
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DOX-loaded pH-sensitive mesoporous silica nanoparticles coated with PDA and PEG induce pro-death autophagy in breast cancer†
Yanhong Duo‡
 a,
Yang Li‡d,
Changke Chenc,
Baiyun Liua,
Xinyu Wang*a,
Xiaowei Zeng*c and
Hongbo Chen*b
a,
Yang Li‡d,
Changke Chenc,
Baiyun Liua,
Xinyu Wang*a,
Xiaowei Zeng*c and
Hongbo Chen*b
aKey Laboratory of Plant Cell Activities and Stress Adaptation, Ministry of Education, School of Life Sciences, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: wangxy@lzu.edu.cn
bSchool of Pharmaceutical Sciences (Shenzhen), Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: chenhb7@mail.sysu.edu.cn
cThe Shenzhen Key Lab of Gene and Antibody Therapy, Division of Life and Health Sciences, Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, P. R. China. E-mail: zeng.xiaowei@sz.tsinghua.edu.cn
dDepartment of Hepatobiliary and Pancreas Surgery, Second Clinical Medical College of Jinan University, Shenzhen People's Hospital, Shenzhen 518000, P. R. China
First published on 14th August 2017
Abstract
The development of multifunctional nano drug delivery carriers has been one of the most effective and prevailing approaches to overcome drug non-selectivity, low cell uptake efficiency and various side effects of traditional chemotherapy drugs. Herein, we report a novel doxorubicin (DOX)-loaded mesoporous silica nanoparticle (MSN) coated with polydopamine (PDA) and polyethylene glycol (PEG) (MSNs-DOX@PDA-PEG) for the treatment of breast cancer. In this system, PDA functions as a pH-sensitive gatekeeper to control the release of DOX from MSNs in response to pH-stimulus and PEG was further grafted on the surface of PDA to increase the stability and biocompatibility under physiological conditions. The in vitro release results suggested that MSNs-DOX@PDA-PEG exhibits a high sensitivity to low pH. A cellular uptake assay showed a high cellular uptake efficiency of MSNs-DOX@PDA-PEG compared to free DOX. Furthermore, MSNs-DOX@PDA-PEG also demonstrated an improved anti-cancer efficacy compared to free DOX both in vivo and vitro breast cancer experiments. Mechanistic studies revealed that MSNs-DOX@PDA-PEG causes a stronger pro-death autophagy compared to free DOX via inhibition of the AKT-mTOR-p70S6K signaling pathway. Taken in concert, our results suggest that the novel material MSNs-DOX@PDA-PEG may represent a promising nanoformulation for breast cancer treatment.
Introduction
Breast cancer represents a leading cause of death among women.1 Presently, chemotherapy is one of the main available treatment options for breast cancer. However, traditional chemotherapeutic treatments often result in drug resistance and frequently cause harmful side effects to non-cancerous tissues due to the lack of selective tumor targeting.2Doxorubicin (DOX) is one of the most efficient anticancer drugs that is widely used for the treatment of multiple cancer types including breast, bladder, lung, hematological malignancies, and others.3 As an anthracycline antibiotic, DOX can interfere with DNA synthesis, induce DNA damages, produce reactive oxygen species, and destroy membrane structures.4,5 Recently, the development of a safe and efficient drug delivery system to deliver DOX to the tumor site attracted a significant amount of attention in the scientific community. With the development of nanotechnology, nanoformulations have been widely employed for the delivery of anticancer drugs. However, drug release from polymer-based nanoparticles takes place mainly through diffusion and/or polymer degradation.6 The latter represents a slow process and the therapeutic drug levels may therefore not promptly reach an effective drug concentration. Recently, mesoporous silica nanoparticles (MSNs) have attracted enormous attention in the field of drug delivery due to their unique physiochemical properties, including large surface area and pore volume, tunable particle/pore size, high drug loading efficiency, easy surface modification and remarkable stability and biocompatibility.7,8 By modifying the outer surface of MSNs with various functional groups such as polymers,9 supramolecules,10 quantum dots,11 ligands,12 or/and by using a combination with other nanomaterials,13,14 stimuli-responsive and active targeting nanosystems can be designed for the targeted delivery of anticancer drugs.
In this study, we developed DOX-loaded mesoporous silica nanoparticles (MSN) and the nanoparticle surface was further coated with polydopamine (PDA) and polyethylene glycol (PEG). In this system, PDA coating not only protects DOX leakage under physiological conditions (pH 7.4) but also allows for the sustained-release of the drugs in an acidic environment (pH 5.0).15,16 PEG on the surface of MSNs ensures water solubility and further prevents nonspecific interactions with biomacromolecules.17 Previous reports published in the literature show that the polymer-based nanoparticles may be taken up by cells through the endocytosis-lysosome pathway.18,19 Thus, we anticipated that DOX may be rapidly released when MSNs-DOX@PDA-PEG enters to lysosomes where the pH value is about 5.020. Furthermore, our results suggested that MSNs-DOX@PDA-PEG exhibits improved anti-cancer abilities compared to free DOX both in vitro and in vivo. Furthermore, mechanistic studies revealed that MSNs-DOX@PDA-PEG may cause a stronger pro-death autophagy compared to free DOX via the AKT-mTOR-p70s6K signaling pathway.
Results and discussion
Synthesis and physicochemical characterization of MSNs-DOX@PDA-PEG
MSNs-DOX@PDA-PEG was synthesized as described below in the section “Materials and methods”. The particle size and surface properties of the nanoparticles play important roles in drug release, cellular uptake and pharmacokinetics.21 Fig. 1A shows a schematic representation of the MSNs-DOX@PDA-PEG synthesis. In order to access the morphology of MSNs, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG, TEM analysis was performed. Fig. 1B, C and D show representative images of MSNs, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG, respectively. As shown in Fig. 1B, the MSNs exhibit a nearly spherical shape and porous surfaces. Compared with MSNs (cf. Fig. 1B), MSNs-DOX@PDA (cf. Fig. 1C) and MSNs-DOX@PDA-PEG (cf. Fig. 1D) show the PDA and PEG coating on the MSNs surface, with a clear layer found on the periphery of the particles. | ||
| Fig. 1 (A) Schematic representation of MSNs-DOX@PDA-PEG synthesis. (B) TEM image of MSNs. (C) TEM image of MSNs-DOX@PDA. (D) TEM image of MSNs-DOX@PDA-PEG. | ||
The sizes of MSNs, MSNs-DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG were determined using dynamic light scattering (DLS). As shown in Table 1, the diameters of the MSNs, MSNs-DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG were 125.34 ± 4.42, 130.21 ± 3.37, 170.32 ± 2.49 and 198.75 ± 2.56 nm, respectively. Although the sizes of MSNs-DOX@PDA and MSNs-DOX@PDA-PEG were slightly larger than MSNs, they were also found to be smaller than the cut-off size of tumor neovasculature pores,22,23 falling within the range of easy accumulation of the enhanced permeation and retention (EPR) effect.
| Polymer | Sizea (nm) | ZP (mV) | BET surface area (m2 g−1) | Pore volumeb (cm3 g−1) | Pore sizec (nm) |
|---|---|---|---|---|---|
| a NPs size was measured by dynamic light scattering.b BJH cumulative pore volume for pores between 1.7 and 300 nm in width.c Most probable pore size. | |||||
| MSNs | 125.34 ± 4.42 | −19.43 ± 5.21 | 264.72 | 0.62 | 2.89 |
| MSNs-DOX | 130.21 ± 3.37 | −8.56 ± 2.19 | 121.32 | 0.43 | 2.31 |
| MSNs-DOX@PDA | 170.32 ± 4.49 | −12.44 ± 3.29 | 46.73 | 0.15 | — |
| MSNs-DOX@PDA-PEG | 198.75 ± 2.56 | −2.11 ± 0.97 | 49.78 | 0.11 | — |
The zeta potentials were also determined as shown in Table 1. The zeta potentials of MSNs, MSNs-DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG were −19.43 ± 5.21, −8.56 ± 2.19, −12.44 ± 3.29 and −2.11 ± 0.97 mV, respectively. Since most cellular membranes are negatively charged, the zeta potential can affect the tendency of the nanoparticles to permeate membranes, with cationic particles exhibiting a higher toxicity with cell membrane disruption. In general, nanoparticles with a zeta potential between −10 and +10 mV are considered neutral. The specific surface area, pore volume and the most probable pore size of MSNs, MSNs-DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG are also shown in Table 1. Compared to MSNs and MSNs-DOX, all pore parameters of MSNs-DOX@PDA and MSNs-DOX@PDA-PEG were found to be significantly decreased. The BET surface area was 264.72 m2 g−1, the pore volume was 0.62 cm3 g−1 and, as evaluated by the BJH method, the most probable pore size was about 2.89 nm. Moreover, the pore size distribution of MSNs was rather narrow. With loading of DOX, the BET surface area and the most probable pore size decreased to 121.32 m2 g−1 and 2.31 nm, respectively. The BET surface area of MSNs-DOX@PDA and MSNs-DOX@PDA-PEG was 46.73 and 49.78 m2 g−1, respectively. However, the pore volumes were 0.15 and 0.11 cm3 g−1 due to the coating of PDA and PEG onto the surface of DOX-loaded MSNs. Overall, the structural parameters of MSNs, MSNs-DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG suggested that DOX occupied the pore space of MSNs and DOX loaded MSNs were coated with PDA and PEG. The encapsulation efficiency of DOX in MSNs was about 95.63 ± 2.32.
In addition, as a result of the slightly negative charge of MSNs-DOX@PDA-PEG, the overall clearance by the reticuloendothelial system (RES) such as liver was found to be reduced.24,25 These data suggested that MSNs-DOX could be successfully synthesized and modified by introduction of PDA and PEG films.
A pH-sensitive drug release profile in vitro
Previously published research data shows that the polymer-based nanoparticles are taken up by cells through the endocytosis-lysosome pathway.18,19 The pH value of lysosomes is about 5.0, which is maintained by proton pumps on the lysosome membrane.20 In addition, low pH conditions are also considered a hallmark of malignant solid tumor tissues. In order to verify the drug blocking potency and pH sensitivity of PDA and PEG coating, we performed a drug release experiment at pH 5.0 and pH 7.4 at different time intervals. As shown in Fig. 2A, without PDA and PEG coating, DOX was quickly released at all tested pH values, freely diffusing from the pores of MSNs into solution. However, compared to the rapid release of MSNs-DOX at pH 7.4, MSNs-DOX@PDA-PEG exhibited a sustained DOX release rate at pH 7.4, suggesting that PDA-PEG coating effectively protects DOX leakage from the MSNs (cf. Fig. 2B). In particular, MSNs-DOX@PDA-PEG exhibited a significantly more rapid release rate at pH 5.0 compared to the slower release rate at pH 7.4 (cf. Fig. 2B), a finding that could be attributed to the pH sensitivity of PDA under acidic conditions.26 | ||
| Fig. 2 (A) In vitro release profile of MSNs-DOX (B) in vitro release profile of MSNs-DOX@PDA-PEG. | ||
Effective internalization of MSNs-DOX@PDA-PEG by cells
To determine the specific recognition and uptake capacity of MSNs-DOX@PDA-PEG towards target cells, we carried out confocal microscopy analysis to compare the cellular uptake efficiencies of free DOX and MSNs-DOX@PDA-PEG (Fig. 3A and B). The results suggested that cellular uptake of MSNs-DOX@PDA-PEG in MCF7 cells was significantly higher than that of free DOX. Furthermore, cellular uptake of MSNs-DOX@PDA-PEG in MCF7 cells was remarkably higher than in MCF10A cells. Flow cytometry analysis also confirmed the enhanced cellular uptake of MSNs-DOX@PDA-PEG in MCF7 cells (Fig. 3C). Most likely, the increase of cellular uptake was caused by PDA and PEG. Previous studies have reported similar results in terms of PDA and PEG being able to increase the cellular uptake of nanoparticles.21,27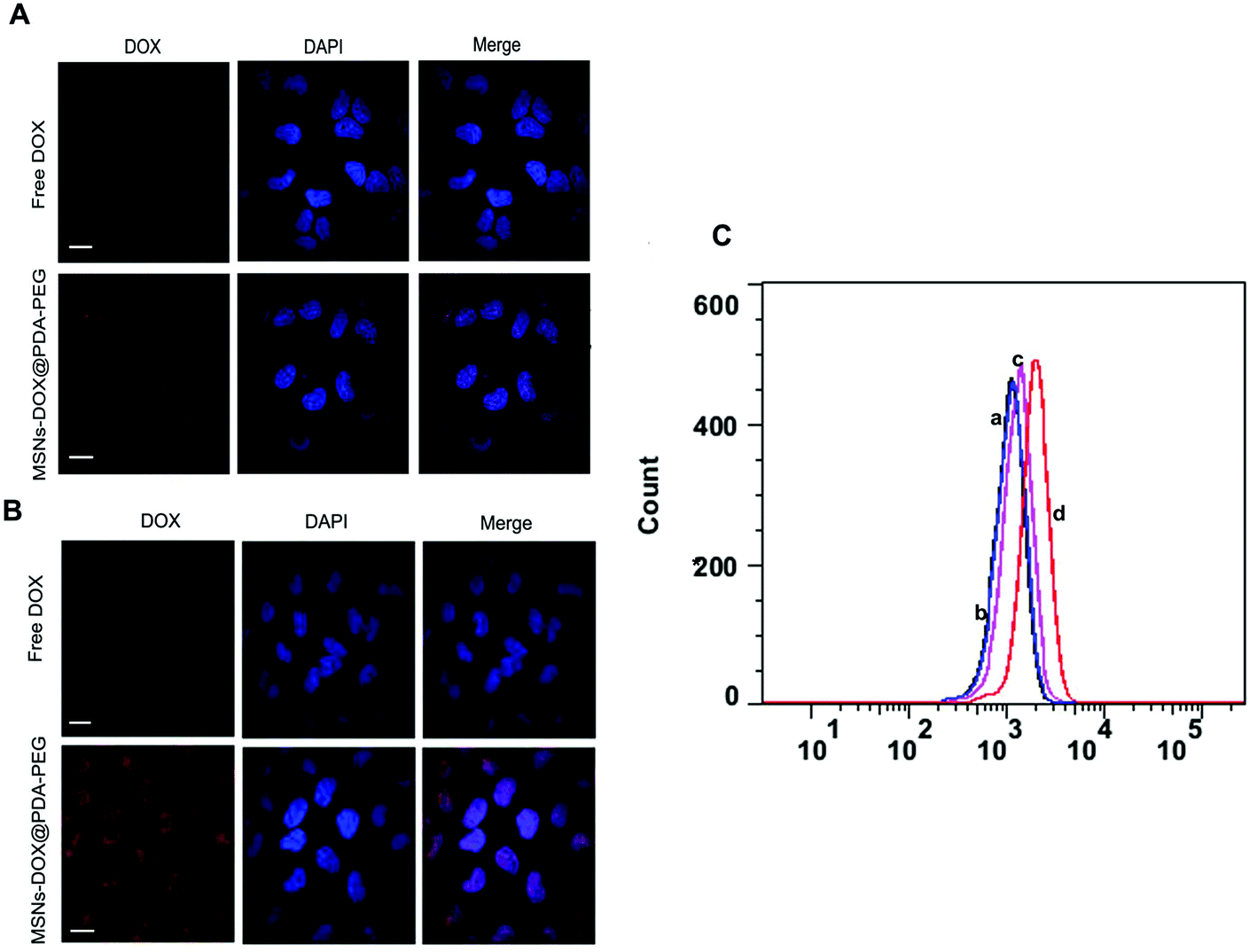 | ||
| Fig. 3 (A) Uptake of free DOX and MSNs-DOX@PDA-PEG detected by confocal microscopy in MCF10A cells (red: DOX; blue: DAPI, scale bar = 10 μm). (B) Same treatment as (A) but using MCF7 cells. (C) Flow cytometry (a) free DOX in MCF10A, (b) free DOX in MCF7, (c) MSNs-DOX@PDA-PEG in MCF10A and (d) MSNs-DOX@PDA-PEG in MCF7 (a – black, b – blue, c – rose red, d – red). | ||
Inhibition of cell growth and proliferation by MSNs-DOX@PDA-PEG
MCF7 and MDA-MB-231 cells (2.6 × 104) were used to study the in vitro cytotoxicity of MSNs-DOX@PDA-PEG. Fig. 4A and B show the in vitro cell viability of the drug formulated in MSNs-DOX@PDA-PEG and DOX at equivalent concentrations of 1, 5, 10, 25 and 35 μg mL−1, respectively. The percentage of viable cells was quantitatively assessed by an MTT method. MSNs or MSNs@PDA-PEG did not exhibit a significant cytotoxicity against MCF7 and MDA-MB-231 cells in vitro. As reported previously, extremely high concentrations of MSNs (about 25 mg mL−1) at some sizes exhibit cytotoxicity.28 However, the required maximum concentration of MSNs herein was 500 μg mL−1, a concentration at which nearly no cytotoxicity could be observed. Similarly to our result, PDA and PEG coating was found to be nontoxic in various cell models and in various in vivo studies.29 | ||
| Fig. 4 (A, B) Viability of MCF7 and MDA-MB-231 cells cultured with MSNs@PDA-PEG, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG, respectively, with different NPs concentrations after 24 h. Data were expressed as mean ± SD (*p < 0.05, **p < 0.01). (C, D) Cell proliferation evaluation of MSNs@PDA-PEG, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG by MTT assay after treatments for 12, 24 and 48 h in MCF7 and MDA-MB-231 cells, respectively. Data were expressed as mean ± SD (*p < 0.05, **p < 0.01). (E) MTT assay of normal breast cell line MCF10A, similar treatment as C and D. (F) BrdU incorporation assay by confocal microscopy (green: BrdU; blue: DAPI, scale bar = 10 μm). (G) Clone formation evaluation of MSNs@PDA-PEG, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG. | ||
MCF7 and MDA-MB-231 cells (2.6 × 104) treated with 10 μg mL−1 MSNs@PDA-PEG, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG for 12, 24 and 48 h, respectively. As shown in Fig. 4C and D, MSNs@PDA-PEG exhibited no significant effect on the cell viability compared with the PBS control, providing further evidence that MSNs@PDA-PEG represents a safe and biocompatible nanocarrier. Both free DOX and MSNs-DOX@PDA exhibited a significant cell growth inhibition while the MSNs-DOX@PDA-PEG demonstrated a stronger inhibition ability to MCF7 and MDA-MB-231 compared to free DOX and MSNs-DOX@PDA. However, for the normal breast cell line MCF10A, both free DOX and MSNs-DOX@PDA exhibited almost no cell growth inhibition as shown in Fig. 4E.
5-Fluorouridine (BrdU), a thymidine analogue, has been crucial in the identification of DNA synthesis and the assessment of cell proliferation. In the present research, a BrdU assay was performed. The MCF7 cells (6 × 106) were treated with 10 μg mL−1 MSNs@PDA-PEG, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG for 6 h and the BrdU incorporation was detected by confocal microscopy. As shown in Fig. 4F, MSNs-DOX@PDA-PEG exhibited the best inhibition effect on BrdU incorporation.
A colony formation assay was also performed as described previously.30 MCF7 cells (1 × 103) were treated with 10 μg mL−1 MSNs@PDA-PEG, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG for 7 days and the obtained colonies were stained with 0.1% crystal violet. As shown in Fig. 4G, similar to the MTT assay, MSNs-DOX@PDA-PEG exhibited a stronger inhibition effect on the colony formation of MCF7 cells compared to other treatments.
Induction of pro-death autophagy through suppression of AKT-mTOR-p70S6K pathway by MSNs-DOX@PDA-PEG
Previously conducted research studies have shown that DOX can induce pro-death autophagy.31 Autophagy primarily represents a degradation pathway that clears malfunctioning cellular components, including intracellular pathogens, in response to various types of stress. LC3 proteins play a critical role in autophagy. Beclin 1 is considered to be related to the initiation and progression of autophagy, albeit the underlying mechanism remains unknown. The autophagy receptor and substrate SQSTM1/p62 inhibits the E3 ligase ubiquitination of histone in response to DNA double-strand breaks. Dysregulation of this process leads to a reduced ability to repair DNA. Some studies have shown evidence for the accumulation of beclin 1 and LC3B-II, but degradation of p62. Meanwhile, the percentage of cells with characteristic LC3B-GFP puncta structure, a characteristic marker of autophagy, was increased following some autophagy induced drug treatments.32 In the present study, we investigated the effects of MSNs@PDA-PEG, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG on autophagy in MCF7 cells. As shown in Fig. 5A, the number of autophagic vesicles (referred to LC3 dotted green fluorescence) was significantly higher in MSNs-DOX@PDA-PEG-treated cells compared to the other groups. Consistently, MSNs-DOX@PDA-PEG treatment induced an increase in LC3-II and p62 levels, however, a decrease in beclin 1 level in MCF7 cells was also observed (cf. Fig. 5B). | ||
| Fig. 5 (A) MCF7 cells were treated with PBS, MSNs@PDA-PEG, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG for 12 h and autophagic vehicles were observed. The cells were imaged under a confocal microscope (blue: DAPI, red: DOX, green: LC3, scale bar = 10 μm). (B) MCF7 cells were treated similarly to (A) and Western blotting was performed with LC3, beclin 1, p62 and β-actin antibodies. (C) MCF7 cells were treated similarly to (A) and Western blotting was performed with the indicated antibodies. | ||
The AKT/mammalian target of rapamycin (mTOR)/p70 ribosomal protein S6 kinase (p70S6K) signaling pathway is known to regulate autophagy.33 To investigate whether MSNs-DOX@PDA-PEG-induced autophagy is involved in the AKT-mTOR-p70S6K signaling pathway, the phosphorylation levels of the associated proteins were studied by Western blotting. As shown in Fig. 5C, all of the treatments with free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG significantly inhibited the phosphorylation levels of AKT, mTOR, and p70S6K, however, MSNs-DOX@PDA-PEG exhibited the best inhibition effect.
Significant inhibition of the MCF7 subcutaneous xenograft tumor growth by MSNs-DOX@PDA-PEG
The anti-tumor efficacy of MSNs-DOX@PDA-PEG was further investigated in nude mice bearing subcutaneous MCF7 tumors. The administration of free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG resulted in significant growth suppression of MCF7 xenografts compared to the PBS control group. As shown in Fig. 6A, tumor growth in the treatment groups was significantly slower than that in the PBS-control group. MSNs-DOX@PDA-PEG demonstrated the best inhibition ability on the xenograft growth. At the end of the experiment, the average tumor weight in the treatment groups was significantly lower than that in the PBS-control group and the MSNs-DOX@PDA-PEG group featured the lowest tumor weight (cf. Fig. 6B and C). Furthermore, an immunohistochemical study showed that the tumor tissues from the treatment of MSNs-DOX@PDA-PEG exhibited the fewest tumor cells and the highest level of tumor necrosis compared to the other treatments (cf. Fig. 6D), indicating a higher anti-tumor activity of MSNs-DOX@PDA-PEG. The latter finding is presumably due to the EPR effect and the controlled release at tumor sites.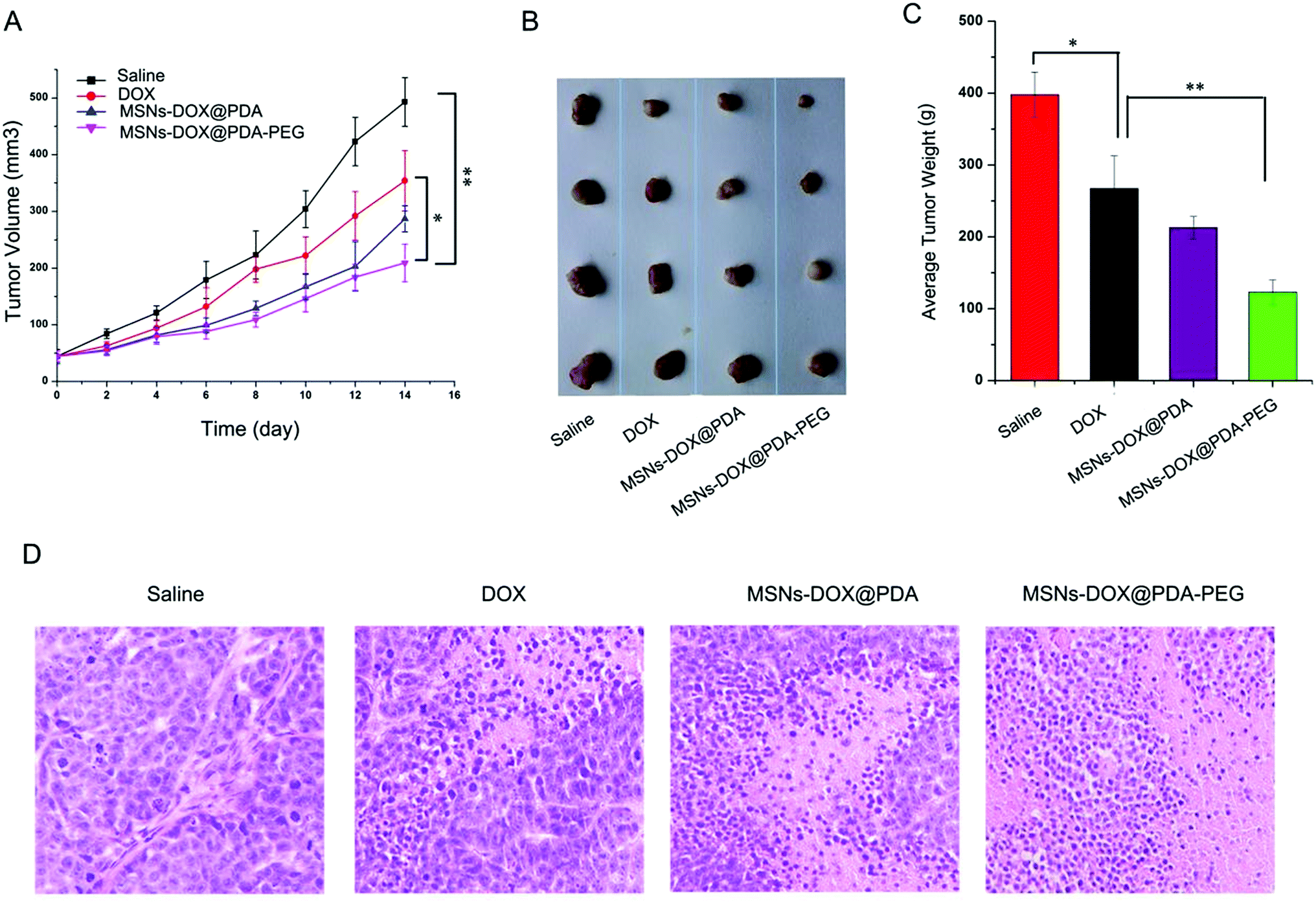 | ||
| Fig. 6 (A) Tumor growth curves of MCF7 xenografts treated with saline, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG, respectively. (B) Xenograft tumors were removed and (C) weighted. Values are provided as mean ± standard error (*p < 0.05, **p < 0.01). (D) Representative H&E staining of xenograft tumors. | ||
The systematic toxicity of MSNs-DOX@PDA-PEG in vivo was evaluated by body weight monitoring and H&E tissue section staining of major organs including heart, liver, spleen, lung, and kidney. No statistically significant differences in body weight could be observed between the MSNs-DOX@PDA-PEG-treated group and other groups (p > 0.05, data not shown) (cf. Fig. 7A) and no noticeable histopathological abnormalities (cf. Fig. 7B) could be observed in MSNs-DOX@PDA-PEG groups suggesting that MSNs-DOX@PDA-PEG featured a good biocompatibility and a low general toxicity in vivo.
 | ||
| Fig. 7 (A) Body weight changes of mice treated with saline, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG. Data is expressed as mean value ± standard error. (B) Representative H&E staining of heart, liver, spleen, lung, kidney. | ||
Conclusions
In summary, a pH-sensitive drug delivery system involving mesoporous silica nanoparticles coated with PDA and PEG was successfully designed for the controlled release of cationic amphiphilic drug DOX. Furthermore, MSNs-DOX@PDA-PEG exhibited a good anti-cancer efficacy through the induction of pro-death autophagy. The obtained results demonstrate that the DOX-loaded MSNs-DOX@PDA-PEG featured competitive advantages, including an excellent pH-sensitivity, suitable cellular uptake and therapeutic efficacy with low side effects (cf. Fig. 8).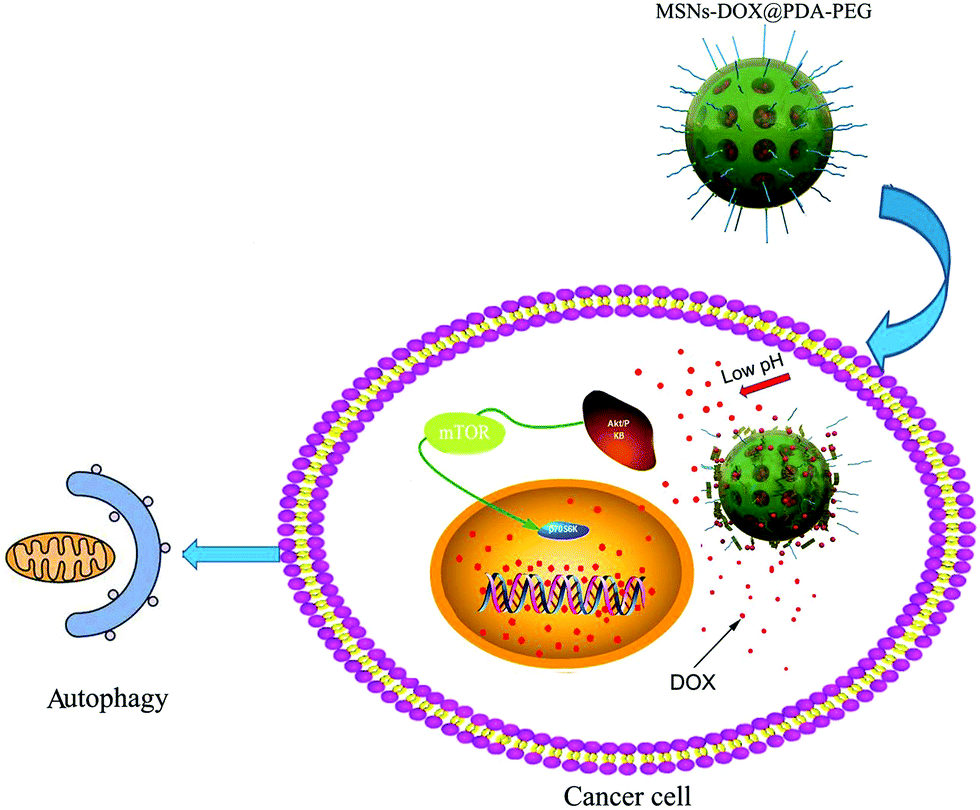 | ||
| Fig. 8 Schematic illustration of the pH-sensitive release of DOX from MSNs-DOX@PDA-PEG triggering pro-death cell autophagy. | ||
Materials and methods
Materials
3-Mercaptopropyltrimethoxysilane (MPTMS, 95%), cetyltrimethyl ammonium bromide (CTAB), tetraethylorthosilicate (TEOS), amino-terminated poly(ethylene glycol) (H2N-PEG, Mw = 2000), hydrochloride dopamine and doxorubicin (DOX) were purchased from J&K Scientific Ltd (Beijing, China). Ammonium fluoride (NH4F) was obtained from Aladdin Industrial Co., Ltd. (Shanghai, China). Acetonitrile was purchased from EM Science (HPLC grade, Mallinckrodt Baker, USA). Dulbecco's modified eagle medium (DMEM), trypsin–EDTA solution (0.25%), fetal bovine serum (FBS) and penicillin-streptomycin were obtained from GIBCO, Invitrogen Co. (Carlsbad, NM, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and bovine serum albumin (BSA) were purchased from Amresco (Solon, OH, USA). 5-Fluorouridine (BrdU) was obtained from Sigma-Aldrich (St. Louis, MO, USA). 6-Diamidino-2-phenylindole (DAPI) was obtained from Biyuntian Co., Ltd (Nanjing, China). BrdU mouse monoclonal antibodies were purchased from Abcam (Cambridge, MA, USA). LC3 antibody (rabbit source) was purchased from Cell Signaling Technology (Beverly, MA, USA). P62 antibody was purchased from Abmart Inc. (Shanghai, China). Beclin 1 was purchased from Cell Signaling Technology (Beverly, MA, USA) AKT, p-AKT, mTOR, p-mTOR, p70s6k, p-p70s6k antibodies were purchased from Santa Cruz (Santa Cruz, CA, USA). Human breast carcinoma cell line MCF7, MDA-MB-231 and the normal breast cell line MCF10A were purchased from American Type Culture Collection (ATCC). Water used throughout the studies was obtained from an ultrapure MilliQ water purification system (resistance > 18 MΩ cm). All other chemicals of the highest available quality were commercially obtained and used as received.Synthesis of MSNs
MSNs were synthesized according to our previously reported procedure with slight modification.34 Briefly, 1.82 g CTAB (5 mM) and 3 g NH4F (81 mM) were dissolved in 500 mL of water and heated to 80 °C in a 1000 mL flask. Under vigorous stirring, 9 mL (8.41 g) TEOS was added dropwise to the mixture and the temperature was then kept at 80 °C for 6 h. The solid product was centrifuged (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 10 min), washed with water and ethanol three times and dried at 40 °C in vacuo. To remove the surfactant template (CTAB), the product was dispersed in 400 mL of ethanol containing 8 mL of hydrochloric acid (37%) and refluxed at 80 °C for 24 h. This procedure was repeated twice to ensure that the surfactant was completely removed. The obtained MSNs were centrifuged and washed with deionized water.
000 rpm, 10 min), washed with water and ethanol three times and dried at 40 °C in vacuo. To remove the surfactant template (CTAB), the product was dispersed in 400 mL of ethanol containing 8 mL of hydrochloric acid (37%) and refluxed at 80 °C for 24 h. This procedure was repeated twice to ensure that the surfactant was completely removed. The obtained MSNs were centrifuged and washed with deionized water.
Preparation of MSNs-DOX@PDA-PEG
For DOX loading, 50 mg MSNs were added to a water solution containing doxorubicin hydrochloride (2.5 mL, 10 mg mL−1) and the mixture was stirred for 24 h. The solution was centrifuged and washed with water to move the remaining DOX from the surface of MSNs. DOX-loaded MSNs (MSNs-DOX) were dried at 40 °C in vacuo. Polydopamine-coated NPs (MSNs-DOX@PDA) were synthesized by incubating 50 mg of MSNs-DOX in 0.5 mg mL−1 dopamine hydrochloride in Tris buffer (10 mM, pH 8.5) for 6 h at room temperature with shaking. Then, MSNs-DOX@PDA was centrifuged (12000 rpm, 10 min) and washed with water to remove any unpolymerized dopamine. The mixture was then added to 2.5 mg NH2-PEG and the resulting material was stirred for 3 h in the dark at room temperature. PEG was employed to coat MSNs in order to ensure high physiological stability.35 Finally, PEG and PDA-coated MSNs-DOX (MSNs-DOX@PDA-PEG) were centrifuged, washed three times with water and dried at 40 °C in vacuo.
The sample was dropped onto a copper grid coated with a carbon membrane and allowed to dry. Then, the surface morphology of MSNs-DOX@PDA-PEG was observed by transmission electron microscopy (TEM, Tecnai G2 20, FEI Company, USA). Drug release from MSNs-DOX@PDA-PEG was determined as described previously.36,37
Cell uptake of MSNs-DOX@PDA-PEG
MCF7 cells were cultured in DMEM supplemented with 10% (v/v) FBS and antibiotics (100 U mL−1 penicillin and 100 μg mL−1 streptomycin) in a humidified 5% CO2 atmosphere at 37 °C. MCF7 cells were incubated with free DOX and MSNs-DOX@PDA-PEG (equal quantity of DOX) at 37 °C for 0.5 h, washed with cold PBS three times, and then fixed by 4% paraformaldehyde for 20 min. Then, the cells were washed with PBS, stained with 4,6-diamidino-2-phenylindole (DAPI) for 15 min and observed by confocal laser scanning microscopy (CLSM, Olympus Fluoview FV-1000, Japan) with imaging software. The images of the cells were determined with differential interference contrast channel, blue channel (DAPI) excitation at 358 nm and red channel (DOX) excitation at 543 nm.38,39Immunofluorescence observations
MCF7 cells seeded on coverslips were treated with MSNs@PDA-PEG, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG, respectively. Cells were fixed with 4% formaldehyde in PBS for 15 min and then permeabilized in PBS containing 0.1% Triton X100 for another 7 min. The cells were blocked with 3% BSA for 2 h at RT and probed with an appropriate primary antibody overnight at 4 °C. The coverslips were then incubated with rhodamine- and fluorescein-conjugated secondary antibodies for 2 h at RT. After staining with 0.5 μg mL−1 of DAPI for 10 min, the cells were observed and imaged using an Olympus FV1000 confocal microscope.MTT cell proliferation assay
Briefly, 1 × 105 MCF7 cells per well seeded in triplicate into a 96-well plate were treated with certain concentrations of free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG. At the appropriate time-points, 20 μL MTT (5 mg mL−1) was added for 4 h at 37 °C and all liquid was carefully removed. Optical density (OD) values were measured with a spectrophotometer at 490 nm following continuous agitation for 20 min with 130 μL DMSO.Colony formation assay
MCF7 cells were seeded into a six-well plate (1 × 103 cells per well) and cultured in DMEM with 10% bovine serum albumin. MCF7 cells were treated with MSNs@PDA-PEG, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG, respectively. After 7 days, the resulting colonies were stained with 0.01% of crystal violet and images were captured.BrdU cell proliferation assay
MCF7 cells seeded in 12-well plates were treated with MSNs@PDA-PEG, free DOX, MSNs-DOX@PDA and MSNs-DOX@PDA-PEG for 6 h, respectively. Cells were incubated with 200 mM 5-fluorouridine for 30 min. The medium was removed and the cells were fixed with 4% formaldehyde in PBS for 15 min and permeabilized in PBS containing 0.1% Triton X100 for 7 min. The cells were probed with a BrdU antibody after blocking with 3% BSA and assessed for BrdU incorporation by confocal microscopy.Western blotting
The cells were lysed with lysis buffer and equal amounts of proteins were separated by 10% SDS-PAGE and transferred onto a PVDF membrane. The membrane was blocked with 5% non-fat milk in Tris-buffered saline containing Tween 20 for 2 h at room temperature and incubated overnight at 4 °C with specific primary antibodies. After washing for a total of three times with TBST, the membrane was incubated with appropriate HRP-linked secondary antibodies for 2 h at room temperature and then detected with ECL reagent.Xenograft tumor growth assay
This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication no. 85-23 Rev. 1985) and was approved by the Institutional Animal Care and Use Committee of Tsinghua University. 4–5 week old female nude mice were purchased from the Medical Experimental Animal Centre of Guangdong Province. All animal experiments were approved by the Institutional Animal Care and Use Committee of Tsinghua University. The mice were housed in a specific-pathogen-free environment maintained at 25 ± 1 °C with 55% relative humidity and food as well as water were provided. The mice were randomly allocated into 4 groups with 4 animals per group. MCF7 cells (5 × 105 in 150 μL PBS) in sub-confluent condition were subcutaneously injected into the mice. Six days after tumor inoculation, the mice were administered a daily intraperitoneal injection of DOX (0.5 mg kg−1). Tumor volumes were measured every three days with a caliper and calculated according to the formula: V = 1/2(L × W2), where L and W represent the length and width, respectively. All mice were sacrificed 20–30 days after tumor inoculation and the tumors were excised and weighed.Statistical analysis
Values are expressed as the mean ± standard deviation of three independent experiments. Comparisons were performed using a two-tailed paired Student's t-test. Differences with p < 0.05 were considered significant.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 81670141), Guangdong Natural Science Foundation (No. 2014A030313758) and Science, Technology & Innovation Commission of Shenzhen Municipality (No. JCYJ20160422170206664).References
- R. L. Siegel, K. D. Miller, S. A. Fedewa, D. J. Ahnen, R. G. Meester, A. Barzi and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 177–193 CrossRef PubMed.
- M. A. Firer and G. Gellerman, J. Hematol. Oncol., 2012, 5, 70 CrossRef CAS PubMed.
- Y. Oh, J.-O. Jin and J. Oh, Nanotechnology, 2017, 28, 125101 CrossRef PubMed.
- O. Tacar, P. Sriamornsak and C. R. Dass, J. Pharm. Pharmacol., 2013, 65, 157–170 CrossRef CAS PubMed.
- A. K. Jain, N. K. Swarnakar, M. Das, C. Godugu, R. P. Singh, P. R. Rao and S. Jain, Mol. Pharm., 2011, 8, 1140–1151 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- Y. Chen, X. Deng, C. Li, F. He, B. Liu, Z. Hou, Z. Cheng, B. Xing and J. Lin, Nanomedicine: Nanotechnology, Biology and Medicine, 2017, 13, 875–883 CrossRef CAS PubMed.
- Y. Guo, J. Sun, S. Bai and X. Jin, J. Biomater. Appl., 2016, 0885328216653287 Search PubMed.
- N. Song and Y.-W. Yang, Chem. Soc. Rev., 2015, 44, 3474–3504 RSC.
- X. Yao, X. Niu, K. Ma, P. Huang, J. Grothe, S. Kaskel and Y. Zhu, Small, 2017, 13 Search PubMed.
- N. Li, N. Li, Q. Yi, K. Luo, C. Guo, D. Pan and Z. Gu, Biomaterials, 2014, 35, 9529–9545 CrossRef CAS PubMed.
- W. Fang, J. Yang, J. Gong and N. Zheng, Adv. Funct. Mater., 2012, 22, 842–848 CrossRef CAS.
- W. Gao, W. Cao, Y. Sun, X. Wei, K. Xu, H. Zhang and B. Tang, Biomaterials, 2015, 69, 212–221 CrossRef CAS PubMed.
- W. Tao, X. Zeng, J. Wu, X. Zhu, X. Yu, X. Zhang, J. Zhang, G. Liu and L. Mei, Theranostics, 2016, 6, 470 CrossRef CAS PubMed.
- J. Hu, X. Zhang, Z. Wen, Y. Tan, N. Huang, S. Cheng, H. Zheng and Y. Cheng, Oncotarget, 2016, 7, 73681 Search PubMed.
- L. Palanikumar, E. S. Choi, J. Y. Cheon, S. H. Joo and J. H. Ryu, Adv. Funct. Mater., 2015, 25, 957–965 CrossRef CAS.
- M. Costanzo, F. Carton, A. Marengo, G. Berlier, B. Stella, S. Arpicco and M. Malatesta, Eur. J. Histochem., 2016, 60 Search PubMed.
- X. Zhang, Y. Dong, X. Zeng, X. Liang, X. Li, W. Tao, H. Chen, Y. Jiang, L. Mei and S.-S. Feng, Biomaterials, 2014, 35, 1932–1943 CrossRef CAS PubMed.
- T. S. Hnasko and R. H. Edwards, Annu. Rev. Physiol., 2012, 74, 225–243 CrossRef CAS PubMed.
- X. Zhu, Y. Xu, L. M. Solis, W. Tao, L. Wang, C. Behrens, X. Xu, L. Zhao, D. Liu and J. Wu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7779–7784 CrossRef CAS PubMed.
- C. Zhang, W. Wang, T. Liu, Y. Wu, H. Guo, P. Wang, Q. Tian, Y. Wang and Z. Yuan, Biomaterials, 2012, 33, 2187–2196 CrossRef CAS PubMed.
- S. K. Hobbs, W. L. Monsky, F. Yuan, W. G. Roberts, L. Griffith, V. P. Torchilin and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4607–4612 CrossRef CAS.
- W. Ngamcherdtrakul, J. Morry, S. Gu, D. J. Castro, S. M. Goodyear, T. Sangvanich, M. M. Reda, R. Lee, S. A. Mihelic and B. L. Beckman, Adv. Funct. Mater., 2015, 25, 2646–2659 CrossRef CAS PubMed.
- T. Li, X. Shen, Y. Geng, Z. Chen, L. Li, S. Li, H. Yang, C. Wu, H. Zeng and Y. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 13748–13758 CAS.
- M. Zhang, J. Liu, Y. Kuang, Q. Li, D.-W. Zheng, Q. Song, H. Chen, X. Chen, Y. Xu and C. Li, Int. J. Biol. Macromol., 2017, 98, 691–700 CrossRef CAS PubMed.
- Q. Zheng, T. Lin, H. Wu, L. Guo, P. Ye, Y. Hao, Q. Guo, J. Jiang, F. Fu and G. Chen, Int. J. Pharm., 2014, 463, 22–26 CrossRef CAS PubMed.
- Q. He, Z. Zhang, Y. Gao, J. Shi and Y. Li, Small, 2009, 5, 2722–2729 CrossRef CAS PubMed.
- J. Park, T. F. Brust, H. J. Lee, S. C. Lee, V. J. Watts and Y. Yeo, ACS Nano, 2014, 8, 3347–3356 CrossRef CAS PubMed.
- H. Chen, Y. Duo, B. Hu, Z. Wang, F. Zhang, H. Tsai, J. Zhang, L. Zhou, L. Wang and X. Wang, Oncotarget, 2016, 7, 78747 Search PubMed.
- B. Sivaraman, G. Swaminathan, L. Moore, J. Fox, D. Seshadri, S. Dahal, I. Stoilov, M. Zborowski, R. Mecham and A. Ramamurthi, Acta Biomater., 2017, 52, 171–186 CrossRef CAS PubMed.
- Q. Zhang, M. Yang, Z. Qu, J. Zhou and Q. Jiang, Biochem. Biophys. Res. Commun., 2016, 480, 334–340 CrossRef CAS PubMed.
- B. Cao, J. Li, X. Zhou, J. Juan, K. Han, Z. Zhang, Y. Kong, J. Wang and X. Mao, Sci. Rep., 2014, 4, 5749 CrossRef CAS PubMed.
- D. Chang, Y. Gao, L. Wang, G. Liu, Y. Chen, T. Wang, W. Tao, L. Mei, L. Huang and X. Zeng, J. Colloid Interface Sci., 2016, 463, 279–287 CrossRef CAS PubMed.
- Y. Cui, H. Dong, X. Cai, D. Wang and Y. Li, ACS Appl. Mater. Interfaces, 2012, 4, 3177–3183 CAS.
- Z. Zhang and S.-S. Feng, Biomaterials, 2006, 27, 4025–4033 CrossRef CAS PubMed.
- X. Liang, Y. Yang, L. Wang, X. Zhu, X. Zeng, X. Wu, H. Chen, X. Zhang and L. Mei, J. Mater. Chem. B, 2015, 3, 9383–9396 RSC.
- M. Zheng, C. Yue, Y. Ma, P. Gong, P. Zhao, C. Zheng, Z. Sheng, P. Zhang, Z. Wang and L. Cai, ACS Nano, 2013, 7, 2056–2067 CrossRef CAS PubMed.
- C. Zheng, J. Xu, X. Yao, J. Xu and L. Qiu, J. Colloid Interface Sci., 2011, 355, 374–382 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05135b |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |