
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipase-mediated synthesis of sugar–PEG-based amphiphiles for encapsulation and stabilization of indocyanine green†
Vinod Khatria,
Sumati Bhatiab,
Katharina Achazib,
Satyanarayan Deepac,
Ekta Kohlic,
Sunil K. Sharma a,
Rainer Haagb and
Ashok K. Prasad*a
a,
Rainer Haagb and
Ashok K. Prasad*a
aBioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi 110 007, India. E-mail: ashokenzyme@gmail.com
bInstitute for Chemistry and Biochemistry, Free University Berlin, Takustrasse 3, Berlin 14195, Germany
cDIPAS, DRDO, Lucknow Road, Timarpur, Delhi 110 054, India
First published on 31st July 2017
Abstract
Indocyanine green (ICG) is a near-infrared dye for wide-ranging applications, but its utility for biological studies is limited due to poor aqueous stability and concentration-dependent aggregation. Aqueous polymeric micelles/micellar aggregates of amphiphilic copolymers could be used to protect indocyanine green (ICG) by encapsulation, which is used in laser-mediated photothermal therapy (PTT) and photodynamic therapy (PDT). The sugar–PEG-based amphiphilic copolymers were synthesized using Novozym 435-catalyzed transesterification reaction under bulk conditions for encapsulation of ICG. A dye loading study revealed that micelles derived from copolymers having decanoylated and myristoylated sugar moieties encapsulate 62% and 92% of the ICG, respectively, with a multi-fold increase in its aqueous stability. Furthermore, an efficient internalization of micelles of acylated amphiphilic copolymers in aqueous medium was demonstrated by incubating cells with Nile red-encapsulated amphiphiles.
1. Introduction
Indocyanine green (ICG) is a suitable near-infrared (NIR) light-absorbing compound, which has several diagnostic and medical applications and is used for the treatment of cancer by laser-mediated photothermal (PTT)1 and photodynamic therapies (PDT).2,3 However, the application of ICG for clinical imaging and therapeutic applications has several limitations like a short plasma half-life and poor aqueous stability.4 Various nanoformulations of ICG have been developed to overcome these limitations.5–8 It is difficult for the human body to degrade most of the established nanoplatforms involved in the nanoformulations, i.e. silica nanoparticles,9,10 gold nanoparticles,11,12 and other inorganic materials.13–16 Therefore, micelles and vesicles derived from polyethylene glycol (PEG) are used for encapsulating ICG.17–20 Polymeric micelles are capable of a high drug loading for drug delivery applications and possess excellent colloidal stability compared to other types of nanocarriers, such as liposomes and nanoparticles.21–24In recent years, sugar-based delivery vehicles have caught significant attention due to their inherent biocompatibility and bioadhesive/targeting properties.25–28 Specifically, there have been reports of lectin proteins and other biomolecules with sugar molecules on the surface of micelles.29,30 Water-soluble, sugar-based polymers are now being routinely used to prolong drug circulation and residence time within affected cells. Further, it has been shown that nano-sized particles have a great potential in cancer therapy due to enhanced permeability and retention (EPR) effect.31 Herein, we report the synthesis of novel sugar–PEG-based amphiphilic copolymers in solvent-free conditions using Novozym 435 as a catalyst. The encapsulation and aqueous stability of the encapsulated ICG dye was determined in the micelles of amphiphilic copolymers by fluorescence measurements at 20 °C and at 37 °C. The Nile red fluorescent dye was used for the cellular uptake study of micelles in A549 cells.
2. Experimental
2.1. Materials
Dry reactions were carried out under a nitrogen atmosphere. Analytical TLCs were performed on pre-coated Merck silica-gel 60F254 plates, and silica gel (100–200 mesh) was used for column chromatography. All materials and reagents were commercially available and used without further purification. For polymerization reaction poly[ethylene glycol bis(carboxymethyl)ether]diethyl ester and sugar monomers 2,5-anhydro-D-mannitol (1) and 2,5-anhydro-3,4-di-O-benzyl-D-mannitol (2) were dried under vacuum for 10 h prior to use. Dialysis membrane (MWCO 1.2–2 kDa), Novozym 435 and Nile red were purchased from Sigma-Aldrich Pvt. Ltd. The cell line A549 was purchased from DMSZ, Germany (Catalogue no. ACC107). Milli-Q water was used for encapsulation and physicochemical characterization experiments.2.2. Instrumentations and methods
Molecular weight and molecular-weight distribution of copolymers were recorded by gel permeation chromatography (GPC) equipped with an Agilent 1100 pump, refractive index detector, and Suprema columns. THF was used as eluent with a flow rate of 1 mL min−1. Calibration of molecular weights was done using polystyrene as a standard. Surface tension of copolymers was determined by the pendent drop tensiometer OCA 20 for calculating the critical aggregation concentration (CAC) values. Samples were prepared in Milli-Q water 24 h before measurement. For the surface tension measurement, first a stock solution of 1 mM concentration was prepared and then samples were prepared by the half-dilution method. The aggregation behavior of copolymers was studied over a concentration range of 10−3 to 10−7 M. Measurement was stopped when surface tension did not change. Equilibrium was maintained generally for 40–60 min at 20 °C. The size of micelles/micellar aggregates was determined by dynamic light scattering (DLS) using a Zetasizernano ZS analyzer, a Malvern instrument equipped with a 633 nm laser at a scattering angle of 173°. The measurement was performed at 25 °C after diluting the samples at an appropriate concentration in Milli-Q water. Solutions were filtered via 0.2 μm polytetrafluoroethylene (PTFE) filters and the samples were allowed to equilibrate for 4 h at 20 °C. All experiments were done with disposable transparent cuvettes. Images of micelles formed were obtained by transmission electron microscope (TEM) using a Jeol JEM-2100F instrument that accelerated at 200 kV. Samples were prepared by dipping a carbon-coated copper grid into a copolymer solution. The grid was blotted with filter paper, which was followed by staining with 1% phosphotungustic acid (PTA). The copper grid loaded with micelles was dried at room temperature before the images were taken. Absorbance spectra were recorded between 250–900 nm using a Scinco S-3150 UV-visible spectrophotometer. All measurements were carried out with a disposable, transparent UV cuvette. The fluorescence spectra were determined on both native ICG and formulated ICG using a Jasco FP-6500 spectro-fluorometer equipped with a thermostatic cell holder and a DC-powered 150 W xenon lamp. For fluorescence, excitation was done at 780 nm and emission was recorded from 790–900 nm. Origin 6.1 software was used for plotting the graphs.2.3. Synthesis of copolymers 3–7
Copolymer 3 was obtained as viscous oil in 70% yield; 1H NMR (500 MHz, CD3OD, δ (ppm)): 1.26 (t, J = 7.0 Hz, 3H), 3.62–3.70 (m, 474H), 3.76–3.99 (m, 34H), 4.15–4.41 (m, 75H); 13C NMR (125 MHz, CD3OD, δ (ppm)): 64.2, 67.9, 70.2, 70.5, 77.6, 81.2, 170.8; IR (thin film, cm−1): 3326, 2872, 1752, 1204, 1110; GPC (THF: 1 mL min−1): Mw = 9258 g mol−1, Mn = 7893 g mol−1, PDI = 1.17.
Copolymer 4 was obtained as viscous oil in 80% yield; 1H NMR (500 MHz, CDCl3, δ (ppm)): 0.82 (t, J = 7.1 Hz, 3.4H), 1.21–1.23 (m, 15H), 1.54–1.57 (m, 2H), 2.29 (t, J = 7.4 Hz, 2H), 3.59–3.69 (m, 44H), 4.10–4.15 (m, 4H), 4.18–4.34 (m, 4H), 5.06–5.19 (m, 1.1H); 13C NMR (125 MHz, CDCl3, δ (ppm)): 14.1, 22.7, 24.7, 29.0, 29.2, 29.3, 29.4, 31.8, 33.9, 63.2, 68.3, 68.6, 70.6, 70.9, 77.8, 81.2, 170.2, 172.7; IR (thin film, cm−1): 3491, 2923, 2868, 1744, 1350, 1248, 1111, 950, 849; GPC (THF: 1 mL min−1): Mw = 9200 g mol−1, Mn = 5757 g mol−1, PDI = 1.59.
Copolymer 5 was obtained as viscous oil in 76% yield; 1H NMR (500 MHz, CDCl3, δ (ppm)): 0.85 (t, J = 6.7 Hz, 3.2H), 1.22–1.25 (m, 22H), 1.56–1.59 (m, 2H), 2.30 (t, J = 7.1 Hz, 2H), 3.61–3.71 (m, 44H), 4.14–4.28 (m, 8H), 5.08–5.20 (m, 1.1H); 13C NMR (125 MHz, CDCl3, δ (ppm)): 14.1, 22.7, 24.7, 29.0, 29.2, 29.3, 29.4, 29.6, 29.7, 31.9, 33.9, 63.1, 68.3, 68.6, 70.5, 70.9, 77.8, 81.2, 170.1, 170.9, 172.7; IR (thin film, cm−1): 3512, 2922, 2854, 1753, 1461, 1350, 1249, 1198, 1113, 950, 849; GPC (THF: 1 mL min−1): Mw = 5239 g mol−1, Mn = 4264 g mol−1, PDI = 1.22.
Copolymer 6 was obtained as viscous oil in 72% yield; 1H NMR (500 MHz, CDCl3, δ (ppm)): 1.26 (t, J = 7.1 Hz, 3H), 3.61–3.69 (m, 891H), 3.97 (d, J = 1.7 Hz, 20H), 4.11–4.14 (m, 49H), 4.17–4.22 (m, 48H), 4.24–4.26 (m, 24H), 4.50–4.53 (m, 48H), 7.27–7.35 (m, 134H); 13C NMR (125 MHz, CDCl3, δ (ppm)): 64.1, 68.4, 70.5, 70.9, 72.0, 80.9, 84.0, 127.8, 128.1, 128.5, 128.6, 137.2, 170.2; IR (thin film, cm−1): 2872, 1751, 1455, 1350, 1250, 1205, 1105, 950, 849; GPC (THF: 1 mL min−1): Mw = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 987 g mol−1, Mn = 12485 g mol−1, PDI = 1.20.
987 g mol−1, Mn = 12485 g mol−1, PDI = 1.20.
Copolymer 7 was obtained as viscous oil in 77% yield; 1H NMR (400 MHz, CDCl3, δ (ppm)): 1.24 (t, J = 7.0 Hz, 3H), 3.59–3.66 (m, 567H), 3.91 (bs, 22H), 4.09–4.11 (m, 44H), 4.15–4.22 (m, 60H), 4.48–4.51 (m, 42H), 7.24–7.33 (m, 124H); 13C NMR (100.6 MHz, CDCl3, δ (ppm)): 64.2, 68.4, 70.6, 71.0, 72.1, 80.9, 84.1, 127.8, 128.1, 128.6, 137.2, 170.2; GPC (THF: 1 mL min−1): Mw = 10573 g mol−1, Mn = 8502 g mol−1, PDI = 1.24.
2.4. ICG encapsulation in sugar–PEG-based copolymeric micelles
The film method was implemented to encapsulate ICG within micelles. For encapsulation, a 0.25 mM stock solution of ICG was prepared by dissolving 2 mg of the dye in 10 mL of dry dichloromethane. Aliquots (100 μL) were added to 5 mL vials and the solvent was allowed to evaporate. Additionally, an aqueous solution of copolymers (10 mg mL−1) that had been stirred for 2 h was added to a sample vial containing ICG. The solutions were stirred overnight, after which a dialysis against water was done to remove the excess ICG. In the end, ICG-loaded aqueous micellar solutions of copolymer 4, 5, and 6 were afforded.2.5. ICG encapsulation efficiency and loading capacity
To determine the encapsulation efficiency (EE) of ICG and loading capacity (LC) of micelles, sugar–PEG-based copolymer (10 mg) and ICG (1 mg) were dissolved in DMSO (3 mL) at room temperature.18 The mixture was dialyzed with a dialysis tube against water for 3 days. A certain volume of the dialyzed encapsulated ICG solution was diluted 20 times with DMSO to destroy the micelles. The concentration of ICG in a diluted solution was determined by comparing the absorbance at 780 nm to a standard curve of ICG within a linear range of 0–10 μM in DMSO. The EE and LC were determined according to the following formulas:| EE (%) = (weight of encapsulated ICG/weight of initially added ICG) × 100. |
| LC (%) = (weight of encapsulated ICG/total weight of ICG and copolymer) × 100. |
2.6. Nile red encapsulation protocol
For encapsulation of Nile red, a 5 mM stock solution of Nile red was prepared by dissolving 1.59 mg of Nile red in 1 mL of dry dichloromethane. A small aliquot (200 μL) from the stock solution was added to the sample vial and the solvent was allowed to evaporate. Additionally, an aqueous solution of copolymer (8 mg) was first stirred for 2 h and then added to the sample vial with the Nile red. The solutions were allowed to stir overnight, which was followed by filtration to remove the non-encapsulated Nile red dye.2.7. Cytotoxicity protocol
The xCELLigence real-time cell analyzer (RTCA) SP from Roche (Mannheim, Germany) was used to investigate the cytotoxicity of all the synthesized copolymers. A549 cells grown on a 96 well E-plate were placed in the RTCA. The impedance was measured every 15 min. After 24 h, the plate was removed from the RTCA, and three water-soluble copolymers were added at final concentrations of 10, 100, and 500 μg mL−1. The non-treated and doxorubicin-treated cells were used for the control experiment. The plate was replaced in the RTCA and real time impedance measurements was continued for another 72 h. GraphPad prism 5.01 software was used to plot the analysis of the end points obtained from the RTCA after 24, 48, and 72 h post-treatment.2.8. Cellular uptake protocol
Briefly, 0.2–0.3 × 10−6 cells per well were placed in a 12 well plate and allowed to attach overnight. To assess the cellular uptake of encapsulated micelles, the growth medium was completely replaced with the serum and an antibiotic-free medium that contained Nile red-loaded polymeric micelles at two different concentrations (10 and 100 μg mL−1) for two different time points (2 and 4 h). For the internalization studies, 0.2 mL solution was added to each well, which had been prepared from 0.1 mL of 10% FBS–DMEM and 0.1 mL of distilled water with Nile red-loaded fluorescent polymeric micelles that led to the respective final micelle concentrations. After the desired incubation period, the cells were washed three times with ice-cold PBS and then incubated for 20 min with DAPI (360 ± 20 nm and 460 ± 25 nm, blue channel). Cells that were incubated in the media alone without any formulations served as the negative control for auto-fluorescence. At the end of each time period, the cells were washed, fixed (using 4% paraformaldehyde), and examined under a confocal microscope.3. Results and discussion
3.1. Synthesis of sugar–PEG-based copolymers 3–7 and its characterization
During the transesterification reaction, some of the lipases like Candida antarctica lipase recognized a primary hydroxyl group over the secondary hydroxyl group.32 The copolymerization of 2,5-anhydro-D-mannitol (1) and poly[ethylene glycol bis(carboxymethyl)ether]diethyl ester (PEG-600 diethyl ester) was achieved by transesterification reaction catalyzed by Candida antarctica lipase B immobilized on polyacrylate (Lewatite), commonly known as Novozym 435 under solvent-free conditions. Thus, Novozym 435 was added to the homogeneous mixture of PEG-600 diethyl ester and 2,5-anhydro-D-mannitol (1) at 70 °C; the mixture was stirred for 48 h under reduced pressure to obtain the dihydroxy copolymer 3 in 70% yield (Scheme 1). The PEG-600 diethyl ester and the 2,5-anhydro-D-mannitol (1) were prepared from PEG-600 and ethanol under acidic condition and from D-glucosamine, respectively by following a literature procedure.33–35 The post-polymerization functionalization of copolymer 3, i.e. its acylation with decanoyl chloride and myristoyl chloride was done in dichloromethane in the presence of potassium carbonate base to obtain decanoylated and myristoylated copolymers 4 and 5 in 80% and 76% yields, respectively. The study of 1H NMR spectra of copolymers 4 and 5 revealed 50–55% installation of acyl group on the secondary hydroxyl groups of dihydroxy copolymer 3. The reaction of 2,5-anhydro-D-mannitol (1) with PEG 1000 diethyl ester36 under the developed bulk reaction condition formed a gel (crosslinked product), which was not soluble in any solvent and therefore was not used for further studies. The copolymers 6 and 7 were prepared under solvent free conditions at 70 °C by Novozym 435-catalyzed transesterification reaction of 2,5-anhydro-3,4-di-O-benzyl-D-mannitol (2) with PEG-1000 and PEG-600 diethyl esters in 72% and 77% yields, respectively. Reaction of PEG-1000 diethyl ester with dibenzylated anhydro-mannitol 2 did not form a gel (crosslinked product) as earlier, which may have been due to the masking of the secondary hydroxyl groups of the sugar. The transesterification reaction between dibenzylated anhydro-mannitol 2 and PEG-600 diethyl ester led to the water insoluble copolymer 7, which was not used for further studies (Scheme 1). | ||
| Scheme 1 Synthesis of sugar–PEG based copolymers 3–7. | ||
All five copolymers 3–7 were well characterized by their IR, 1H-, 13C-, DEPT-135, COSY, and HSQC NMR spectral data analysis. The number-average molecular weight (Mn) for all five copolymers was determined by GPC. The Mn of copolymers 3, 6, and 7 was further confirmed by an end-group analysis by comparing three protons of methyl group of PEG ethyl ester that resonated at 1.26 ppm with protons of polyethylene glycol units in 1H NMR spectra (Table S1, ESI†). The Mn of copolymers 4 and 5 was not determined by end-group analysis due to the overlapping of protons of acyl chain with the three protons of methyl group of PEG ethyl ester. The Mn determined by GPC and 1H NMR indicated the presence of approximately ten monomeric units in copolymers 3, 4, and 5 and twelve monomeric units in copolymers 6 and 7.
The integration for three protons of the methyl group of acyl chains resonating at 0.88 ppm in comparison to the 44 protons of polyethylene glycol chain of one repeating unit resonating at 3.62–3.70 ppm clearly indicated 50–55% acylation of the secondary hydroxyl groups in copolymers 4 and 5.
3.2. Aggregation behavior study of copolymers 4–6 in aqueous solution
Aggregation behavior and size of the aggregates of copolymers 4, 5, and 6 in aqueous solution were investigated by using surface tension and dynamic light scattering measurements, respectively. Surface tension data were plotted as logarithm function of copolymer concentration, where a break in the curve occurred at a critical aggregation concentration (CAC) (Fig. 1).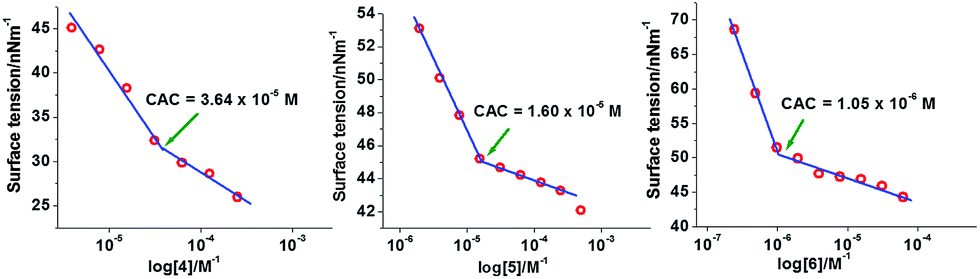 | ||
| Fig. 1 Surface tension (γ) versus log concentration plots of copolymers 4, 5, and 6 in aqueous solution at 20 °C. | ||
The CAC values obtained by surface tension measurement for copolymers 4, 5, and 6 are 36.4, 16.0, and 1.0 μM, respectively (Table 1). The CAC data revealed that the installation of more hydrophobic groups decreased the CAC value. CAC measurements and further studies on copolymers 3 and 7 could not be done, because the former copolymer was not amphiphilic and the latter one was insoluble in water. The size of aggregates of copolymers 4, 5 and 6 was determined by using the dynamic light scattering (DLS) method. The concentration of samples in aqueous solution to determine their size was kept above CAC for all three copolymers 4–6, i.e., 5 mg mL−1 for copolymers 4 and 5 and 2 mg mL−1 for copolymer 6. The hydrodynamic diameters of copolymers 4, 5, and 6 determined by DLS were found to be 10.9 ± 0.3, 13.4 ± 0.8, and 219.7 ± 4.9 nm, respectively (Fig. S19, ESI†). The DLS data showed that the size of micelles rose with the increase of hydrophobic character in the copolymers. The particle size of ICG-loaded micelles was also investigated by DLS and observed that the size of the micelles of copolymer 4 and 5 slightly increased after encapsulation of ICG but the size of micellar aggregates of copolymer 6 dramatically increased to 1.2 μm after encapsulation of ICG (Table 1). The morphology of micelles derived from copolymers 4, 5, and 6 in aqueous medium was determined by transmission electron microscope (TEM). The TEM analysis of copolymers 4 and 5 showed the hydrodynamic diameter of 9.0 ± 1.0 nm and 15.5 ± 1.0 nm, respectively (Table 1). The TEM images revealed uniform small micelles for copolymers 4 and 5. The TEM images of the supramolecular structure for the copolymer 6 revealed that the size of the aggregates was 50 ± 5 nm, which indicated that micelle aggregates had formed. Furthermore, the TEM images of the distribution of micellar aggregates of copolymer 6 were not uniform (Fig. 2).
| Polymer | CAC (μM) | Before ICG encapsulation | After ICG encapsulation | LC (%) | ||
|---|---|---|---|---|---|---|
| Dh (nm) | PDI | Dh (nm) | PDI | |||
| 4 | 36.4 | 10.9 ± 0.3 | 0.2 | 11.4 ± 0.4 | 0.2 | 5.8 ± 0.4 |
| 5 | 16.0 | 13.4 ± 0.8 | 0.2 | 20.5 ± 4.5 | 0.3 | 8.3 ± 0.5 |
| 6 | 1.0 | 219.7 ± 4.9 | 0.3 | 1267 ± 96.9 | 0.7 | 1.8 ± 0.2 |
 | ||
| Fig. 2 TEM images of micelles/micellar aggregates of copolymer 4 (A), copolymer 5 (B), and copolymer 6 (C) with a scale bar of 100 nm. | ||
3.3. Study of encapsulation of ICG by copolymers 4–6
ICG has extensively been used in PTT, PDT, and tumor localization studies in-spite of the fact that it quickly degrades in aqueous medium.37–40 The first step towards developing a nanoformulation for sustainable delivery of the ICG in physiological condition was to encapsulate the dye in the micelles and micellar aggregates of copolymers 4, 5, and 6. The aqueous solutions of ICG-encapsulated micelles of copolymers 4, 5, and 6 were then analyzed by a UV-visible and fluorescence spectrophotometer. The λmax of UV-visible spectrum of free ICG taken in Milli-Q water was 780 nm. The λmax of UV-visible spectra of ICG encapsulated in copolymers 4, 5, and 6 exhibited bathochromic shifts at 795, 798, and 792 nm, respectively (Fig. 3A). The λmax of emission in the fluorescence spectrum of an aqueous solution of ICG was observed at 802 nm. Bathochromic shifts of 26, 24, and 8 nm were observed when the fluorescence emission spectra of ICG encapsulated in micelles/micellar aggregates that had been derived from copolymers 4, 5 and 6, respectively were recorded (Fig. 3B). These bathochromic shifts clearly indicated that ICG had been encapsulated in micelles/micellar aggregates of copolymers 4, 5, and 6, which afforded the nanoformulation. Both the absorbance and fluorescence spectra revealed that the micellar aggregates derived from copolymer 6 had considerably less encapsulation. | ||
| Fig. 3 (A) UV-visible, (B) emission spectra, (C) fluorescence stability test at 20 °C, (D) fluorescence stability test at 37 °C of free ICG and encapsulated ICG in copolymers 4, 5, and 6. | ||
3.4. ICG loading and aqueous stability of the formulation
The ICG-loading efficiency in micelles/micellar aggregates of copolymers 4, 5, and 6 were determined with the help of a standard curve obtained from plotting the absorbance of the solution of different concentrations of ICG in DMSO. The ICG encapsulation efficiency in the micelles formed by copolymers 4 and 5 was much higher (62 and 92%) than the micellar aggregate formed by copolymer 6 (19%). The loading capacities of micelles obtained from copolymers 4 and 5 were 5.8 and 8.3%, respectively, which was far better than the micellar aggregates formed by copolymer 6 (1.8%).To compare the stability of ICG encapsulated in micelles/micellar aggregates of copolymers 4, 5, and 6 with native ICG dissolved in aqueous solution, the fluorescence intensity of the encapsulated and the native ICG was recorded for a period of 21 days at 20 °C and 19 days at 37 °C. It was revealed that nanoformulated ICG had better fluorescence stabilization while free ICG lost approximately 50% fluorescence intensity in 24 h and had only 5% after 3 weeks at 20 °C (Fig. 3C). The formulation of ICG with copolymer 6 showed better stabilization up to 2 days but rapidly lost the fluorescence after that and had only 2% after 3 weeks at 20 °C. However, the nanoformulation in micelles derived from copolymers 4 and 5 had better fluorescence stabilization, because approximately 100% fluorescence intensity was retained, even after 3 weeks, at 20 °C. DLS showed that the size of ICG-encapsulated micelles of copolymer 6 increased many times and no particle size was observed after 5 days, which indicated that the ICG-loaded micelles of copolymer 6 were not stable. However, the size of ICG encapsulated in the micelles of copolymers 4 and 5 showed little change in size, even on the 5th day; these micelles were thus stable and remained intact during the experiment.
Fluorescence intensity measurements of native ICG and its nanoformulation in micelles/micellar aggregates of copolymers 4, 5, and 6 at 37 °C revealed the native dye degraded up to 97% and the dye in the micellar aggregates of copolymer 6 decreased to 70% within 5 days (Fig. 3D). ICG encapsulated in the micelles of copolymers 4 and 5 exhibited almost 100% stability for 5 days. Comparing the fluorescence intensity beyond 5 days revealed that ICG-loaded micelles of copolymer 5 were more stable than copolymer 4. Thus, there was only 10% loss of the ICG encapsulated in the micelles of copolymer 5, whereas ICG in micelles of copolymer 4 lost its 50% fluorescence on the 11th day. About 40% fluorescence of ICG remained in the micelles of copolymer 5 after 19 days compared to 20% activity in the micelles of copolymer 4 (Fig. 3D). This shows that micelles of sugar–PEG copolymer with longer C14 acyl chains were more stable than the one with C10 acyl chains.
3.5. In vitro cytotoxicity assay
A good carrier should be non-toxic to cells. Therefore, the cytotoxicity of three micelles/micellar aggregates of sugar–PEG-based copolymers 4, 5, and 6 was investigated by real-time cell analysis using A549 cells for 72 h. All the copolymers showed good compatibility, because the cell viability was observed to be above 80%, even after 72 h of incubation up to a concentration of 500 μg mL−1 for copolymers 5 and 6 and up to a concentration of 100 μg mL−1 for copolymers 4. For copolymers 4 and 5, a concentration-dependent decrease in the cell's viability was monitored indicating a slight toxicity at the higher test concentrations. A time-dependent decrease in the cell's viability to a value below 60% was detected for copolymer 4 at the highest test concentration of 500 μg mL−1 (Fig. 4A). The cytotoxicity of ICG-loaded micelles was also tested using A549 cells for 72 h. The ICG-loaded copolymers 4 and 5 showed good compatibility up to the concentration of 100 μg mL−1 and the ICG-loaded copolymer 6 showed better compatibility up to a concentration of 500 μg mL−1 (Fig. 4B). | ||
| Fig. 4 (A) Cytotoxicity data of copolymers 4, 5, and 6, (B) cytotoxicity data of ICG-loaded copolymers. | ||
3.6. Cellular uptake study of micelles of copolymers 4 and 5
The cellular internalization of micelles of copolymer 4 and 5 was assessed by incubation of A549 cells with Nile red loaded micelles followed by the measurement of fluorescence intensity of incubated cells. The micellar aggregates of compound 6 were not tested for cellular internalization due to its poor Nile red-loading capability. The Nile red loaded micelles of copolymer 4 and 5 indicated the DLS size of 27.1 ± 2.9 and 12.5 ± 1.3 nm, respectively which is comparable with the ICG loaded micelles. The result of confocal images of micelles of copolymer 4 loaded with Nile red showed that 2 h incubation of cells with 10 μg mL−1 of Nile red-loaded micelles had a lower fluorescence intensity than the 4 h incubation (Fig. 5B and C) of the same micellar solution. But the incubation of cells with 100 μg mL−1 of Nile red-encapsulated micellar solution for 2 h had as much fluorescence intensity as the 4 h incubation (Fig. 5D and E). This suggests that there was an optimum concentration and time for the cellular uptake of Nile red-encapsulated micellar solution. The micelles of copolymer 5 loaded with Nile red also showed the cellular internalization of Nile red at different time intervals. The fluorescence intensity at a 2 h incubation of polymeric micelles loaded with Nile red had a weaker intensity than the 4 h incubation (Fig. 6B and C) at 10 μg mL−1 concentration. Similarly, the results of the confocal images showed that the 2 h incubation of cells at a concentration of 100 μg mL−1 had a lower fluorescence intensity than 4 h incubation (Fig. 6D and E) of cell with 100 μg mL−1 Nile red-encapsulated micellar solution.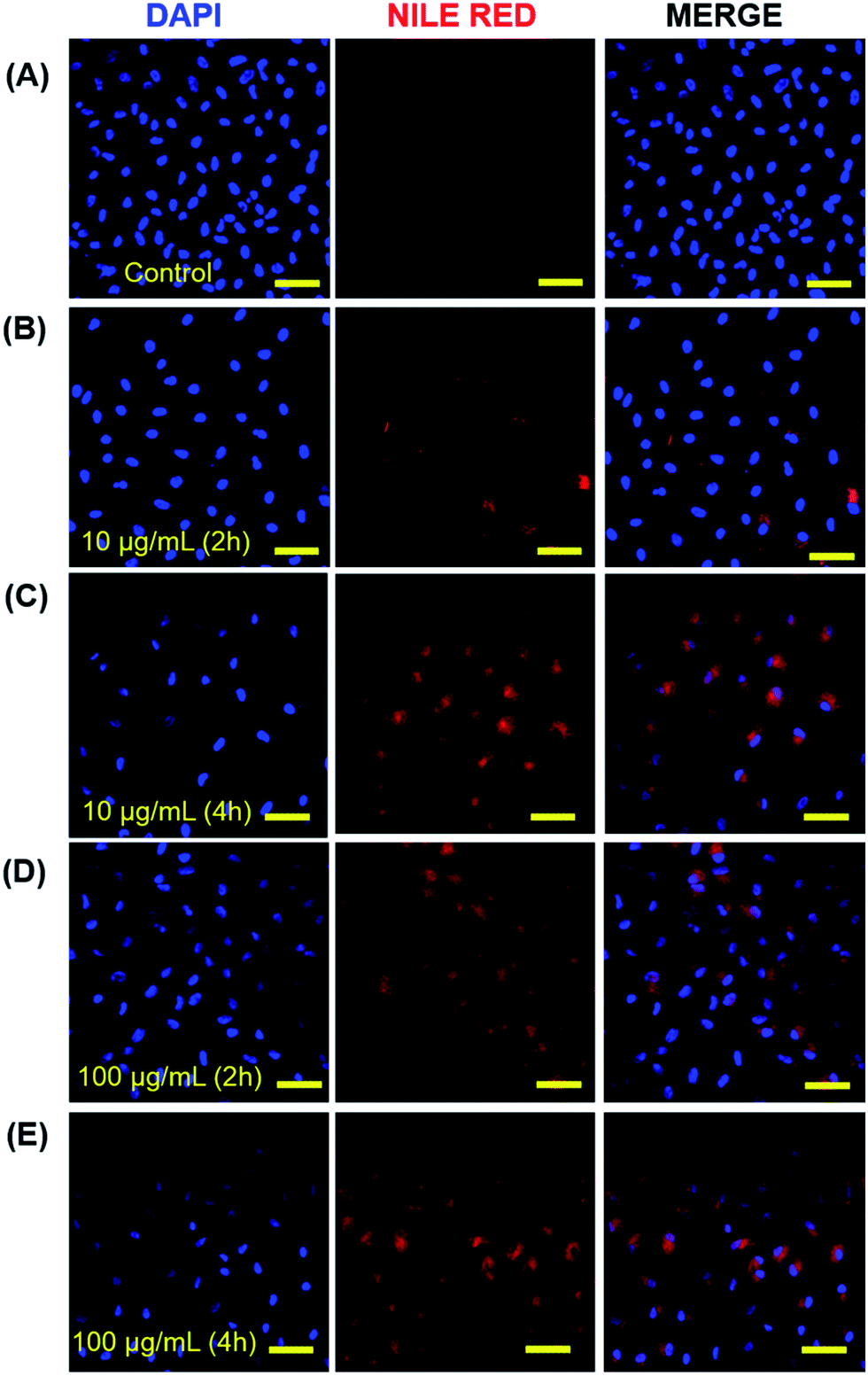 | ||
| Fig. 5 Confocal laser scanning fluorescence micrograph of A549 cells incubated with 10 and 100 μg mL−1 concentration of copolymer 4 loaded with Nile red at 2 h and 4 h (A–E) with a scale bar of 100 μm. | ||
 | ||
| Fig. 6 Confocal laser scanning fluorescence micrograph of A549 cells incubated with 10 and 100 μg mL−1 concentration of copolymer 5 loaded with Nile red at 2 h and 4 h (A–E) with a scale bar of 100 μm. | ||
4. Conclusions
Environment friendly methodology has been developed for the synthesis of five sugar–PEG-based copolymers via Novozym 435-mediated transesterification reaction between 2,5-anhydro-D-mannitol/2,5-anhydro-3,4-di-O-benzyl-D-mannitol and PEG-600/PEG-1000 diethyl ester under bulk conditions. It has been discovered that three water-soluble amphiphilic copolymers form micelles and micellar aggregates in aqueous conditions. In particular, two micelles derived from acylated amphiphilic copolymers 4 and 5 encapsulated the dye indocyanine green (ICG) 62% and 92%, respectively. Furthermore, micellar solutions of copolymers 4 and 5 efficiently stabilized the encapsulated ICG for 21 days at 20 °C and 5 days at 37 °C. The two micelles that efficiently encapsulated ICG also exhibited good cellular uptake, which was demonstrated by incubating A549 cells with Nile red-encapsulated micelles. Since these amphiphilic copolymers were not toxic, they can be used to develop a suitable drug carrier agent.Acknowledgements
We gratefully acknowledge the financial support from the Indo-German Science and Technology Centre (IGSTC), Gurugram, and University of Delhi for providing financial support under DU-DST Purse grant for strengthening research and development. We are also thankful to the CIF-USIC, University of Delhi, for providing the NMR spectral and HRMS recording facilities. V. K. thanks CSIR, New Delhi, for the award of Junior/Senior research fellowships.Notes and references
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869 CrossRef CAS PubMed.
- A. Topete, M. A. Meda, P. Iglesias, E. M. Villar-Alvarez, S. Barbosa, J. A. Costoya, P. Taboada and V. Mosquera, ACS Nano, 2014, 8, 2725 CrossRef CAS PubMed.
- A. Yuan, X. Tang, X. Qiu, K. Jiang, J. Wu and Y. Hu, Chem. Commun., 2015, 51, 3340 RSC.
- Z. Sheng, D. Hu, M. Xue, M. He, P. Gong and L. Cai, Nano-Micro Lett., 2013, 5, 145 CrossRef.
- A. K. Kirchherr, A. Briel and K. Mader, Mol. Pharmaceutics, 2009, 6, 480 CrossRef CAS PubMed.
- Y. Chen and X. Li, Biomacromolecules, 2011, 12, 4367 CrossRef CAS PubMed.
- W. H. Jian, T. W. Yu, C. J. Chen, W. C. Huang, H. C. Chiu and W. H. Chiang, Langmuir, 2015, 31, 6202 CrossRef CAS PubMed.
- X. Tan, J. Wang, X. Pang, L. Liu, Q. Sun, Q. You, F. Tan and N. Li, ACS Appl. Mater. Interfaces, 2016, 8, 34991 CAS.
- J. Yu, M. A. Yaseen, B. Anvari and M. S. Wong, Chem. Mater., 2007, 19, 1277 CrossRef CAS.
- M. Cao, P. Wang, Y. Kou, J. Wang, J. Liu, Y. Li, J. Li, L. Wang and C. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 25014 CAS.
- Y. Zheng, D. Zhang, M. Wu, Y. Liu, X. Zhang, L. Li, Z. Li, X. Han, X. Wei and X. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 14266 Search PubMed.
- Y. Li, T. Wen, R. Zhao, X. Liu, T. Ji, H. Wang, X. Shi, J. Shi, J. Wei, Y. Zhao, X. Wu and G. Nie, ACS Nano, 2014, 8, 11529 CrossRef CAS PubMed.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889 CrossRef CAS PubMed.
- K. Deng, Z. Hou, X. Deng, P. Yang, C. Li and J. Lin, Adv. Funct. Mater., 2015, 25, 7280 CrossRef CAS.
- L. Jing, J. Shi, D. Fan, Y. Li, R. Liu, Z. Dai, F. Wang and J. Tian, ACS Appl. Mater. Interfaces, 2015, 7, 22095 CAS.
- L. Han, Y. Zhang, X. W. Chen, Y. Shu and J. H. Wang, J. Mater. Chem. B, 2016, 4, 105 RSC.
- T. H. Kim, C. W. Mount, B. W. Dulken, J. Ramos, C. J. Fu, H. A. Khant, W. Chiu, W. R. Gombotz and S. H. Pun, Mol. Pharmaceutics, 2012, 9, 135 CrossRef CAS PubMed.
- L. Wu, S. Fang, S. Shi, J. Deng, B. Liu and L. Cai, Biomacromolecules, 2013, 14, 3027 CrossRef CAS PubMed.
- M. Zheng, P. Zhao, Z. Luo, P. Gong, C. Zheng, P. Zhang, C. Yue, D. Gao, Y. Ma and L. Cai, ACS Appl. Mater. Interfaces, 2014, 6, 6709 CAS.
- A. Zhu, K. Miao, Y. Deng, H. Ke, H. He, T. Yang, M. Guo, Y. Li, Z. Guo, Y. Wang, X. Yang, Y. Zhao and H. Chen, ACS Nano, 2015, 9, 7874 CrossRef CAS PubMed.
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600 CAS.
- K. Yasugi, Y. Nagasaki, M. Kato and K. Kataoka, J. Controlled Release, 1999, 62, 89 CrossRef CAS PubMed.
- R. Savic, L. B. Luo, A. Eisenberg and D. Maysinger, Science, 2003, 300, 615 CrossRef CAS PubMed.
- C. Deng, Y. Jiang, R. Cheng, F. Meng and Z. Zhong, Nano Today, 2012, 7, 467 CrossRef CAS.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650 CrossRef CAS PubMed.
- S. Bhatia, A. Mohr, D. Mathur, V. S. Parmar, R. Haag and A. K. Prasad, Biomacromolecules, 2011, 12, 3487 CrossRef CAS PubMed.
- Y. Zhang, J. W. Chan, A. Moretti and K. E. Uhrich, J. Controlled Release, 2015, 219, 355 CrossRef CAS PubMed.
- J. A. Galbis, M. G. Garcia-Martin, M. V. de Paz and E. Galbis, Chem. Rev., 2016, 116, 1600 CrossRef CAS PubMed.
- Y. Nagasaki, K. Yasugi, Y. Yamamoto, A. Harada and K. Kataoka, Biomacromolecules, 2001, 2, 1067 CrossRef CAS PubMed.
- B. Pandey, J. Mahato, K. B. Cotta, S. Das, D. K. Sharma, S. S. Gupta and A. Chowdhury, ACS Omega, 2016, 1, 600 CrossRef CAS.
- J. Khandare, M. Calderon, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824 RSC.
- J. Maity, G. Shakya, S. K. Singh, V. T. Ravikumar, V. S. Parmar and A. K. Prasad, J. Org. Chem., 2008, 73, 5629 CrossRef CAS PubMed.
- S. Gupta, M. K. Pandey, K. Levon, R. Haag, A. C. Watterson, V. S. Parmar and S. K. Sharma, Macromol. Chem. Phys., 2010, 211, 239 CrossRef CAS.
- D. Horton and K. D. Philips, Carbohydr. Res., 1973, 30, 367 CrossRef CAS.
- M. Chaumontet, V. Pons, K. Marotte and J. Prandi, Tetrahedron Lett., 2006, 47, 1113 CrossRef CAS.
- M. Kumari, A. K. Singh, S. Kumar, K. Achazi, S. Gupta, R. Haag and S. K. Sharma, Polym. Adv. Technol., 2014, 25, 1208 CrossRef CAS.
- J. Yu, D. Javier, M. A. Yaseen, N. Nitin, R. R. Kortum, B. Anvari and M. S. Wong, J. Am. Chem. Soc., 2010, 132, 1929 CrossRef CAS PubMed.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869 CrossRef CAS PubMed.
- B. Zheng, H. Chen, P. Zhao, H. Pan, X. Wu, X. Gong, H. Wang and J. Chang, ACS Appl. Mater. Interfaces, 2016, 8, 21603 CAS.
- N. Li, T. Li, C. Hu, X. Lei, Y. Zuo and H. Han, ACS Appl. Mater. Interfaces, 2016, 8, 15013 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04994c |
| This journal is © The Royal Society of Chemistry 2017 |