
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tea polyphenols ameliorate hydrogen peroxide- and constant darkness-triggered oxidative stress via modulating the Keap1/Nrf2 transcriptional signaling pathway in HepG2 cells and mice liver
Guoyuan Qi,
Yashi Mi,
Rong Fan,
Runnan Li,
Yiwen Wang,
Xingyu Li,
Shuxian Huang and
Xuebo Liu *
*
Laboratory of Functional Chemistry and Nutrition of Food, College of Food Science and Engineering, Northwest A&F University, 28 Xi-nong Road, Yangling, Shaanxi 712100, China. E-mail: xueboliu@aliyun.com; Fax: +86-029-87092325; Tel: +86-029-87092325
First published on 23rd June 2017
Abstract
Tea polyphenols, which are natural plant flavonoids found in the leaves of tea plants, possess bioactivities that affect the pathogenesis of several chronic diseases. However, the beneficial effects of tea polyphenols in balancing redox status and the intervention of apoptosis in the liver of mice housed in constant darkness and H2O2-stimulated HepG2 cells remain to be elucidated. The results demonstrated that TP significantly reversed decreases in H2O2-elicited cell viability, mitochondrial dysfunction, activation of NFκB and MAPK stress pathways via balancing cellular redox status. Moreover, pretreatment with TP could modulate the nuclear translocation of Nrf2 by stimulating the ERK1/2 pathway and thus transcriptionally regulate the downstream expression of antioxidant enzymes including HO-1 and NQO-1 in HepG2 cells. In addition, in vivo studies revealed that mice exposed to constant darkness, which simulated disruption due to shift work in humans, had remarkably elevated levels of H2O2 and reduced nuclear translocation of Nrf2 and its downstream phase II detoxification enzymes HO-1 and NQO-1 in liver tissue. Notably, supplementation with TP via drinking water eliminated these changes. Overall, our results indicated that TP ameliorated oxidative stress triggered by H2O2 and constant darkness via mediating Keap1/Nrf2 transcriptional pathways and regulating the expression of downstream enzymes, which indicated that treatment with TP could represent a nutritional preventive strategy in liver pathology related to oxidative stress.
Introduction
Population-based studies have indicated that oxidative stress is correlated with a wide spectrum of diseases, including diabetes, chronic hepatitis, neurodegenerative diseases, and cancer.1–3 Reactive oxygen species (ROS) produced in the process of aerobic metabolism, including O2˙−, ˙OH, H2O2, HOCl, NO, O3 and singlet oxygen, have toxic effects on the majority of cells.4 Growing evidence has revealed that ROS act as important redox signaling molecules that regulate many signal transduction pathways and play a crucial role in oxidative stress.5 The overproduction of ROS disrupts the balance between oxidation and antioxidant defense systems, which induces progressive damage to cellular lipids, DNA, and cellular integrity and function and eventually results in irreversible cell damage and death.6 Shift work is defined as work that is primarily outside normal daytime working hours.7 Mounting evidence suggests that oxidative stress indices increase significantly after continuous shift work.8–12 Disruptions of the normal light/dark cycle in wild-type mice can simulate disruption due to shift work in humans.13Intervention in oxidative status in the liver can reduce the risk of many liver diseases such as chronic hepatitis, cholestasis, and alcoholic and non-alcoholic fatty liver disease.14,15 H2O2, as a second messenger generated by the redox-regulating activity of the mitochondria, regulates multiple cell signaling pathways and a range of cell functions.16 Evidence that has accumulated during recent years has revealed that oxidative stress induced by H2O2 is closely associated with mitochondrial dysfunction via the regulation of MAPKs and PI3K/AKT pathways.17,18 Moreover, nuclear factor-erythroid 2-related factor 2 (Nrf2), which is a predominant regulator of one defense mechanism against damage due to oxidative stress, plays an essential role in cellular defense against various inflammatory and oxidative stress-induced diseases.19,20 During basal conditions, the transcription factor Nrf2 is repressed by binding to its negative regulator Kelch-like ECH-associated protein-1 (Keap1).21,22 However, under oxidative conditions Nrf2 dissociates from Keap1 and is translocated into the nucleus to bind to the antioxidant response element (ARE), which regulates the production of various antioxidant enzymes including heme oxygenase 1 (HO-1), NAD(P)H:quinone oxidoreductase 1 (NQO-1), superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase (CAT).21 On the basis of previous research, food signals exert cytoprotective potentials to maintain the dynamic balance of ROS and normal physiological processes via activation of the Nrf2/ARE signaling pathway.23 More specifically, polyphenolic compounds, such as chlorogenic acid, can activate the PI3K/Nrf2/ARE signaling pathway and elevate the expression of HO-1 stimulated by H2O2.6 Additional research has demonstrated that pinocembrin upregulated the expression of Nrf2 and HO-1, which limited the production of superoxide radical anions (O2−˙) and nitric oxide (NO˙) in SH-SY5Y cells induced by H2O2.24
Tea represents the most popular beverage consumed in the world besides water,25 and its content in the human diet is relatively high. Recently, extensive research using various in vitro and animal models has demonstrated that tea polyphenols have multiple health benefits such as improving insulin resistance, treating neurodegenerative diseases, and decreasing oxidative stress and inflammation and thus treating cancer and cardiovascular diseases.26,27 The major components of TP, namely, gallic acid, epigallocatechin (EGC), epigallocatechin-3-gallate (EGCG), and epicatechin-3-gallate (ECG), all significantly inhibit the production of 2,2-diphenyl-1-picrylhydrazyl (DPPH˙) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS˙+) free radicals.28–30 TP have been reported to protect against apoptosis triggered by the neurotoxin 6-hydroxydopamine in SH-SY5Y cells via suppression of the accumulation of ROS.31 However, whether the beneficial effects of TP involve balancing redox status and intervention in apoptosis in the liver remains to be revealed. Therefore, in the current study, we investigated the beneficial effects of TP on maintaining the balance of redox status in the liver and HepG2 cells and demonstrated possible cellular mechanisms that involved mitochondrial function and the activation of Keap1/Nrf2 antioxidant defense pathways via the regulation of ERK1/2 and the activities of intracellular antioxidant enzymes. The cytoprotective functions of TP may act as a therapeutic target for imbalances in redox status and apoptosis and thus improve the quality of life.
Materials and methods
Tea polyphenols composition
Tea polyphenols (TP) (purity of >98%) were purchased from Yuanye Biotechnology, Ltd. (Shanghai, China). The composition of TP was as follows: gallic acid (67.0 ± 2.2 mg g−1), epigallocatechin (EGC) (84.6 ± 0.3 mg g−1), epigallocatechin-3-gallate (EGCG) (431.0 ± 2.2 mg g−1), and epicatechin-3-gallate (ECG) (394.3 ± 4.0 mg g−1).Animals and diet
Three-month-old male C57BL/6J mice were purchased from Xi'an Jiaotong University (Xi'an, Shaanxi, China). Mice were randomly assigned to three groups (n = 10/group): a control group housed in a 12/12 light/dark cycle, a DD (constant darkness) group kept in constant darkness, and a DD plus TP group, which were housed in constant darkness and fed with 2 g L−1 tea polyphenols in drinking water for 8 weeks (approximately 176–224 mg per kg per day). On the basis of allometric scaling, the presently used dose of 2 g L−1 in drinking water in mice approximately corresponds to 7 cups of green tea (containing 2 g tea leaves per cup) per day for an average person who requires 2000 kcal d−1. All three groups were fed with the AIN-93M standard diet. All experimental procedures conformed to the Guide for the Care and Use of Laboratory Animals: Eighth Edition, ISBN-10: 0-309-15396-4, and the animal protocol was approved by the animal ethics committee of Xi'an Jiaotong University. All surgical procedures were performed under anesthesia and all efforts were made to minimize suffering.Cell culture
An HepG2 human hepatoblastoma cell line was provided by the cell bank of the Fourth Military Medical University (Xi'an, China) and cultured in RPMI 1640 (Hyclone Co., USA) with 10% fetal bovine serum (FBS), 100 IU mL−1 penicillin, and 100 μg mL−1 streptomycin at 37 °C in a humidified atmosphere with 5% CO2. HepG2 cells were pretreated with TP for 12 h and then with H2O2 for 12 h after being washed with PBS. After treatment, the cells were collected for further analysis.Cell viability assay
HepG2 cells were subcultured in a 96-well plate at a density of 2 × 104 cells per mL and incubated overnight at 37 °C with 5% (v/v) CO2. After 100 μL medium was removed from each well, the cells were pretreated with TP for 12 h and then with H2O2 for 12 h after being washed with PBS. Subsequently, a final concentration of 0.5 mg mL−1 MTT was added and the mixture was incubated for 4 h. Then, 100 μL dimethyl sulfoxide was added to each well after the medium was removed. The formazan crystals that were formed by live cells were solubilized with 100 μL DMSO, and the absorbance at 560 nm was measured with a microplate reader (Bio-Rad Laboratories, Ltd., China).Cell morphology
HepG2 cells were seeded in a 6-well plate at a density of 1 × 104 cells per well at 37 °C with 5% (v/v) CO2 overnight. Cells were pretreated with 40 μg mL−1 TP for 12 h and then treated with H2O2 (200 μM) for 12 h. The cell morphologies were observed using an inverted fluorescence microscope.Mitochondrial membrane potential (MMP) assay
Disruption of the mitochondrial membrane potential was measured using a mitochondrial membrane potential assay kit with a JC-1 probe (Beyotime Institute of Biotechnology, China). Cells were subcultured in a 96-well plate at a density of 2 × 104 cells per mL and incubated overnight at 37 °C with 5% (v/v) CO2. After treatment, cells were stained with 10 μg mL−1 JC-1 for 30 min at 37 °C in the dark. After being washed with PBS twice, the cell fluorescence was measured by a fluorescence microplate reader (Molecular Devices Co., Sunnyvale, CA) with settings of FL1 (green) at 538 nm and FL2 (red) at 585 nm.32Detection of reactive oxygen species (ROS) and H2O2 production
The production of ROS was measured using a DCFH-DA (2′,7′-dichlorodihydrofluorescein diacetate) assay kit (Beyotime Institute of Biotechnology, China). DCFH-DA can freely diffuse through the cell membrane; after entering cells, it can be hydrolyzed to DCFH by esterases. Intracellular ROS can convert DCFH into fluorescent DCF. Detection of the fluorescence of DCF can represent the level of active oxygen in cells. Cells were washed with PBS twice (pH 7.4), lysed in lysis buffer containing 1% phenylmethanesulfonyl fluoride (PMSF) and 1% NaF for 10 min on ice, and centrifuged for 10 min (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g) at 4 °C. The fluorescence intensity of DCF in the supernatant was measured by a fluorescence microplate reader at an excitation wavelength of 485 nm and an emission wavelength of 535 nm.
000g) at 4 °C. The fluorescence intensity of DCF in the supernatant was measured by a fluorescence microplate reader at an excitation wavelength of 485 nm and an emission wavelength of 535 nm.
Extracellular H2O2 from HepG2 cells and liver tissue was determined by an Amplex Red hydrogen peroxide/peroxidase assay kit (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. In the presence of horseradish peroxidase (HRP), Amplex Red reacts with H2O2 in a stoichiometry of 1:1 to produce highly fluorescent resorufin.33
RNA preparation and quality control
Total RNA was extracted from liver tissue and cells using an RNA extraction kit (TaKaRa, MiniBEST Universal RNA Extraction Kit, Dalian, China), as previously described.33 For quality control, the purity and integrity of RNA were evaluated using a Quawell 5000 UV-vis spectrophotometer (Quawell Technology, San Jose, CA, USA). RNA was stored at −80 °C prior to further analysis by microarray and real-time quantitative PCR (RT-qPCR).RT-qPCR
Total RNA was reverse-transcribed into cDNA using a PrimeScript RT Master Mix reverse transcription kit (TaKaRa PrimeScript RT Master Mix, Dalian, China). The expression of mRNA was quantified by RT-qPCR using an SYBR Green PCR kit (TaKaRa SYBR Premix Ex Taq II, Dalian, China) and a CFX96 real-time system (Bio-Rad, Hercules, CA). Gene-specific mouse primers were used, as shown in Table 1. Ct values were normalized to GAPDH and the relative expressions of genes were calculated by the 2−ΔΔCt method.| Forward primer | Reverse primer | |
|---|---|---|
| ho-1 (human) | GGAACTTTCAGAAGGGCCAG | GTCCTTGGTGTCATGGGTCA |
| nqo-1 (human) | CCATTCTGAAAGGCTGGTTTG | CTAGCTTTGATCTGGTTGTC |
| β-Actin (human) | TCCACCTTCCAGCAGATGTG | GCATTTGCGGTGGACGAT |
| ho-1 (mouse) | ATGTGGCCCTGGAGGAGGAGA | CGCTGCATGGCTGGTGTGTAG |
| nqo-1 (mouse) | GGATTGGACCGAGCTGGAA | AATTGCAGTGAAGATGAAGGCAAC |
| γgcsr (mouse) | AGGAGCTTCGGGACTGTATT | TGGGCTTCAATGTCAGGGAT |
| mnsod (mouse) | CAAGCGTGACTTTGGGTCTT | GCGACCTTGCTCCTTATTGA |
| gpx-1 (mouse) | AAGGCTCACCCGCTCTTTAC | ACACCGGAGACCAAATGATG |
| gapdh (mouse) | TGGAGAAACCTGCCAAGTATGA | TGGAAGAATGGGAGTTGCTGT |
Measurement of the activities of antioxidant enzymes
The activities of GSH, CAT, and SOD in vivo and in vitro were determined using a commercially available kit from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China) according to the manufacturer's protocol.Western blot analysis
Liver tissue homogenates or cell lysates were solubilized in SDS sample buffer as previously described.34 Samples were separated by Laemmli SDS/PAGE and transferred onto PVDF membranes. Using appropriate antibodies, the immunoreactive bands were visualized with an ECL commercial kit (Millipore, USA) using a Molecular Imager ChemiDoc XRS system (Bio-Rad, Shanghai, China). Antibodies against PARP (SC-7150), Keap1 (SC-33569), GAPDH (SC-25778), lamin B (SC-6217), HO-1 (SC-1796), NQO-1 (SC-16464), and Nrf2 (SC-722) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against p-NFκB p65 (3033), NFκB p65 (8242), IκB (9242), p-IκBα (Ser32/36) (5A5), p-p44/42 MAPK (ERK1/2) (9101), p44/42 MAPK (ERK1/2) (9102), p-SAPK/JNK (Thr183/Tyr185) (9251), SAPK/JNK (9252), p-p38 MAPK (9211), p38 MAPK (9212), p-AKT (9271), and AKT (9272). Densitometric analysis of western blots was performed with Quantity One 4.6.2 software (Bio-Rad Company, Hercules, CA, USA).Data analysis
Data were reported as the mean ± SD from three biological replicates for one representative experiment in vitro and were reported as the mean ± SEM in vivo (n = 6–10). Significant differences between measurements for the control and treated samples were analyzed using one-way factorial analysis of variance (ANOVA), followed by Tukey's test (SPSS 16.0). Means were considered to be statistically significant if p < 0.05.Results
TP reversed cell death and mitochondrial dysfunction initiated by exposure to H2O2 in HepG2 cells
To assess whether supplementation with TP is effective in preventing loss of cell viability, HepG2 cells were pretreated with TP for 12 h and then with H2O2 for 12 h. As shown in Fig. 1A, H2O2 (200 μM) substantially (p < 0.01) decreased cell viability to 33.1%, whereas TP (40 μg mL−1) had no toxic effect on HepG2 cells. In comparison with the group stimulated with H2O2, pretreatment with TP for 12 h (20, 40, and 60 μg mL−1) significantly (p < 0.01) increased cell viability to 66.8%, 95.7% and 95.3%, respectively, which indicated that TP restored the viability of HepG2 cells that had been impaired by H2O2 in a dose-dependent manner. Similarly, optical microscopy observations also demonstrated a protective effect of TP on damage induced by H2O2 and showed a decrease in cell shrinkage in cells pretreated with TP (Fig. 1C).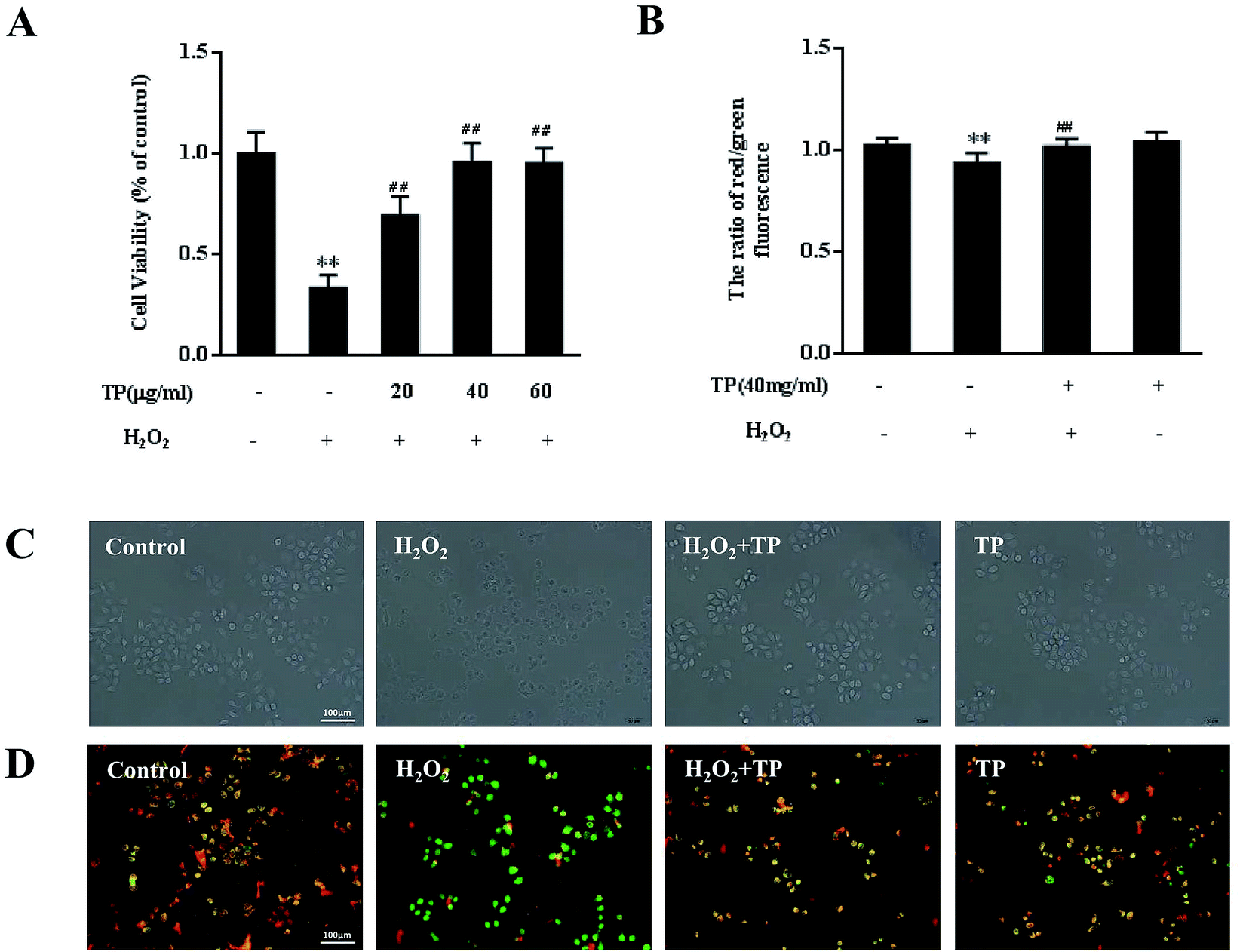 | ||
| Fig. 1 TP reversed loss of cell viability and mitochondrial dysfunction initiated by exposure to H2O2 in HepG2 cells. (A) Effect of tea polyphenols (TP) on HepG2 cells stimulated by H2O2. HepG2 cells were treated with H2O2 (200 μM) for 12 h with or without pretreatment with TP (20, 40, or 80 μg mL−1) for 12 h. Cell viability was measured by an MTT assay. HepG2 cells were pretreated with 40 μg mL−1 TP for 12 h and then exposed to H2O2 (200 μM) for 12 h (B). (C) Effect of TP on H2O2-induced HepG2 morphology changes. (D) The mitochondrial membrane potential was examined by staining with JC-1 (×200). Data are presented as the mean ± SD (n = 3). (*) p < 0.05 and (**) p < 0.01 versus the control group; (#) p < 0.05 and (##) p < 0.01 versus the H2O2 group. | ||
Evidence has suggested that mitochondria are not only major cellular “energy factories” but are also responsible for regulating the production of free radicals such as H2O2 to maintain the dynamic balance of cellular redox status.17 Loss of mitochondrial membrane potential (Δψ) is an early event that leads to alterations in mitochondrial function. H2O2 could induce loss of mitochondrial membrane potential, whereas TP significantly restored defective MMP (p < 0.01) (Fig. 1B and D).
TP alleviated cellular redox imbalance triggered by H2O2 in HepG2 cells
Because TP alleviate the loss of cell viability and mitochondrial function, we further tested whether they could protect against mitochondrially regulated imbalances in cellular redox status. ROS have been confirmed to be signaling molecules in various pathways that regulate cell inflammation and death.35,36 As shown in Fig. 2A and B, H2O2 led to increases in the levels of intracellular ROS, whereas TP effectively reduced intracellular ROS to normal levels (p < 0.01). The release of H2O2 in cell culture media stimulated by exposure to H2O2 was suppressed by pretreatment with TP in HepG2 cells (p < 0.01) (Fig. 2C).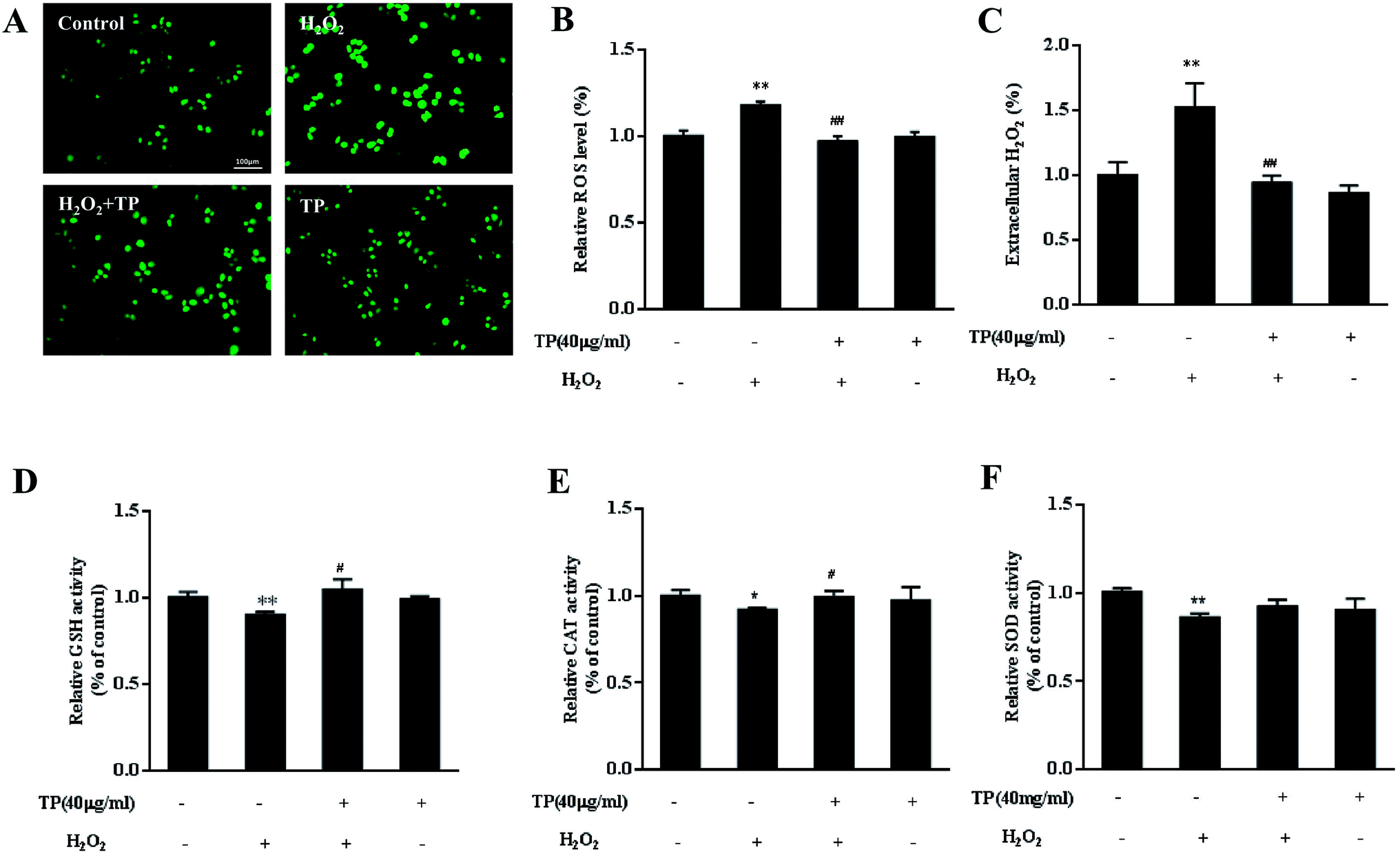 | ||
| Fig. 2 TP alleviated an imbalance in cellular redox status triggered by H2O2 in HepG2 cells. HepG2 cells were treated with H2O2 (200 μM) for 12 h with or without pretreatment with TP (40 μg mL−1) for 12 h. (A and B) The cellular oxidation status was detected by staining with H2DCFDA. (C) The production of H2O2 was determined using an Amplex Red hydrogen peroxide/peroxidase assay kit. (D–F) The levels of glutathione (GSH) and the activities of catalase (CAT) and superoxide dismutase (SOD) were determined using assay kits. Data are presented as the mean ± SD (n = 3). (*) p < 0.05 and (**) p < 0.01 versus the control group; (#) p < 0.05 and (##) p < 0.01 versus the H2O2 group. | ||
Antioxidant enzymes are mainly responsible for clearing excess ROS, preventing oxidative stress, and maintaining the dynamic balance of ROS and normal physiological processes. Treatment of HepG2 cells with H2O2 caused a significant decrease in levels of GSH, which plays a pivotal role in balancing cellular redox status together with its oxidized form glutathione disulfide (GSSG). Interestingly, TP restored the level of GSH (p < 0.05) (Fig. 2D). As expected, pretreatment with TP also dramatically reversed the decrease in the activity of catalase (CAT) relative to the H2O2 group (Fig. 2E). Taken together, these results implied that TP reduced the levels of ROS and H2O2 to maintain a balanced cellular redox status, which is associated with the activity of antioxidant enzymes.
N-Acetyl-L-cysteine (NAC), which is a precursor of GSH and a redox modulator, was used to further investigate the impact of TP on cellular redox status in apoptosis.37 TP inhibited the upregulation of the expression of the apoptosis marker PARP triggered by H2O2 and cleaved caspase-3 (Fig. 3A and B). In addition, several signaling pathways were involved in regulating apoptosis and cellular redox status. Pretreatment of cells with NAC further enhanced the survival signal inherent in the activation (phosphorylation) of Akt without changes in total Akt levels and increased the activation of ERK in the presence of TP (Fig. 3C–F). Reciprocally, NAC had a strong inhibitory effect on the activation of both JNK and p38 and the expression of p-NFκB (Fig. 3E–H).
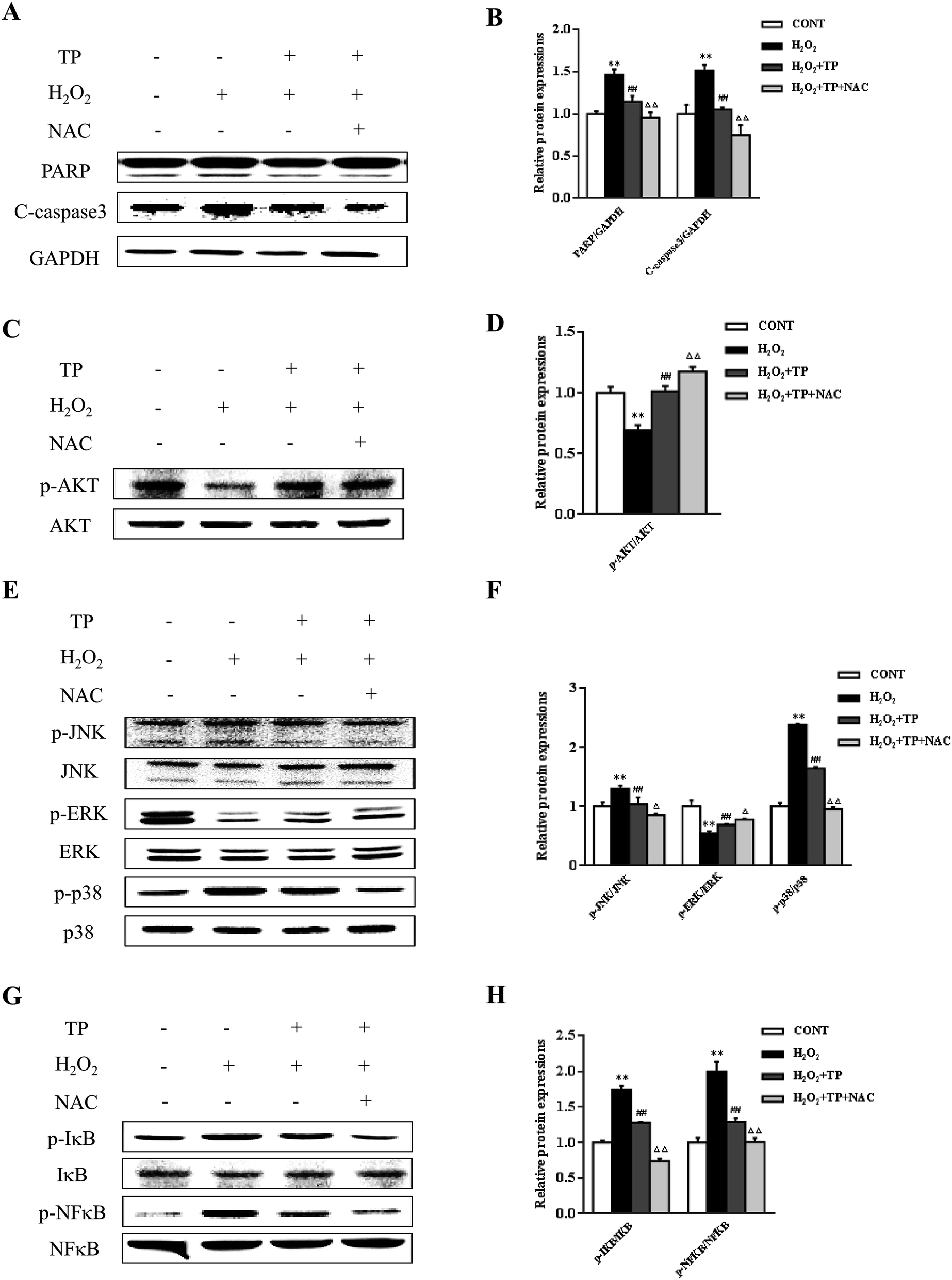 | ||
| Fig. 3 TP mediated changes in redox-sensitive apoptosis signaling in HepG2 cells. HepG2 cells were pre-incubated for 30 min with or without N-acetyl-L-cysteine (NAC) (10 μM) and were then treated with H2O2 (200 μM) for 12 h with or without pretreatment with TP (40 μg mL−1) for 12 h. Expressions of (A and B) the apoptosis marker PARP and c-caspase-3, (C and D) phosphorylated and total forms of AKT, (E and F) phosphorylated and total forms of ERK1/2, JNK, and p38, and (G and H) phosphorylated and total forms of IκB and NFκB. Data are presented as the mean ± SD (n = 3). (*) p < 0.05 and (**) p < 0.01 versus the control group; (#) p < 0.05 and (##) p < 0.01 versus the H2O2 group; (△) p < 0.05 and (△△) p < 0.01 versus the H2O2 plus TP group. | ||
Role of ERK1/2 in the Keap1/Nrf2 antioxidant defense pathway activated by TP in HepG2 cells
Emerging evidence has revealed that the Keap1/Nrf2 antioxidant defense pathway regulates the expression of antioxidant-responsive genes and the induction of phase II detoxifying enzymes, including HO-1 and NQO-1, which are considered to be major regulators of resistance to oxidants and are involved in the maintenance of cellular redox homeostasis.20,38,39 As shown in Fig. 4A and B, in comparison with the group triggered by H2O2, the nuclear translocation of Nrf2 and decline in Keap1 were significantly increased by pretreatment with TP prior to incubation with H2O2 in HepG2 cells (p < 0.01). Similarly, pretreatment with TP markedly elevated the expression of HO-1 and NQO-1 in terms of levels of both mRNA and protein (p < 0.01) (Fig. 4A and B).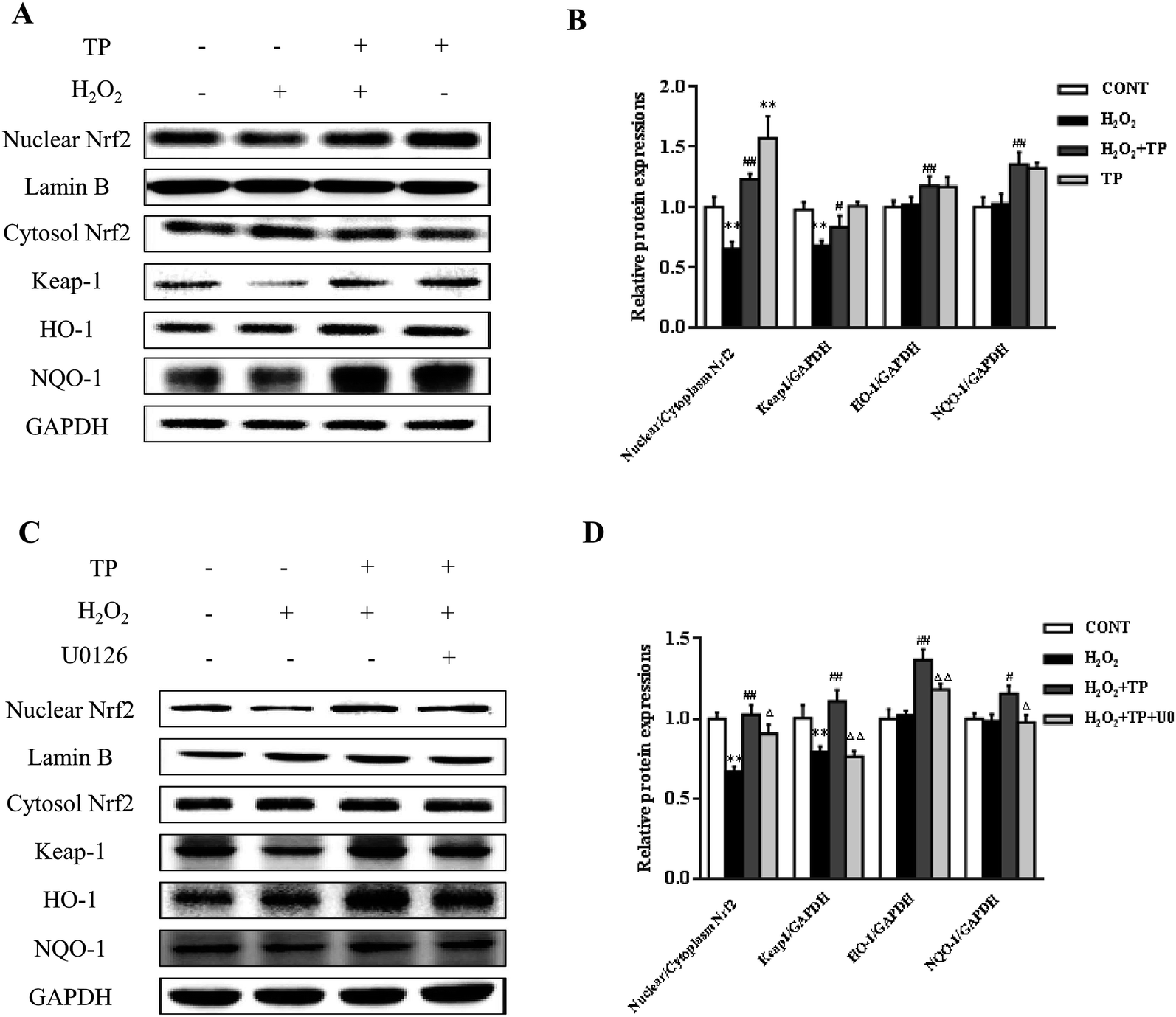 | ||
| Fig. 4 Role of ERK1/2 on TP-activated Keap1/Nrf2 antioxidant defense pathway in HepG2 cells. HepG2 cells were treated with H2O2 (200 μM) for 12 h with or without pretreatment of TP (40 μg mL−1) for 12 h. (A) Expressions of Nrf2, Keap1, HO-1, and NQO-1 were measured by western blotting. Densitometry analysis is shown in (B). (C) Expressions of Nrf2, Keap1, HO-1, and NQO-1 were measured by western blotting. Densitometry analysis is shown in (D). Data are presented as the mean ± SD, n = 3. (*) p < 0.05 and (**) p < 0.01, versus control group; (#) p < 0.05 and (##) p < 0.01, versus H2O2 group; (Δ) p < 0.05 and (ΔΔ) p < 0.01, versus H2O2 plus TP group. | ||
The ERK1/2 pathway, which is a key signaling pathway in the modulation of the cellular defense system against oxidative injury, plays a vital role in the antioxidant response mediated by Nrf2.22 U0126 (Selleck, Shanghai, China), which is an inhibitor of ERK1/2, diminished the protective effect of TP against the inhibition by H2O2 of the nuclear translocation of Nrf2 and the expression of Keap1 (p < 0.01) (Fig. 4C and D). Consistently, the results showed that the expression of HO-1 and NQO-1 stimulated by TP was markedly suppressed by U0126 (p < 0.01) (Fig. 4C and D).
TP protected C57BL/6J mice from liver oxidative stress triggered by DD
To confirm whether supplementation with TP had an effect on redox status homeostasis in the liver of mice exposed to DD, in vivo studies were performed. C57BL/6J mice were randomly assigned to three groups (n = 10/group): a control group housed in a 12/12 light/dark cycle, a DD group kept in constant darkness, and a DD plus TP group, which were housed in constant darkness and fed with 2 g L−1 tea polyphenols in drinking water for 8 weeks. There were no changes in weight in the body and tissues such as the liver, kidney, and brain.The results of the Amplex Red hydrogen peroxide/peroxidase assay revealed that long-term darkness substantially increased the levels of H2O2 in liver tissue. Notably, supplementation with TP markedly suppressed the formation of H2O2 to the normal level (Fig. 5A). The levels of expression of components of the Nrf2/Keap1 antioxidant defense pathway in mouse liver homogenates are shown in Fig. 5C–E. The expressions of the nuclear transcription factor Nrf2, Keap1, and their downstream phase II detoxification enzymes HO-1 and NQO-1 were dramatically downregulated in the liver of mice exposed to constant darkness with respect to the control group. Supplementation with TP in drinking water reversed these changes triggered by constant darkness in terms of levels of both mRNA and protein (p < 0.01) (Fig. 5B–E). Consistently, the results confirmed that exposure to DD dramatically reduced the mRNA levels of γ-glutamylcysteine synthetase (γGCS), which is a rate-limiting enzyme in the biosynthesis of GSH (Fig. 6A). Intriguingly, TP significantly restored levels of γGCS and GSH in the liver that had been reduced by DD. In addition, the mice exposed to DD exhibited a marked decrease in the mRNA levels of other antioxidant enzymes, such as MnSOD and GPX1 (Fig. 6B and C), in comparison with levels in the control group, which was consistent with the activities of SOD and CAT (Fig. 6E and F), whereas the intake of TP restored the level of these antioxidant enzymes in the liver.
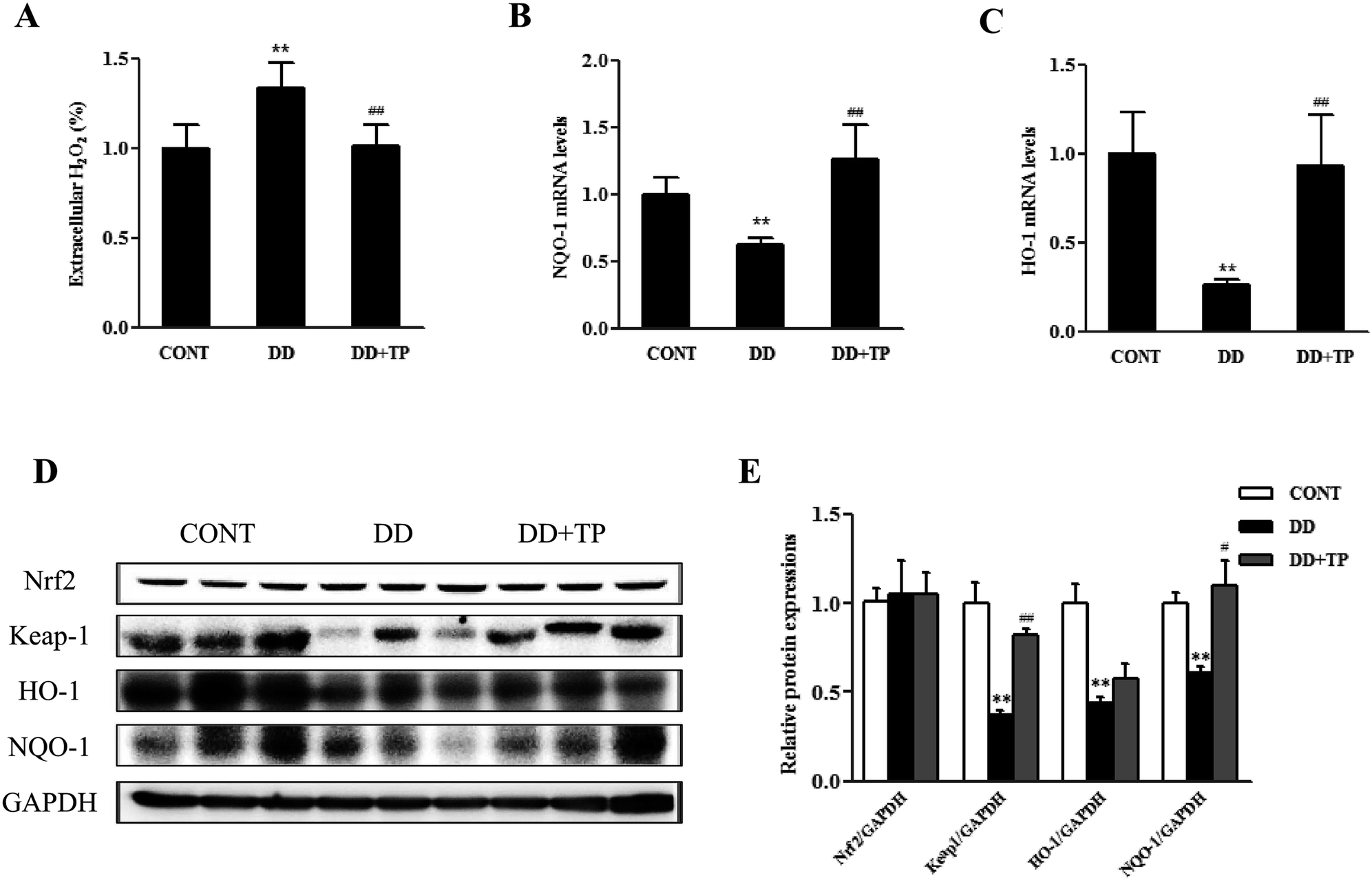 | ||
| Fig. 5 Activating effects of TP on the Keap1/Nrf2 signaling pathway in the liver of mice exposed to DD. Three-month-old C57BL/6J mice were randomly assigned to three groups (n = 10/group): a control group housed in a 12/12 light/dark cycle, a DD group kept in constant darkness, and a DD plus TP group, which were housed in constant darkness and fed with 2 g L−1 tea polyphenols in drinking water for 8 weeks. Liver samples were collected and (A) the production of H2O2 was determined using an Amplex Red hydrogen peroxide/peroxidase assay kit. (B and C) The mRNA levels of NQO-1 and HO-1. (D) The expressions of Nrf2, Keap1, HO-1, and NQO-1 were measured by western blotting. The results of densitometry analysis are shown in (E). Data are presented as the mean ± SEM (n = 6–10). (*) p < 0.05 and (**) p < 0.01 versus the control group; (#) p < 0.05 and (##) p < 0.01 versus the H2O2 group. | ||
 | ||
| Fig. 6 TP protected C57BL/6J mice from liver oxidative stress triggered by DD. The animal experiments were performed as described in the legend to Fig. 5. The mRNA levels of (A) γGCS, (B) MnSOD, and (C) GPX1 were determined by RT-PCR. The relative levels of (D) GSH and activities of (E) SOD and (F) CAT in liver homogenates were determined using assay kits. Data are presented as the mean ± SEM (n = 6–10). (*) p < 0.05 and (**) p < 0.01 versus the control group; (#) p < 0.05 and (##) p < 0.01 versus the H2O2 group. | ||
Discussion
Coordination between mitochondrial function and the activities of antioxidant enzymes helped maintain redox status homeostasis. Growing evidence has demonstrated that alterations or imbalances in the redox status system and apoptosis interfere with each other.4 In this study, we revealed that TP exerted beneficial pleiotropic effects in terms of modulating mitochondrial function, activating the Keap1/Nrf2 antioxidant defense pathway, and regulating the activities of intracellular antioxidant enzymes: (i) pretreatment with TP resulted in improvements in the viability of HepG2 cells and mitochondrial function, which had been impaired by H2O2; (ii) treatment with TP alleviated imbalances in redox status, including the levels of antioxidant enzymes and the production of ROS in mouse livers and HepG2 cells; (iii) treatment with TP promoted the Keap1/Nrf2 antioxidant defense signaling pathway by regulating the ERK1/2 signaling pathway; and (iv) TP dramatically alleviated redox-sensitive signaling pathways such as ERK1/2 and MAPKs and transcriptional pathways such as NFκB. All the above-mentioned factors lay the foundation for further research into TP in treating imbalances in human redox status.Dietary polyphenols have received extensive attention among nutritionists, food scientists and consumers owing to their regulatory roles in human health.40 Polyphenols are known to be strong antioxidants that complement and add to the functions of antioxidant vitamins and enzymes as a defense against oxidative stress caused by excess ROS.41 Numerous studies suggest that a high intake of vegetables and fruits, which are rich in polyphenols, has been linked to a reduction in the risks of many chronic diseases related to oxidative stress, including cardiovascular disease, cancer, and chronic inflammation.42–44 Isoorientin, which is a flavonoid compound, can protect BRL-3A cells against apoptosis induced by H2O2 by inhibiting mitochondrial dysfunction, inactivating MAPK kinases, activating Akt, and scavenging ROS and NO.18 It has been demonstrated that the consumption of tea polyphenols alleviates glomerulonephritis owing to their anti-inflammatory and antioxidant properties.45 Other studies indicated that tea polyphenols exerted an excellent inhibitory effect on cognitive impairment induced by chronic cerebral hypoperfusion via modulating oxidative stress.46 Additional studies have shown that polyphenolic components extracted from green tea leaves are effective antioxidants against the peroxidation of rat liver microsomes induced by AAPH.47 Collectively, supplementation with TP might serve as a potential strategy for combating liver pathology induced by oxidative stress.
Shift work usually involves between 20% and 25% of the work force in industrialized countries.48 Shift work is accompanied by a greater incidence of many medical disorders, such as cancer and cardiovascular, gastrointestinal, and neurological disorders.49–51 The established mechanism was that oxidative stress could induce cellular damage and result in such disorders.8 Recently, studies have shown that levels of markers of oxidative stress were significantly increased in healthcare staff after prolonged working hours.9 Similarly, at the end of day and night shifts parameters of oxidative stress were enhanced in all nurses.12 Circadian misalignment is one possible mechanism that could explain these results. Shift workers, night workers, individuals experiencing jet lag, and sleep-deprived individuals were exposed to constant circadian disruption.52 In several studies, mice housed in constant darkness for two weeks exhibited reductions in the levels of expression of Bmal1 and Sirt1 proteins in both liver and muscle tissues. Yechoor et al. revealed that Bmal1 directly regulates the expression of the key regulator of antioxidant response Nrf2.53 In addition, Jacobi et al. indicated that the circadian regulator Bmal1 alleviates oxidative stress by controlling rhythmic mitochondrial dynamics in the liver.54 In the present study, we simulated disruption due to shift work in humans by disrupting the normal light/dark cycle in wild-type mice. Exposure to DD reduced the activity of Bmal1, which led to decreases in the expression of antioxidant genes modulated by Nrf2 via the ARE in proximal promoters, such as HO-1 and NQO-1 (Fig. 5D and E), and ultimately triggered oxidative stress in the liver of mice (Fig. 6A–F). Besides, the dysregulation of mitochondrial dynamics and oxidative metabolism induced by the deficiency in Bmal1 was responsible for the elevated levels of oxidative stress in mice kept in constant darkness. Supplementation with tea polyphenols alleviated oxidative stress via mechanisms related to the circadian clock, which need to be further investigated.
Nrf2, which is the central regulator involved in maintaining cellular redox homeostasis, can interact with the ARE in the nucleus under oxidative conditions and modulate the expression of phase II enzymes such as HO-1 and NQO-1, which protects cells from oxidative damage, inflammation, and apoptosis.55 Previous studies indicated that EGCG, which is a major antioxidant in green tea, potentiates the cellular defense capacity against UV and oxidative stress via the de novo synthesis of antioxidant or phase II detoxifying enzymes mediated by Nrf2.56 In accordance with a previous study, TP activated the Keap1/Nrf2 antioxidant defense pathway in the liver and HepG2 cells (Fig. 4 and 5). HO-1 and NQO-1, which are the pivotal cytoprotective enzymes in cellular defense, play essential roles in the cellular defense against imbalances in cellular redox status induced by pathological events.19,20 Evidence has shown that the upregulation of HO-1 contributes to the cellular defense mechanism in response to oxidative insults.57 In addition, NQO-1 reduces the production of ROS by catalyzing the reduction of quinones to hydroquinones within the cell.58 The current study revealed that TP can dramatically promote the expression of HO-1 and NQO-1 by regulating the Nrf2 antioxidant defense pathway.
In addition, the Keap1/Nrf2 antioxidant defense pathway can be activated directly by targeting the Nrf2–Keap1 complex or indirectly by the phosphorylation of Nrf2 mediated by ERK1/2 in response to oxidative stress and different stimuli. More specifically, metallothionein-III alleviated oxidative stress induced by 6-hydroxydopamine via the Nrf2/HO-1 signaling pathway regulated by PI3K/Akt and ERK1/2.59 Lutein markedly enhanced the expression of Nrf2/HO-1 via upregulation of the ERK1/2 pathway to alleviate oxidative stress and inflammatory responses triggered by LPS in BV-2 microglial cells.22 The current experiments were designed to assess whether ERK1/2 is a potential regulator of the Keap1/Nrf2 antioxidant defense pathway activated by TP and prevents imbalances in cellular redox status in HepG2 cells induced by H2O2. In fact, we demonstrated that pretreatment with TP reduced the suppression of ERK1/2 triggered by H2O2 in HepG2 cells, which indicated that activation of the phosphorylation of ERK1/2 may contribute to the Keap1/Nrf2 antioxidant defense pathway regulated by TP (Fig. 3E). Moreover, pretreatment with the ERK1/2 inhibitor U0126 reduced the activation of Nrf2 upregulated by TP and the expression of HO-1 and NQO-1 (Fig. 4C and D). Notably, the results revealed that ERK1/2 plays a crucial role in the mechanism of activation of Nrf2/HO-1 triggered by TP.
In addition, the induction of antioxidant enzyme defense systems plays an essential role in scavenging ROS and protecting cells against damage by free radicals. It is well known that the overexpression of GSH, SOD and CAT can provide cytoprotective effects against ROS in BRL-3A rat liver cells, human umbilical vein endothelial cells (HUVECs), and HepG2 human hepatoma cells.18,60,61 Similar results were also found in this study: oxidative stress triggered by H2O2 and constant darkness decreased the activities of GSH, SOD and CAT and led to an increase in ROS levels, which could be efficiently reversed by treatment with TP (Fig. 2 and 6). At the expense of levels of H2O2 or organic peroxides, CAT converts H2O2 into H2O to help maintain redox status homeostasis. Similarly, GPx catalyzes the oxidation of GSH to oxidized glutathione (GSSG) and GR recycles GSSG to form reduced glutathione using NADPH. The recovery of GSH levels achieved by TP may be explained by the upregulation of γGCS, which is a rate-limiting enzyme in the biosynthesis of GSH. Such findings suggest that TP protected HepG2 cells from apoptosis triggered by H2O2 in association with their antioxidant properties.
As discussed below, the activation of cytosol signaling is regulated by cellular redox status. Recent research has revealed that H2O2 induces apoptosis via regulating the PI3K/AKT, MAPK, and NFκB signaling pathways. In fact, the PI3K pathway was thought to be a key component in governing the cellular defense system against oxidative injury and apoptosis. In addition, the activation of JNK and p38 kinase is pro-apoptotic in response to stress and cellular damage, whereas ERK1/2 is required for survival signaling. In accordance with previous studies, our results showed that H2O2 markedly inhibited the phosphorylation of Akt and ERK and meanwhile activated the JNK and p38 signaling pathways, which could be partly reversed by pretreatment with TP (Fig. 3C–F). Furthermore, TP significantly inhibited the phosphorylation of NFκB triggered by H2O2 (Fig. 3G and H); NFκB is one of the categories of “rapid-acting” primary transcription factors and plays a central regulatory role in a complicated network of inflammatory cytokines, cell proliferation, apoptosis and autophagy. Previous studies indicated that the elevated expression of ROS in cells acted as one of the principal stimuli that modulated the PI3K/AKT, MAPK, and NFκB signaling pathways.17 In addition, pretreatment with TP reduced the production of ROS triggered by H2O2. Moreover, NAC, which scavenges ROS, exhibited a synergistic effect in the presence of TP (Fig. 3), which implies that ROS may act as pivotal upstream signaling molecules that modulate the PI3K/AKT, MAPK, and NFκB signaling pathways.
Our findings demonstrated that TP alleviated oxidative stress and apoptosis induced by H2O2 in HepG2 cells. Specifically, TP partially normalized cellular redox homeostasis in C57BL/6J mouse liver and HepG2 cells via regulating the Keap1/Nrf2 antioxidant defense pathway. Therefore, TP could be an effective functional factor for combating damage to the liver related to oxidative stress.
Conflict of interest
The authors declare that there are no conflicts of interest.Abbreviations
| TP | Tea polyphenols |
| NFκB | Transcription factors of the nuclear factor κB |
| MAPK | Mitogen-activated protein kinase |
| Nrf2 | Nuclear factor-erythroid 2-related factor 2 |
| ROS | Reactive oxygen species |
| Keap1 | Kelch-like ECH-associated protein-1 |
| HO-1 | Heme oxygenase 1 |
| NQO-1 | NAD(P)H:quinone oxidoreductase 1 |
| GSH | Glutathione |
| SOD | Superoxide dismutase |
| GPx | Glutathione peroxidase |
| CAT | Catalase |
| EGC | Epigallocatechin |
| EGCG | Epigallocatechin-3-gallate |
| ECG | Epicatechin-3-gallate |
| NAC | N-Acetyl-L-cysteine |
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 31571842) and the National Key Research and Development Program of China (No. 2016YFD0400601).References
- A. Owen, A. Schapira, P. Jenner and C. Marsden, Ann. N. Y. Acad. Sci., 1996, 786, 217–223 CrossRef CAS PubMed.
- S. Rosales-Corral, D.-X. Tan, L. Manchester and R. J. Reiter, Oxid. Med. Cell. Longevity, 2015, 2015 Search PubMed.
- X. Xu, H. Li, X. Hou, D. Li, S. He, C. Wan, P. Yin, M. Liu, F. Liu and J. Xu, Mediators Inflammation, 2015, 2015 Search PubMed.
- Y. Mi, C. Xiao, Q. Du, W. Wu, G. Qi and X. Liu, Free Radical Biol. Med., 2016, 90, 230–242 CrossRef CAS PubMed.
- L. Ling, K. Tan, H. Lin and G. Chiu, Cell Death Dis., 2011, 2, e129 CrossRef PubMed.
- D. Han, W. Chen, X. Gu, R. Shan, J. Zou, G. Liu, M. Shahid, J. Gao and B. Han, Oncotarget, 2017, 8, 14680–14692 Search PubMed.
- A. K. Pati, A. Chandrawanshi and A. Reinberg, Curr. Sci., 2001, 81, 32–52 Search PubMed.
- A. Sharifian, S. Farahani, P. Pasalar, M. Gharavi and O. Aminian, J. Circadian Rhythms, 2005, 3, 15 CrossRef PubMed.
- H. Buyukhatipoglu, I. Kirhan, O. F. Dag, M. N. Turan, M. Vural, A. Taskin, N. Aksoy and Y. Sezen, Am. J. Med. Sci., 2010, 340, 462–467 CrossRef PubMed.
- A. Casado, A. Castellanos, M. López-Fernández, R. Ruiz, E. L. Imedio, C. Castillo and A. M. Fernández-Nieto, Clin. Chem. Lab. Med., 2011, 49, 471–477 CrossRef CAS PubMed.
- V. Y. Kulikov, Y. M. Fridman and A. Fomin, Alaska Medicine, 2006, 49, 81–84 Search PubMed.
- T. Ulas, H. Buyukhatipoglu, I. Kirhan, M. Dal, M. Eren, A. Hazar, M. Demir, T. Aydogan, F. Karababa and A. Uyanikoglu, Eur. Rev. Med. Pharmacol. Sci., 2012, 16, 594–599 CAS.
- S.-q. Shi, T. S. Ansari, O. P. McGuinness, D. H. Wasserman and C. H. Johnson, Curr. Biol., 2013, 23, 372–381 CrossRef CAS PubMed.
- Q. Xu, P. Ma, W. Yu, C. Tan, H. Liu, C. Xiong, Y. Qiao and Y. Du, Mar. Biotechnol., 2010, 12, 292–298 CrossRef CAS PubMed.
- E. Albano, Mol. Aspects Med., 2008, 29, 9–16 CrossRef CAS PubMed.
- F. Yin, H. Sancheti, Z. Liu and E. Cadenas, J. Physiol., 2016, 594, 2025–2042 CrossRef CAS PubMed.
- Q. Liu, Y. Hu, Y. Cao, G. Song, Z. Liu and X. Liu, J. Agric. Food Chem., 2017, 65, 338–347 CrossRef CAS PubMed.
- L. Yuan, X. Ren, Y. Wu, J. Wang, H. Xiao and X. Liu, Biomed. Aging Pathol., 2013, 3, 153–159 CrossRef CAS.
- Y. P. Hwang and H. G. Jeong, Toxicol. Appl. Pharmacol., 2010, 242, 18–28 CrossRef CAS PubMed.
- L. Ibáñez, M. L. Ferrándiz, R. Brines, D. Guede, A. Cuadrado and M. J. Alcaraz, Oxid. Med. Cell. Longevity, 2014, 2014 Search PubMed.
- H. E. de Vries, M. Witte, D. Hondius, A. J. Rozemuller, B. Drukarch, J. Hoozemans and J. van Horssen, Free Radical Biol. Med., 2008, 45, 1375–1383 CrossRef CAS PubMed.
- W. Wu, Y. Li, Y. Wu, Y. Zhang, Z. Wang and X. Liu, Mol. Nutr. Food Res., 2015, 59, 1663–1673 CAS.
- S. Qin and D. X. Hou, Mol. Nutr. Food Res., 2016, 60, 1731–1755 CAS.
- M. R. de Oliveira, G. da Costa Ferreira, F. B. Brasil and A. Peres, Mol. Neurobiol., 2017, 1–15 Search PubMed.
- J. Kanwar, I. M. Mujtaba Taskeen, C. Huo, T. H. Chan and Q. P. Dou, Front. Biosci., Elite Ed., 2012, 4, 111 CrossRef.
- M. A. Keske, H. L. H. Ng, D. Premilovac, S. Rattigan, J. Kim, K. Munir, P. Yang and M. J. Quon, Curr. Med. Chem., 2015, 22, 59–69 CrossRef CAS PubMed.
- E. Lecumberri, Y. M. Dupertuis, R. Miralbell and C. Pichard, Clin. Nutr., 2013, 32, 894–903 CrossRef CAS PubMed.
- M. K. Roy, M. Koide, T. P. Rao, T. Okubo, Y. Ogasawara and L. R. Juneja, Int. J. Food Sci. Nutr., 2010, 61, 109–124 CrossRef CAS PubMed.
- A. E.-M. M. Afify, E. A. Shalaby and H. S. El-Beltagi, Not. Bot. Horti Agrobot. Cluj-Napoca, 2011, 39, 117 CAS.
- Y.-H. Hong, E. Y. Jung, K.-S. Shin, T. Y. Kim, K.-W. Yu, U. J. Chang and H. J. Suh, Appl. Biochem. Biotechnol., 2012, 166, 165–175 CrossRef CAS PubMed.
- S. Guo, E. Bezard and B. Zhao, Free Radical Biol. Med., 2005, 39, 682–695 CrossRef CAS PubMed.
- F. Nie, X. Zhang, Q. Qi, L. Yang, Y. Yang, W. Liu, N. Lu, Z. Wu, Q. You and Q. Guo, Toxicology, 2009, 260, 60–67 CrossRef CAS PubMed.
- Z. Liu, G. Song, C. Zou, G. Liu, W. Wu, T. Yuan and X. Liu, Free Radical Biol. Med., 2015, 84, 42–53 CrossRef CAS PubMed.
- Z. Liu, Q. Qiao, Y. Sun, Y. Chen, B. Ren and X. Liu, Mol. Nutr. Food Res., 2017 DOI:10.1002/mnfr.201600717.
- H.-U. Simon, A. Haj-Yehia and F. Levi-Schaffer, Apoptosis, 2000, 5, 415–418 CrossRef CAS PubMed.
- M. B. Azad, Y. Chen and S. B. Gibson, Antioxid. Redox Signaling, 2009, 11, 777–790 CrossRef CAS PubMed.
- M. Zafarullah, W. Li, J. Sylvester and M. Ahmad, Cell. Mol. Life Sci., 2003, 60, 6–20 CrossRef CAS PubMed.
- T. Nguyen, P. Nioi and C. B. Pickett, J. Biomed. Sci., 2009, 284, 13291–13295 CAS.
- S. Yong-Xin, X. Ai-Hua, Y. Yang and J. Li, J. Biomed. Sci., 2015, 22 DOI:10.1186/s12929-015-0212-5.
- R. Tsao, Nutrients, 2010, 2, 1231–1246 CrossRef CAS PubMed.
- M.-H. Pan and C.-T. Ho, Chem. Soc. Rev., 2008, 37, 2558–2574 RSC.
- M.-L. Hu, Chang Gung Medical Journal, 2011, 34, 449–460 Search PubMed.
- J. Dai and R. J. Mumper, Molecules, 2010, 15, 7313–7352 CrossRef CAS PubMed.
- V. Habauzit and C. Morand, Ther. Adv. Chronic Dis., 2012, 3, 87–106 CrossRef CAS PubMed.
- A. Peng, T. Ye, D. Rakheja, Y. Tu, T. Wang, Y. Du, J. K. Zhou, N. D. Vaziri, Z. Hu and C. Mohan, Kidney Int., 2011, 80, 601–611 CrossRef CAS PubMed.
- Y. Xu, J.-j. Zhang, L. Xiong, L. Zhang, D. Sun and H. Liu, J. Nutr. Biochem., 2010, 21, 741–748 CrossRef CAS PubMed.
- Y.-J. Cai, L.-P. Ma, L.-F. Hou, B. Zhou, L. Yang and Z.-L. Liu, Chem. Phys. Lipids, 2002, 120, 109–117 CrossRef CAS PubMed.
- B. Faraut, V. Bayon and D. Léger, Sleep Med. Rev., 2013, 17, 433–444 CrossRef PubMed.
- E. L. Haus and M. H. Smolensky, Sleep Med. Rev., 2013, 17, 273–284 CrossRef PubMed.
- S. Puttonen, M. Härmä and C. Hublin, Scand. J. Work, Environ. Health, 2010, 96–108 Search PubMed.
- C. L. Drake, T. Roehrs, G. Richardson, J. K. Walsh and T. Roth, Sleep, 2004, 27, 1453–1462 CrossRef PubMed.
- J. Bass and J. S. Takahashi, Science, 2010, 330, 1349–1354 CrossRef CAS PubMed.
- J. Lee, M. Moulik, Z. Fang, P. Saha, F. Zou, Y. Xu, D. L. Nelson, K. Ma, D. D. Moore and V. K. Yechoor, Mol. Cell. Biol., 2013, 33, 2327–2338 CrossRef CAS PubMed.
- D. Jacobi, S. Liu, K. Burkewitz, N. Kory, N. H. Knudsen, R. K. Alexander, U. Unluturk, X. Li, X. Kong and A. L. Hyde, Cell Metab., 2015, 22, 709–720 CrossRef CAS PubMed.
- I. Buendia, P. Michalska, E. Navarro, I. Gameiro, J. Egea and R. León, Pharmacol. Ther., 2016, 157, 84–104 CrossRef CAS PubMed.
- H.-K. Na and Y.-J. Surh, Food Chem. Toxicol., 2008, 46, 1271–1278 CrossRef CAS PubMed.
- P. Wang, X. Peng, Z.-F. Wei, F.-Y. Wei, W. Wang, W.-D. Ma, L.-P. Yao, Y.-J. Fu and Y.-G. Zu, Biochim. Biophys. Acta, 2015, 1850, 1751–1761 CrossRef CAS PubMed.
- V. Vasiliou, D. Ross and D. W. Nebert, Hum. Genomics, 2006, 2, 329 CrossRef CAS PubMed.
- Y. P. Hwang, H. G. Kim, E. H. Han and H. G. Jeong, Toxicol. Appl. Pharmacol., 2008, 231, 318–327 CrossRef CAS PubMed.
- H. H. Kwok, W. Y. Ng, M. S. M. Yang, N. K. Mak, R. N. S. Wong and P. Y. K. Yue, Free Radical Biol. Med., 2010, 48, 437–445 CrossRef CAS PubMed.
- S. R. Subramaniam and E. M. Ellis, Toxicol. Appl. Pharmacol., 2011, 250, 130–136 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |