera system. EDS mapping was conducted on a S–3400N Type II scanning electron microscope (Hitachi High-Technologies Corp.). X-Ray photoelectron spectroscopy (XPS) was tested on an AXIS ULTRA X-ray Photoelectron Spectrometer with Al monochromatic X-ray by Kratos Analytical. C 1s peak was used as a reference.2.3 Electrochemical characterization
The glassy carbon electrode was polished with 5, 0.3, and 0.05 μm Al2O3 solution (Allied) in sequence. It was then cleaned by sonication and dried with compressed air. To prepare catalyst ink, 10 mg Pd/Nb–TiO2–C was dissolved in 2 ml 0.05 wt% Nafion solution, which was ∼5 wt% Nafion® 117 solution (Sigma-Aldrich) diluted 100 times by DI water. After sonication, 20 μl catalyst ink was dropped onto the glassy carbon electrode of 5 mm diameter with micropipette. The loading of Pd for both catalysts on the electrode is around 0.035 mg cm−2. The electrode was dried at room temperature overnight. Electrochemical tests were carried out with MSR Rotator, PINE research and CHI 600 electrochemical workstation. Three electrodes setup, i.e. working electrode, Pt coil counter electrode and silver chloride Ag/AgCl (4 M) reference electrode were used at room temperature.3. Results and discussion
3.1 Structure and morphology
Fig. 1 shows the XRD pattern of the as-synthesized Pd/Nb–TiO2–C. Peaks of face centered cubic Pd and Anatase TiO2 can be indexed by PDF#98-000-0344 and PDF#98-000-0081, respectively. No peaks of niobium oxides or metallic Nb can be found. Lattice parameters of TiO2 are a = 3.798 Å and c = 9.547 Å. Since the ionic size of Nb4+ is 82 pm, 10.1% bigger than that of Ti4+, the enlarged lattice parameters (+0.34% for a and +0.35% for c) compared with ref. 37 indicate Nb incorporation into TiO2. Pd (111), (200) and (220) surfaces reveal face centered cube (fcc) structure. Using Sherrer's equation on (111) plane shows that Pd in Pd/Nb–TiO2–C has a crystallite size of ∼6.0 nm, with lattice parameter of a = 3.88 Å, close to 3.86 Å in ref. 38. | ||
| Fig. 1 XRD pattern of Pd/Nb–TiO2–C and PDF cards of anatase TiO2 and fcc Pd. | ||
Morphology and Pd particle size were investigated by TEM. Fig. 2 shows the TEM images of Nb–TiO2–C support, Pd/Nb–TiO2–C, and Pd/C. In Fig. 2(a) Nb–TiO2–C shows multilayer structure with thickness around 40 nm. It can be seen that the Pd nanoparticles exhibit uniform dispersion on both supports Nb–TiO2–C and C, with average diameter of 2.8 nm and 3.2 nm (14% bigger), respectively. The finer Pd nanoparticle formation in the former sample can be explained by the three-phase junction, (three-phase means Pd, TiO2 and C) which prevents the catalyst from agglomeration.39 HRTEM, Energy-dispersive X-ray spectroscopy (EDS) and elemental mapping of Pd/Nb–TiO2–C were conducted to further investigate the microstructure and element distribution, as shown in Fig. S1–S3.† The EDS mapping demonstrates that Pd, Nb, Ti, O, and C elements were uniformly distributed in the sample and the weight percentages of elements are listed in Table 1. The numbers are consistent with theoretical values of 6.7 wt% Pd and 33 wt% Nb–TiO2. Metal oxide with carbon not only facilitate Pd deposition on the catalyst support during preparation,29 but also hinder catalyst migration in electrochemical reactions.40
 | ||
| Fig. 2 TEM images of (a) Nb–TiO2–C support, (b) Pd/Nb–TiO2–C, and (c) Pd/C (scale bars: 5 nm, 20 nm, and 20 nm, respectively). | ||
| Element line | Element wt% | wt% error |
|---|---|---|
| C K | 60.77 | ±6.77 |
| O K | 14.37 | ±0.83 |
| Ti K | 13.26 | ±0.72 |
| Nb L | 4.47 | ±0.45 |
| Pd L | 7.13 | ±1.04 |
X-ray photoelectron spectroscopy was carried out to evaluate the functional groups on carbon and to better understand the synergistic effect between TiO2 and Pd. Fig. 3(a) is C 1s XPS spectrum with peak fitting in Pd/Nb–TiO2–C. Peaks from the low to the high binding energies can be attributed to C–C sp2, C–C sp3, C–OH, C–O–C, 〉C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and COOH functional groups.41 The hydrophilic groups not only increase solubility, but also give more anchoring and reactive sites for the catalyst. Fig. 3(b and c) show XPS spectra of Pd 3d of Pd/Nb–TiO2–C and Pd/C, each deconvoluted into three pairs of doublets, i.e. metallic Pd0, Pd2+ and Pd4+ species. Pd/C shows 25.1% Pd0 at 335.0 eV, 13.4% Pd2+ at 335.8 eV and 61.4% Pd4+ at 336.6 eV, while 40.8% Pd0, 27.7% Pd2+ and 31.4% Pd4+ are found for Pd/Nb–TiO2–C. The 30% decrease of Pd4+ and 15.7% increase of metallic Pd0 shift Pd 3d5/2 peaks to weaker binding energy levels, resulting in the improved metallic catalyst surface area and the decreased adsorption of carbonaceous poisoning chemicals on catalyst surface.42
O and COOH functional groups.41 The hydrophilic groups not only increase solubility, but also give more anchoring and reactive sites for the catalyst. Fig. 3(b and c) show XPS spectra of Pd 3d of Pd/Nb–TiO2–C and Pd/C, each deconvoluted into three pairs of doublets, i.e. metallic Pd0, Pd2+ and Pd4+ species. Pd/C shows 25.1% Pd0 at 335.0 eV, 13.4% Pd2+ at 335.8 eV and 61.4% Pd4+ at 336.6 eV, while 40.8% Pd0, 27.7% Pd2+ and 31.4% Pd4+ are found for Pd/Nb–TiO2–C. The 30% decrease of Pd4+ and 15.7% increase of metallic Pd0 shift Pd 3d5/2 peaks to weaker binding energy levels, resulting in the improved metallic catalyst surface area and the decreased adsorption of carbonaceous poisoning chemicals on catalyst surface.42
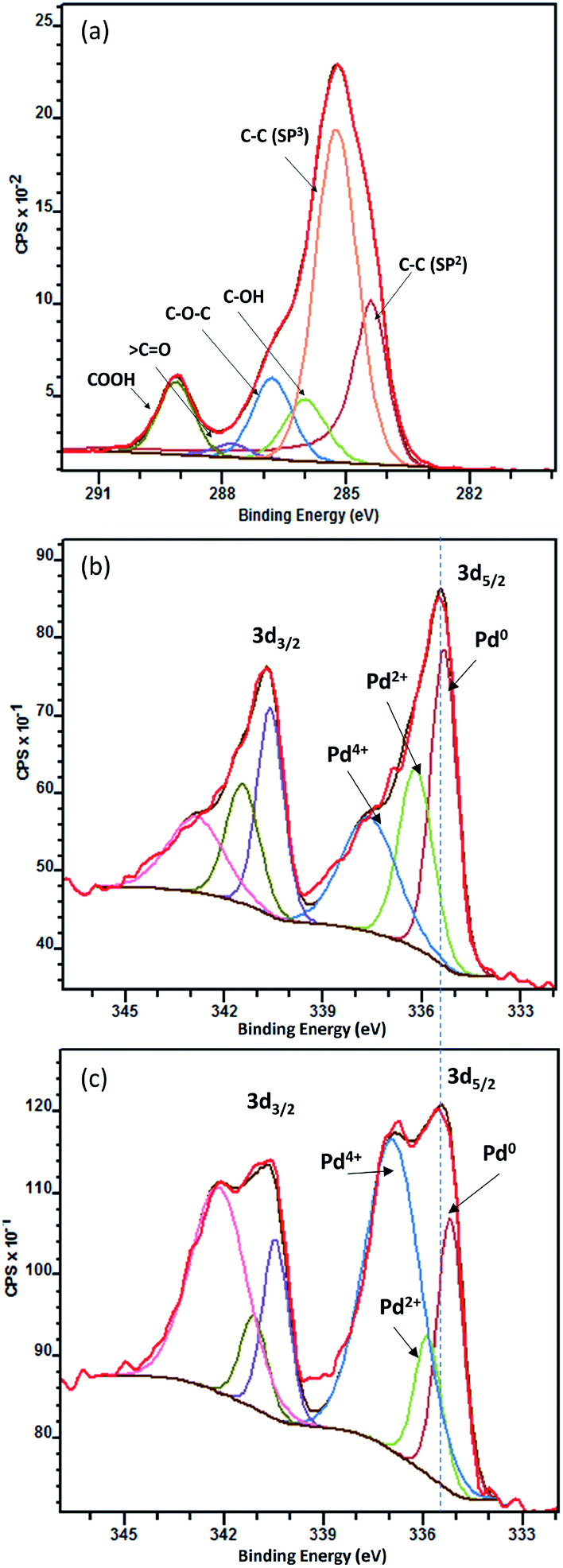 | ||
| Fig. 3 XPS of C 1s of Pd/Nb–TiO2–C (a), Pd 3d of Pd/Nb–TiO2–C (b) and Pd/C (c). | ||
3.2 Electrochemical performance
Cyclic voltammetry (CV) was performed in 1 M KOH solution for Pd/Nb–TiO2–C and Pd/C. The similar CV patterns in Fig. 4(a) for both samples show typical Pd activity in alkaline media.43 In the cathodic scan, peak at −0.35 V (vs. Ag/AgCl) reflects the reduction of palladium oxide into Pd; the stronger reduction peak for Pd/Nb–TiO2–C suggests higher Pd catalyst activity and larger electrochemical active surface area (ECSA) due to the reduced Pd oxidation state and smaller particle size of catalyst, consistent with XPS analysis and TEM observation. Increased ECSA is fundamental for high power energy device considering density of reaction sites. The small peaks at −0.18 V for both catalysts could be attributed to the reduction of dissolved oxygen by carbon. With the small reduction peak taken into consideration, electrochemical active surface areas of Pd/C and Pd/Nb–TiO2–C are calculated by the charge integration of the reduction peak in CV as 0.96 mC cm−2 and 1.17 mC cm−2, respectively.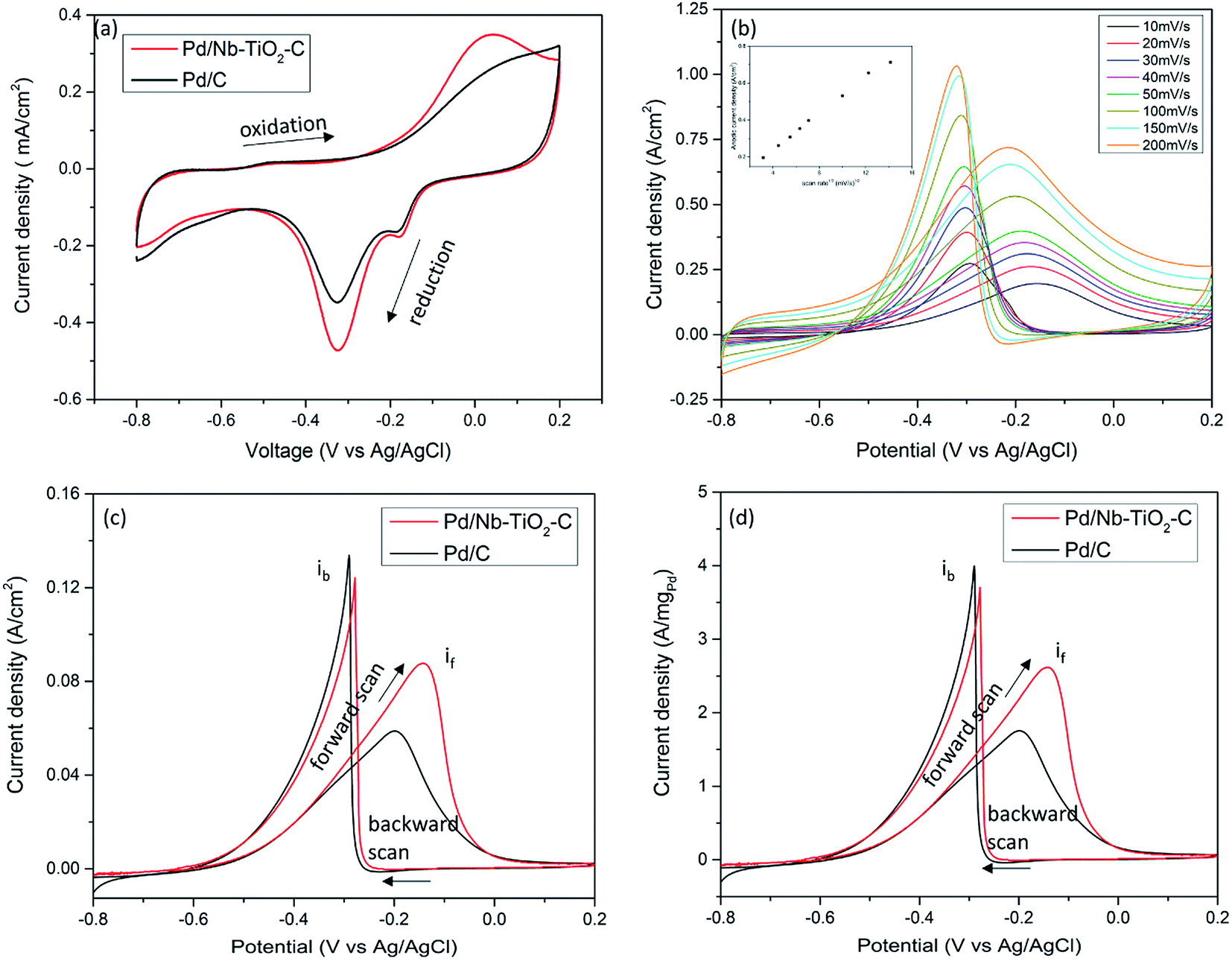 | ||
| Fig. 4 (a) CV of Pd/Nb–TiO2–C and Pd/C in 1 M KOH with a scan rate of 20 mV s−1; (b) CV of Pd/Nb–TiO2–C in 1 M KOH and 1 M ethanol solution with different scan rates. Insert of (b) is anodic scan current density vs. scan rate1/2; (c) ECSA and electrode area normalized CV of Pd/Nb–TiO2–C and Pd/C in 1 M KOH and 1 M ethanol solution with a scan rate of 20 mV s−1; and (d) ECSA and palladium mass normalized CV of Pd/Nb–TiO2–C and Pd/C in 1 M KOH and 1 M ethanol solution with a scan rate of 20 mV s−1. | ||
To unveil ethanol electro-oxidation activity, CVs for catalysts in 1 M KOH in ethanol were tested. As a comparison, CVs of functionalized carbon, Nb–TiO2–C support and ethanol concentration effect on Pd/Nb–TiO2–C are presented in Fig. S4.† No oxidation peak was observed in the low concentration of ethanol of 0.001 M. Peaks can be identified in the 0.01 M ethanol and peaks are of similar shape in 0.1, 1 and 2 M ethanol solutions, indicative of practical ethanol electro-oxidation range from 0.1 to 2 M. In the following experiments, 1 M KOH and 1 M ethanol solution was chosen. First, scan rate influence of ethanol electro-oxidation was investigated on Pd/Nb–TiO2–C catalyst. As shown in Fig. 4(b), both the anodic current density and cathodic current density increase when the scan rate increases from 10 to 200 mV s−1. The insert figure demonstrates a linear relationship between the anodic current density and the square root of scan rate, indicative of diffusion controlled reaction kinetics. Further ethanol electro-oxidation results are shown in ECSA and electrode area normalized CV in Fig. 4(c) and ECSA and Pd mass normalized CV in Fig. 4(d). In the forward scan, ethanol chemisorbed on the catalyst surface is oxidized, whereas in the reverse sweep, oxidation of carbonaceous species occurs. The ratio of the forward sweep peak current if to the reverse scan peak current ib represents the tolerance of catalyst poisoning, and a higher if/ib indicates more complete oxidation of ethanol and less accumulation of carbonaceous chemicals.43 Though with a 0.04 V positively shifted ethanol oxidation peak, Pd/Nb–TiO2–C shows if of 0.088 A cm−2, 47% higher than 0.06 A cm−2 of Pd/C. The massive increase in ethanol oxidation current density is mainly explained by 23% higher ECSA of metallic Pd catalyst and the synergistic effect between Pd and Nb–TiO2. Strong metal-support interaction is responsible for higher metallic catalyst content and immobilized catalyst during electro-oxidation. Besides, the if/ib ratio of Pd/Nb–TiO2–C reaches 0.7, while the value of Pd/C is only 0.44. The ratio raise can be explained by the reduced noble metal bonding energy with adsorbed carbonaceous chemicals,44 which is the reason of 0.02 V positively shifted backward scan peak in Pd/Nb–TiO2–C. A lower if/ib ratio in Pd/C causes accumulation of poisoning species in electrolyte, leading to inefficient oxidation of irrelevant organics, degradation of catalyst, power loss and energy waste. Due to varying operating conditions such as catalyst content and electrolyte concentration, it is difficult to directly compare if/ib ratio of catalysts among different works. Based on similar Pd/C catalysts, comparing the if/ib ratio could help to eliminate effects of interfering factors. In our work, Pd/Nb–TiO2–C has ∼60% if/ib increase, much higher than 23% from Pd on carbon-doped TiO2,43 46% from Ni@Pd core–shell nanoparticles on multi-walled carbon nanotubes,45 and few works with decreased if/ib.16,46 The high if/ib ratio increase could be attributed to the synergistic effect with high TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd molar ratio around 6.2:1 in our work, which reduces Pd2+ and Pd4+ dramatically. In Fig. 4(d), Pd/Nb–TiO2–C reaches 2.6 A mgPd−1 in the forward scan, 45% higher than that of Pd/C, which is consistent with the result in Fig. 4(c), and the current density scale matches that in literature.43 It is noted that the same origin was reported for the forward oxidation peak and backward oxidation peak by methanol electro-oxidation reaction with Pt-based catalysts in acidic media.47 In this work ethanol electro-oxidation with Pd-based catalysts was conducted in alkaline media. Since similar principles might apply even in different conditions, the origin of the forward scan current and backward scan current in ethanol and alkaline media needs detailed investigation.
:Pd molar ratio around 6.2:1 in our work, which reduces Pd2+ and Pd4+ dramatically. In Fig. 4(d), Pd/Nb–TiO2–C reaches 2.6 A mgPd−1 in the forward scan, 45% higher than that of Pd/C, which is consistent with the result in Fig. 4(c), and the current density scale matches that in literature.43 It is noted that the same origin was reported for the forward oxidation peak and backward oxidation peak by methanol electro-oxidation reaction with Pt-based catalysts in acidic media.47 In this work ethanol electro-oxidation with Pd-based catalysts was conducted in alkaline media. Since similar principles might apply even in different conditions, the origin of the forward scan current and backward scan current in ethanol and alkaline media needs detailed investigation.
Stability of Pd/C and Pd/Nb–TiO2–C was tested by chronoamperometry in a solution containing 1 M KOH and 1 M ethanol at −0.2 V vs. Ag/AgCl. Results indicate that Pd/C lost 50% initial current density after 150 s, while Pd/Nb–TiO2–C lasted another 100 s before it lost half of its catalytic ability. Besides, Pd/Nb–TiO2–C catalyst could possibly degrade faster than Pd/C considering much higher initial current density at −0.2 V in Fig. 5(a), because intensive surface reactions usually cause severe particle aggregation on the working electrode. After 3000 s, ∼94% ethanol oxidation current lost for Pd/C catalyst in Fig. 5(b), and ∼5% higher current retention was observed for Pd/Nb–TiO2–C. The better reserved activity of Pd/Nb–TiO2–C after long period of reaction can be attributed to the synergistic effect between the catalyst and the support. It was assumed that the size of Pd nanoparticles gradually grows after reactions.48 Loss of electrochemical active surface area during reaction and dissolution of active materials are two direct reasons for the slow oxidation rate. It is inferred that TiO2 and functionalized carbon around Pd nanoparticles prevent catalyst from agglomeration, which helps to preserve Pd catalytic ability in the durability test.
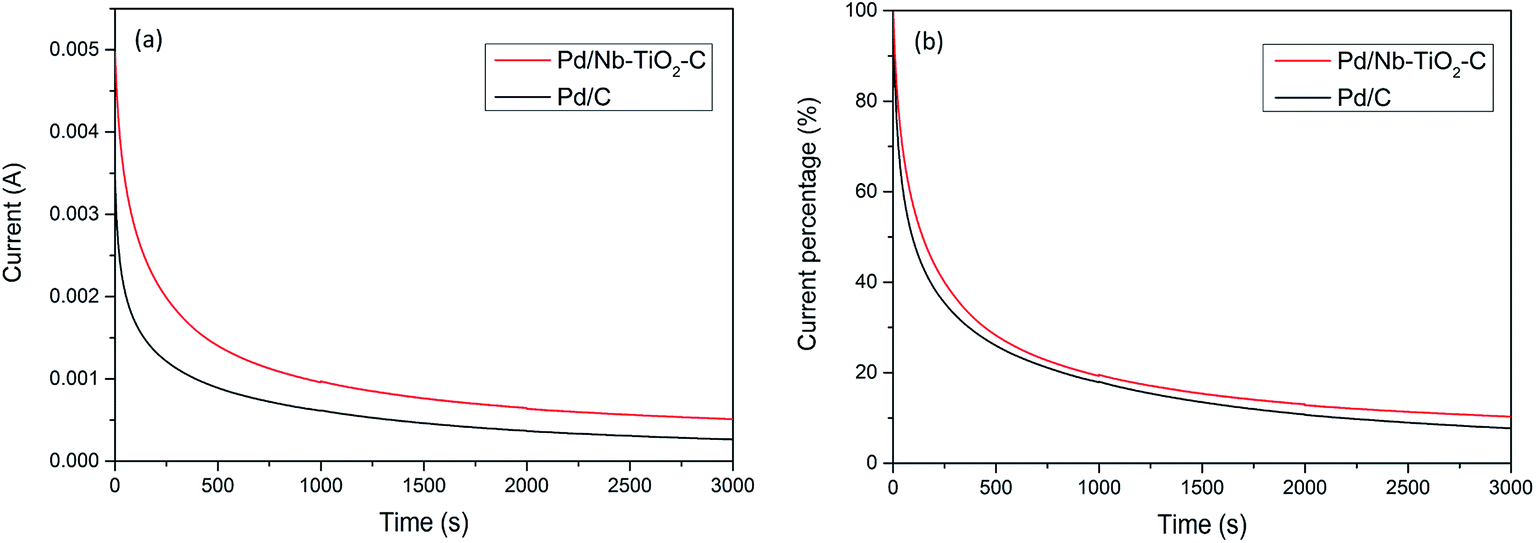 | ||
| Fig. 5 Chronoamperometry (CA) of Pd/Nb–TiO2–C and Pd/C in 1 M KOH and 1 M ethanol solution at −0.2 V for 3000 seconds for (a) current–time curve and (b) current percentage–time curve. | ||
4. Conclusions
In this work, Nb-doped TiO2 on functionalized carbon supported Pd catalyst is synthesized and tested against Pd/C. Pd/Nb–TiO2–C catalyst shows 14% smaller Pd particle size, 23% higher electrochemical active surface area, 75% higher ethanol electro-oxidation current density and 5% more catalyst stability after chronoamperometry. The synergistic effect including electron transfer from TiO2 to Pd and the three-phase junction by C, TiO2 and Pd account for higher metallic catalyst ECSA, finer catalyst size, and less sample degradation leading to better electrochemical performance.Acknowledgements
Dr Luo thanks the support from New Mexico EPSCoR (NSF-1301346) and the USDA National Institute of Food and Agriculture, HSI Collaboration: Integrating Food Science/Engineering and Education Network (IFSEEN, award number: 2015-38422-24059).References
- D. Collister, G. Duff, W. Palatnick, P. Komenda, N. Tangri and J. Hingwala, Am. J. Kidney Dis., 2017, 69, 696–700 CrossRef PubMed.
- C. Toquero and S. Bolado, Bioresour. Technol., 2014, 157, 68–76 CrossRef CAS PubMed.
- W. Zhuang, L. He, J. Zhu, R. An, X. Wu, L. Mu, X. Lu, L. Lu, X. Liu and H. Ying, Int. J. Hydrogen Energy, 2015, 40, 3679–3688 CrossRef CAS.
- Z. Bedolla-Valdez, Y. Verde-Gomez, A. Valenzuela-Muniz, Y. Gochi-Ponce, M. Oropeza-Guzmán, G. Berhault and G. Alonso-Núñez, Electrochim. Acta, 2015, 186, 76–84 CrossRef CAS.
- L. Yan, K. Huang, Y. Chen and Y. Xing, ECS Electrochem. Lett., 2014, 3, F27–F29 CrossRef CAS.
- C. Xu, L. Cheng, P. Shen and Y. Liu, Electrochem. Commun., 2007, 9, 997–1001 CrossRef CAS.
- C. Xu, H. Wang, P. K. Shen and S. P. Jiang, Adv. Mater., 2007, 19, 4256–4259 CrossRef CAS.
- N. Kakati, J. Maiti, S. H. Lee, S. H. Jee, B. Viswanathan and Y. S. Yoon, Chem. Rev., 2014, 114, 12397–12429 CrossRef CAS PubMed.
- C.-C. Kung, P.-Y. Lin, Y. Xue, R. Akolkar, L. Dai, X. Yu and C.-C. Liu, J. Power Sources, 2014, 256, 329–335 CrossRef CAS.
- I. A. Rutkowska, A. Wadas and P. J. Kulesza, Electrochim. Acta, 2016, 210, 575–587 CrossRef CAS.
- E. J. Lim, Y. Kim, S. M. Choi, S. Lee, Y. Noh and W. B. Kim, J. Mater. Chem. A, 2015, 3, 5491–5500 CAS.
- V. Baglio, D. Sebastián, C. D'Urso, A. Stassi, R. Amin, K. El-Khatib and A. Aricò, Electrochim. Acta, 2014, 128, 304–310 CrossRef CAS.
- X. Fang, L. Wang, P. K. Shen, G. Cui and C. Bianchini, J. Power Sources, 2010, 195, 1375–1378 CrossRef CAS.
- Y. Wang, S. Zou and W.-B. Cai, Catalysts, 2015, 5, 1507–1534 CrossRef CAS.
- J. Tayal, B. Rawat and S. Basu, Int. J. Hydrogen Energy, 2012, 37, 4597–4605 CrossRef CAS.
- S. T. Nguyen, Y. Yang and X. Wang, Appl. Catal., B, 2012, 113, 261–270 CrossRef.
- P. A. Delgado, J. P. Brutman, K. Masica, J. Molde, B. Wood and M. A. Hillmyer, J. Appl. Polym. Sci., 2016, 133, 43926 CrossRef.
- A. Bhatnagar, W. Hogland, M. Marques and M. Sillanpää, Chem. Eng. J., 2013, 219, 499–511 CrossRef CAS.
- L. Assaud, N. Brazeau, M. K. Barr, M. Hanbücken, S. Ntais, E. A. Baranova and L. Santinacci, ACS Appl. Mater. Interfaces, 2015, 7, 24533–24542 CAS.
- H. Hua, C. Hu, Z. Zhao, H. Liu, X. Xie and Y. Xi, Electrochim. Acta, 2013, 105, 130–136 CrossRef CAS.
- P. C. Sahoo, K. Kim, J. H. Lee, J.-I. Han and Y.-K. Oh, ACS Sustainable Chem. Eng., 2015, 3, 1764–1770 CrossRef CAS.
- D.-Y. Zhang, Z.-F. Ma, G. Wang, K. Konstantinov, X. Yuan and H.-K. Liu, Electrochem. Solid-State Lett., 2006, 9, A423–A426 CrossRef CAS.
- X. He, C. Hu, Q. Yi, X. Wang, H. Hua and X. Li, J. Electrochem. Soc., 2013, 160, F566–F572 CrossRef CAS.
- H. Wang and Q. Ma, J. Electrochem. Soc., 2014, 161, F1202–F1207 CrossRef.
- P. Justin and G. R. Rao, Catal. Today, 2009, 141, 138–143 CrossRef CAS.
- J. Datta, A. Dutta and M. Biswas, Electrochem. Commun., 2012, 20, 56–59 CrossRef CAS.
- C. Xia and M. Liu, Adv. Mater., 2002, 14, 521 CrossRef CAS.
- B. Y. Xia, H. B. Wu, J. S. Chen, Z. Wang, X. Wang and X. W. D. Lou, Phys. Chem. Chem. Phys., 2012, 14, 473–476 RSC.
- Y. Qu, Y. Gao, F. Kong, S. Zhang, L. Du and G. Yin, Int. J. Hydrogen Energy, 2013, 38, 12310–12317 CrossRef CAS.
- X. Wang, Y. Li, S. Liu and L. Zhang, Nanomaterials, 2016, 6, 40 CrossRef PubMed.
- H. Yoshitake, T. Sugihara and T. Tatsumi, Chem. Mater., 2002, 14, 1023–1029 CrossRef CAS.
- N. Yamada, T. Hitosugi, N. L. H. Hoang, Y. Furubayashi, Y. Hirose, T. Shimada and T. Hasegawa, Jpn. J. Appl. Phys., 2007, 46, 5275 CrossRef CAS.
- G. Chen, S. R. Bare and T. E. Mallouk, J. Electrochem. Soc., 2002, 149, A1092–A1099 CrossRef CAS.
- H. Chhina, S. Campbell and O. Kesler, J. Electrochem. Soc., 2009, 156, B1232–B1237 CrossRef CAS.
- S. Wang, L. Pan, J.-J. Song, W. Mi, J.-J. Zou, L. Wang and X. Zhang, J. Am. Chem. Soc., 2015, 137, 2975–2983 CrossRef CAS PubMed.
- L. Yan, X. Rui, G. Chen, W. Xu, G. Zou and H. Luo, Nanoscale, 2016, 8, 8443–8465 RSC.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- W. P. Davey, Phys. Rev., 1925, 25, 753 CrossRef CAS.
- S. Maheswari, P. Sridhar and S. Pitchumani, Electrochem. Commun., 2013, 26, 97–100 CrossRef CAS.
- Y. Zhao, Y. Wang, J. Zang, J. Lu and X. Xu, Int. J. Hydrogen Energy, 2015, 40, 4540–4547 CrossRef CAS.
- L. Zhang, L. Ji, P.-A. Glans, Y. Zhang, J. Zhu and J. Guo, Phys. Chem. Chem. Phys., 2012, 14, 13670–13675 RSC.
- X. Yue, W. Yang, X. Liu, Y. Wang, C. Liu, Q. Zhang and J. Jia, Electrochim. Acta, 2015, 174, 667–671 CrossRef CAS.
- Y.-H. Qin, Y. Zhuang, R.-L. Lv, T.-L. Wang, W.-G. Wang and C.-W. Wang, Electrochim. Acta, 2015, 154, 77–82 CrossRef CAS.
- Q. Lv, M. Yin, X. Zhao, C. Li, C. Liu and W. Xing, J. Power Sources, 2012, 218, 93–99 CrossRef CAS.
- Y. Zhao, X. Yang, J. Tian, F. Wang and L. Zhan, Int. J. Hydrogen Energy, 2010, 35, 3249–3257 CrossRef CAS.
- J. Zhu, X. Zhao, M. Xiao, L. Liang, C. Liu, J. Liao and W. Xing, Carbon, 2014, 72, 114–124 CrossRef CAS.
- A. M. Hofstead-Duffy, D. J. Chen, S. G. Sun and Y. J. Tong, J. Mater. Chem., 2012, 22, 5205–5208 RSC.
- F. Hu, F. Ding, S. Song and P. K. Shen, J. Power Sources, 2006, 163, 415–419 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HRTEM, energy-dispersive X-ray spectroscopy, SEM with EDS mapping of Pd/Nb–TiO2–C. CV of functionalized carbon and niobium-doped titanium dioxide on functionalized carbon (Nb–TiO2–C) in 1 M KOH and 1 M ethanol solution. The ethanol electro-oxidation by Pd/Nb–TiO2–C in 1 M KOH solution with different ethanol concentrations with a scan rate of 20 mV s−1. See DOI: 10.1039/c7ra05208a |
| This journal is © The Royal Society of Chemistry 2017 |