
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydroquinone derivatives from the marine-derived fungus Gliomastix sp.†
Mohamed S. Elnaggarab,
Weaam Ebrahimac,
Attila Mándid,
Tibor Kurtánd,
Werner E. G. Müller e,
Rainer Kalscheuera,
Abdelnasser Singabb,
Wenhan Linf,
Zhen Liu*a and
Peter Proksch*a
e,
Rainer Kalscheuera,
Abdelnasser Singabb,
Wenhan Linf,
Zhen Liu*a and
Peter Proksch*a
aInstitute of Pharmaceutical Biology and Biotechnology, Heinrich-Heine-Universität Düsseldorf, 40225 Düsseldorf, Germany. E-mail: zhenfeizi0@sina.com; proksch@uni-duesseldorf.de
bDepartment of Pharmacognosy, Faculty of Pharmacy, Ain-Shams University, Cairo 11566, Egypt
cDepartment of Pharmacognosy, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
dDepartment of Organic Chemistry, University of Debrecen, Debrecen 4032, Hungary
eInstitute of Physiological Chemistry, Universitätsmedizin der Johannes Gutenberg-Universität Mainz, 55128 Mainz, Germany
fState Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing 100191, P. R. China
First published on 13th June 2017
Abstract
Eight new hydroquinone derivatives, gliomastins A–D (1–4), 9-O-methylgliomastin C (5), acremonin A 1-O-β-D-glucopyranoside (6), gliomastin E 1-O-β-D-glucopyranoside (7), and 6′-O-acetyl-isohomoarbutin (8), together with seven known analogues were isolated from the marine-derived fungus Gliomastix sp. Their structures were elucidated by extensive spectroscopic analysis including 1D and 2D NMR measurements aided by DFT NMR calculations as well as MS data. TDDFT-ECD and OR calculations were performed to determine the absolute configurations of 1 and the aglycones of 6 and 7. Compound 1 features a novel skeleton, biogenetically derived from a Diels–Alder reaction between derivatives of 11 and 13. Compound 2 represents a rare sulfur-containing alkaloid derived from the known hydroquinone 13. Compounds 1, 10 and 12 showed strong cytotoxicity against the L5178Y mouse lymphoma cell line with IC50 values of 1.8, 1.0 and 1.1 μM, respectively. Compound 3 exhibited moderate antitubercular activity against Mycobacterium tuberculosis with a MIC value of 12.5 μM.
Introduction
Marine-derived microorganisms are attracting considerable attention owing to their high potential for producing new bioactive secondary metabolites.1,2 Unique and stressful marine habitats, such as those characterized by extreme pressure, changing levels of salinity or temperature, may have a great impact on fungal biological activity.3 One of the ecologically most diverse marine ecosystems are coral reefs.4,5 In recent years, numerous bioactive secondary metabolites derived from coral-associated fungi have been reported.3 Examples include the prenylated polyketides territrems A–C, which were isolated from the fungus Aspergillus terreus obtained from the gorgonian Echinogorgia aurantiaca, which exhibit pronounced acetylcholinesterase inhibitory activity.6 Hydroanthraquinones and anthraquinone dimers, which were isolated from the soft coral-derived fungus Alternaria sp., showed cytotoxicity against PC-3 and HCT-116 tumor cell lines.7 In addition, the structurally unique 14-membered resorcylic acid lactones cochliomycins A–C, which exhibit antifouling activity, were isolated from the gorgonian-derived fungus Cochliobolus lunatus.8As a part of our ongoing exploration of bioactive metabolites from fungi,9–12 examination of the Red Sea derived hard coral Stylophora sp. collected in Egypt, afforded different fungal strains from the coral tissues,9 one of which was identified as Gliomastix sp. So far only few secondary metabolites have been reported from the genus Gliomastix, including the macrolides gliomasolide A–E,13 the dihydroxanthenones muroxanthenone A–E,14,15 as well as quinone/hydroquinone meroterpenoids.16 The crude extract of the solid rice culture of Gliomastix sp. in this study showed cytotoxic activity against the L5178Y mouse lymphoma cell line with inhibition rate of 69.1% at a dose of 10 μg mL−1. Chromatographic workup of the fungal extract afforded fifteen hydroquinone derivatives including eight new natural products (1–8). Herein, we report the isolation, structure elucidation as well as cytotoxic and antimicrobial activity of the isolated compounds.
Results and discussion
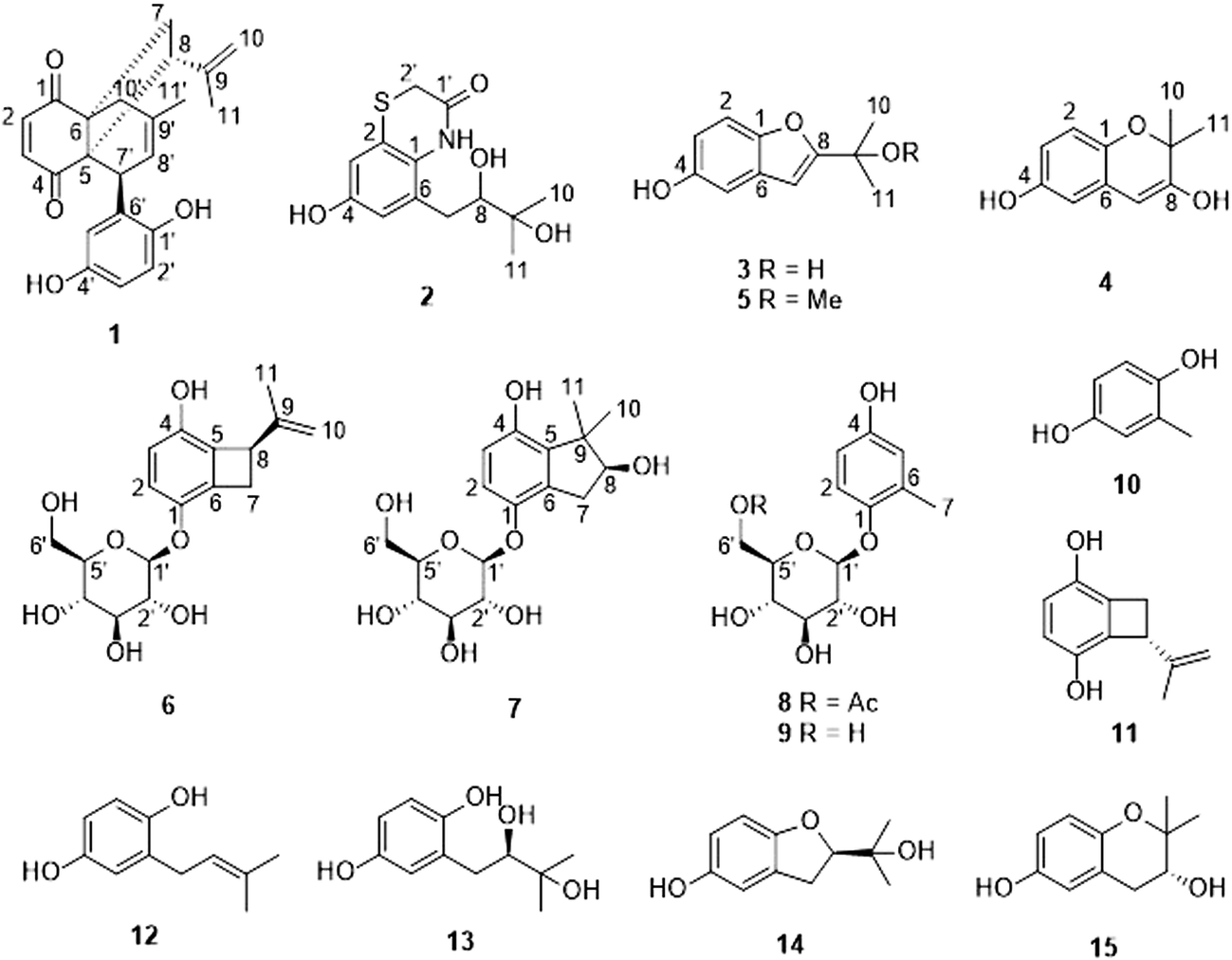
Compound 1 was isolated as a yellow amorphous powder. Its molecular formula was determined to be C22H22O4 by HRESIMS, indicating twelve degrees of unsaturation. The 13C NMR data of 1 (Table 1) together with HSQC and HMBC spectra revealed the presence of 22 carbons, including two carbonyl groups (δC 202.3 and 200.5), twelve olefinic or aromatic carbons (δC 110–151) and eight aliphatic carbons (two quaternary carbons at δC 59.8 and 47.6, two methines at δC 48.0 and 46.8, two methylenes at δC 36.9 and 36.5, and two methyls at δC 24.5 and 23.5). The 1H NMR spectrum of 1 (Table 1) displayed five olefinic protons at 6.73 (d, H-2), 6.50 (d, H-3), 5.68 (br s, H-8′), 4.83 (br s, H-10a) and 4.66 (br s, H-10b), representing a disubstituted double bond, a trisubstituted double bond and a terminal double bond, respectively. In addition, three aromatic protons of an ABX ring system at δH 6.40 (d, H-2′), 6.34 (dd, H-3′) and 6.21 (d, H-5′) as well as two methyls at 1.84 (s, Me-11′) and 1.77 (d, Me-11) were observed. These data accounted for nine degrees of unsaturation. Thus, compound 1 must possess a tricyclic skeleton in addition to an aromatic ring. The presence of a cyclohexenedione ring in 1 was established by the COSY correlation between H-2 and H-3 along with the HMBC correlations from H-2 to C-4 (δC 200.5) and C-6 (δC 47.6) and from H-3 to C-1 (δC 202.3) and C-5 (δC 59.8) (Fig. 1). The COSY correlations between H-7a (δH 2.66, dd)/H-8 (δH 3.31) and H-7b (δH 1.97, dd)/H-8 together with the HMBC correlations from H-7a to C-1, from H-7b to C-5, from H-8 to C-4 and C-5, and from Me-11 to C-8 (δC 48.0), C-9 (δC 145.8) and C-10 (δC 111.7) revealed that the cyclohexenedione ring was fused with a cyclobutane ring at the C-5 and C-6 positions and a isopropenyl group was attached at the C-8 position. In addition, the COSY correlation between H-7′ (δH 3.97)/H-8′ and the HMBC correlations from H-8′ to C-5, from Me-11′ to C-8′ (δC 122.7), C-9′ (δC 133.8) and C-10′ (δC 36.9), from H-10′a (δH 2.97, d) to C-1, and from H-10′b (δH 2.28, d) to C-5 and C-7 (δC 36.5) indicated a cyclohexene ring with a methyl substituent at the C-9′ position, which was fused with the cyclohexenedione ring through the C-5/C-6 bridge. Moreover, the presence of a benzene ring substituted at the C-7′ position was confirmed by the previous observation of an ABX aromatic spin system (H-2′, 3′ and 5′) and the HMBC correlations from H-2′ to C-4′ (δC 150.9) and C-6′ (δC 130.8), from H-3′ to C-1′ (δC 148.3) and C-5′ (δC 117.3), from H-5′ to C-3′ (δC 114.5), and from H-7′ to C-5′. Two hydroxy groups were suggested to be located at C-1′ and C-4′ as indicated by the chemical shifts of C-1′ and C-4′. Thus, the planar structure of 1 was elucidated as shown, for which the trivial name gliomastin A is proposed.| Position | 1a | 2a | ||
|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, typeb | δH (J in Hz) | |
| a Recorded at 600 MHz for 1H and 150 MHz for 13C in CD3OD.b Data extracted from HSQC and HMBC spectra.c Overlapped with water peak. | ||||
| 1 | 202.3, C | 129.8, C | ||
| 2 | 140.5, CH | 6.73, d (10.4) | 124.4, C | |
| 3 | 142.2, CHb | 6.50, d (10.4) | 112.8, CH | 6.65, d (2.6) |
| 4 | 200.5, C | 155.0, C | ||
| 5 | 59.8, C | 117.0, CH | 6.57, d (2.6) | |
| 6 | 47.6, C | 132.6, C | ||
| 7 | 36.5, CH2 | 2.66, dd (11.9, 10.6) | 35.1, CH2 | 2.80, dd (14.1, 1.6) |
| 1.97, dd (11.9, 8.8) | 2.66, dd (14.1, 10.2) | |||
| 8 | 48.0, CH | 3.31c | 81.6, CH | 3.46, dd (10.2, 1.6) |
| 9 | 145.8, C | 73.4, C | ||
| 10 | 111.7, CH2 | 4.83, s | 25.9, CH3 | 1.25, s |
| 4.66, s | ||||
| 11 | 23.5, CH3 | 1.77, s | 24.1, CH3 | 1.24, s |
| 1′ | 148.3, C | 167.6, C | ||
| 2′ | 117.1, CH | 6.40, d (8.5) | 30.8, CH2 | 3.45, d (14.5) |
| 3.22, d (14.5) | ||||
| 3′ | 114.5, CH | 6.34, dd (8.5, 2.9) | ||
| 4′ | 150.9, C | |||
| 5′ | 117.3, CHb | 6.21, d (2.9) | ||
| 6′ | 130.8, Cb | |||
| 7′ | 46.8, CHb | 3.97, br s | ||
| 8′ | 122.7, CHb | 5.68, br s | ||
| 9′ | 133.8, C | |||
| 10′ | 36.9, CH2 | 2.97, d (17.7) | ||
| 2.28, d (17.7) | ||||
| 11′ | 24.5, CH3 | 1.84, s | ||
 | ||
| Fig. 1 Key COSY, HMBC and ROESY correlations of compound 1. | ||
The relative configuration of 1 was determined by ROESY data (Fig. 1). The NOE correlations from H-7′ to H-8 and Me-11 suggested H-7′ and the cyclobutane ring to be on the same face of the cyclohexene ring while the NOE relationships between H-8/H-7b and H-7b/H-10′b indicated these protons to be orientated on the same side of the cyclobutane ring. In addition, DFT NMR calculations were performed on the arbitrarily chosen (5S,6R,8R,7′R)-1 and (5S,6R,8S,7′R)-1 epimers to decide between the (5S*,6R*,8R*,7′R*) and (5S*,6R*,8S*,7′R*) relative configuration of 1. Merck Molecular Force Field (MMFF) conformational searches of the two epimers resulted in 14 and 14 low-energy conformers in a 21 kJ mol−1 energy window, respectively. These conformers were reoptimized at the B3LYP/6-31+G(d,p) level and NMR shift values were computed for conformers over 1% Boltzmann population at the mPW1PW91/6-311+G(d,p) level.17 In accordance with the experimental NOE data, computed 13C-NMR chemical shifts of most carbons suggest 8S* relative configuration, while good agreement between experimental and computed 1H-NMR chemical shifts of the 8S* epimer compared to that of the 8R* epimer allowed unambiguous assignment of the relative configuration of 1 as (5S*,6R*,8S*,7′R*).
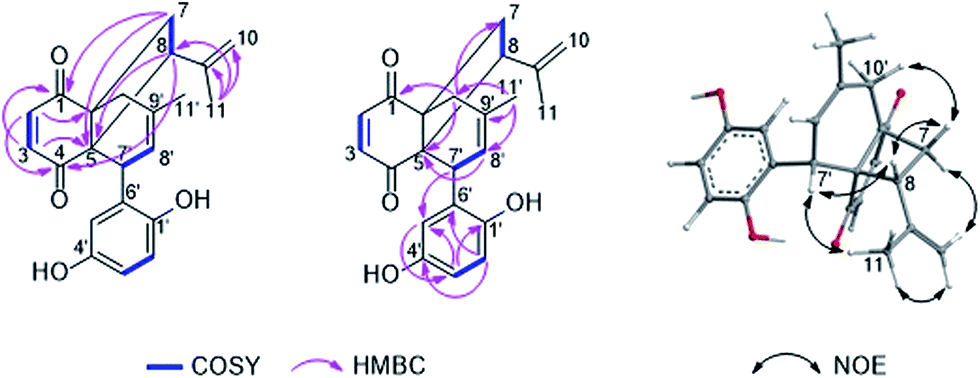
To determine the absolute configuration of 1, the solution TDDFT-ECD method was applied on the arbitrarily chosen (5S,6R,8S,7′R)-1 enantiomer.18,19 The 14 MMFF conformers obtained in the conformational search of the NMR calculations were reoptimized at the B3LYP/6-31G(d), B97D/TZVP20,21 PCM/MeCN and CAM-B3LYP/TZVP22,23 PCM/MeCN levels and ECD computations were performed for the low-energy conformers with various functionals (B3LYP, BH&HLYP, CAM-B3LYP, PBE0) combined with the TZVP basis set. Computed ECD spectra obtained at all of the applied combination of levels gave moderate to good mirror-image agreement with the experimental spectrum, allowing determination of the absolute configuration of 1 as (5R,6S,8R,7′S) (Fig. 2).
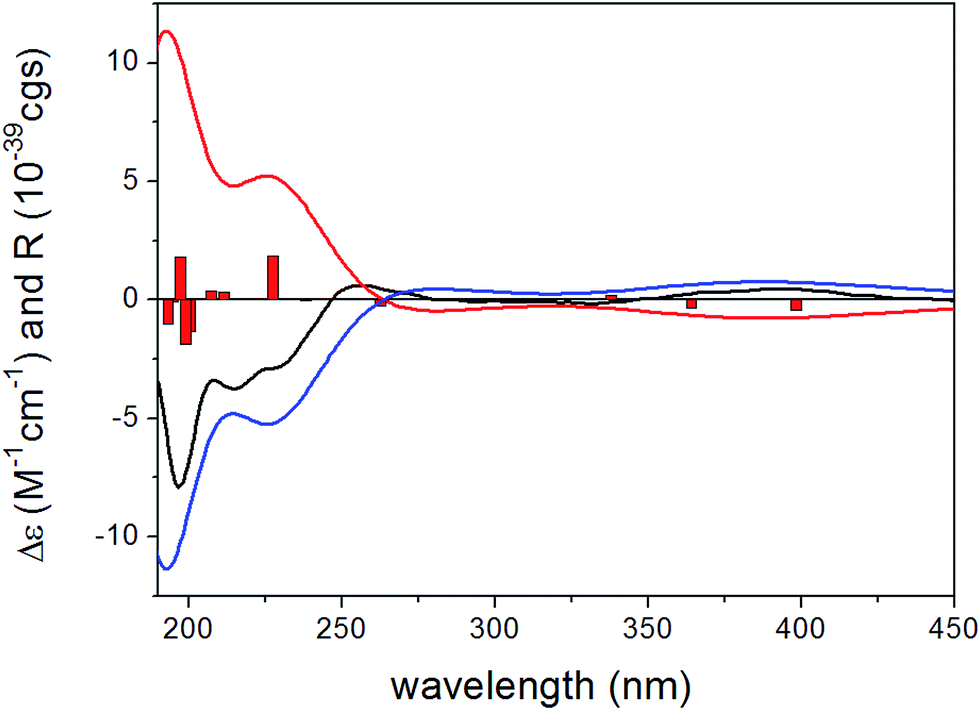 | ||
| Fig. 2 Experimental ECD spectrum of 1 in MeCN (black line) compared with the B3LYP/TZVP ECD spectra computed for the B3LYP/6-31G(d) in vacuo optimized low-energy conformers of (5S,6R,8S,7′R)-1 (average of 4 conformers, red line) and (5R,6S,8R,7′S)-1 (average of 4 conformers, blue line). Bars represent computed rotational strength values of the lowest-energy conformer of (5S,6R,8S,7′R)-1. | ||
Gliomastin A (1) features a new carbon skeleton, which could be derived from two co-isolated known hydroquinone derivatives acremonin A (11)24 and F-11334A1 (13).25,26 A biosynthetic pathway from the latter two compounds to 1 via Diels–Alder reaction is proposed (Fig. 3).
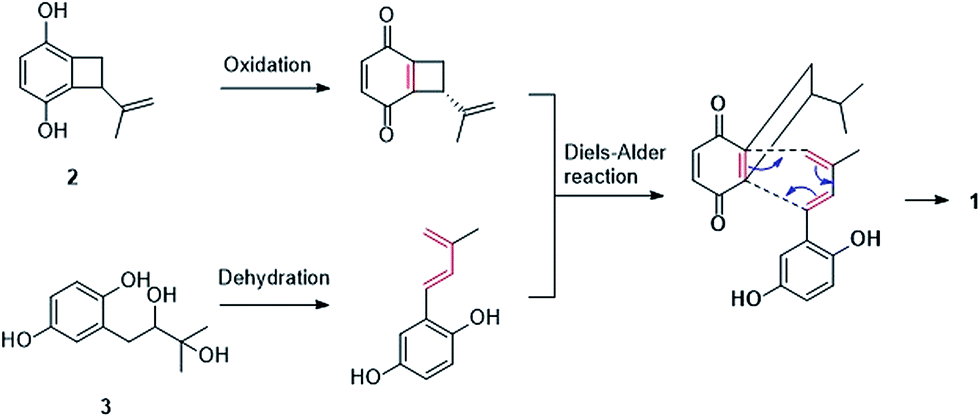 | ||
| Fig. 3 A plausible biosynthetic pathway for compound 1. | ||
The molecular formula of compound 2 was established as C13H17NO4S by HRESIMS, requiring six degrees of unsaturation. In the 1H NMR spectrum of 2 (Table 1), two meta-coupled aromatic protons at δH 6.65 (d, J = 2.6 Hz, H-3) and 6.57 (d, J = 2.6 Hz, H-5), one oxygenated methine at δH 3.46 (dd, J = 10.2, 1.6 Hz, H-8), one isolated methylene at δH 3.45 (d, J = 14.5 Hz, H-2′a) and 3.22 (d, J = 14.5 Hz, H-2′b), one methylene at δH 2.80 (dd, J = 14.1, 1.6 Hz, H-7a) and 2.66 (dd, J = 14.1, 10.2 Hz, H-7b), and two methyl groups at δH 1.25 (s, Me-10) and 1.24 (s, Me-11) were observed. The COSY correlations between H-7a/H-8 and H-7b/H-8 as well as the HMBC correlations from Me-10 and Me-11 to C-8 (δC 81.6) and C-9 (δC 73.4) indicated the presence of a 2,3-dihydroxy-3-methyl-butyl moiety in compound 2 (Fig. 4). The attachment of this moiety to a meta-coupled benzene ring at the C-6 position was confirmed by the HMBC correlations from H-7a and H-7b to C-1 (δC 129.8), C-5 (δC 117.0) and C-6 (δC 132.6), from H-3 to C-1, C-4 (δC 155.0) and C-5, and from H-5 to C-1, C-3 (δC 112.8), C-4 and C-7 (δC 35.1). C-4 was suggested to be substituted with a hydroxy group due to its high chemical shift. The HMBC correlations from H-2′a and H-2′b to C-2 (δC 124.4) and C-1′ (δC 167.6), combined with its molecular formula, indicated that the isolated methylene CH2-2′ was linked to the benzene ring via an amide bond and a sulfur atom at the C-1 and C-2 positions to form an additional ring. Thus, the structure of 2 was determined, representing a rare sulfur-containing alkaloid, of which the co-isolated known hydroquinone derivative F-11334A1 (13) could be the precursor.
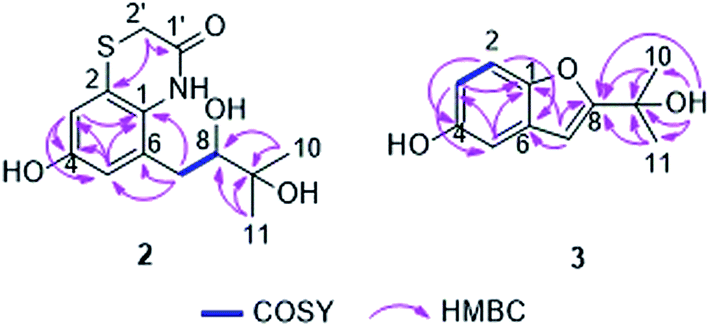 | ||
| Fig. 4 Key COSY and HMBC correlations of compounds 2 and 3. | ||
Gliomastin C (3) was recovered as a white amorphous powder, possessing the molecular formula C11H12O3 as determined by HRESIMS. Its 1H NMR data (Table 2) exhibited three aromatic protons at δH 7.25 (d, J = 8.8 Hz, H-2), 6.95 (d, J = 2.5 Hz, H-5) and 6.76 (dd, J = 8.8, 2.5 Hz, H-3), which are characteristic signals of an ABX ring system. Together with the HMBC correlations from H-2 to C-4 (δC 154.1) and C-6 (δC 130.3), from H-3 to C-1 (δC 149.9) and C-5 (δC 106.4), and from H-5 to C-1 and C-3 (δC 113.0), a benzene ring in 3 was established (Fig. 4). Furthermore, the HMBC correlations from Me-10/11 to C-8 (δC 166.3) and C-9 (δC 69.1), from OH-9 (δH 4.32) to C-8, C-9 and C-10/11 (δC 29.4), and from H-7 (δH 6.52, s) to C-1, C-6 and C-8 indicated the nature of a 3-hydroxy-3-methyl-butenyl moiety and its attachment to C-6. A hydroxy group at C-4 and an oxygen bridge between C-1 and C-8 were assigned in consideration of the molecular formula and the chemical shifts of the corresponding carbons. Thus, the structure of gliomastin C (3) was elucidated as shown.
| Position | 3a | 4a | 5b | |||
|---|---|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | |
| a Recorded at 600 MHz for 1H and 150 MHz for 13C in CD3COCD3.b Recorded at 600 MHz for 1H and 150 MHz for 13C in CD3OD. | ||||||
| 1 | 149.9, C | 150.1, C | 150.8, C | |||
| 2 | 111.7, CH | 7.25, d (8.8) | 112.0, CH | 7.29, d (8.8) | 112.1, CH | 7.25, d (8.8) |
| 3 | 113.0, CH | 6.76, dd (8.8, 2.5) | 113.6, CH | 6.80, dd (8.8, 2.5) | 114.0, CH | 6.74, dd (8.8, 2.5) |
| 4 | 154.1, C | 154.2, C | 154.3, C | |||
| 5 | 106.4, CH | 6.95, d (2.5) | 106.4, CH | 6.98, d (2.5) | 106.7, CH | 6.91, d (2.5) |
| 6 | 130.3, C | 129.9, C | 130.2, C | |||
| 7 | 100.7, CH | 6.52, s | 104.4, CH | 6.63, s | 105.0, CH | 6.61, s |
| 8 | 166.3, C | 161.9, C | 161.5, C | |||
| 9 | 69.1, C | 73.8, C | 74.9, C | |||
| 10 | 29.4, CH3 | 1.57, s | 25.6, CH3 | 1.55, s | 25.6, CH3 | 1.59, s |
| 11 | 29.4, CH3 | 1.57, s | 25.6, CH3 | 1.55, s | 25.6, CH3 | 1.59, s |
| OH-4 | 8.03, s | 8.10, s | ||||
| OH-9 | 4.32, s | |||||
| OMe-9 | 51.3, CH3 | 3.10, s | ||||
Gliomastin D (4) shared the same molecular formula as gliomastin C (3). The UV spectra and NMR data (Table 2) of 4 were also comparable to those of 3. Three aromatic protons belonging to an ABX coupling system at δH 7.29 (d, H-2), 6.98 (d, H-5) and 6.80 (dd, H-3) were found in addition to a singlet olefinic methine at δH 6.63 (H-7) and two singlet methyls at δH 1.55 (Me-10 and 11). Analysis of the 1D and 2D NMR spectra of 4 revealed that the two compounds were structurally similar except for the position of the hydroxy group and the oxygen bridge. The upfield shifted C-8 (−4.4 ppm) and downfield shifted C-9 (+4.7 ppm) in 4 compared to those of 3 suggested the presence of a pyran ring and a hydroxy group at C-8 in 4 instead of a furan ring.
Compound 5 was isolated as a white amorphous powder with the molecular formula C12H14O3 as established by HRESIMS, containing an additional CH2 unit compared to gliomastin C (3). The UV absorption spectrum and NMR data (Table 2) of 5 resembled those of 3 except for the appearance of an additional methoxy group at δC 51.3 and δH 3.10, which was further confirmed to be located at C-9 by the HMBC correlation from the protons of the methoxy group to C-8 (δC 74.9). Thus, compound 5 was elucidated as 9-O-methylgliomastin C.
The HRESIMS of compound 6 displayed a pseudomolecular ion peak at m/z 361.1255 [M + Na]+, corresponding to the molecular formula C17H22O7. Positive and negative ESIMS showed fragment peaks at m/z 177 [M + H − 162]+ and 175 [M − H − 162]+, respectively, which suggested the presence of a hexose residue in the molecule. The UV and NMR data (Table 3) of 6 were similar to those of acremonin A 4-O-β-D-glucopyranoside.24 Acid hydrolysis of 6 yielded two products, one of which was confirmed to be the co-isolated hydroquinone acremonin A (11) by comparison of their NMR data and optical rotation values.24 The other product was determined as β-D-glucopyranose by TLC analysis and optical rotation measurement compared to a known standard as well as by the J value (7.7 Hz) of the anomeric proton (H-1′) in 6. Along with the 1D and 2D NMR spectra, compound 6 was unambiguously determined to contain an acremonin A moiety and a glucopyranose residue (Fig. 5). However, the HMBC correlations from H-1′ (δH 4.88, d) to C-1 (δC 146.6) rather than to C-4 (δC 147.2) indicated that the glucopyranose residue was attached to C-1 instead of C-4. Thus compound 6 was elucidated as acremonin A 1-O-β-D-glucopyranoside.
| Position | 6a | 7a | 8a | |||
|---|---|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, typeb | δH (J in Hz) | δC, typeb | δH (J in Hz) | |
| a Recorded at 600 MHz for 1H and 150 MHz for 13C in CD3OD.b Data extracted from HSQC and HMBC spectra. | ||||||
| 1 | 146.6, C | 148.6, C | 150.4, C | |||
| 2 | 118.2, CH | 6.69, d (8.8) | 116.8, CH | 6.80, d (8.7) | 119.2, CH | 6.92, d (8.7) |
| 3 | 116.2, CH | 6.54, d (8.8) | 115.4, CH | 6.50, d (8.7) | 113.7, CH | 6.52, dd (8.7, 3.0) |
| 4 | 147.2, C | 151.0, C | 153.8, C | |||
| 5 | 132.8, C | 136.9, C | 118.1, CH | 6.58, d (3.0) | ||
| 6 | 129.8, C | 131.4, C | 131.0, C | |||
| 7 | 36.8, CH2 | 3.55, dd (13.3, 5.7) | 36.3, CH2 | 3.23, dd (16.1, 7.2) | 16.7, CH3 | 2.21, s |
| 2.87, dd (13.3, 2.5) | 2.70, dd (16.1, 7.2) | |||||
| 8 | 48.9, CH | 3.99, dd (5.7, 2.5) | 82.2, CH | 3.99, t (7.2) | ||
| 9 | 146.8, C | 48.4, C | ||||
| 10 | 110.6, CH2 | 4.84, s | 26.0, CH3 | 1.36, s | ||
| 4.78, s | ||||||
| 11 | 20.4, CH3 | 1.78, s | 20.0, CH3 | 1.20, s | ||
| 1′ | 102.1, CH | 4.88, d (7.7) | 104.1, CH | 4.69, d (7.6) | 104.3, CH | 4.66, d (7.6) |
| 2′ | 75.0, CH | 3.36, m | 75.0, CH | 3.41, m | 75.1, CH | 3.43, m |
| 3′ | 77.9, CH | 3.42, m | 78.2, CH | 3.42, m | 78.0, CH | 3.44, m |
| 4′ | 71.4, CH | 3.36, m | 71.5, CH | 3.35, m | 71.7, CH | 3.35, m |
| 5′ | 78.2 CH | 3.37, m | 78.0 CH | 3.32, m | 75.2 CH | 3.51, m |
| 6′ | 62.6, CH2 | 3.89, dd (12.2, 1.8) | 62.7, CH2 | 3.87, dd (12.0, 2.1) | 64.7, CH2 | 4.37, dd (11.8, 2.3) |
| 3.69, dd (12.2, 5.2) | 3.68, dd (12.0, 5.5) | 4.25, dd (11.8, 6.6) | ||||
| OAc-6′ | 172.7, C | 2.03, s | ||||
| 20.7, CH3 | ||||||
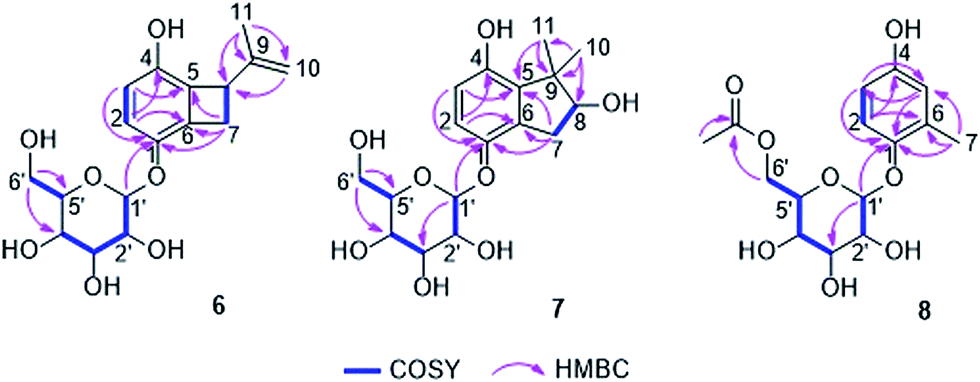 | ||
| Fig. 5 Key COSY and HMBC correlations of compounds 6, 7 and 8. | ||
Since the absolute configuration of acremonin A (11) is unknown in literature, ECD calculations were carried out for the arbitrarily chosen (S) enantiomer of the aglycone 11. DFT reoptimization of the initial 15 MMFF conformers yielded 8 low-energy conformers at both the B3LYP/6-31G(d) in vacuo and the B97D/TZVP PCM/MeCN levels of theory. ECD spectra computed at various levels for these conformers gave nice mirror-image agreement with the experimental spectrum (Fig. 6). In some of the higher-energy conformers (e.g. conformers C and D, Fig. 7), the isopropenyl adopted a different orientation from that of the lowest-energy conformer, which is reflected in a near mirror image ECD of the conformers. Considering the relatively small computed energy differences of the conformers, this might be a possible source of error (energy difference between conformers A and C is less than 1 kJ mol−1).18,21 Therefore OR calculations were also performed on the low-energy gas-phase conformers and on those reoptimized at the B97D/TZVP PCM/MeOH level (8 conformers over 2% also at this level).27,28 The OR calculations performed at various levels for both sets of conformers were in line with the ECD results confirming the previous assignment. However, the sign of the OR values of the individual conformers were influenced by the orientation of isopropenyl group such as in the case of ECD. The (R) absolute configuration of the calculations was also confirmed by the biosynthetic relationship of 1 and 11.
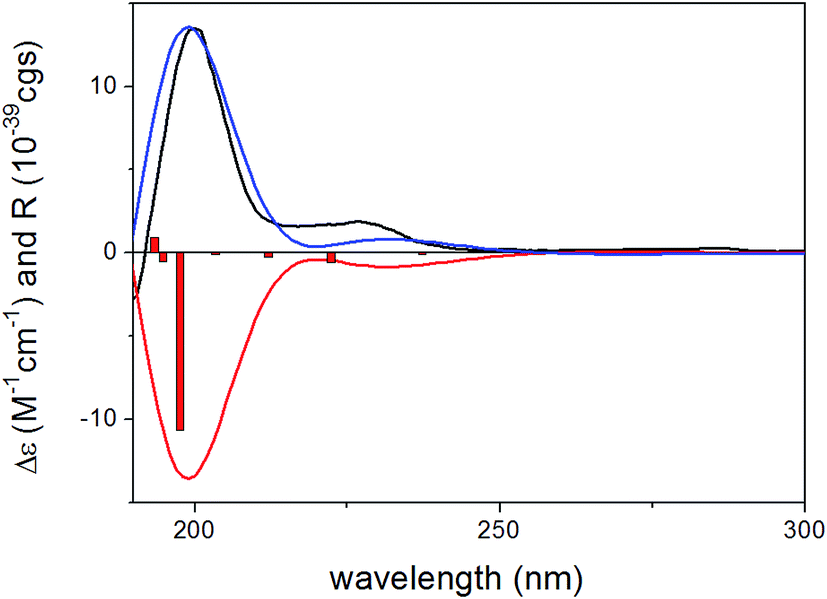 | ||
| Fig. 6 Experimental ECD spectrum of 11 in MeCN (black line) compared with the PBE0/TZVP PCM/MeCN ECD spectra computed for the B97D/TZVP PCM/MeCN optimized low-energy conformers of (S)-11 (average of 8 conformers/2, red line) and (R)-11 (average of 8 conformers/2, blue line). Bars represent computed rotational strength values of (S)-11 (conformer A/2). | ||
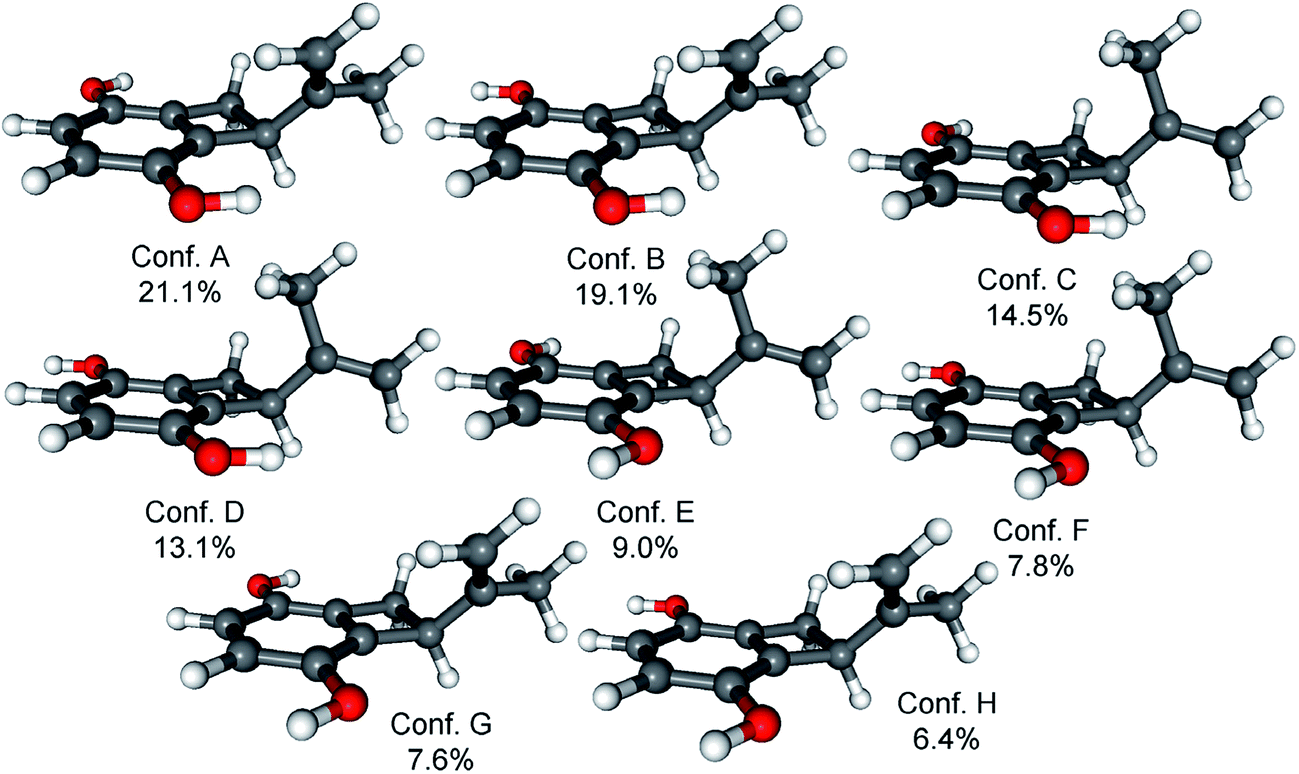 | ||
| Fig. 7 Structures and populations of the low-energy (≥2%) B97D/TZVP PCM/MeCN conformers of (S)-11. | ||
Compound 7 was obtained as a white solid. It possesses the molecular formula C17H24O8 as established by analysis of the HRESIMS data. Similar to 6, the NMR data (Table 3) of 7 exhibited two ortho-coupled aromatic protons at 6.80 (d, J = 8.7 Hz, H-2) and 6.50 (d, J = 8.7 Hz, H-3), signals of a methylene group at δH 3.23 (dd, J = 16.1, 7.2 Hz, H-7a) and 2.70 (dd, J = 16.1, 7.2 Hz, H-7b) and signals of a hexose residue including an anomeric proton at 4.69 (d, J = 7.6 Hz, H-1′). The nature of the benzene ring was established by the HMBC correlations from H-2 to C-4 (δC 151.0) and C-6 (δC 131.4) and from H-3 to C-1 (δC 148.6) and C-5 (δC 136.9) (Fig. 5). Analysis of the hydrolysis products of 7 confirmed the presence of a glucopyranose residue, which was determined to be attached to C-1 by the HMBC correlation from H-1′ to C-1. However, compared to 6, the signals of the isopropenyl group were replaced by two singlet methyl groups at δH 1.35 (s, Me-10) and 1.20 (s, Me-11) as well as by an oxygenated methine at δC 82.2 and δH 3.99 (CH-8). The COSY correlations between H-7a/H-8 and H-7b/H-8 together with the HMBC correlations from Me-10 and Me-11 to C-5, C-8 and C-9 (δC 48.4), and from H-7a and H-7b to C-1, C-5 and C-6 indicated the presence of a further cyclopentene ring with two methyls at C-9 and a hydroxy group at C-8, which was fused to the benzene ring. Thus, the structure of 7 was elucidated as shown. Its aglycone was obtained following hydrolysis of 7, representing a new hydroquinone derivative, for which the name gliomastin E is proposed.
For the configurational assignment of the aglycone gliomastin E, the solution TDDFT-ECD calculation protocol was carried out on the arbitrarily chosen (S) enantiomer. Reoptimization of the initial 23 MMFF conformers resulted in 13 and 15 low-energy (≥2%) conformers at the B3LYP/6-31G(d) in vacuo and the B97D/TZVP PCM/MeCN levels. ECD spectra computed at various levels for both sets of conformers resembled the 247 and the 198 nm transitions of gliomastin E suggesting (S) absolute configuration (Fig. 8). Since the 273 nm positive Cotton effect (CE) could not be reproduced by any of the applied combination, OR calculations performed in vacuo and in MeOH (similarly to 11) were applied to prove the (S) absolute configuration.28 OR values computed at all of the applied combinations of levels resulted in small positive overall optical rotations in the range from +10.2 to +18.4, while the experimental value was +15.2, which verified the ECD results.
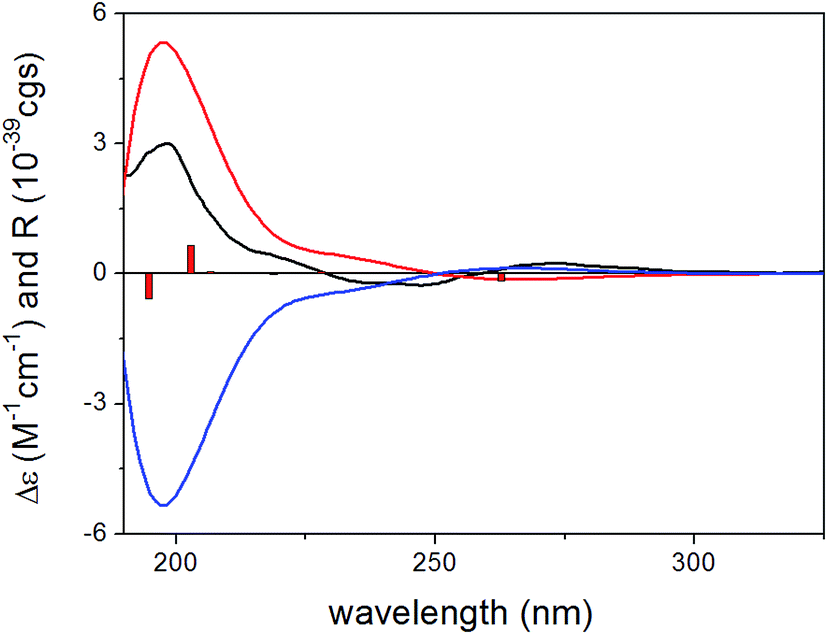 | ||
| Fig. 8 Experimental ECD spectrum of gliomastin E in MeCN (black line) compared with the B3LYP/TZVP ECD spectra computed for the B3LYP/6-31G(d) in vacuo optimized low-energy conformers of (S)-gliomastin E (average of 13 conformers, red line) and (R)-gliomastin E (average of 13 conformers, blue line). Bars represent computed rotational strength values of the lowest-energy conformer of (S)-gliomastin E. | ||
The molecular formula C15H20O8 was deduced for compound 8 from the HRESIMS data. The UV spectrum and NMR data (Table 3) of 8 were almost identical to those of isohomoarbutin (9),29,30 except for the appearance of an additional acetoxy group (δH 2.03, δC 20.7 and 172.7) in 8. This acetoxy group was determined to be located at C-6′ on the basis of the HMBC correlations from H-6′a and H-6′b (δH 4.37 and 4.25) to the carbonyl carbon of the acetoxy group. The remaining structure of 8 was confirmed to be the same as 9 by detailed analysis of the 2D NMR spectra of 8 (Fig. 5). The hexose residue was determined to be β-D-glucopyranose by the coupling constant (7.6 Hz) of the anomeric proton (H-1′) and analysis of the acid hydrolysis products of 8 in comparison with a known standard. Owing to the data mentioned above, compound 8 was elucidated as 6′-O-acetyl-isohomoarbutin.
By comparison with the literature data, the remaining known compounds were identified as isohomoarbutin (9),29,30 2-methyl-1,4-benzenediol (10),31 acremonin A (11),24 prenylhydroquinone (12),32,33 F-11334A1 (13),25,26 (R)-2-(2-hydroxypropan-2-yl)-2,3-dihydro-5-hydroxybenzofuran (14),34 and 2,2-dimethylchroman-3,6-diol (15).24
All isolated compounds (1–15) were evaluated for their cytotoxicity against the L5178Y murine lymphoma cell line (Table 4). Compounds 1, 10, 12 and 13 showed significant cytotoxic activity with IC50 values of 1.8, 1.0, 1.1 and 3.0 μM, respectively, which are below that of the positive control kahalalide F. Compared to the two precursors 11 and 13, compound 1 demonstrated stronger cytotoxicity while the sulfur-containing alkaloid 2 was inactive compared to its precursor 13. Compounds 10 and 11 showed strong cytotoxicity while their glycosides 8, 9 and 6 were inactive. The ether bridge in 14 and 15 led to total loss of cytotoxicity compared to 13.
| Compd | Cytotoxicity IC50 (μM) | Antitubercular and antibacterial MIC (μM) | ||||||
|---|---|---|---|---|---|---|---|---|
| L5178Y | TB | S. aureus ATCC 25923 | S. aureus ATCC 700699 | E. faecalis ATCC 29212 | E. faecalis ATCC 51299 | E. faecium ATCC 35667 | E. faecium ATCC 700221 | |
| a IC50 or MIC > 50 μM.b Kahalalide F as positive control.c Rifampicin as positive control.d Moxifloxacin as positive control.e Ciprofloxacin as positive control. | ||||||||
| 1 | 1.8 | — | — | — | — | — | — | — |
| 3 | —a | 12.5 | — | — | — | — | — | — |
| 10 | 1.0 | 12.5 | 25 | 6.25 | 12.5 | 12.5 | 12.5 | 25 |
| 11 | 9.6 | 25 | — | — | — | — | — | — |
| 12 | 1.1 | 12.5 | 25 | 12.5 | 12.5 | 6.25 | 6.25 | 12.5 |
| 13 | 3.0 | 25 | — | — | — | — | — | — |
| Positive control | 4.3b | <0.64c | 1.56d | 1.56d | 0.008e | 0.008e | 0.008e | 0.008e |
In addition, all compounds were tested for their antitubercular activity against Mycobacterium tuberculosis and for their antibacterial activities against different strains of pathogenic bacteria (Table 4). Only compounds 10 and 12 exhibited antibacterial activity but they were also cytotoxic. Compound 3, which lacked cytotoxicity, showed antitubercular activity with a MIC value of 12.5 μM. Comparison of the antitubercular activity of 3 with those of 4, 5, 14 and 15 indicated the importance of the furan ring and of the free hydroxy group at C-9.
In conclusion, our investigation of secondary metabolites from the marine-derived fungus Gliomastix sp. led to the isolation of fifteen hydroquinone derivatives including eight new natural products (1–8). Among the latter, the dimer 1 possesses a novel carbon skeleton, which could be derived from derivatives of 11 and 13 through a Diels–Alder reaction. Compound 1 also exhibited significant cytotoxicity against the L5178Y mouse lymphoma cell line with an IC50 value of 1.8 μM. Compound 2 is a rare sulfur-containing alkaloid. TDDFT-ECD and OR calculations were performed to determine the absolute configurations of 1 and the aglycones of 6 and 7. All isolated compounds were tested for their cytotoxic and antimicrobial activity and the structure–activity relationships were discussed.
Experimental section
General procedures
Optical rotations were recorded on a PerkinElmer-241 MC polarimeter. 1H, 13C and 2D NMR spectra were measured on a Bruker Avance DMX 600 NMR spectrometer. A Finnigan LCQ Deca XP Thermoquest spectrometer was used to record mass spectra while a FTHRMS-Orbitrap (Thermo Finnigan) mass spectrometer was utilized to obtain HRESIMS data. The Dionex P580 HPLC system was coupled to a photodiode array detector (UVD340S) and the analytical column (125 × 4 mm, L x i.d.) was prefilled with Eurospher-10 C18 (Knauer, Germany). Semi-preparative HPLC separation was performed on a Lachrom-Merck Hitachi HPLC system (Pump L7100, UV detector L7400 and Eurosphere 100 C18 column: 300 × 8 mm) with 5.0 mL min−1 flow rate. Normal phase column chromatography was carried out using Merck MN Silica gel 60 M (0.04–0.063 mm) or Sephadex LH-20. TLC was carried out on precoated Silica Gel 60 F254 plates (Merck, Germany) with UV detection at 254 and 365 nm as well as following spraying with anisaldehyde reagent or with methanol–sulfuric acid (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5).
:5).
Fungal material and identification
The hard coral Stylophora sp. was obtained from the Red Sea in Egypt near the coastline of Ain El-Sokhna area in November 2012. The fungal strain was isolated from the freshly crushed inner tissues of the coral following standard procedures and was identified as Gliomastix sp. according to a molecular biological protocol that was carried out through DNA amplification and sequencing of ITS region as described previously.9,35 The obtained data of sequencing were submitted to GenBank with the accession number KX354951. A voucher strain was deposited in the Institute of Pharmaceutical Biology and Biotechnology, Heinrich-Heine University, Düsseldorf, Germany, with the ID code ST-F3.Cultivation, extraction and isolation
Fungal biomass fermentation was performed in Erlenmeyer flasks (15 × 1 L, each contains 100 g rice of commercially available rice and 110 mL water, which was kept overnight before being autoclaved for 20 min at a temperature of 121 °C) on solid rice medium at 25 °C under static conditions for 30 d. Harvesting process started with soaking the culture of each flask in EtOAc (500 mL), in addition to cutting the culture mass into small pieces using a spatula. The flasks were then kept overnight and subjected to shaking at 150 rpm for 8 h on the next day. The obtained extract was subjected to filtration, and evaporated to obtain a dark brown crude extract (6.5 g).This crude extract was partitioned between n-hexane and 90% aqueous methanol and the resulting methanolic phase was evaporated to give 4.5 g of methanolic extract, which was further chromatographed over silica gel using vacuum liquid chromatography (VLC). Gradient elution with n-hexane–EtOAc (100:0 to 0:100) and CH2Cl2–MeOH (100:0 to 0:100) was used and 500 mL eluting volume for each fraction was collected to obtain fifteen subfractions (F1–F15).
F4 (750 mg) was subjected to a Sephadex LH-20 (100 × 5 cm) column using MeOH as eluent to obtain six subfractions (F4-1–F4-6), of which F4-5 (80 mg) was purified using semi-preparative RP-HPLC with 60% MeOH–H2O as mobile phase to yield compounds 10 (42.3 mg), 11 (3.6 mg) and 12 (13.0 mg).
F5 (120 mg) was further fractionated using a Sephadex LH-20 column (80 × 4 cm) with MeOH as eluent to give five subfractions (F5-1–F5-5). Subfraction F5-1 (34 mg) was subjected to semi-preparative RP-HPLC with 38% MeOH–H2O to yield 15 (14.0 mg) and 14 (3.2 mg). Subfraction F5-2 (14 mg) was purified by semi-preparative RP-HPLC using 50% MeOH–H2O to give 5 (1.5 mg). Subfraction F5-3 (10 mg) was separated by semi-preparative RP-HPLC using 57% MeOH–H2O to afford 3 (0.9 mg) and 4 (2.4 mg). Subfraction F5-4 (15 mg) was purified by semi-preparative RP-HPLC with 64% MeOH–H2O as mobile phase to yield 1 (3.5 mg).
Following similar procedures, compound 13 (7.4 mg) was obtained from fraction F8, by column chromatography over a Sephadex LH-20 column (100 × 5 cm) with MeOH as eluent, followed by purification using semipreparative RP-HPLC with 34% MeOH–H2O.
F10 (235 mg) was separated into five subfractions (F10-1–F10-5) by a Sephadex LH-20 column (80 × 4 cm) using 100% MeOH as eluent. Subfraction F10-3 (35 mg) was then subjected to semi-preparative RP-HPLC eluted with 40% MeOH–H2O to yield 9 (1.8 mg) and 8 (3.0 mg) while subfraction F10-4 (13 mg) was purified by semi-preparative RP-HPLC with 50% MeOH–H2O to afford 2 (0.8 mg).
Compounds 7 (2.2 mg) and 6 (3.0 mg) were obtained from fraction F12 (150 mg) using a Sephadex LH-20 column (80 × 4 cm) with MeOH for elution followed by semi-preparative HPLC separation with 35% MeOH–H2O as eluent.
Acid hydrolysis
Compounds 6–8 (1 mg each) were separately hydrolyzed with 2 N HCl (2.0 mL) at 90 °C for 4 h. After cooling, the solution was partitioned with EtOAc (3 mL × 3) and H2O. The EtOAc phase containing the aglycone was dried under reduced pressure. The water phase containing the sugar moiety, was then examined using TLC analysis with D-glucose as an authentic standard (Sigma-Aldrich, Germany). Two different eluting systems were used including DCM–MeOH–H2O (6:4:1) and EtOAc–MeOH–H2O (10:4:1), along with methanol–sulphuric acid as spraying reagent.
Cytotoxicity assay
The MTT method was performed to test the cytotoxicity of the compounds against the L5178Y mouse lymphoma cell line (European Collection of Authenticated Cell Cultures, Catalogue No. 87111908) as described previously.36 Kahalalide F was used as positive control and media with 0.1% DMSO was used as negative control.Antibacterial assay
The antibacterial assay was performed using the broth microdilution method. Following the recommendations of the Clinical and Laboratory Standards Institute (CLSI), the MIC values against S. aureus ATCC 25923, S. aureus ATCC 700699, E. faecalis ATCC 29212, E. faecalis ATCC 51299, E. faecium ATCC 35667, E. faecium ATCC 700221 and Mycobacterium tuberculosis strain H37Rv were determined.37 Moxifloxacin (for S. aureus strains), ciprofloxacin (for E. faecalis and E. faecium strains) and rifampicin (for Mycobacterium tuberculosis) were used as positive controls.Computational section
Mixed torsional/low-frequency mode conformational searches were carried out by means of the Macromodel 9.9.223 software using the Merck Molecular Force Field (MMFF) with an implicit solvent model for CHCl3.38 Geometry reoptimizations were carried out at the B3LYP/6-31G(d) level in vacuo, the B3LYP/6-31+G(d,p) level in vacuo, the B97D/TZVP20,21 and the CAMB3LYP/TZVP22,23 levels with the PCM solvent model for MeCN or MeOH. TDDFT ECD calculations and OR calculations were run with various functionals (B3LYP, BH&HLYP, CAM-B3LYP, PBE0) and the TZVP basis set as implemented in the Gaussian 09 package with the same or no solvent model as in the preceding DFT optimization step.39 NMR calculations were performed at the mPW1PW91/6-311+G(2d,p) level.17 ECD spectra were generated as sums of Gaussians with 3000 and 3300 cm−1 widths at half-height (corresponding to ca. 12 and 13 nm at 200 nm), using dipole-velocity-computed rotational strength values.40 Computed ECD spectra were shifted by −48 (B3LYP/TZVP for the B3LYP/6-31G(d) in vacuo conformers) for 1, +6 (PBE0/TZVP PCM/MeCN for the B97D/TZVP PCM/MeCN conformers) for 11 and −3 (B3LYP/TZVP for the B3LYP/6-31G(d) in vacuo conformers) for gliomastin E. Computed C-NMR data were corrected with I = 185.4855 and S = −1.0306 and H-NMR data with I = 31.8996 and S = −1.0734.41,42 Boltzmann distributions were estimated from the ZPVE-corrected B3LYP/6-31G(d) energies in the B3LYP/6-31G(d) gas-phase calculations, and from the uncorrected B3LYP/6-31+G(d,p), B97D/TZVP and CAM-B3LYP/TZVP energies in the other cases. The MOLEKEL software package was used for visualization of the results.43Acknowledgements
A scholarship granted and financed by the Egyptian government (Ministry of High Education) to M. S. E. is gratefully acknowledged. Financial support by the DFG (GRK 2158) to P. P. and R. K. is gratefully acknowledged. T. K. and A. M. thank the National Research, Development and Innovation Office (NKFI K120181 and PD121020) for financial support and the Governmental Information-Technology Development Agency (KIFÜ) for CPU time.Notes and references
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2014, 31, 160–258 RSC.
- M. E. Rateb and R. Ebel, Nat. Prod. Rep., 2011, 28, 290–344 RSC.
- X. M. Hou, R. F. Xu, Y. C. Gu, C. Y. Wang and C. L. Shao, Curr. Med. Chem., 2015, 22, 3707–3762 CrossRef CAS PubMed.
- T. A. Richards, M. D. M. Jones, G. Leonard and D. Bass, Annual Review of Marine Science, 2012, 4, 495–522 CrossRef PubMed.
- W. Chen, Y. Li and Y. Guo, Acta Pharm. Sin. B, 2012, 2, 227–237 CrossRef CAS.
- F. He, J. Bao, X. Y. Zhang, Z. C. Tu, Y. M. Shi and S. H. Qi, J. Nat. Prod., 2013, 76, 1182–1186 CrossRef CAS PubMed.
- C. J. Zheng, C. L. Shao, Z. Y. Guo, J. F. Chen, D. S. Deng, K. L. Yang, Y. Y. Chen, X. M. Fu, Z. G. She, Y. C. Lin and C. Y. Wang, J. Nat. Prod., 2012, 75, 189–197 CrossRef CAS PubMed.
- C. L. Shao, H. X. Wu, C. Y. Wang, Q. A. Liu, Y. Xu, M. Y. Wei, P. Y. Qian, Y. C. Gu, C. J. Zheng, Z. G. She and Y. C. Lin, J. Nat. Prod., 2011, 74, 629–633 CrossRef CAS PubMed.
- M. S. Elnaggar, S. S. Ebada, M. L. Ashour, W. Ebrahim, W. E. G. Müller, A. Mándi, T. Kurtán, A. Singab, W. Lin, Z. Liu and P. Proksch, Tetrahedron, 2016, 72, 2411–2419 CrossRef CAS.
- M. S. Elnaggar, S. S. Ebada, M. L. Ashour, W. Ebrahim, A. Singab, W. Lin, Z. Liu and P. Proksch, Fitoterapia, 2017, 116, 126–130 CrossRef CAS PubMed.
- W. Ebrahim, M. El-Neketi, L. I. Lewald, R. S. Orfali, W. Lin, N. Rehberg, R. Kalscheuer, G. Daletos and P. Proksch, J. Nat. Prod., 2016, 79, 914–922 CrossRef CAS PubMed.
- S. Liu, H. Dai, G. Makhloufi, C. Heering, C. Janiak, R. Hartmann, A. Mándi, T. Kurtán, W. E. G. Müller, M. U. Kassack, W. Lin, Z. Liu and P. Proksch, J. Nat. Prod., 2016, 79, 2332–2340 CrossRef CAS PubMed.
- J. Zhang, X. P. Lin, L. C. Li, B. L. Zhong, X. J. Liao, Y. H. Liu and S. H. Xu, RSC Adv., 2015, 5, 54645–54648 RSC.
- H. Y. Yang, D. Y. Niu, L. Wang, S. J. Wang, C. M. Zhang, X. M. Gao, G. Du and Q. F. Hu, J. Asian Nat. Prod. Res., 2015, 17, 319–323 CrossRef CAS PubMed.
- W. Dong, C. Liu, Q. Shen, T. Zhang, Y. Wang, K. Zhou, B. Ji, H. Yang, G. Du, Q. Hu and M. Zhou, Chem. Nat. Compd., 2016, 52, 620–623 CrossRef CAS.
- W. J. He, X. J. Zhou, X. C. Qin, Y. X. Mai, X. P. Lin, S. R. Liao, B. Yang, T. Zhang, Z. C. Tu, J. F. Wang and Y. Liu, Nat. Prod. Res., 2017, 31, 604–609 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- G. Pescitelli and T. Bruhn, Chirality, 2016, 28, 466–474 CrossRef CAS PubMed.
- A. Mándi, I. W. Mudianta, T. Kurtán and M. J. Garson, J. Nat. Prod., 2015, 78, 2051–2056 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- P. Sun, D. X. Xu, A. Mándi, T. Kurtán, T. J. Li, B. Schulz and W. Zhang, J. Org. Chem., 2013, 78, 7030–7047 CrossRef CAS PubMed.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2011, 40, 4603–4625 RSC.
- A. Abdel-Lateff, G. M. König, K. M. Fisch, U. Höller, P. G. Jones and A. D. Wright, J. Nat. Prod., 2002, 65, 1605–1611 CrossRef CAS.
- M. Tanaka, F. Nara, Y. Yamasato, Y. Ono and T. Ogita, J. Antibiot., 1999, 52, 827–830 CrossRef CAS PubMed.
- L. Shen, S. D. Zhao and H. J. Zhu, Gaodeng Xuexiao Huaxue Xuebao, 2011, 32, 2568–2573 CAS.
- P. L. Polavarapu, Chirality, 2008, 20, 664–672 CrossRef CAS PubMed.
- P. Sun, Q. Yu, J. Li, R. Riccio, G. Lauro, G. Bifulco, T. Kurtán, A. Mándi, H. Tang, C. L. Zhuang, W. H. Gerwick and W. Zhang, J. Nat. Prod., 2016, 79, 2552–2558 CrossRef CAS PubMed.
- H. Thieme, Pharmazie, 1970, 25, 129 CAS.
- E. Walewska and H. Thieme, Pharmazie, 1969, 24, 423 CAS.
- H. M. Chawla, S. K. Sharma, K. Chakrabarty and S. Bhanumati, Tetrahedron, 1988, 44, 1227–1234 CrossRef CAS.
- Y. C. Chien, C. H. Lin, M. Y. Chiang, H. S. Chang, C. H. Liao, I. S. Chen, C. F. Peng and I. L. Tsai, Phytochemistry, 2012, 80, 50–57 CrossRef CAS PubMed.
- S. Yamada, F. Ono, T. Katagiri and J. Tanaka, Bull. Chem. Soc. Jpn., 1977, 50, 750 CrossRef CAS.
- W. J. Lan, W. Liu, W. L. Liang, Z. Xu, X. Le, J. Xu, C. K. Lam, D. P. Yang, H. J. Li and L. Y. Wang, Mar. Drugs, 2014, 12, 4188–4199 CrossRef CAS PubMed.
- J. Kjer, A. Debbab, A. H. Aly and P. Proksch, Nat. Protoc., 2010, 5, 479–490 CrossRef CAS PubMed.
- M. Ashour, R. Edrada, R. Ebel, V. Wray, W. Wätjen, K. Padmakumar, W. E. G. Müller, W. H. Lin and P. Proksch, J. Nat. Prod., 2006, 69, 1547–1553 CrossRef CAS PubMed.
- CLSI, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard–Ninth Edition, Clinical and Laboratory Standards Institute, 2012 Search PubMed.
- MacroModel; Schrödinger, LLC, 2012, http://www.schrodinger.com/MacroModel.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- P. J. Stephens and N. Harada, Chirality, 2010, 22, 229–233 CAS.
- CHESHIRE CCAT, the Chemical Shift Repository for computed NMR scaling factors, http://cheshirenmr.info/index.htm.
- M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef CAS PubMed.
- U. Varetto, MOLEKEL, v. 5.4, Swiss National Supercomputing Centre, Manno, Switzerland, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV, HRESIMS and NMR spectra of all the new compounds (1–8), NMR and ECD calculations for compound 1 as well as ECD and OR calculations for the aglycones of 6 and 7. See DOI: 10.1039/c7ra04941b |
| This journal is © The Royal Society of Chemistry 2017 |