
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mn–Fe bi-metal oxides in situ created on metal wire mesh as monolith catalysts for selective catalytic reduction of NO with NH3†
Jie Liu,
Lin Kang,
Hongrui Li,
Phornphimon Maitarad,
Jianping Zhang,
Liyi Shi and
Dengsong Zhang *
*
Research Center of Nano Science and Technology, Shanghai University, Shanghai 200444, China. E-mail: dszhang@shu.edu.cn; Tel: +86-21-66137152
First published on 18th August 2017
Abstract
In this work, we use an Fe wire mesh to provide homogeneous nucleation sites to support a continuous in situ growth of Mn–Fe bi-metal oxides as monolith catalysts for selective catalytic reduction of NO with NH3. The strategy of a “twin iron source” makes the Mn–Fe seeds easily grow on the Fe wire mesh through the surface Fe metal sites rather than free growth. The Fe wire mesh exhibited excellent affinity properties with Mn–Fe hydroxides precursor. Through the calcination treatment, a spinel structure of Mn–Fe bi-metal oxides coated monolithic catalyst was prepared and used for denitrification. By adjusting the ratio of the Mn–Fe precursors, we obtained Mn–Fe bi-metal oxides with various morphologies coated on the surface of Fe wire mesh. Impressively, the cube-like Mn–Fe bi-metal oxides structure on the Fe wire mesh as monolith catalysts exhibited high De-NOx performance, catalytic activity, stability, H2O tolerance, K+ poisoning resistance and regeneration performance. The results showed that the spinel structure of Mn–Fe bi-metal oxides in the coating layer was the critical factor for enhanced adsorption behaviours and reducibilities, which promoted selective catalytic reduction of NO with NH3. The good adhesion between the Mn–Fe spinel and Fe wire mesh contributed to the super stability and the strong adsorption properties of NH3, which made it dominant in the NH3 adsorption process competing with water. The obtained monolith catalysts showed good resistances to K+ poisoning and good regeneration performance, which can be attributed to the structural stability of Mn–Fe spinel and strong synergistic effect between the support and active species. This new kind of monolithic catalyst prepared by an in situ technique can be used as a potential substitute for vanadium based ceramic catalysts.
1. Introduction
Nitrogen oxides (NOx) are a major kind of pollutant emitted from stationary sources that can lead to various environmental problems, such as acid rain, greenhouse effects and photochemical smog.1–4 The selective catalytic reduction of NO with NH3 (NH3-SCR) is an efficient and economic commercial technique for denitrification (De-NOx).5–7 The V-based catalysts are commonly used to eliminate NOx in stationary sources, and they exhibit an excellent catalytic activity and selectivity.8–10 However, the high and narrow operation temperature window (300–400 °C), susceptibility to alkali poisoning, as well as the volatility and toxicity of VOx inhibit their enormous applications.11–15Recently, manganese based catalysts have stimulated great interest by virtue of their excellent oxidation–reduction properties, environmentally friendly features and low-temperature activities, which render their great potential in NH3-SCR application.16–27 Chen et al. investigated the Cr–Mn mixed-oxide catalysts and achieved 98.5% NOx conversion at 120 °C, which can be attributed to the strong interaction between the active components in Cr1.5MnO4 spinel structure.23 Zhan et al. prepared a kind of Mn-FeOx hexagonal microsheet for low temperature NH3-SCR. It was found that this catalyst exhibited above 90% NOx conversion between 150 and 300 °C and excellent H2O resistance, as well as thermal stability.28 Cui et al. also prepared Mn–Ce–Ni/TiO2 catalyst for low temperature NH3-SCR of NO by introducing a co-precipitation method, which exhibited a 88% NO conversion in the presence of alkali metals between 150 and 300 °C (K/Mn = 0.5).29 In our previous studies, the MnxCo3−xO4 nanocage catalyst was developed for the NOx removal and the catalyst exhibited a wide operation window from 120 °C to 350 °C.18 The MnOx–FeOy nanocage achieved 87.8% NOx conversion at 80 °C.30 However, the catalytic performance of the above manganese based catalysts for the practical De-NOx application is still far from satisfactory. This can be ascribed to the following factors. Generally, the powdered catalysts should be coated on the surface of supports to achieve high specific surface area and low pressure drop in industrial utilization. However, the commonly used ceramic honeycomb supports suffer from the poor adhesion stability between the powdered catalysts and the support, which inevitably leading to the leakage of active species on the surface during the catalytic process and the deactivation of the monolithic catalyst.31,32 In addition, the ceramic supports also possess certain defects of poor interphase heat transfer ability, low mechanical strength and random distribution of catalyst powder.33,34
To overcome those disadvantages, some metal supported monolith catalysts have been designed and developed, which have such advantages as strong mechanical stability, plasticity, thermal conductivity and mass transfer ability.26,27 It is favorable for the catalytic processes and suitable for practical applications.35–41 In order to construct a stable bi-metal oxides layer in the support of wire mesh, an in situ growth technique has been effectively introduced. For this method, heterogeneous nucleation and following crystallite intergrowth are indispensable. Therefore, the strategy of “twin metal source” will be a viable solution.42 Good affinity between the same metal precursor and the wire mesh makes the seed crystals easily grow on metal wire mesh through the surface of metal sites rather than free growth.
Herein, we developed a “twin iron source” strategy for the in situ construction of Mn–Fe oxides on Fe wire mesh and the formation route of the Mn–Fe oxides was shown in Fig. 1. First, Fe wire meshes that removed surface oxide were immersed into the Mn–Fe precursor solution for hydrothermal reaction at 90 °C for 6 h. In the course of the decomposition of urea, the Mn–Fe species were heterogeneous on the surface of the Fe wire mesh by the iron sites and then form the seed crystals. The subsequent growth of seed crystals could lead to form different morphologies and uniformity distribution Mn–Fe bi-metal hydroxides particles by adjusting the ratio of the precursors. Finally, stable and uniform distribution Mn–Fe bi-metal oxides formed on the surface of the Fe wire mesh after the calcination process. This in situ coating technology not only realized the stable decoration of Mn–Fe hydroxides precursors on the surface of the Fe wire mesh, but also ensured the uniform dispersion of the active species. The formation of the bi-metal oxide layer mainly contained spinel structure of Mn–Fe bi-metal oxides species, which showed excellent catalytic activity, stability, H2O tolerance, K+ poisoning resistance and regeneration performance.
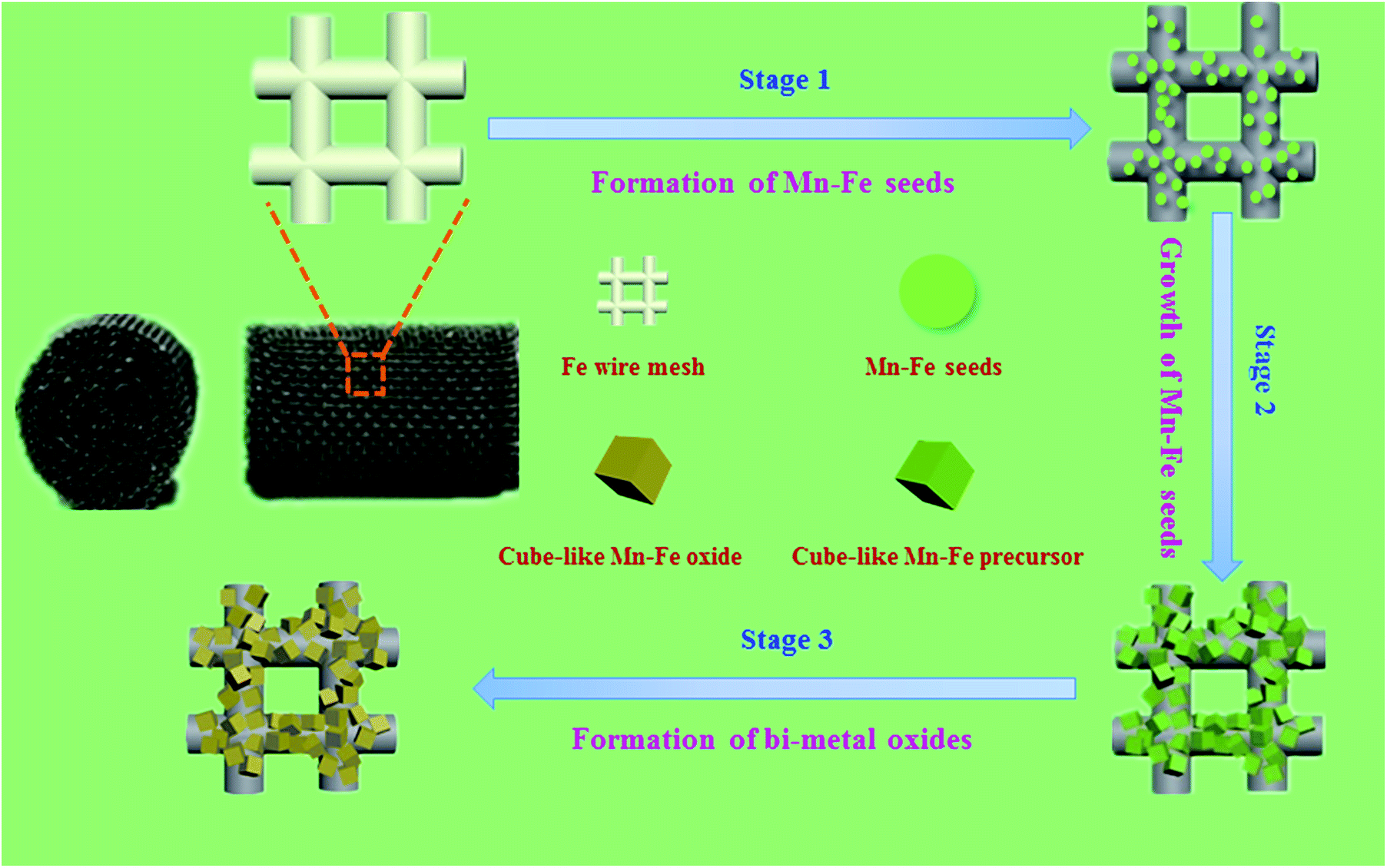 | ||
Fig. 1 Schematic illustration of the fabrication of Mn–Fe(1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)@Fe WM monolithic catalyst. :1)@Fe WM monolithic catalyst. | ||
2. Experimental section
Catalysts preparation
Commercial iron meshes and titanium meshes with the mesh pore size of 0.180 mm were provided by Shanghai Fine Sieving Filtrating Equipment Co., Ltd (China). While nickel foam and copper foam with the porosity of 95–98% were supplied by Ailantian Advanced Technology Materials Co. Ltd (Dalian, China). These metal substrates were separated into desired scales and sizes (3 cm × 10 cm), and then pretreated with 1 M HCl to remove the surface oxides. Afterwards, the metal substrates were washed with deionized water to eliminate residual acids.The Mn–Fe(x:y)@Fe wire mesh monolithic catalysts were fabricated using a water bath deposition method with urea. The x:y was the molar ratio of manganese and iron. A typical example of catalyst preparation was presented as following steps. 0.005 mol Mn(Ac)2·4H2O, 0.005 mol Fe(NO3)3·9H2O and 0.03 mol CO (NH2)2 were dissolved in 70 ml of deionized water with continuous stirring. After completely dissolved, the Fe wire mesh (3 cm × 10 cm) was immersed into the solution and then transferred the reaction system to a 100 ml Teflon-lined stainless steel autoclave and reacted at 90 °C for 6 h. After the reaction, the autoclave was cooled at room temperature. The product was washed by deionized water for several times and dried at 80 °C. The in situ coated Mn–Fe bi-metal oxides wire mesh precursor was calcined at 500 °C for 4 h in muffle furnace with a heating rate of 2 °C min−1. Finally, we got Mn–Fe(1:1)@Fe wire mesh (Mn–Fe(1:1)@Fe WM) monolithic catalyst. As contrast, Mn–Fe(2:1)@Fe wire mesh (Mn–Fe(2:1)@Fe WM), Mn–Fe(1:2)@Fe wire mesh (Mn–Fe(1:2)@Fe WM), Mn@Fe wire mesh (Mn@Fe WM), Fe@Fe wire mesh (Mn@Fe WM), Mn–Fe(1:1)@honeycomb ceramics (Mn–Fe(1:1)@HCCS) and Mn–Fe(1:1)@other common metal substrates (Cu wire mesh, Ti wire mesh, Ni form and Cu form) monolithic catalysts were prepared using the similar conditions.
Catalyst characterization
The morphology and structure of the nanocatalysts were characterized by a scanning electron microscope (SEM, JEOL JEM-200CX). The X-ray diffraction (XRD) patterns were recorded on a Rigaku D/MAS-RB X-ray diffract meter, using Cu-Kα radiation operated at 40 kV and 40 mA. Thermo gravimetric analysis (TGA) was performed on a NETZSCH STA 449 F1. The temperature was increased from 100 to 600 °C with a ramping rate of 10 °C min−1 in N2. H2 temperature-programmed reduction (H2-TPR) was carried out on a Tianjin XQ tp5080 autoadsorption apparatus. In a typical run, 500 mg of monolithic catalyst was used and heated to 300 °C under N2 (30 ml min−1) to remove any adsorbed species for 30 min with a temperature rise rate of 10 °C min−1. After cooling down to 25 °C, the catalyst was exposed to 5% H2/N2 (30 ml min−1) and the temperature was subsequently raised from 25 to 900 °C with a ramping rate of 10 °C min−1. NH3 temperature-programmed desorption (NH3-TPD) was carried out on a Tianjin XQ tp5080 autoadsorption apparatus. In a typical run, 1000 mg of monolithic catalyst was used and heated to 300 °C under He (30 ml min−1) to remove any adsorbed species for 30 min with a rising rate of 10 °C min−1. After cooling down to 100 °C, the catalyst was exposed to 500 ppm NH3 (30 ml min−1) for 1 h, followed by He purging for 0.5 h to remove physisorbed NH3, and the temperature was subsequently raised from 100 to 800 °C with a ramping rate of 10 °C min−1.Catalyst performance tests
The NH3-SCR activity was tested in a fixed-bed quartz reactor (8 mm i.d.) using 3 cm × 10 cm of the catalysts. The feed gas mixture in a N2 stream contained 500 ppm of NO, 500 ppm of NH3, and 3 vol% O2. The total flow rate of feed gases was 260 ml min−1 with the gas hourly space velocity (GHSV) of 20000 h−1. The concentration of NO in the inlet and outlet gases was got continuously by a 4000VM analyzer. All the data points were collected in a steady state for 15 min at the corresponding temperature. NO conversion was calculated according to the following equation:
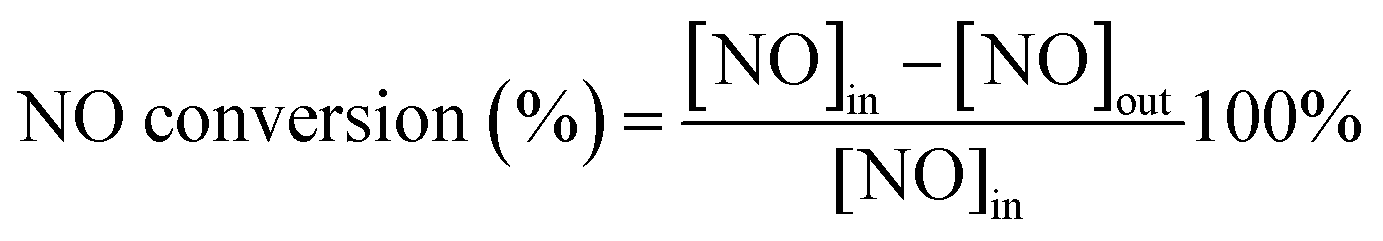 | (1) |
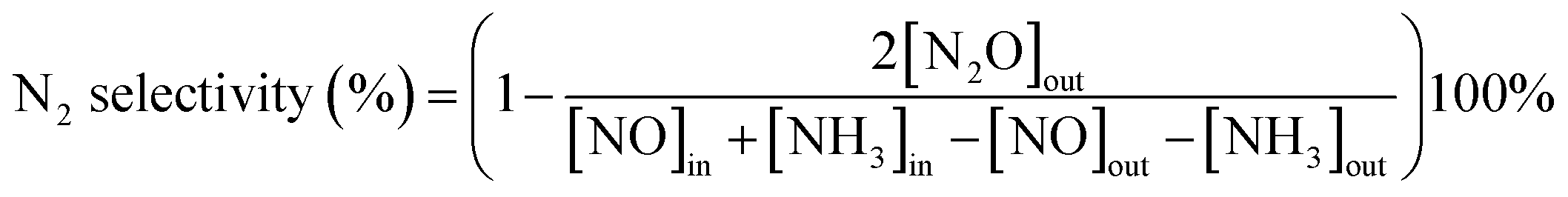 | (2) |
In the water resistance test, the water was pumped into the vaporization chamber before the reactor with a controlled flow water pump. The water was heated and vaporized at 200 °C and then was passed through the reactor. In the K+ resistance test, the catalyst was immersed in potassium nitrate solution containing K+ with 0.3 wt% of the total mass of the monolith catalyst. Then, the solution was steamed to dry and the sample was dried at 80 °C and calcined for 2 h at 500 °C. The catalysts after poisoning were named K-poisoning-Mn–Fe(1:1)@Fe WM, K-poisoning-Mn–Fe(1:2)@Fe WM and K-poisoning-Mn–Fe(2:1)@Fe WM, respectively. For the regeneration, the corresponding catalyst was soaked in a 100 ml water at 60 °C for 30 minutes and repeated three times.
3. Results and discussion
Morphology evolution of Mn–Fe@Fe wire mesh catalysts
In order to confirm the composition of different structures and understand the spatial distribution of the corresponding elements, SEM and EDS mapping analysis were performed to probe surface morphologies of three monolithic catalysts and the results were shown in Fig. 2 and S1.† Through SEM observation, we found that Mn–Fe(1:1)@Fe WM monolithic catalyst was covered with cube-like bi-metal layer. The surface morphologies of Mn–Fe(2:1) and Mn–Fe(1:2) bi-metal oxides were mainly of block-like shape. The morphologies of these particles on the surface of these monolithic catalysts were obviously different due to the different ratio of the manganese and iron precursors. According to the EDS mapping images of Mn–Fe(1:1)@Fe WM, the cubic structure in wire mesh surface were composed of rich Mn and Fe elements, which were highly dispersed and uniform. At the same time, in the EDS mapping images of Mn–Fe(2:1)@Fe WM, it also displayed a highly dispersed and uniform distribution of Mn and Fe elements in the block-like shape structure. But there were more Mn elements than Fe elements. It was worth noting that there was almost no Mn species in the small globular morphology of Mn–Fe(1:2)@Fe WM, which indicated that the deposition of Mn species was very little in this feed ratio of Mn and Fe species. The reason for this proportion of different elements was the different ratio of the manganese and iron precursors.
 | ||
| Fig. 2 (a–c) SEM images of Fe wire mesh and surface structure of Mn–Fe(1:1)@Fe WM monolithic catalyst; (d–f) SEM images and elemental mappings results of Mn–Fe(1:1)@Fe WM monolithic catalyst. | ||
In order to analyse the crystal structure of Mn–Fe precursor and bi-metallic oxides on three monolithic catalysts, we conducted XRD tests. To reduce the impact of the Fe substrate, we obtained the precursor and bi-metal oxide powder on the surface of the wire mesh. In the XRD patterns of Mn–Fe(1:1) precursors (Fig. S2†), we found that the precursor mainly contains Mn(OH)2 (JCPDs: 12-0696), Fe2O3·1.2H2O (JCPDs: 22-1117) and Fe2O3 (JCPDs: 04-0755). The material composition of hydroxides provided an important material basis for the formation of Mn–Fe bi-metal. In the XRD patterns of bi-metal oxide (Fig. 3), it could be observed two crystalline phases. The peaks located at 24.2°, 33.3°, 41.0°, 49.5°, 53.7°, 64.1° and 72.1° correspond to the spinel structure of (Mn0.37Fe0.63)2O3 (JCPDs: 71-0637). The peaks located at 18.2°, 30.3°, 35.7°, 37.2°, 43.3°, 53.7°, 57.2°, 62.9° and 72.1° corresponding to the Fe3O4 (JCPDs: 75-0033). It was visually observable that there were similar characteristic peaks in Mn–Fe(1:1)@Fe WM, Mn–Fe(2:1)@Fe WM and Mn–Fe(1:2)@Fe WM catalysts, indicating the similar features of composition and structure in these catalysts. It is worth mentioning that the intensity of (Mn0.37Fe0.63)2O3 was the highest in Mn–Fe(1:1)@Fe mesh and the lowest in Mn–Fe(1:2)@Fe mesh, which coincided with the activity curve trend. It indicated that the ratio of 1:1 of Mn and Fe was more favourable for the formation of (Mn0.37Fe0.63)2O3 and participated in the NH3-SCR reaction.
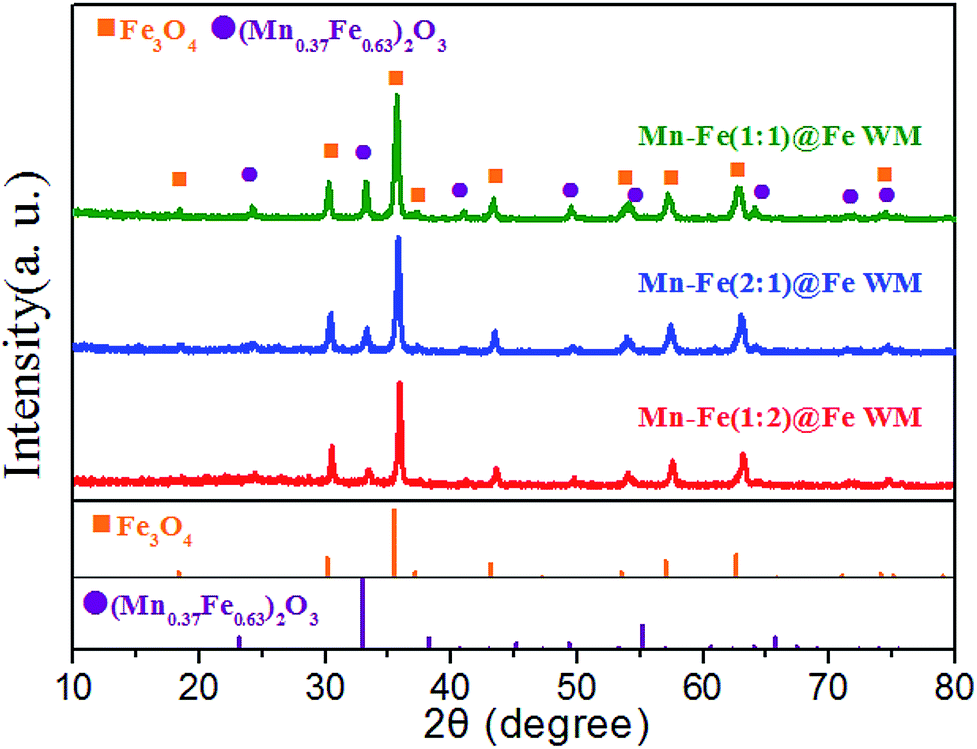 | ||
| Fig. 3 XRD patterns of the powder obtained from the Mn–Fe@Fe WM catalysts. | ||
Surface properties of Mn–Fe@Fe wire mesh catalysts
The TGA was used to analyse the thermal decomposition behaviour of the monolith precursors through hydrothermal reaction. The TGA results showed that the monolith precursors decomposed completely at about 450 °C, which gave us a guideline for the calcination temperature of these monolith precursors (Fig. S3†). Therefore, it could be ensured that the precursors were completely decomposed at 500 °C. In addition, the weight loss ratio of Mn–Fe(1:1)@Fe was the highest, reaching −2.72%. It suggested that this proportion of Mn–Fe species was the most conducive to the growth of bi-metallic oxide precursor on the Fe wire mesh.
NH3-TPD adsorption properties
The absorption of NH3 onto the catalyst plays a significant role in the SCR reaction.29,43 The adsorption behaviour of ammonia on Fe wire mesh monolith catalysts were studied by NH3-TPD method as shown in Fig. 4. It could be observed in Fig. 4 that the three kinds of monolith catalysts showed similar ammonia adsorption sites. The desorption peaks before 250 °C were attributed to the weak adsorption of ammonia in the Brønsted acid sites. At the same time, desorption peaks from 350 to 550 °C were ascribed to the strong adsorption of ammonia in the Lewis acid sites. The area of desorption peaks in the NH3-TPD profile could reflect the amounts of NH3 species coordinated to the surface acid sites.44 Mn–Fe(1:1)@Fe and Mn–Fe(1:2)@Fe WM monolith catalysts showed the best and least amount of ammonia desorption. This trend was positively correlated with the intensity of the peak of (Mn0.37Fe0.63)2O3, which indicated that the main adsorption site of the monolith catalyst was (Mn0.37Fe0.63)2O3 site. It confirmed the superior ammonia storage capacity onto the surfaces of Mn–Fe(1:1)@Fe WM, which was favorable for the NH3-SCR reaction.
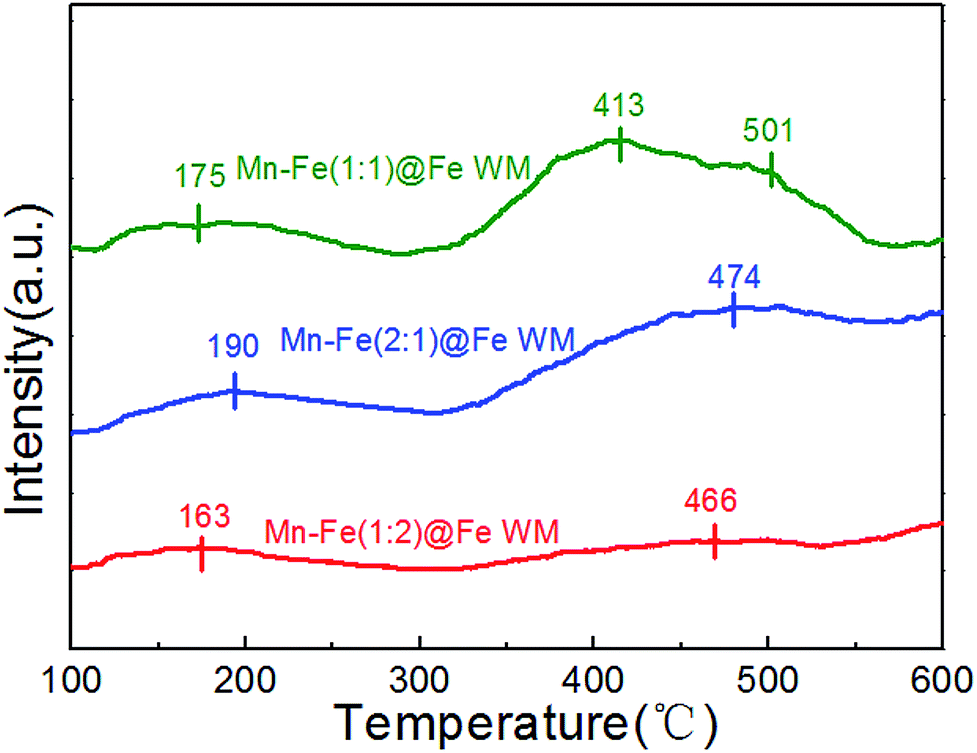 | ||
| Fig. 4 NH3-TPD profiles of Mn–Fe@Fe WM catalysts. | ||
The adsorption behaviours of ammonia on monolith catalysts with other metal supports were different due to the unfavorable formation of active sites on the supports (Fig. S4†). It could be observed that there was hardly any NH3 desorption peak in Mn–Fe(1:1)@Cu WM and Mn–Fe(1:1)@Ti WM catalysts, which was attributed to the unfavorable growth of Mn–Fe species on other metal supports.
H2-TPR reduction performances
The redox properties of the catalysts were crucial to the NH3-SCR reaction. Therefore, we applied H2-TPR to understand the redox process of Mn–Fe@Fe WM monolith catalysts, which were shown in Fig. 5. These reduction steps on the Mn–Fe(1:1)@Fe WM corresponded to the peaks at 386 °C, 433 °C, and 680 °C. While the Mn–Fe(2:1)@Fe WM resulted in the peaks at 352 °C, 430 °C and 649 °C. This represented the reduced of (Mn0.37Fe0.63)2O3, Fe3O4 → FeO, as well as the further reduced of low state of Mn and Fe species.45–47 It was worth noting that the Mn–Fe(1:2)@Fe WM only contained two peaks at 432 °C and 624 °C, which mainly corresponded to Fe3O4 → FeO and FeO → Fe. It was mainly due to the catalyst contains too little Mn species. The redox peak of (Mn0.37Fe0.63)2O3 crystalline was highest in Mn–Fe(1:1)@Fe WM. It could be demonstrated that the ratio of 1:1 of Mn and Fe lead to more reducible species and thereafter enrich the redox cycle in the catalytic process. For comparison, the H2-TPR of Mn–Fe@Cu WM, Mn–Fe@Ti WM, Cu WM and Ti WM were carried out (Fig. S5†). The peaks at 286 and 640 °C represented the reduction of CuO → Cu2O and Cu2O → Cu. The peak at 327 °C was attributed to the reduction of TiO2 → Ti. It was interested that the Mn–Fe@Cu WM, Mn–Fe@Ti WM showed the similar peak positions and types with Cu WM and Ti WM, respectively. It confirms the unfavorable growth of Mn–Fe species on other metal supports.
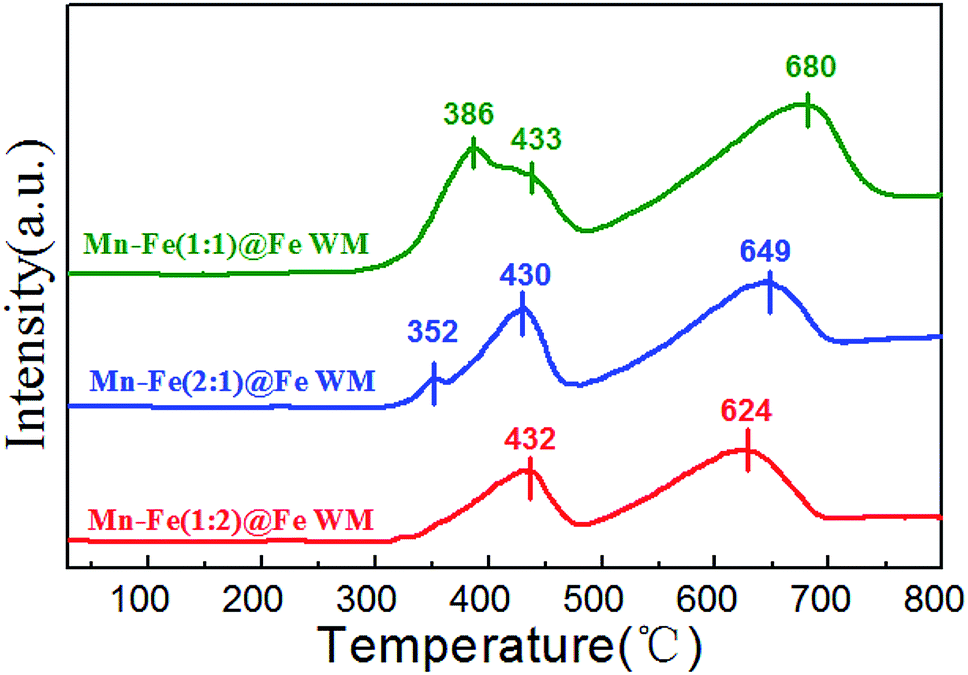 | ||
| Fig. 5 H2-TPR profiles of Mn–Fe@Fe WM catalysts. | ||
Catalytic performance
The NH3-SCR reaction over Mn–Fe@Fe WM monolith catalysts were performed during 100–400 °C as showed in Fig. 6. The Mn–Fe(1:1)@Fe WM monolith catalyst showed the lowest activation temperature and highest NO conversion for a whole temperature window. The NO conversion was over 90% at 175–300 °C for Mn–Fe(1:1)@Fe WM with a high N2 selectivity (Fig. S6†). Especially, the N2 selectivity of Mn–Fe(1:1)@Fe WM was as high as more than 75% until 300 °C. It showed Mn–Fe(1:1)@Fe WM had good N2 selectivity. The excellent catalytic activity was attributed to the formation of (Mn0.37Fe0.63)2O3 in the cubic structure in the coating layer, which acted as the main active species for the NH3-SCR reaction. The Mn–Fe(2:1)@Fe WM showed 10% lower activity than Mn–Fe(1:1)@Fe WM. It contributed less active sites exposure in block-like structure than cubic structure. The Mn–Fe(1:2)@Fe WM exhibited the lowest catalytic activity and the most narrow temperature window than Mn–Fe(1:1)@Fe WM and Mn–Fe(2:1)@Fe WM, which only had near 75% NO conversion from 200–300 °C. It was mainly due to the low content of Mn in the coating layer, which affected the catalytic activity. Moreover, Mn–Fe(1:1)@HCCS showed a poor catalytic performance, which resulted in Mn–Fe undesirable growth onto the honeycomb ceramics. This reflected the irreplaceable role of “twin iron source” in the process of crystal formation.
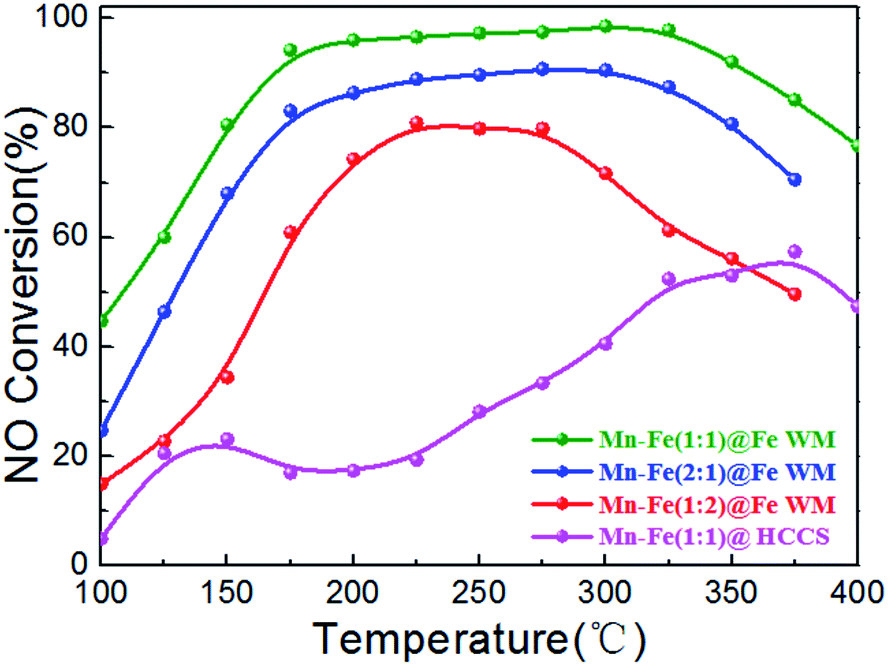 | ||
| Fig. 6 Plots of NO conversion versus reaction temperature over Mn–Fe@Fe WM catalysts. Reaction conditions: [NH3] = [NO] = 500 ppm, [O2] = 3 vol%, N2 as balance gas, GHSV = 20000 h−1. | ||
Furthermore, we studied the growth of Mn–Fe on other common metal supports and tested the catalytic performance (Fig. S7†). The results showed that the catalytic activities of the other metal supports were significantly reduced (less than 60%) under the same conditions, which was mainly attributed to the fact that the Mn–Fe species could not grow well on the surface. It reflected the irreplaceable role of “twin iron source” in the crystal growth process of heterogeneous nucleation and crystal growth process.
In actual working conditions, monolith catalysts should face a longer operation time and resist the adverse influences of water vapor on catalysts.48 Therefore, the stability and water resistance of the catalysts were investigated. The monolith catalysts were tested under the continuous running duration at 200 °C for 24 h (Fig. 7). In the whole reaction time, all of the catalysts kept a constant NO conversion. This result suggested the considerable stability of the catalysts, which could be related to the good catalytic stability of (Mn0.37Fe0.63)2O3 and adhesion between the Mn–Fe bi-metal oxides and Fe wire mesh arising from the in situ growth and calcination procedure. The H2O tolerance was also tested under the same reaction temperature (Fig. 7, inset). As clearly shown, the addition of 10% H2O could barely affect the De-NOx activities of the catalysts during the 8 h testing period. It was well documented that the inhibition effect of H2O did not affect the adsorption of NH3 molecules on the catalyst surface.
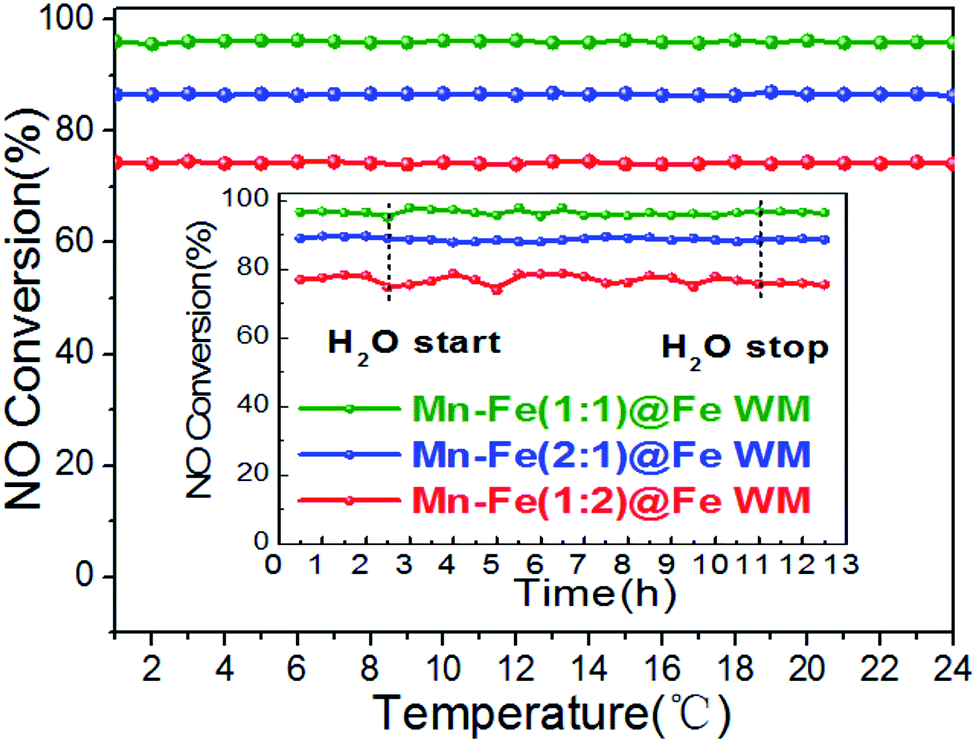 | ||
| Fig. 7 Plots of stability and H2O tolerance (inset) of the Mn–Fe@Fe WM monolith catalysts. Reaction conditions: reaction temperature = 200 °C, [NH3] = [NO] = 500 ppm, [O2] = 3 vol%, [H2O] = 10 vol% (when used), N2 as balance gas, GHSV = 20000 h−1. | ||
In order to understand the role of the active species, single Fe or Mn species monolithic catalysts (Mn@Fe WM and Fe@Fe WM) were prepared by deposition of single manganese or iron species on Fe wire mesh and the catalytic properties were studied. The results were shown in Fig. 8. There were less 40% of the NO conversion of Mn@Fe WM and less 80% of Fe@Fe WM. The results showed that the growth of single manganese or iron on the Fe wire mesh cannot bring high catalytic activity as Mn–Fe@Fe WM monolith catalysts. For Fe@Fe WM, because the catalytic process only contained Fe species, the catalytic activity was not high. But for Mn@Fe WM, it was mainly due to the poor growth of pure Mn species on the Fe wire mesh, which resulted in a low NO conversion. It proved that (Mn0.37Fe0.63)2O3 might be the main active species in the NH3-SCR reaction.
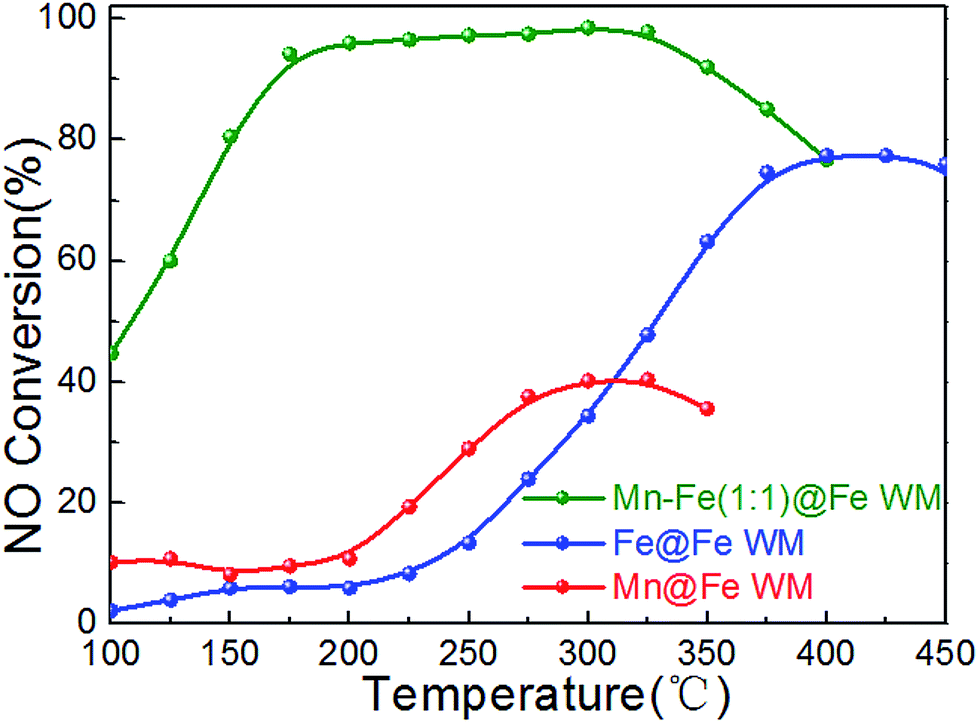 | ||
| Fig. 8 Plots of NO conversion versus reaction temperature over Mn–Fe@Fe WM catalysts. Reaction conditions: [NH3] = [NO] = 500 ppm, [O2] = 3 vol%, N2 as balance gas, GHSV = 20000 h−1. | ||
The catalyst was always deactivated due to the abundant alkali and alkaline earth metal ions contained in stack gases.49 The catalytic performance of Mn–Fe@Fe WM monolith catalysts for alkali resistance and regeneration were shown in Fig. 9. As shown clearly, though poisoned by K+ with 0.3 wt% of the total mass of the monolith catalysts, the results indicated that the catalytic activity of three catalysts decreased obviously after alkali metal poisoning. The highest catalytic activities of K-poisoning-Mn–Fe(1:1)@Fe WM, K-poisoning-Mn–Fe(2:1)@Fe WM and K-poisoning-Mn–Fe(1:2)@Fe WM of catalysts still achieved about 50% with temperature range of 225–400 °C, 225–350 °C and 300–400 °C, respectively. It showed that these monolith catalysts present good alkali resistance properties. After water washing the poisoned catalysts, the three catalysts showed excellent activity recovery. Especially for the K-poisoning-Mn–Fe(1:1)@Fe WM, the catalyst activity achieved above 85% at 225 °C. It can be attributed to the stable Mn–Fe bi-metal oxides structure and strong synergistic effect between the support and active species. The simple and good regeneration performance ensure that the catalyst could be used repeatedly, which effectively improves the utilization efficiency of the catalysts.
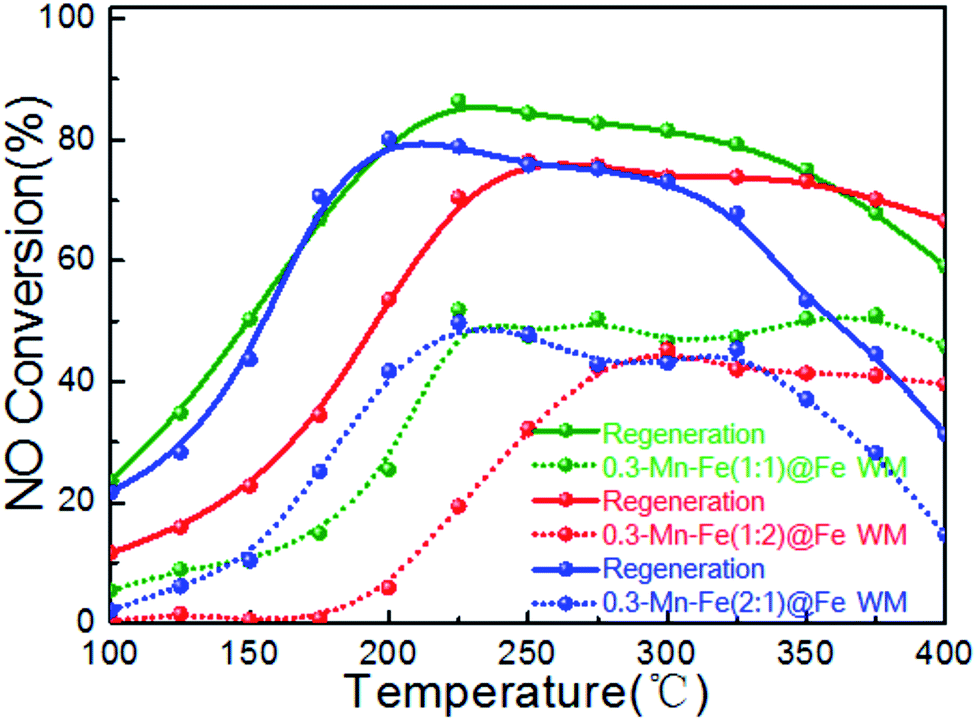 | ||
| Fig. 9 Plots of alkaline tolerance and regeneration properties of Mn–Fe@Fe WM catalysts. Reaction conditions: [NH3] = [NO] = 500 ppm, [O2] = 3 vol%, N2 as balance gas, GHSV = 20000 h−1. | ||
4. Conclusions
In this work, we applied a “twin iron source” growth method for the in situ construction of Mn–Fe bi-metal oxides on the Fe wire mesh and used in NH3-SCR of NO. The results suggest that the cube-like structure on the surface of Fe wire mesh performed above 90% catalytic activity at 175–300 °C. It is mainly attributed to the enhancement of reducibility and adsorption behaviors of (Mn0.37Fe0.63)2O3, which was favorable for the catalytic process. In addition, the catalyst showed good stability and H2O tolerance. It was related to the good adhesion between the Mn–Fe bi-metal oxides and Fe wire mesh and the competitive adsorption of NH3 on the surfaces of the Mn–Fe@Fe WM monolith catalysts. Moreover, the Mn–Fe(1:1)@Fe WM catalyst showed the best resistance to K+ poisoning and the best regeneration performance. It was mainly because the structural stability of spinel structure of (Mn0.37F0.63)2O3 and strong synergistic effect between the support and active species. This simple in situ technique can not only obtain the catalyst directly, but also avoid the use of vanadium harmful to the environment and human health. This new kind of monolithic catalysts prepared by an in situ technique can be used as a potential substitute for vanadium based ceramic catalysts.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the support of the National Natural Science Foundation of China (U1462110). P. M. would like to thank the National Natural Science Foundation of China for International Young Scientists FY 2016 (21650110450).References
- P. Forzatti, I. Nova and E. Tronconi, Angew. Chem., 2009, 48, 8366–8368 CrossRef CAS PubMed
.
- S. Beirle, K. F. Boersma, U. Platt, M. G. Lawrence and T. Wagner, Science, 2011, 333, 1737–1739 CrossRef CAS PubMed
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed
- P. Granger and V. I. Parvulescu, Chem. Rev., 2011, 111, 3155–3207 CrossRef CAS PubMed
- C. Paolucci, A. A. Verma, S. A. Bates, V. F. Kispersky, J. T. Miller, R. Gounder, W. N. Delgass, F. H. Ribeiro and W. F. Schneider, Angew. Chem., 2014, 53, 11828–11833 CrossRef CAS PubMed
- P. Forzatti, I. Nova and E. Tronconi, Ind. Eng. Chem. Res., 2010, 49, 10386–10391 CrossRef CAS
- W. Wang, G. McCool, N. Kapur, G. Yuan, B. Shan, M. Nguyen, U. M. Graham, B. H. Davis, G. Jacobs, K. Cho and X. Hao, Science, 2012, 337, 832–835 CrossRef CAS PubMed
- X. Li, J. H. Li, Y. Peng, W. Z. Si, X. He and J. M. Hao, Environ. Sci. Technol., 2015, 49, 9971–9978 CrossRef CAS PubMed
- P. G. W. A. Kompio, A. Brückner, F. Hipler, G. Auer, E. Löffler and W. Grünert, J. Catal., 2012, 286, 237–247 CrossRef CAS
- P. Forzatti, I. Nova, E. Tronconi, A. Kustov and J. R. Thogersen, Catal. Today, 2012, 184, 153–159 CrossRef CAS
- X. Q. Wang, A. J. Shi, Y. F. Duan, J. Wang and M. Q. Shen, Catal. Sci. Technol., 2012, 2, 1386–1395 CAS
- S. B. Kristensen, A. J. Kunov-Kruse, A. Riisager, S. B. Rasmussen and R. Fehrmann, J. Catal., 2011, 284, 60–67 CrossRef CAS
- J. P. Chen and R. T. Yang, J. Catal., 1990, 125, 411–420 CrossRef CAS
- Y. J. Zheng, A. D. Jensen and J. E. Johnsson, Ind. Eng. Chem. Res., 2004, 43, 941–947 CrossRef CAS
- H. Kamata, K. Takahashi and C. U. I. Odenbr, J. Mol. Catal. A: Chem., 1999, 139, 189–198 CrossRef CAS
- C. Fang, D. S. Zhang, S. X. Cai, L. Zhang, L. Huang, H. R. Li, P. Maitarad, L. Y. Shi, R. H. Gao and J. P. Zhang, Nanoscale, 2013, 5, 9199–9207 RSC
- L. Wang, X. Cheng, Z. Wang, C. Ma and Y. Qin, Appl. Catal., B, 2017, 201, 636–651 CrossRef CAS
- P. G. Smirniotis, D. A. Pena and B. S. Uphade, Angew. Chem., 2001, 40, 2479–2482 CrossRef CAS
- D. M. Meng, W. C. Zhan, Y. Guo, Y. L. Guo, L. Wang and G. Z. Lu, ACS Catal., 2015, 5, 5973–5983 CrossRef CAS
- Z. M. Liu, J. Z. Zhu, J. H. Li, L. L. Ma and S. I. Woo, ACS Appl. Mater. Interfaces, 2014, 6, 14500–14508 CAS
- L. Zhang, L. Shi, L. Huang, J. Zhang, R. Gao and D. Zhang, ACS Catal., 2014, 4, 1753–1763 CrossRef CAS
- S. X. Cai, H. Hu, H. R. Li, L. Y. Shi and D. S. Zhang, Nanoscale, 2016, 8, 3588–3598 RSC
- Z. H. Chen, Q. Yang, H. Li, X. H. Li, L. F. Wang and S. C. Tsang, J. Catal., 2010, 276, 56–65 CrossRef CAS
- F. F. Cao, J. H. Chen, C. L. Lyu, M. J. Ni, X. Gao and K. F. Cen, Catal. Sci. Technol., 2015, 5, 1267–1279 CAS
- M. Y. Qiu, S. H. Zhan, H. B. Yu, D. D. Zhu and S. Q. Wang, Nanoscale, 2015, 7, 2568–2577 RSC
- S. X. Cai, J. Liu, K. W. Zha, H. R. Li, L. Y. Shi and D. S. Zhang, Nanoscale, 2017, 9, 5648–5657 RSC
- J. Xu, H. R. Li, Y. Liu, L. Huang, J. P. Zhang, L. Y. Shi and D. S. Zhang, RSC Adv., 2017, 7, 36319–36325 RSC
- Y. Li, Y. Wan, Y. P. Li, S. H. Zhan, Q. X. Guan and Y. Tian, ACS Appl. Mater. Interfaces, 2016, 8, 5224–5233 CAS
- L. J. Zhang, S. P. Cui, H. X. Guo, X. Y. Ma and X. G. Luo, J. Mater. Chem. A, 2014, 390, 14–21 CAS
- L. J. Yan, Y. Y. Liu, K. W. Zha, H. R. Li, L. Y. Shi and D. S. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 5224–5233 Search PubMed
- Y. L. Ye, M. Jin and D. C. Wan, J. Mater. Chem. A, 2015, 3, 13519–13525 CAS
- A. V. Boix, S. G. Aspromonte and E. E. Miro, Appl. Catal., A, 2008, 341, 26–34 CrossRef CAS
- H. R. Li, D. S. Zhang, P. Maitarad, L. Y. Shi, R. H. Gao, J. P. Zhang and W. G. Cao, Chem. Commun., 2012, 48, 10645–10647 RSC
- Y. Shu, T. Aikebaier, X. Quan, S. Chen and H. Yu, Appl. Catal., B, 2014, 150–151, 630–635 CrossRef CAS
- A. Montebelli, C. G. Visconti, G. Groppi, E. Tronconi, C. Cristiani, C. Ferreira and S. Kohler, Catal. Sci. Technol., 2014, 4, 2846–2870 CAS
- Q. F. Zhang, X. P. Wu, G. F. Zhao, Y. K. Li, C. Z. Wang, Y. Liu, X. Q. Gong and Y. Lu, Chem. Commun., 2015, 51, 12613–12616 RSC
- J. Han, J. Meeprasert, P. Maitarad, S. Nammuangruk, L. Y. Shi and D. S. Zhang, J. Phys. Chem. C, 2016, 120, 1523–1533 CAS
- L. Huang, X. Zhao, L. Zhang, L. Y. Shi, J. P. Zhang and D. S. Zhang, Nanoscale, 2015, 7, 2743–2749 RSC
- H. Sun, Y. B. Zhang, X. Quan, S. Chen, Z. P. Qu and Y. L. Zhou, Catal. Today, 2008, 139, 130–134 CrossRef CAS
- Y. Liu, J. Xu, H. R. Li, S. X. Cai, H. Hu, C. Fang, L. Y. Shi and D. S. Zhang, J. Mater. Chem. A, 2015, 3, 11543–11553 CAS
- S. X. Cai, D. S. Zhang, L. Y. Shi, J. Xu, L. Zhang, L. Huang, H. R. Li and J. P. Zhang, Nanoscale, 2014, 6, 7346–7353 RSC
- H. L. Guo, G. S. Zhu, I. J. Hewitt and S. L. Qiu, J. Am. Chem. Soc., 2009, 131, 1646–1647 CrossRef CAS PubMed
- T. Yu, T. Hao, D. Fan, J. Wang, M. Shen and W. Li, J. Phys. Chem. C, 2014, 118, 6565–6575 CAS
- J. H. Kwak, R. Tonkyn, D. Tran, D. Mei, S. J. Cho, L. Kovarik, J. H. Lee, C. H. F. Peden and J. Szanyi, ACS Catal., 2012, 2, 1432–1440 CrossRef CAS
- F. D. Liu and H. He, J. Phys. Chem. C, 2010, 114, 16929–16936 CAS
- W. X. Tang, X. F. Wu, S. Li, W. H. Li and Y. F. Chen, Catal. Commun., 2014, 56, 134–138 CrossRef CAS
- M. H. Castaño, R. Molina and S. Moreno, Appl. Catal., A, 2015, 492, 48–59 CrossRef
- Z. G. Lei, B. Han, K. Yang and B. H. Chen, Chem. Eng. J., 2013, 215, 651–657 CrossRef
- L. Zheng, M. J. Zhou, Z. W. Huang, Y. X. Chen, J. Y. Gao, Z. Ma, J. M. Chen and X. F. Tang, Environ. Sci. Technol., 2016, 50, 11951–11956 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: TGA curves, N2 selectivity, and catalytic experimental results of the catalysts. See DOI: 10.1039/c7ra05007k |
| This journal is © The Royal Society of Chemistry 2017 |