
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved overall hydrogen storage properties of a CsH and KH co-doped Mg(NH2)2/2LiH system by forming mixed amides of Li–K and Cs–Mg†
Jiaxun Zhanga,
Yiqi Wanga,
Min Zhanga,
Zihan Lenga,
Mingxia Gao a,
Jianjiang Hub,
Yongfeng Liu*ac and
Hongge Pana
a,
Jianjiang Hub,
Yongfeng Liu*ac and
Hongge Pana
aState Key Laboratory of Silicon Materials, Key Laboratory of Advanced Materials and Applications for Batteries of Zhejiang Province, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: mselyf@zju.edu.cn; Fax: +86 571 87952615
bLaboratory for Energetics and Safety of Solid Propellants, Hubei Institute of Aerospace Chemotechnology, Xiangyang 441003, China
cKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, China
First published on 12th June 2017
Abstract
A CsH and KH co-doped Mg(NH2)2/2LiH composite was prepared with a composition of Mg(NH2)2/2LiH–(0.08 − x)CsH–xKH, and the hydrogen storage characteristics was systematically investigated. The results showed that the presence of KH further improved the reaction thermodynamics and kinetics of hydrogen storage in a CsH-containing Mg(NH2)2/2LiH system. A sample with 0.04 mol CsH and 0.04 mol KH had optimal hydrogen storage performance; its dehydrogenation could proceed at 130 °C and hydrogenation at 120 °C with 4.89 wt% of hydrogen storage capacity. At 130 °C, a 25-fold increase in the dehydrogenation rate was achieved for the CsH and KH co-doped sample. More importantly, the CsH and KH co-doped sample also had good cycling stability because more than 97% of the hydrogen storage capacity (4.34 wt%) remained for the Mg(NH2)2/2LiH–0.04CsH–0.04KH sample after 30 cycles. A structural characterization revealed that added CsH and KH participated in the dehydrogenation and hydrogenation reactions by reversibly forming mixed amides of Li–K and Cs–Mg, which caused the improved hydrogen storage thermodynamics and kinetics.
Introduction
Hydrogen is an attractive energy carrier for mobile applications because it has the highest energy density and produces only water when consumed in a fuel cell.1 However, efficient hydrogen storage, which is one of the most challenging technical barriers to the implementation of widespread hydrogen energy, must be developed. Various methods, including high pressure, cryogenic liquid storage, physisorption on high-surface-area adsorbents, and chemisorption, have been studied and evaluated in recent decades.2,3 Of these, hydrogen storage in solid-state materials, especially in complex hydrides, such as alanates, borohydrides, and amides, has recently attracted intense interest.4–13 Xiong et al.14 and Luo15 reported nearly at the same time that approximately 5.6 wt% hydrogen can be reversibly stored in a Mg(NH2)2/2LiH system via the following reaction, which comprises a new Li–Mg–N–H hydrogen storage system.| Mg(NH2)2 + 2LiH ↔ Li2MgN2H2 + 2H2 | (1) |
Following this study, considerable effort has been devoted to improving the hydrogen sorption properties of the Li–Mg–N–H system, including a reduction of the particle size, an adjustment of the composition and the introduction of additives.16–25 Specifically, alkali metal compounds, including hydrides, halides and hydroxides, were found to be very effective in reducing the operating temperature and enhancing the reaction kinetics of hydrogen storage in a Mg(NH2)2/2LiH system.23–28 A first attempt to improve hydrogen storage properties of the Mg(NH2)2/2LiH system by partially substituting LiH with NaH was conducted by Liu et al. in 2007.29 These researchers observed a 14 °C reduction in the dehydrogenation peak temperature for the Mg(NH2)2/1.6LiH–0.4NaH system. In 2009, Wang et al. reported that the peak temperature for hydrogen release from a Mg(NH2)2/1.9LiH–0.1KH sample was greatly reduced from 186 °C to 132 °C, achieving a 54 °C reduction compared to a pristine sample.25 More importantly, a full cycle of hydrogen uptake and release for a K-doped sample could be carried out at a temperature as low as 107 °C in a pressure–composition–temperature (PCT) model. Liu et al. subsequently evaluated the effects of potassium halides (KF, KCl, KBr and KI) on the hydrogen storage characteristics of a Mg(NH2)2/2LiH system.23 The results showed that introducing a minor amount of KF could significantly improve hydrogen storage performance, while the hydrogen storage characteristics of samples with the addition of KCl, KBr and KI remained almost constant. This was mainly attributed to the reaction of KF with LiH producing KH, which became the actual active species that decreased the activation energy of the reaction and the desorption enthalpy change, but KCl, KBr and KI did not. A similar phenomenon was also observed for a Rb- and Cs-containing Li–Mg–N–H system.24,30–32 The onset temperature of dehydrogenation for a Mg(NH2)2/2LiH–0.08CsH composite was reduced to 70 °C, a 62 °C reduction compared to a pristine sample.30 More interestingly, the fully dehydrogenated Mg(NH2)2/2LiH–0.08CsH sample absorbed more than 4 wt% hydrogen at 120 °C and 100 bar of H2 within 20 min, which was superior to the K- and Rb-containing samples. Unfortunately, the available hydrogen capacity was reduced to approximately 4.62 wt% for a Mg(NH2)2/2LiH–0.08CsH sample, lower than that for K- (5.1 wt%) and Rb-containing (4.8 wt%) samples, because the molecular weight of Cs (132.9) is greater than that of K (39.1) or Rb (85.5). Therefore, retaining the high available hydrogen storage capacity while reducing the operating temperature is still a challenge for the practical use of a Li–Mg–N–H system as a hydrogen storage medium.
In this study, we attempted to balance the reduction of the operating temperature and the loss of storage capacity of a Cs-doped Mg(NH2)2/2LiH system by partially replacing the CsH with KH. The Mg(NH2)2/2LiH–(0.08 − x)CsH–xKH (x = 0, 0.02, 0.04, 0.06, 0.08) samples were prepared by ball milling, and the hydrogen storage characteristics and mechanism of the resultant samples were systematically studied and discussed. The results showed that the Mg(NH2)2/2LiH–0.04CsH–0.04KH sample had optimal hydrogen storage properties because its dehydrogenation occurred at 130 °C and hydrogenation at 120 °C with 4.89 wt% of hydrogen capacity.
Experimental section
Commercial LiH (purity 98%) was purchased from Alfa-Aesar and used as received. KH (30 wt% dispersion in mineral oil) was obtained from Alfa-Aesar and the mineral oil was removed before use. Mg(NH2)2 and CsH were produced in our laboratory by reacting pre-milled Mg powder (Sinopharm, purity 99%) with 7 bar of NH3 at 300 °C and by ball milling metallic Cs (Alfa-Aesar, purity 99.8%) under 50 bar of H2 at 500 rpm on a planetary ball mill (QM-3SP4, Nanjing) for 12 h, respectively. Five samples with the composition Mg(NH2)2/2LiH–(0.08 − x)CsH–xKH (x = 0, 0.02, 0.04, 0.06, 0.08) were prepared by ball milling the corresponding chemicals at 500 rpm for 36 h. During the ball milling process, the milling vessels were filled with 50 bar of hydrogen pressurized to prevent the release of the hydrogen. All sample handling was carried out in an MBRAUN glove box filled with high purity argon to prevent contamination with air and moisture (O2: <1 ppm, H2O: <1 ppm).The dehydrogenation/hydrogenation characteristics were qualitatively and quantitatively evaluated via a temperature-programmed desorption (TPD) technique and the volumetric method, respectively. TPD measurements were performed on a custom-designed system coupled with a QIC-20 mass spectrometer (Hiden, England), which could simultaneously measure hydrogen (m/z: 2) and ammonia (m/z: 17). Approximately 50 mg of sample was loaded and tested each time. Pure Ar was passed through the sample as the carrier gas during testing, and the sample was gradually heated to a preset temperature at a ramping rate of 2 °C min−1. The dehydrogenation/hydrogenation quantities were determined using a custom-designed Sievert-type apparatus in both isothermal and nonisothermal modes. Samples weighing approximately 120 mg were used for each experiment. For the non-isothermal experiments, the temperature was gradually increased from room temperature at a rate of 2 °C min−1 for dehydrogenation and 1 °C min−1 for hydrogenation. For the isothermal examination, the samples were quickly heated to and held at a given temperature during the entire measurement. The sample temperature was measured and controlled with an automatic temperature controller and a thermocouple that was inserted into the interior of the sample. The reactor was evacuated prior to making the measurements. For dehydrogenation, the gas was initially desorbed into a calibrated volume of 1 × 10−3 Torr and the pressure was increased by approximately 0.5 bar after hydrogen desorption was completed. The starting hydrogen pressure for hydrogenation was 100 bar. The dehydrogenation/hydrogenation quantities were calculated in accordance with the pressure changes in the calibration volume using the equation of state. Differential scanning calorimetry (DSC) measurements were performed with a Netzsch DSC 200 F3 unit. Approximately 3 mg of sample was gradually heated from room temperature to 350 °C at a rate of 2 °C min−1 under a flow of pure Ar.
The phase structure was characterized using a MiniFlex 600 (Rigaku, Japan) X-ray diffractometer (XRD) with Cu Kα radiation operating at 40 kV and 15 mA. XRD data were collected over the 2θ range of 10 to 90° with step increments of 0.02°. A custom-designed container was used to prevent the samples from contacting moisture in the air during sample transfer and testing. Infrared measurements were performed using a Bruker Tensor 27 Fourier transform infrared spectrometer (FTIR, Germany) in transmission mode. Powdered samples and potassium bromide (KBr) powder in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 weight ratio were first cold-pressed in the glove box to form pellets, and the pellets were quickly transferred to the FTIR apparatus for testing. Each spectrum was created using an average of 16 scans with a 4 cm−1 resolution. The sample morphology was observed with scanning electron microscopy (SEM, Hitachi S-4800), with an equipped energy-dispersive X-ray spectrometer (EDS) to analyse the distribution of elemental Mg, K, and Cs in the sample.
:30 weight ratio were first cold-pressed in the glove box to form pellets, and the pellets were quickly transferred to the FTIR apparatus for testing. Each spectrum was created using an average of 16 scans with a 4 cm−1 resolution. The sample morphology was observed with scanning electron microscopy (SEM, Hitachi S-4800), with an equipped energy-dispersive X-ray spectrometer (EDS) to analyse the distribution of elemental Mg, K, and Cs in the sample.
Results and discussion
To balance the reduction of the dehydrogenation temperature and the loss of available storage capacity, KH was introduced into Mg(NH2)2/2LiH–0.08CsH to replace CsH partially, and five samples with a composition of Mg(NH2)2/2LiH–(0.08 − x)CsH–xKH (x = 0, 0.02, 0.04, 0.06, 0.08) were prepared. Fig. 1 shows the XRD patterns and FTIR spectra of the resultant samples. Fig. 1(a) shows that only two constituent phases, Mg(NH2)2 and LiH, were identified in the XRD profile of the milled samples although their peak intensities gradually weakened with an increasing addition of KH. The FTIR results (Fig. 1(b)) confirmed the presence of Mg(NH2)2 because its typical doublet N–H vibration at 3272 and 3326 cm−1 was clearly discernible.33 However, no information related to Cs- and K-containing species was detected by XRD and FTIR, possibly due to poor crystallization. To understand the chemical state of Cs and K, we took the Mg(NH2)2/2LiH–0.04CsH–0.04KH sample as an example to recrystallize at 130 °C under 100 bar of H2 pressure for 12 h. As shown in Fig. 1(c), the characteristic reflections of Mg(NH2)2 and LiH were unambiguously identified at a considerable intensity in the XRD profile after recrystallization. Additionally, two new peaks were also detected at 21.8 and 27.2° (2θ), which correspond to the strongest diffraction peaks of CsNH2 and KH, respectively. We therefore deduced that KH retained its structure, while CsH was converted to CsNH2, possibly by reacting with Mg(NH2)2 during the ball milling process. This hypothesis was further evidenced by the generation of CsNH2 during the first ball milling of the three chemicals, Mg(NH2)2, LiH and CsH, at a molar ratio of 1:2:0.08, then heating at 130 °C under 100 bar of H2 for 12 h (Fig. 1(c)). Additionally, the typical N–H vibration assignable to MgNH at 3251 cm−1 was detected with FTIR while reacting Mg(NH2)2 with CsH at a molar ratio of 1:1, as shown in the inset of Fig. 1(c). As a consequence, we suggest that the following reaction occurred during ball milling and recrystallization.| Mg(NH2)2 + CsH → CsNH2 + MgNH + H2 | (2) |
 | ||
| Fig. 1 XRD patterns (a) and FTIR spectra (b) of the Mg(NH2)2–2LiH–(0.08 − x)CsH–xKH samples; XRD patterns (c) of the Mg(NH2)2–2LiH–0.04CsH–0.04KH and Mg(NH2)2–2LiH–0.08CsH sample after recrystallization. The inset of (c) is the FTIR spectra of the Mg(NH2)2–CsH sample after recrystallization. | ||
The milled Mg(NH2)2/2LiH–(0.08 − x)CsH–xKH samples were then subjected to qualitative and quantitative measurements to determine the thermal dehydrogenation characteristics by TPD and volumetric release techniques, respectively. Fig. 2 shows the TPD-MS curves of the Mg(NH2)2/2LiH–(0.08 − x)CsH–xKH samples as a function of temperature. The presence of CsH and KH partially changed the dehydrogenation process and significantly reduced the dehydrogenation temperature of the Mg(NH2)2/2LiH system (Fig. 2(a)). Unlike the pristine sample, the CsH- and KH-containing samples started to release hydrogen below 75 °C and terminated at 215 °C with a shoulder at 130 °C and a distinct peak at 160 °C. Specifically, the dehydrogenation onset temperature was reduced by more than 60 °C as compared to a pristine sample (135 °C), and the dehydrogenation process clearly exhibited two-step characteristics. A careful comparison revealed that an increase of the addition of KH slightly increased the dehydrogenation onset temperature and two peaks at approximately 130 and 160 °C became considerably more obvious with increased intensities, especially for the x = 0.08 sample, which suggested an increase in the available hydrogen storage capacity. Further investigation of the NH3-signal indicated that the presence of CsH and KH remarkably retarded the evolution of NH3 during the dehydrogenation process, because almost no NH3 signal was detected for the CsH and KH-containing samples below 250 °C (Fig. 2(b)).
 | ||
| Fig. 2 H2 (a) and NH3 (b) TPD-MS signals of the Mg(NH2)2–2LiH–(0.08 − x)CsH–xKH samples as a function of temperature. | ||
Fig. 3(a) shows the quantitative dehydrogenation curves for the Mg(NH2)2/2LiH–(0.08 − x)CsH–xKH samples as a function of temperature obtained with a volumetric method. The results further confirm that the dehydrogenation temperature was significantly reduced for the CsH and KH-containing samples.
 | ||
| Fig. 3 Nonisothermal (a) and isothermal (b) dehydrogenation curves of the Mg(NH2)2–2LiH–(0.08 − x)CsH–xKH samples. | ||
Moreover, because the addition of KH was increased from x = 0 to 0.08, the available dehydrogenation capacity was increased from 4.65 to 5.2 wt% along with an additional 6 °C reduction in the mid-point temperature. The increased dehydrogenation capacity was mainly attributed to the lower molecular weight of K (39.1) compared to Cs (132.9), as previously mentioned. The isothermal dehydrogenation showed that the CsH and KH-containing samples released 3.5–3.8 wt% hydrogen within 400 min at 130 °C, whereas a pristine sample under identical conditions released less than 1.0 wt% (Fig. 3(b)). It should be noted that the presence of KH not only increased the dehydrogenation amount but also enhanced the dehydrogenation kinetics of the Mg(NH2)2/2LiH–(0.08 − x)CsH–xKH samples. Fig. 3(b) shows that it took 150 min to release 3.5 wt% H2 for the Mg(NH2)2/2LiH–0.08KH sample, however, 400 min was required for the Mg(NH2)2/2LiH–0.08CsH sample. An analysis of the tangent slope of the linear region of the isothermal dehydrogenation curve indicated that the rate constant of the 0.08CsH- and 0.08KH-containing samples were respectively, 0.132 wt% min−1 and 0.193 wt% min−1, which was 18- and 28-fold greater than that of pristine sample (0.007 wt% min−1). Moreover, the 0.04CsH and 0.04KH co-doped samples also showed a dehydrogenation rate of 0.174 wt% min−1, 25-fold faster than that of a pristine sample.
The fully dehydrogenated samples were exposed at 100 bar H2 to hydrogenate in both isothermal and non-isothermal modes. The results are shown in Fig. 4. In the non-isothermal mode, the dehydrogenated Mg(NH2)2/2LiH sample started to absorb hydrogen at approximately 85 °C and finished at 210 °C, with a hydrogen uptake amount of 5.3 wt%, in good agreement with the previous amount of dehydrogenation. In the presence of CsH and KH, the onset temperature for hydrogen uptake decreased to 57 °C, and full hydrogenation was achieved at 150 °C, which was much lower than for a pristine sample. Similar to the dehydrogenation process, the amount of hydrogen uptake also increased from 4.65 wt% to 5.2 wt% as the addition of KH was increased from x = 0 to 0.08, More interestingly, the temperature dependent hydrogenation curves remained nearly unchanged for the x = 0.02 and 0.04 samples, in addition to the slightly greater hydrogen capacity. In contrast, the hydrogenation curves of the x = 0.06 and 0.08 samples moved to higher temperatures, especially for the final temperature. Further isothermal hydrogenation experiments confirmed this change in the hydrogenation kinetics. Fig. 4(b) shows that the hydrogen uptake of the x = 0.06 and 0.08 samples was, respectively, 3.6 wt% and 3.5 wt% at 110 °C and 100 bar for 150 min, lower than for the x = 0–0.04 samples. This indicates that the partial substitution of KH for CsH decreased the hydrogenation kinetics in the Mg(NH2)2/2LiH–(0.08 − x)CsH–xKH samples.
 | ||
| Fig. 4 Nonisothermal (a) and isothermal (b) hydrogenation curves of the dehydrogenated Mg(NH2)2–2LiH–(0.08 − x)CsH–xKH samples. | ||
The above discussion leads to the conclusion that partially replacing the CsH with KH in the Mg(NH2)2/2LiH–(0.08 − x)CsH–xKH system further reduced the dehydrogenation temperature and increased the available hydrogen capacity, but decreased the hydrogenation kinetics. The effects on both the available hydrogen capacity and the hydrogenation/dehydrogenation temperature and kinetics suggest that the Mg(NH2)2/2LiH–0.04CsH–0.04KH sample is the optimal composition and has relatively better overall hydrogen storage properties. This sample can reversibly store 4.89 wt% of hydrogen with a dehydrogenation/hydrogenation onset temperatures of 75 and 57 °C, respectively, and approximately 3.5 wt% of hydrogen can be rapidly charged into the dehydrogenated 0.04CsH and 0.04KH co-containing sample within 50 min at 110 °C, as shown in Fig. 3 and 4.
To understand the effects of the co-addition of CsH and KH on the dehydrogenation kinetics, the apparent activation energy (Ea) was determined via Kissinger's approach,34 and the results are shown in Fig. 5(a). In the present study, the Ea values were, respectively, calculated to be 87.3 ± 3.4 and 100.6 ± 1.9 kJ mol−1 for the first and second steps of dehydrogenation of Mg(NH2)2/2LiH–0.04CsH–0.04KH. Here it should be noted that the Ea for the first-step of dehydrogenation was remarkably lower than that of the pristine Mg(NH2)2/2LiH sample (107.2 ± 2.8 kJ mol−1), implying that adding CsH and/or KH as a catalyst lowered the reaction barrier for hydrogen desorption from the Mg(NH2)2/2LiH system. This is an important reason for the reduced dehydrogenation temperature of the CsH and KH-containing Mg(NH2)2/2LiH system.
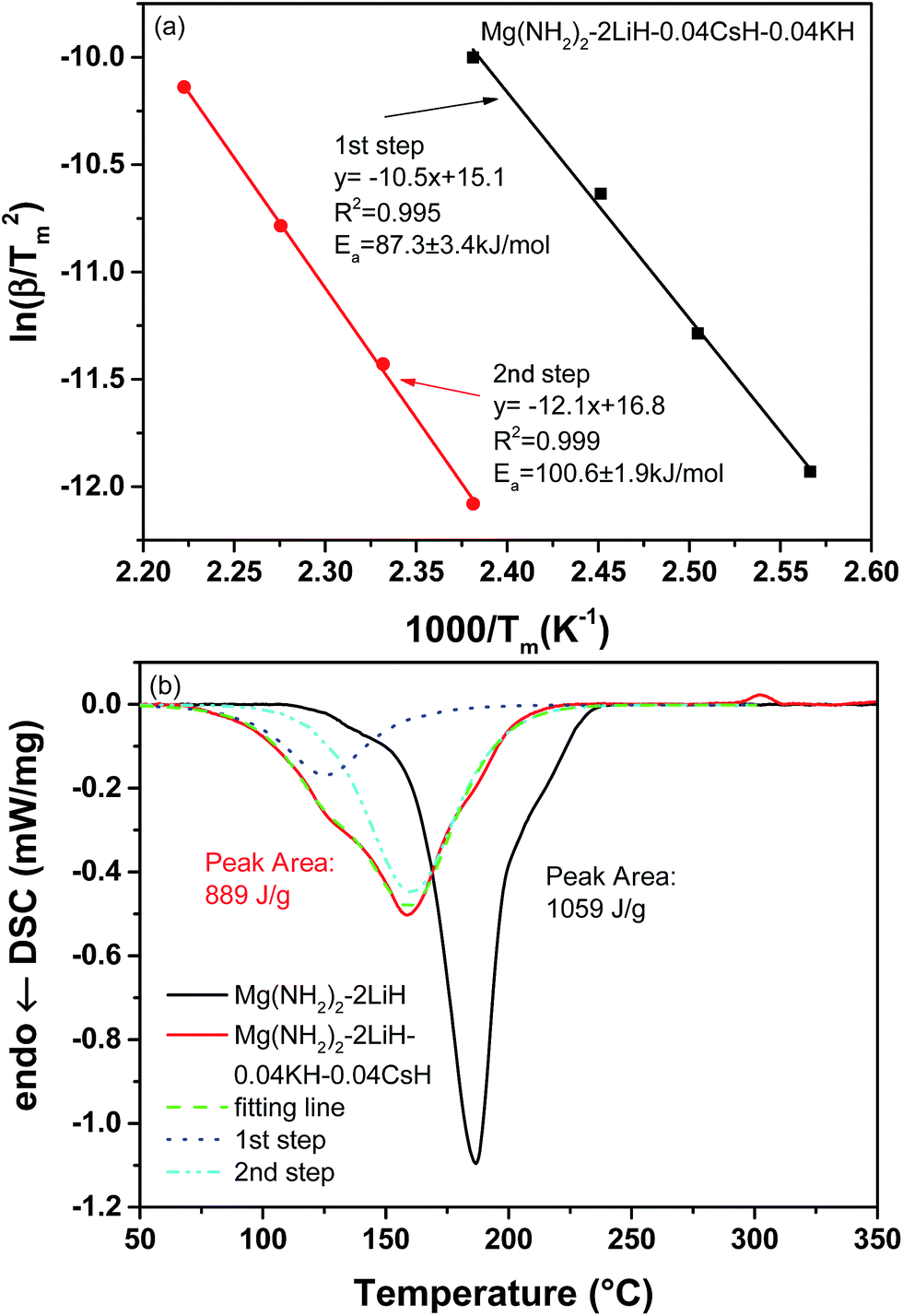 | ||
| Fig. 5 Kissinger's plots (a) and DSC curves (b) of the Mg(NH2)2–2LiH–0.04CsH–0.04KH sample. | ||
Fig. 5(b) shows the DSC curves of pristine and 0.04CsH–0.04KH-containing samples with temperature. It is clear that the heat flow curve of the Mg(NH2)2/2LiH–0.04CsH–0.04KH sample displayed two adjacent peaks in the temperature range of 70–220 °C, and the peak position underwent an appreciable low-temperature shift compared to the Mg(NH2)2/2LiH sample. This further confirmed the occurrence of two dehydrogenation stages with different thermodynamics and/or kinetics for the CsH and KH co-added sample. Moreover, an additional exothermic peak appeared at 280–300 °C in the Mg(NH2)2/2LiH–0.04CsH–0.04KH sample, which should correspond to a polymorphic transformation of Li2MgN2H2 from a cubic structure to an orthorhombic structure, as previously reported.27 By integrating the endothermic peak, a heat effect of 1059 J g−1 was obtained for the Mg(NH2)2/2LiH sample, which is equivalent to 39.8 kJ mol−1-H2. Furthermore, the enthalpy change for the two steps of dehydrogenation of the 0.04CsH–0.04KH-containing sample was determined to be 39.3 and 34.7 kJ mol−1-H2. Obviously, a 12% decrease was observed for the second step, which is the most likely reason for the lower temperature of the second step. Thus, the presence of CsH and KH changed the reaction pathway of hydrogen desorption in the Mg(NH2)2/2LiH system and made it thermodynamically more favourable.27,30
The chemical events occurring during the dehydrogenation process and the roles played by CsH and KH were further elucidated by characterizing the compositional and structural changes in the Mg(NH2)2/2LiH–0.04CsH–0.04KH sample at different dehydrogenation stages with a dynamic heating mode. The results are shown in Fig. 6. The 130 °C-dehydrogenated sample exhibited a nearly identical pattern to the recrystallized sample in addition to a slightly decreased intensities for Mg(NH2)2, LiH and CsNH2 in the XRD profile (Fig. 6(a)). However, a new broad absorption peak appeared at 3190 cm−1 in the FTIR spectrum although its intensity was weak (Fig. 6(b)). This new absorbance can be assigned to a ternary imide, Li2Mg2N3H3, as previously reported.35 After dehydrogenation at 160 °C, the reflections of Mg(NH2)2 and LiH were remarkably weaker and that of CsNH2 was invisible. Interestingly, KH remained almost unchanged at 160 °C. In the FTIR spectrum, the newly appearing absorbance at 3190 cm−1 was further intensified, and the typical doublet N–H vibration of LiNH2 was observed at 3315 and 3258 cm−1. When dehydrogenation ceased at 180 °C, the characteristic reflections of Mg(NH2)2 nearly disappeared and three new peaks of considerable intensity emerged at 30.3, 51.0 and 60.7°, which were very similar to the XRD pattern of Li2Mg2N3H3. Meantime, the diffraction peak of KH was distinctly weaker. FTIR indicated that the broad absorption peak at 3190 cm−1 moved to 3183 cm−1, which was much closer to the typical absorption peak of the N–H vibration in cubic Li2MgN2H2.35 While heating the sample to 200 °C, the XRD profile clearly exhibited the diffraction characteristics of cubic Li2MgN2H2. This property was further confirmed by the FTIR result by the strong absorbance at 3178 cm−1. A further increase in the operating temperature to 220 °C, produced an XRD pattern of the dehydrogenation product that could be assigned to orthorhombic Li2MgN2H2, and the FTIR absorbance moved further to 3174 cm−1. After full dehydrogenation at 280 °C, the reflections of orthorhombic Li2MgN2H2 remarkably intensified, and simultaneously, four additional phases, Li2NH, Mg3N2, CsMg(NH)(NH2) and Li3K(NH2)4 were also detected. The FTIR result also indicated that the characteristic absorbance of orthorhombic Li2MgN2H2 at 3159/3181 cm−1 dominated the FTIR spectrum. Moreover, two additional peaks at 3250 and 3308 cm−1 also appeared, which were very similar to the IR characteristics of CsMg(NH)(NH2) and Li3K(NH2)4,23,26,30 respectively.
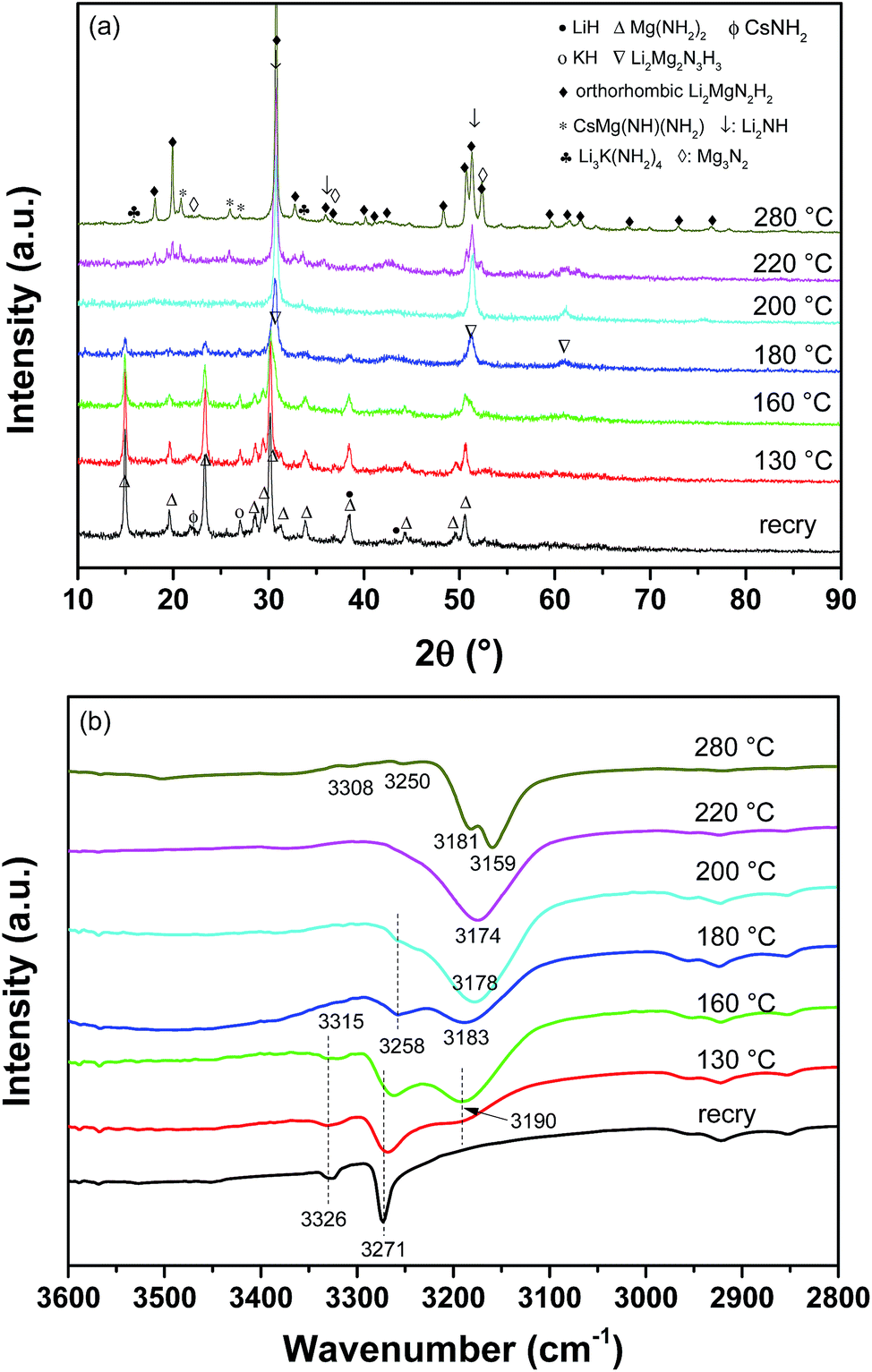 | ||
| Fig. 6 XRD patterns (a) and FTIR spectra (b) of the dehydrogenated Mg(NH2)2–2LiH–0.04CsH–0.04KH samples at different stages. | ||
According to the above discussion, we can describe the chemical process for hydrogen desorption from Mg(NH2)2/2LiH–0.04CsH–0.04KH upon heating. As mentioned above, the milled sample was mainly composed of Mg(NH2)2, LiH, CsNH2 and MgNH as indicated by the occurrence of a metathesis reaction between Mg(NH2)2 and CsH initiated by an energetic mechanical collision. As the sample temperature increased from room temperature to 180 °C, Mg(NH2)2 reacted with LiH, CsNH2 and MgNH to release hydrogen and produced Li2Mg2N3H3, LiNH2 and CsMg(NH)(NH2), simultaneously, KH remained.
| Mg(NH2)2 + 1.5LiH + 0.04CsNH2 + 0.04MgNH → 0.5Li2Mg2N3H3 + 0.5LiNH2 + 0.04CsMg(NH)(NH2) + 1.5H2 | (3) |
During further heating of the sample from 180 to 280 °C, KH took part in the dehydrogenation reaction with LiNH2, Li2Mg2N3H3 and LiH to become Li2MgN2H2, Li3K(NH2)4, Li2NH and Mg3N2 along with an additional release of hydrogen, as previously reported.23
| LiNH2 + Li2Mg2N3H3 + 0.84LiH + 0.16KH → 1.04Li2MgN2H2 + 0.16Li3K(NH2)4 + 0.64Li2NH + 0.32Mg3N2 + H2 | (4) |
Reaction (4) was further evidenced by directly heating the mixture of LiNH2–Li2Mg2N3H3–0.84LiH–0.16KH because four phases, Li2MgN2H2, Li2NH, Li3K(NH2)4 and Mg3N2, were also identified in the XRD profile of 280 °C-dehydrogenation sample (Fig. S1, ESI†). Thus, the overall reaction process for hydrogen desorption from the Mg(NH2)2/2LiH–0.04CsH–0.04KH sample upon ball milling and heating can be described by the following reaction:
| Mg(NH2)2 + 2LiH + 0.04KH + 0.04CsH → 0.72Li2MgN2H2 + 0.16Li2NH + 0.08Mg3N2 + 0.04Li3K(NH2)4 + 0.04CsMg(NH)(NH2) + 0.12LiH + 1.96H2 | (5) |
As seen in reactions (3) and (4), it is clear that in the first step of dehydrogenation, only CsNH2 takes part in the reaction and KH retains its structure, acting as a catalyst. In the second step of dehydrogenation, KH also takes part in the reaction. These results provide a reasonable explanation of the changes in the thermodynamics and kinetics, as shown in Fig. 5.
Fig. 7 illustrates the cycling dehydrogenation/hydrogenation curves of the Mg(NH2)2/2LiH–0.04CsH–0.04KH system. In this instance, to avoid the high-temperature failure of CsH and KH and maximize the hydrogen capacity,28 the cycling measurements were performed at 180 °C for dehydrogenation and 140 °C and 100 bar of H2 for hydrogenation. As shown in Fig. 7, hydrogen release amounted to 4.47 wt% while operating at 180 °C at the first cycle. After 30 cycles, the hydrogen storage capacity was maintained at 4.34 wt%, which corresponds to 97% of capacity retention. This is greatly superior to the pristine Mg(NH2)2/2LiH and Mg(NH2)2/2LiH–0.08KH samples,27,30 suggesting that a synergetic effect occurred with the co-addition of CsH and KH.
 | ||
| Fig. 7 Hydrogen desorption/absorption cycle curves of the Mg(NH2)2–2LiH–0.04CsH–0.04KH sample. | ||
After 30 cycles, the hydrogenation product of the Mg(NH2)2/2LiH–0.04KH–0.04CsH sample was collected for XRD, FTIR and SEM. As shown in Fig. 8, the hydrogenation sample mainly consisted of Mg(NH2)2 and LiH because Li2MgN2H2, Li2NH and Mg3N2 were consumed. Moreover, the strongest reflections of CsNH2 and KH were also identified at 21.9 and 26.9°, respectively, although the intensity was very weak. It indicates that the dehydrogenation product can return to its initial state, providing a fully reversible hydrogen storage process. Fig. 9 shows the SEM images of the Mg(NH2)2/2LiH–0.04CsH–0.04KH sample before and after cycling. A distinct enlargement of the particle size was observed for the 30-cycled sample, which is believed to be one of the most important reasons for the cycling capacity degradation.
 | ||
| Fig. 8 XRD patterns (a) and FTIR spectra (b) of the Mg(NH2)2–2LiH–0.04CsH–0.04KH samples after 30 dehydrogenation/hydrogenation cycles. | ||
 | ||
| Fig. 9 SEM images of the Mg(NH2)2–2LiH–0.04CsH–0.04KH samples before (a) and after (b and c) cycling. | ||
Conclusions
Partially replacing CsH with KH, improved the hydrogen storage properties of a Mg(NH2)2/2LiH–0.08CsH system. The optimal composition was determined to be Mg(NH2)2/2LiH–0.04CsH–0.04KH, which could reversibly store 4.89 wt% of hydrogen through a two-step reaction. The reaction enthalpy changes for the first and second dehydrogenation steps were, respectively, calculated to be 39.3 and 34.7 kJ mol−1-H2 for the Mg(NH2)2/2LiH–0.04CsH–0.04KH sample, and activation energy was, respectively, 87.3 ± 3.4 and 100.6 ± 1.9 kJ mol−1. XRD and FTIR analyses revealed that during the ball milling process, a metathesis reaction first occurred between Mg(NH2)2 and the added CsH to convert into CsNH2 and MgNH. Upon heating, Mg(NH2)2 reacted with LiH with KH as a catalyst to release H2 and produce Li2Mg2N3H3 and LiNH2 below 180 °C. A further increase of the sample temperature allowed the newly formed Li2Mg2N3H3 and LiNH2 to react with the KH and the remaining LiH to release additional hydrogen and form Li2MgN2H2, Li3K(NH2)4, Li2NH and Mg3N2. These are responsible for the improved thermodynamics and kinetics of hydrogen storage in the Mg(NH2)2/2LiH–0.04CsH–0.04KH sample. After 30 cycles, the hydrogen capacity of the Mg(NH2)2/2LiH–0.04CsH–0.04KH sample remained at 4.34 wt% while operating at 180 °C for dehydrogenation and 140 °C and 100 bar of H2 for hydrogenation, which corresponds to 97% capacity retention. This is superior to the cycling stability of pristine Mg(NH2)2/2LiH and CsH-doped samples.Acknowledgements
We gratefully acknowledge the financial support received from the Zhejiang Provincial Natural Science Foundation of China (LR16E010002), the National Natural Science Foundation of China (51671172, U1601212) and the National Youth Top-Notch Talent Support Program.Notes and references
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- M. Baricco, M. Bang, M. Fichtner, B. Hauback, M. Linder, C. Luetto, P. Moretto and M. Sgroi, J. Power Sources, 2017, 342, 853–860 CrossRef CAS.
- X. B. Yu, Z. W. Tang, D. L. Sun, L. Z. Ouyang and M. Zhu, Prog. Mater. Sci., 2017, 88, 1–48 CrossRef.
- R. Mohtadi and S. Orimo, Nat. Rev. Mater., 2017, 2, 16091 CrossRef.
- Y. F. Liu, Y. X. Yang, M. X. Gao and H. G. Pan, Chem. Rec., 2016, 16, 189–204 CrossRef CAS PubMed.
- I. P. Jain, P. Jain and A. Jain, J. Alloys Compd., 2010, 503, 303–339 CrossRef CAS.
- Y. G. Yan, A. Remhof, D. Rentsch and A. Züttel, Chem. Commun., 2015, 51, 700–702 RSC.
- R. Y. Wu, K. Wang, Z. Y. Wang, M. X. Gao, H. G. Pan and Y. F. Liu, J. Power Sources, 2016, 327, 519–525 CrossRef CAS.
- H. J. Cao, Y. Zhang, J. H. Wang, Z. T. Xiong, G. T. Wu and P. Chen, Prog. Nat. Sci.: Mater. Int., 2012, 22, 550–560 CrossRef.
- K. P. Brooks, M. E. Bowden, A. J. Karkamkar, A. Y. Houghton and S. T. Autrey, J. Power Sources, 2016, 324, 170–178 CrossRef CAS.
- H. W. Li, Y. G. Yan, S. Orimo, A. Züttel and C. M. Jensen, Energies, 2011, 4, 185–214 CrossRef CAS.
- P. Chen, Z. Xiong, J. Luo, J. Lin and K. L. Tan, Nature, 2002, 420, 302–304 CrossRef CAS PubMed.
- Z. T. Xiong, G. T. Wu, H. J. Hu and P. Chen, Adv. Mater., 2004, 16, 1522–1525 CrossRef CAS.
- W. F. Luo, J. Alloys Compd., 2004, 381, 284–287 CrossRef CAS.
- Y. F. Liu, Z. T. Xiong, J. J. Hu, G. T. Wu, P. Chen, K. Murata and K. Sakata, J. Power Sources, 2006, 159, 135–138 CrossRef CAS.
- L. P. Ma, H.-B. Dai, Y. Liang, X. D. Kang, Z. Z. Fang, P. J. Wang, P. J. Wang and H.-M. Cheng, J. Phys. Chem. C, 2008, 112, 18280–18285 CAS.
- R. R. Shahi, T. P. Yadav, M. A. Shaz and O. N. Srivastva, Int. J. Hydrogen Energy, 2010, 35, 238–246 CrossRef CAS.
- H. Cao, Y. Zhang, J. Wang, Z. Xiong, G. Wu, J. Qiu and P. Chen, Dalton Trans., 2013, 42, 5524–5531 RSC.
- J. Hu, Y. Liu, G. Wu, Z. Xiong, Y. S. Chua and P. Chen, Chem. Mater., 2008, 20, 4398–4402 CrossRef CAS.
- B. Li, Y. Liu, J. Gu, M. Gao and H. Pan, Chem.–Asian J., 2013, 8, 374–384 CrossRef CAS PubMed.
- R. F. Bill, D. Reed, D. Book and P. A. Anderson, J. Alloys Compd., 2015, 645, S96–S99 CrossRef CAS.
- Y. Liu, C. Li, B. Li, M. Gao and H. Pan, J. Phys. Chem. C, 2013, 117, 866–875 CAS.
- C. Li, Y. Liu, Y. Gu, M. Gao and H. Pan, Chem.–Asian J., 2013, 8, 2136–2143 CrossRef CAS PubMed.
- J. Wang, T. Liu, G. Wu, W. Li, Y. Liu, C. M. Araujo, R. H. Scheicher, A. Blomqvist, R. Ahuja, Z. Xiong, P. Yang, M. Gao, H. Pan and P. Chen, Angew. Chem., Int. Ed., 2009, 48, 5828–5832 CrossRef CAS PubMed.
- F. Torre, A. Valentoni, C. Milanese, C. Pistidda, A. Marini, M. Dornheim, S. Enzo, G. Mulas and S. Garroni, J. Alloys Compd., 2015, 645, S284–S287 CrossRef CAS.
- C. Li, Y. Liu, Y. Pang, Y. Gu, M. Gao and H. Pan, Dalton Trans., 2014, 43, 2369–2377 RSC.
- C. Li, Y. Liu, Y. Yang, M. Gao and H. Pan, J. Mater. Chem. A, 2014, 2, 7345–7353 CAS.
- Y. Liu, J. Hu, Z. Xiong, G. Wu and P. Chen, J. Mater. Res., 2007, 22, 1339–1345 CrossRef CAS.
- J. Zhang, Y. Liu, X. Zhang, Y. Yang, Q. Zhang, T. Jin, Y. Wang, M. Gao, L. Sun and H. Pan, Int. J. Hydrogen Energy, 2016, 41, 11264–11274 CrossRef CAS.
- C. Li, Y. Liu, R. Ma, X. Zhang, Y. Li, M. Gao and H. Pan, ACS Appl. Mater. Interfaces, 2014, 6, 17024–17033 CAS.
- J. Hayes and A. Goudy, Int. J. Hydrogen Energy, 2015, 40, 12336–12342 CrossRef CAS.
- W. F. Luo and S. Sickafoose, J. Alloys Compd., 2006, 407, 274–281 CrossRef CAS.
- H. E. Kissinger, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- J. Hu, Y. Liu, G. Wu, Z. Xiong and P. Chen, J. Phys. Chem. C, 2007, 111, 18439–18443 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1. See DOI: 10.1039/c7ra05166b |
| This journal is © The Royal Society of Chemistry 2017 |