
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A polysalen based on polyacylamide stabilized palladium nanoparticle catalyst for efficient carbonylative Sonogashira reaction in aqueous media†
Yan Wang‡
 ab,
Xiaolong Yang‡c and
Jianqiang Yu*a
ab,
Xiaolong Yang‡c and
Jianqiang Yu*a
aLaboratory of Clean Energy Chemistry and Materials, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, No. 18 Tianshui Middle Road, Lanzhou 730000, China. E-mail: yjq@licp.cas.cn
bQingdao Center of Resource Chemistry & New Materials, No. 36 Jinshui Road, Qingdao 266000, China
cKey Laboratory of Marine Chemistry Theory and Technology, Ministry of Education, Ocean University of China, 238 Songling Road, Qingdao 266100, China
First published on 21st June 2017
Abstract
A highly cross-linked polymer matrix polysalen based on acrylamide and N,N′-methylene-bisacrylamide (NNMBA) was successfully prepared. It has been proved that this type of polysalen is an effective support for immobilizing palladium complex. The polysalen may provide coordination sites for conjugation with a palladium catalyst to produce polysalen–Pd catalyst. The polysalen–Pd was firstly applied in phosphine-free carbonylative Sonogashira coupling reactions of aryl iodides with terminal alkynes to produce α,β-alkynyl ketones in 81–95% yields in aqueous media, which was much higher than that of homogeneous Pd–salen in 52% yield. Furthermore, the catalyst showed excellent recyclability without any significant loss in its activity.
Introduction
Alkynyl ketones are key structural motifs in organic synthesis as they provide an easy access to various natural products and heterocyclic derivatives1 such as pyrimidine,2 quinolone,3 furan,4 pyrazole,5 pyrrole,6 and flavone.7 Under physiological conditions, alkynyl ketones exist in various biological active molecules. So the synthesis of alkynyl ketones has attracted considerable interest. Traditionally, alkynyl ketones were prepared by the coupling of acid chlorides with alkynyl organometallic reagents such as copper, cadmium, lithium, silicon, sodium, silver, tin and zinc.8 However, these methods require dry solvents under an inert atmosphere.Since Tanaka9 reported the first example of Pd-catalyzed carbonylative Sonogashira reaction of aryl halides with terminal alkynes in the presence of carbon monoxide, this atom economic route has become an alternative for the synthesis of α,β-alkynones. Due to its great importance, promising development has been driven on and the scope of this reaction has also been extended for the synthesis of various heterocycles.10 In the early reports, considerable efforts have been made towards this reaction, and the most restricting aspect of these studies was that they focused on homogeneous and heterogeneous catalytic systems together with phosphine ligands.11 However, these phosphines were not considered to be the most desired class of ligands for catalysis because of their high costs, environmentally unfriendly properties, complex synthesis, air and moisture sensitivity, inconvenient handing, and other problems. Thus, an array of N-based ligands, such as N-heterocyclic carbenes,12 and amines,13 has been employed successfully in phosphine-free carbonylative Sonogashira reactions.
Salen ligands, one of the N-based ligands, have received particular attention mainly due to their simple synthesis and high stability. Salen can be tuned easily by selecting suitable condensing aldehydes and amines. The salen ligands play an important role in the development of catalysis not only due to varity of transition metals that can be incorporated, but also the varity of ligand derivative from classics salen. Most of the reported metal salen complexes belong to the first transition series,14 and the second series of these complexes are still rare. Palladium(II) salen complexes have been reported as catalysts for different C–C cross-coupling reactions,15 hydrogenation of imines,16 and styrene polymerization.17 Hence, at first we used a simple Pd–salen (salicylaldehydoethylenediamine) in the carbonylation of iodobenzene with phenylacetylene. Unfortunately, the Pd–salen just gave the desired carbonylative coupling product in 52% yield. As previously reported,18 the drawback of using metal salens in homogeneous solutions is the formation of μ-oxo dimers and other polymeric species leading to the irreversible catalyst deactivation. In principle, this problem can be solved by immobilizing salen on supports to isolate the metal–salen complexes from each other. In recent years, the immobilization of catalysts on suitable supports has been a topic of special interest. The catalytic systems containing immobilized organometallic compounds have several advantages, e.g. the simple recycling of the catalysts by filtration, preventing the loss of both ligands and metal, thus considerably decreasing the environmental problems of waste materials, compared with homogeneous catalysis.19 Much effort has been focused on the immobilization of these salen ligands through a variety of strategies till now.20 Immobilization of catalyst onto salen containing polymers not only shows higher thermal stability than corresponding salen monomer metal complexes because of their polymeric network structure, but also more active centers. Therefore, it is of interest to study the catalytic activity of salen containing polymers. Tamami21 and co-works synthesized copolymer polyacrylamide from acrylamide with N,N′-methylene-bisacrylamide (NNMBA), which was used for the immobilization of cobalt complex for selective oxidation of olefins and alkyl halides. However, to the best of our knowledge, the carbonylation reactions catalyzed by salen containing polymers supported palladium catalysts has not been reported up to now. In continuation to our previous study on heterogeneous carbonylation,22 in this paper, the noble metal Pd was loaded on a polysalen support that was derived from the co-polymerization of acrylamide and N,N′-methylene-bisacrylamide (NNMBA). The polysalen–Pd was formed by post-synthesis modification in the incorporation of Pd(II) in the polymer networks.
Results and discussion
The designing procedure of the polysalen–Pd catalyst is shown in Scheme 1. Polyacrylamide crosslinked with N,N′-methylene-bisacryl-amide (NNMBA) (5%) was prepared by polymerization of acrylamide monomer in ethanol using K2S2O8 as an initiator. The polar nature of the crosslinking agent makes it compatible with the polymer backbone as well as the reaction medium. We found that the polymer with less than 5% NNMBA was sticky and not physically proper to be used in the further reactions. The polysalen was synthesized by the addition of hydrazine hydrate and then by aldimine condensation route with salicylaldehyde. Followed by the addition of Pd(OAc)2, polysalen–Pd was produced.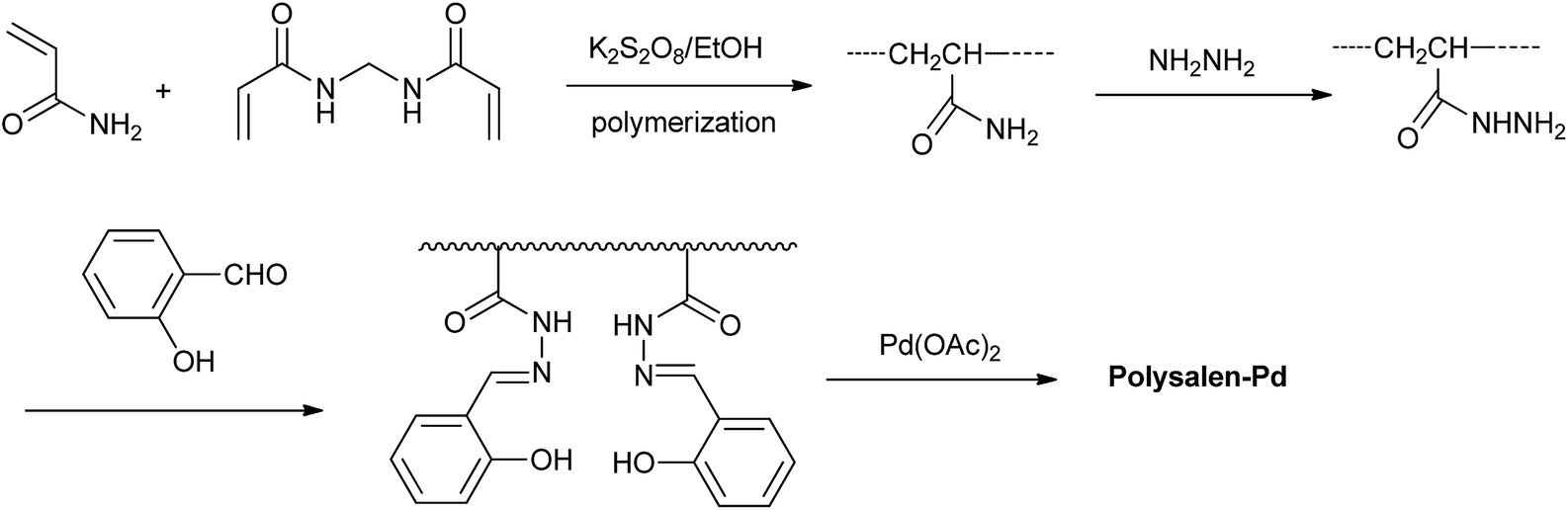 | ||
| Scheme 1 The synthesis of polysalen–Pd catalyst. | ||
In order to confirm the components of the heterogeneous catalyst, the chemical structure of intermediate polymer was analyzed by FTIR and the results are shown in Fig. 1. FT-IR spectrum of the crosslinked polyacrylamide showed the characteristic absorbance of amide (N–H) group at 3384 cm−1, and carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 1666 cm−1 (Fig. 1a). Polyacrylohydrazide was obtained by transamidation reaction of crosslinked polyacrylamide with excess of hydrazine hydrate. The resultant polymer had a higher content of amino group of the polymer than polyacrylamide (Fig. 1b). IR spectrum of the polyacrylohydrazide showed the characteristic absorbance of amino (N–H) group at 3378 cm−1. Crosslinked polyacrylohydrazide-imino-salicylaldehyde was prepared by the reaction of polyacrylohydrazide with salicylaldehyde in ethanol. The IR spectrum of the polymer showed peaks at 1636 cm−1, corresponding to the formation of imine group (CN) and at 1268 cm−1 and 750 cm−1 due to C–OH stretching of aromatic ring, respectively (Fig. 1c). The imine content was evaluated from CHN analysis and was found to the 2.6 mmol g−1.
O) at 1666 cm−1 (Fig. 1a). Polyacrylohydrazide was obtained by transamidation reaction of crosslinked polyacrylamide with excess of hydrazine hydrate. The resultant polymer had a higher content of amino group of the polymer than polyacrylamide (Fig. 1b). IR spectrum of the polyacrylohydrazide showed the characteristic absorbance of amino (N–H) group at 3378 cm−1. Crosslinked polyacrylohydrazide-imino-salicylaldehyde was prepared by the reaction of polyacrylohydrazide with salicylaldehyde in ethanol. The IR spectrum of the polymer showed peaks at 1636 cm−1, corresponding to the formation of imine group (CN) and at 1268 cm−1 and 750 cm−1 due to C–OH stretching of aromatic ring, respectively (Fig. 1c). The imine content was evaluated from CHN analysis and was found to the 2.6 mmol g−1.
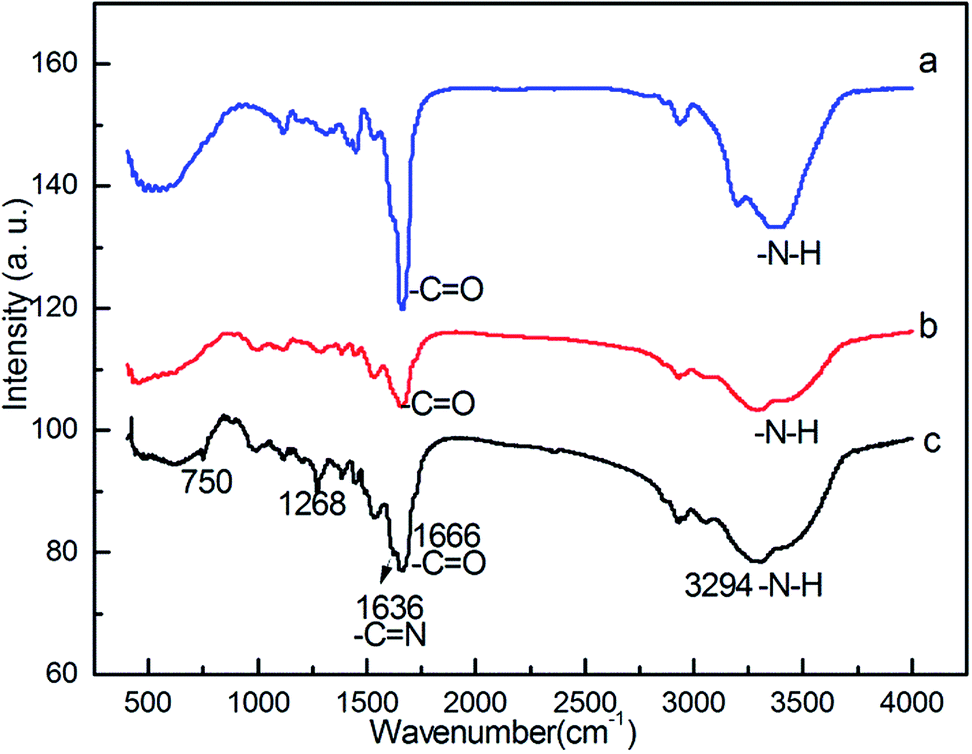 | ||
| Fig. 1 FT-IR spectra of (a) crosslinked polyacylamide; (b) crosslinked polyacrylohydrazide; (c) crosslinked polyacrylohydrazide-imino-salicyaldehyde. | ||
Treating the solution of Pd(OAc)2 with the polysalen ligand results in the formation of polysalen supported palladium complex. In order to make the process completely heterogeneous, the catalyst was conditioned by washing thoroughly with solvents to remove any loose Pd species. The metal content of the catalyst found by ICP was 1.98% of the resin. The IR spectra of the polymer before and after palladium loading (Fig. 2) showed a shift in frequency from 1636 to 1603 cm−1 for ν(CN) and from 1268 to 1283 cm−1 for ν(C–O). In addition, IR spectrum of the polysalen–Pd showed two new absorption bands at 478 and 544 cm−1, which were attributed to the formation of Pd–N and Pd–O, respectively. These variations confirmed the coordination of palladium ion with the azomethine nitrogen and phenolic oxygen of the polymeric salen ligand.
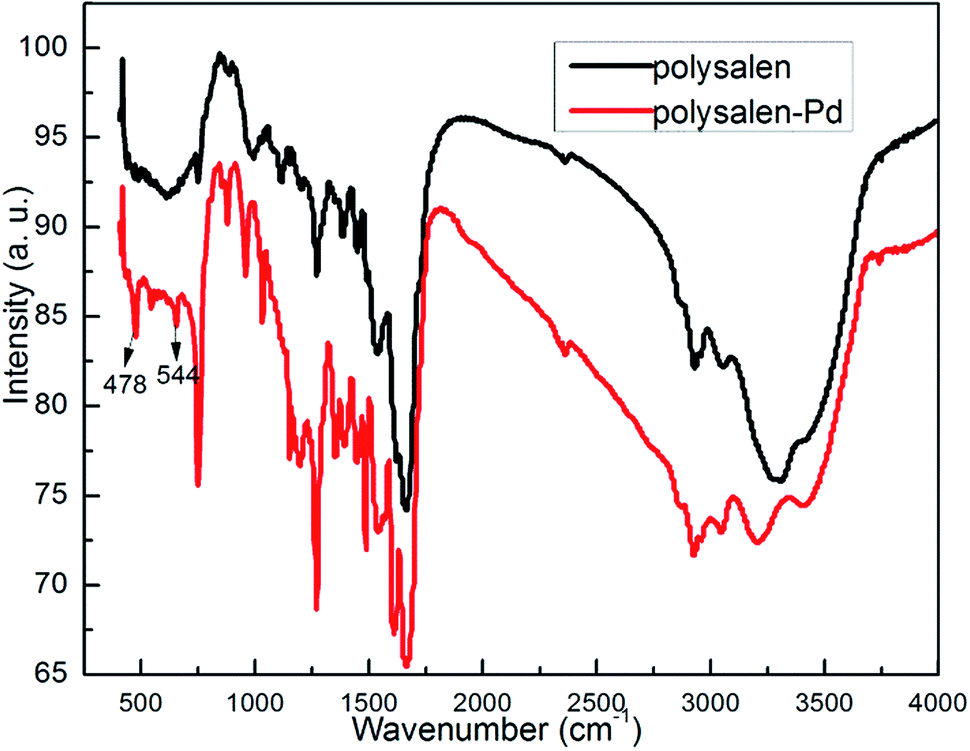 | ||
| Fig. 2 FT-IR spectra of polysalen and polysalen–Pd. | ||
The X-ray photoelectron spectroscopy (XPS) measurements were carried out to investigate the chemical environment and surface stoichiometry of the elements of samples. The catalyst polysalen–Pd was also characterized by XPS before reaction, and the results are shown in Fig. 3. It can be seen that the Pd 3d spectrum was resolved into two spin–orbit pairs with binding energies of 335.9 eV, 338.4 eV and 341.4 eV, 343.7 eV, respectively, which were assigned to electron transitions of 3d5/2 and 3d3/2 of Pd2+ (Fig. 3a). The results indicated that the polysalen–Pd catalyst contains two types of Pd(II) species. In addition, compared with the BE of Pd 3d5/2 of Pd(OAc)2 (338.6 eV), the relatively large negative BE shift of Pd2+ indicated that the cross-linked polysalen support act as ligands stabilizing the Pd species with a strong interaction. The XPS survey spectra of the polysalen–Pd as shown in Fig. 3b–d, indicated the binding energies of the C 1s, N 1s and O 1s correspond to different components. The C 1s spectra can be deconvoluted into six components which appeared at 284.1, 284.8, 285.4, 285.9, 286.7 and 288.1 eV, respectively, and were associated with C–OH, C–C, C–N,23 C(C6H5–), C–O, CO.24
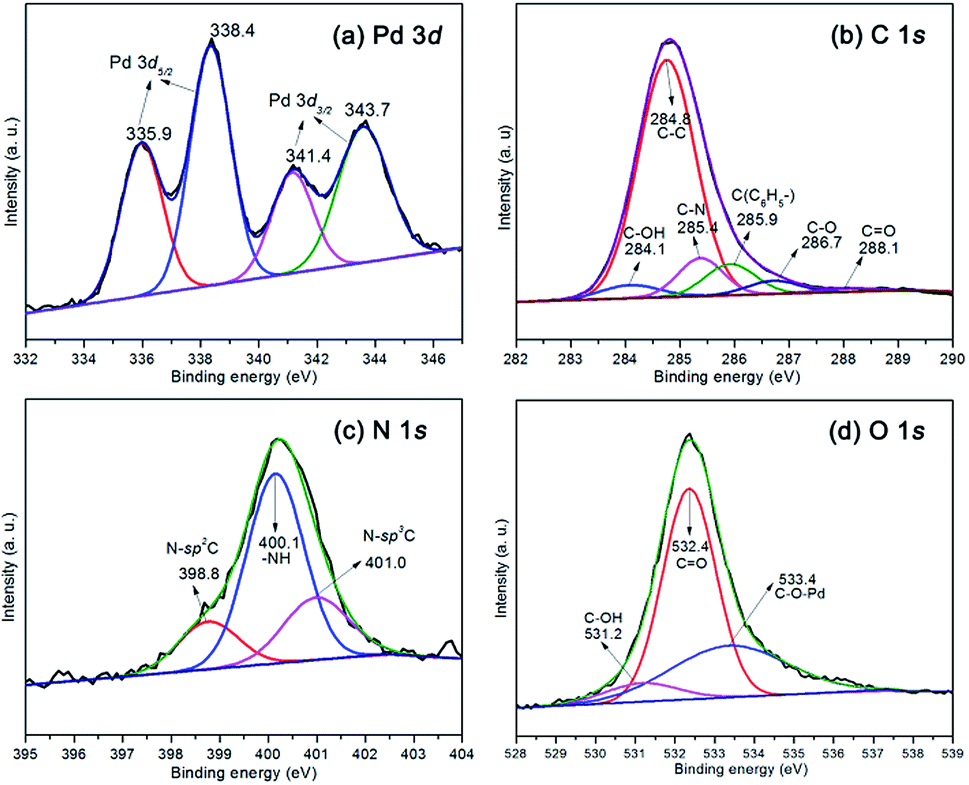 | ||
| Fig. 3 XPS spectrum of the polysalen–Pd: Pd 3d (a), C 1s (b), N 1s (c), O 1s (d). | ||
The N 1s spectra can be decovoluted into three peaks (Fig. 3c) which appear at 398.8, 400.1 and 401.0 eV. The main peak at 400.1 eV was attributed to –NH–, the lower binding energy tail at 398.8 eV and the higher binding energy tail at 401.1 eV, which were assigned to N–sp2C and N–sp3C bonds.25 Deconvolution of the O 1s spectrum results in three peaks located at 531.2, 532.4, and 533.4 eV in Fig. 3d, respectively. The main peak at 532.4 eV which was consistent with standard spectrum O of OC.26 The component at 531.2 eV and 533.4 eV were attributed to C–OH bonding of polysalen and C–O–Pd of polysalen–Pd.
Based on the XPS analysis above, we can speculate there may be an electrostatic interaction between the Pd atoms with N, O of polyacrylohydrazide-imino-salicyaldehyde (polysalen) and the oxygen atoms of acetate. In other words, the Pd atom formed bond with the nitrogen atom, oxygen atom of the support, the Pd particle was immobilized on the cross-linked polymer through covalent bonding as supported palladium salen complex.
The morphology and structure of the polysalen and polysalen–Pd were characterized by transmission electronic microscopy (TEM). Selected area electron diffraction (SAED) pattern of the polysalen is shown in the insert of Fig. 4b.
 | ||
| Fig. 4 TEM and HRTEM images of the polysalen (a, b) and fresh polysalen–Pd (c–e), recovered catalyst (f). Insert in (b) was selected-area diffraction (SEAD) pattern. Insert in (d) was histogram of Pd particles' size. Insert in (e) was HRTEM image. | ||
The appearance of the diffused rings indicated that the polysalen was in polycrystalline phase. From the TEM micrographs of the polysalen–Pd, it was clearly observed that Pd species are dispersed granularly and uniformly on the surface of polysalen, the average size of Pd nanoparticles was measured about 5.2 nm. The histogram of Pd particles' size was shown in insert in (d). From the HRTEM image of Fig. 4e, it can be seen that the interplanar space is 0.225 nm, corresponding to the (111) plane of face centered cubic Pd (JCPDS 46-1043).
The crystalline nature of polysalen–Pd was further confirmed by powder X-ray diffraction (XRD) (Fig. 5). The peaks at about 40.0°, 46.4° and 68.0° correspond to the (111), (200) and (220) lattice planes of the face center cubic crystalline structure of Pd, respectively. The peak positions and relative intensities match well with the standard XRD pattern for face centered cubic Pd (JCPDS 46-1043), agreeing well with that observed by HRTEM. Obviously, both TEM and XRD results confirm that the Pd nanoparticles in a narrow size range are successfully prepared. The formation of the small Pd nanoparticles might be attributed to the following facts: (1) the N–, O– on the surface of polysalen coordinate to Pd cations are beneficial for the dispersion of Pd nanoparticles, and (2) the interactions between the polymeric support with palladium and the salen ligands stabilized the Pd nanoparticles also contributes to the dispersibility.
 | ||
| Fig. 5 XRD patterns of polysalen–Pd. | ||
To assess the catalytic performance of the prepared catalyst, the carbonylative Sonogashira coupling reaction between iodobenzene and phenylacetylene in the presence of CO was chosen as model reaction. The effects of various reaction parameters such as catalyst loading, solvent, base, temperature, reaction time and CO pressure on the catalytic performance were thoroughly examined. The solvent plays an important role in the reaction: when the reaction was performed in acetonitrile, acetone and THF, the isolated yields reached 57%, 39%, 78% (Table 1, entries 1–3), respectively. It is found that when toluene and H2O were used as solvent, α,β-alkynyl ketones will be formed in an excellent yield.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | T (°C) | t (h) | Yielda (%) |
| a Iodobenzene 1.0 mmol, phenylacetylene 1.2 mmol, solvent 5.0 mL, base 2.0 mmol, CO 2.0 MPa, polysalen–Pd 0.5 mol%, isolated yield.b Pd–salen (salicylaldehydoethylenediamine) 0.5 mol%. | |||||
| 1 | CH3CN | NEt3 | 130 | 6 | 57 |
| 2 | Acetone | NEt3 | 130 | 6 | 39 |
| 3 | THF | NEt3 | 130 | 6 | 78 |
| 4 | Toluene | NEt3 | 130 | 6 | 91 |
| 5 | H2O | NEt3 | 130 | 6 | 90 |
| 6 | H2O | NaOH | 130 | 6 | 21 |
| 7 | H2O | Pyridine | 130 | 6 | 46 |
| 8 | H2O | NEt3 | 120 | 6 | 85 |
| 9 | H2O | NEt3 | 100 | 6 | 56 |
| 10 | H2O | NEt3 | 130 | 2 | 70 |
| 11 | H2O | NEt3 | 130 | 3 | 82 |
| 12b | H2O | NEt3 | 130 | 6 | 52 |

Therefore, H2O was chosen as a non-toxic solvent to be the best mediate. Using H2O as the solvent, we further investigated the influence of alkalinity on the carbonylative Sonogashira coupling reaction. Noteworthy, Et3N was the most suitable base for the water system while the use of NaOH and pyridine delivered the product in low yields (Table 1, entries 6, 7). Increasing the reaction temperature and time also had a positive effect on the catalytic activity. For comparison, when homogeneous Pd–salen (salicylaldehydoethylenediamine) was used as the catalyst, only 52% yield was obtained, although the reaction is proceeded in a homogeneous catalysis (Scheme 2). The improvement in the activity using polysalen–Pd as catalyst might be attributed to the interactions between the polymeric support with palladium, and the polysalen ligands stabilized the Pd nanoparticles to isolate the metal–salen complexes from each other.
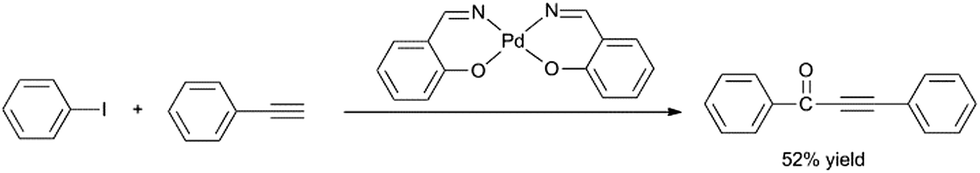 | ||
| Scheme 2 Homogeneous Pd–salen catalyzed carbonylative Sonogashira coupling of iodobenzene with phenylacetylene. | ||
As a result of these studies, we were encouraged to examine the reaction with a broad range of substrates to determine the specificity and scope of substrates for this system. Thus, using water as the solvent, 0.5 mmol% Pd catalyst loading, starting with terminal alkynes and aryl iodides, we carried out the carbonylative Sonogashira coupling reaction under 2.0 MPa CO with Et3N as the base. To explore the generality and scope of the carbonylative coupling reaction, we evaluated various aryl iodides and terminal alkynes as substrates. The results are given in Table 2. Generally, a wide range of aryl iodides bearing an electron-withdrawing group or an electron-donating group all gave corresponding carbonyl products in excellent yields when react with phenylacetylene (entry 1–13). The aryl iodides with electron-donating groups, such as methoxy, methyl and ethyl, gave excellent yields (Table 2, entries 2–4, 7–9). Even the reaction of sterically hindered 2-iodoanisole with phenylacetylene gave the desired coupling product in 81% yield (Table 1, entry 3). The reactions of 4-chloro-iodobenzene and 4-bromo-iodobenzene with phenylacetylene also afforded the corresponding products in satisfactory yield (Table 2, entry 5–6). The reaction of α-iodonaphthalene with phenylacetylene afforded the corresponding alkynyl ketone in 85% yield (Table 2, entry 11). We also investigated three acetylenes containing different groups for the reaction. The reactions of the substituted phenylacetylenes, such as 4-methoxy-phenylacetylene, 4-tertiary-butyl-phenylacetylene and 4-bromo-phenylacetylene with iodobenzene, also proceeded effectively under the same conditions to yield 94%, 90% and 87% of the desired α,β-alkynyl ketones, respectively (Table 2, entries 12–14).
|
||||
|---|---|---|---|---|
| Entry | R | R′ | Product | Yieldb (%) |
| a Reaction condition: aryl iodide (1 mmol), terminal alkyne (1 mmol), polysalen–Pd (30 mg, 0.005 mmol Pd), Et3N (0.4 mL), distilled water (5 mL), temperature: 130 °C, time: 6 h, CO pressure: 2.0 MPa.b Isolated yield. | ||||
| 1 | H | H | 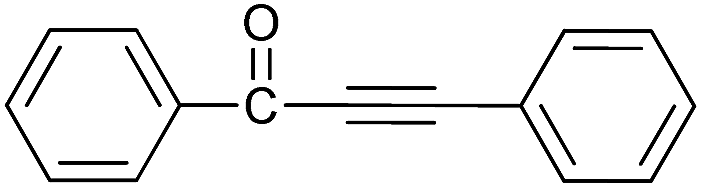 |
90 |
| 2 | 4-CH3O | H | 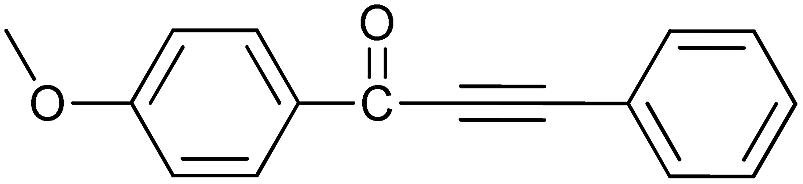 |
95 |
| 3 | 2-CH3O | H | 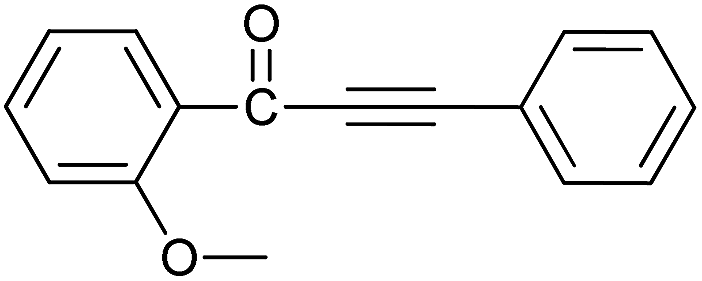 |
81 |
| 4 | 3-CH3O | H | 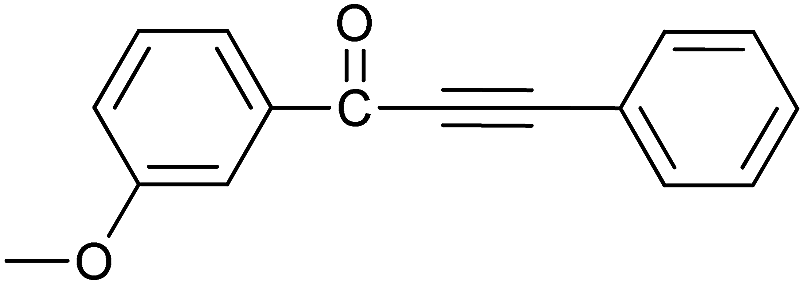 |
88 |
| 5 | 4-Cl | H | 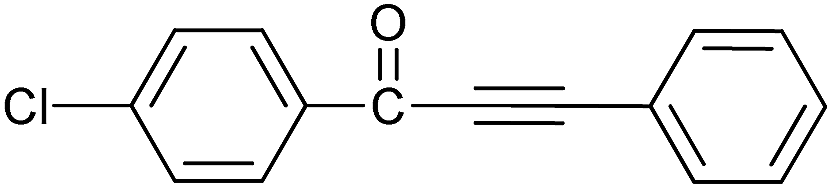 |
92 |
| 6 | 4-Br | H | 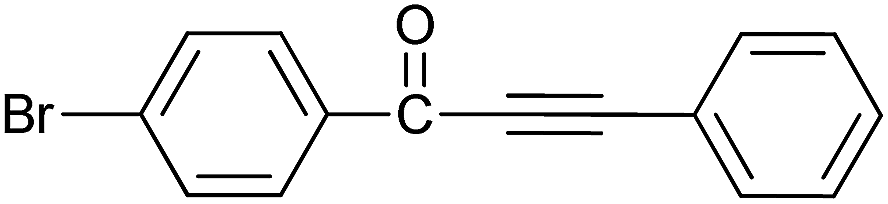 |
92 |
| 7 | 4-CH3 | H |  |
93 |
| 8 | 2-CH3 | H | 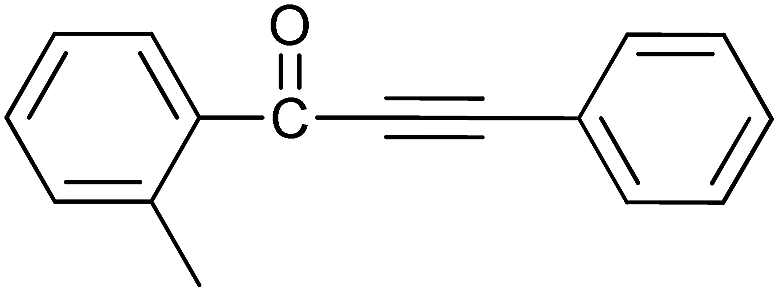 |
89 |
| 9 | 4-C2H5 | H | 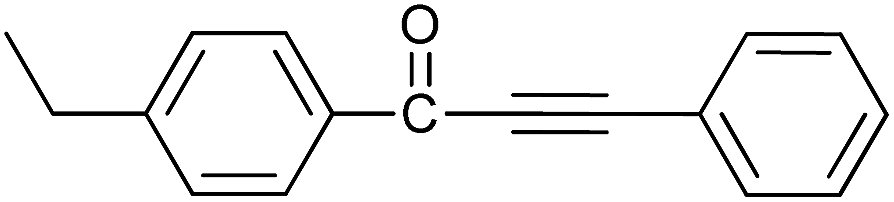 |
91 |
| 10 | 4-CH3CO | H | 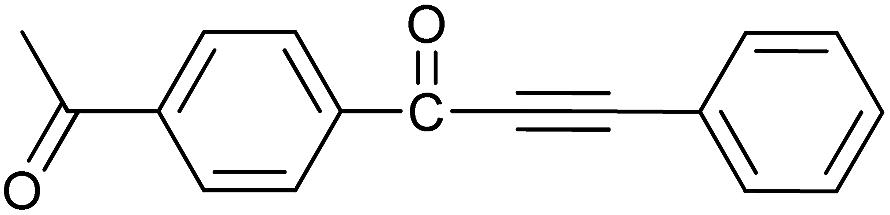 |
82 |
| 11 | Naphthyl | H | 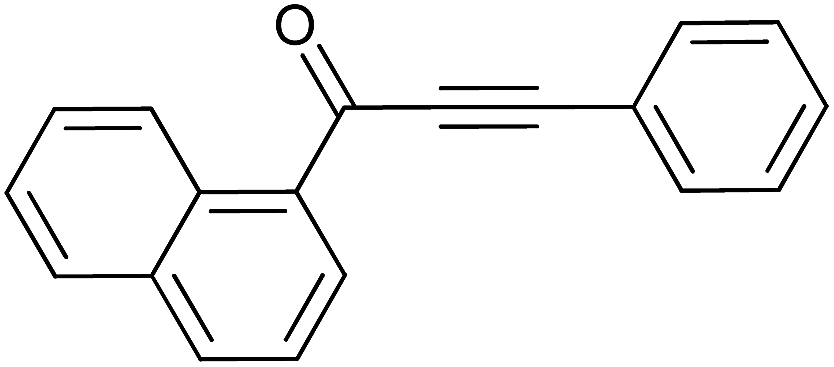 |
85 |
| 12 | H | 4-CH3O | 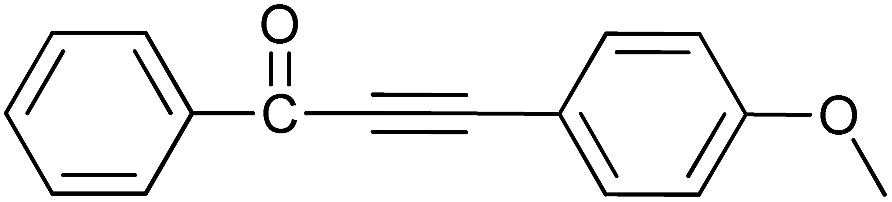 |
94 |
| 13 | H | 4-tBu | 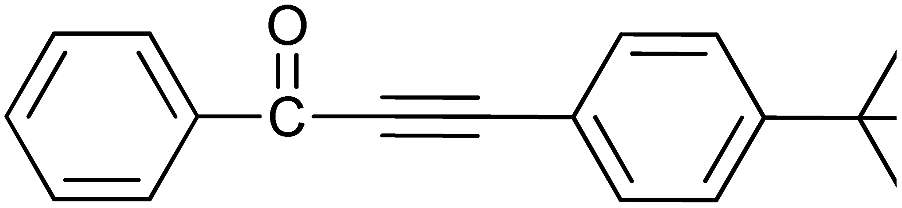 |
90 |
| 14 | H | 4-Br |  |
87 |

Besides the efficient catalytic activity, the stable recyclability is also crucial for an outstanding heterogeneous catalyst from the viewpoint of both academic research and industrial applications. To evaluate the recyclability of polysalen–Pd in carbonylative Sonogashira coupling reaction, the continuous carbonylative coupling reaction of iodobenzene with phenylacetylene in water was investigated and the results are presented in Fig. 6. The catalyst could be reused five times only with a slight loss of activity (the 5th recycle yield still can obtain 84%). The aggregation of the Pd particles in the used catalyst was not obvious from the TEM photograph in Fig. 4f. The content of Pd in the catalyst was 1.76 wt% (0.229 mmol g−1), as determined by the AAS analysis. Accordingly, the coordination bond of polysalen–Pd(II) lead to its better catalytic activity and recycle stability.
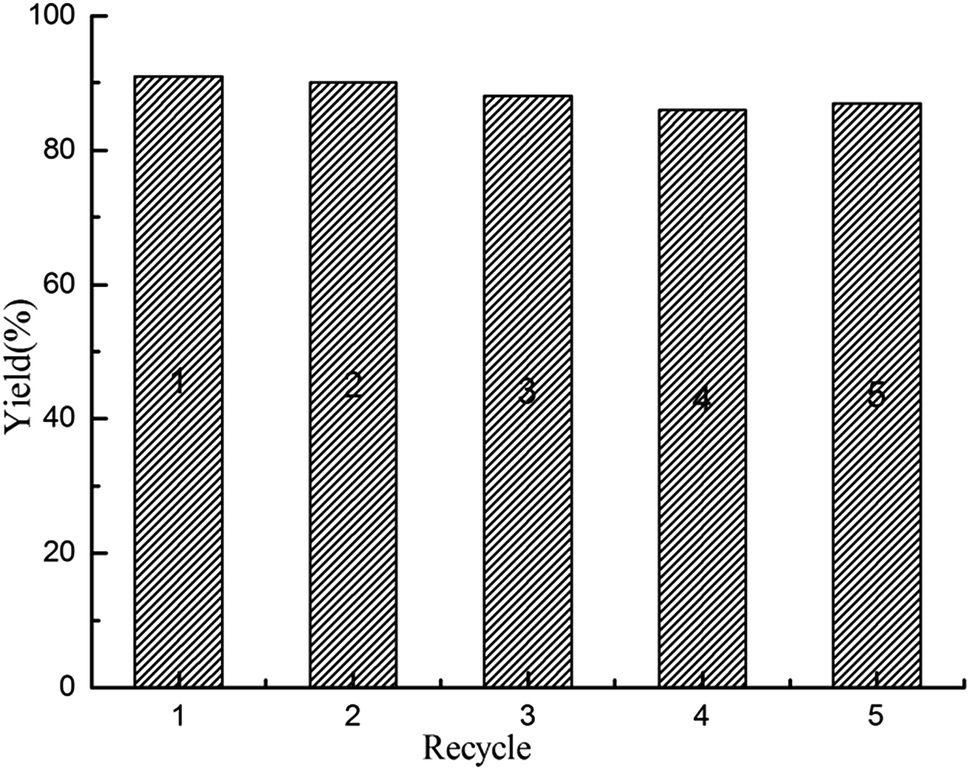 | ||
| Fig. 6 Recycle test of polysalen–Pd in the carbonylative Sonogashira coupling reaction. | ||
To get the deeper insight into the active sites for the supported polysalen–Pd catalyzed the carbonylative Sonogashira reaction, the hot filtration test was performed. After 1 h of reaction, the catalyst was isolated by filtration without cooling the reaction mixture and the conversion of iodobenzene was detected by in 43% GC. Moreover, the filtrate was maintained under the reaction condition and the mixture was analyzed after an additional hour by GC. The conversion of iodobenzene was detected in 48%. The hot filtration test proved that the catalytic activity could be attributed to the heterogeneous catalyst and the leached species in the solution. The TEM images of the reaction mixture and the catalyst after 1 h of reaction by filtration was also investigated in Fig. 7. Fig. 7a was the TEM image of the reaction mixture by filtration and no Pd particle was found in the mixture, this result showed that the leached species in the solution was the water-soluble Pd2+. Fig. 7b was the TEM image of the catalyst after 1 h reaction, it was clearly observed that Pd species are dispersed granularly and uniformly on the surface of polysalen. The histogram of Pd particles' size was shown in insert in (b), the average size of Pd nanoparticles was measured about 5.2 nm. The nanoparticles size was the same as the fresh catalyst. According to the result above, we proposed that the polysalen–Pd complex was corresponding to the Pd nanoparticles with the Pd5/2 binding energy 335.9 eV, and the released water-soluble Pd was corresponding to the Pd(OAc)2 attracted to the surface with the Pd5/2 binding energy 338.4 eV. The catalytic activity could be attributed to the Pd nanoparticles and the leached species in the solution. Meanwhile, we use ESI-MS to examine the liquid phase of the reaction of iodobenzene and phenylacetylene catalysed by polysalen–Pd (Fig. S1†). The Pd containing anionic species, [PdPhCOI]- and [PhCOPd(OAc)CCPh]-were detected in the solution. The Pd anionic species desorbed from the supports can explain the Pd loss of polysalen–Pd in a certain extent.
 | ||
| Fig. 7 TEM images of reaction mixture (a) and catalyst (b) by after 1 h reaction. Insert in (d) was histogram of Pd particles' size. | ||
Conclusions
In summary, a highly cross-linked polymer matrix polysalen based on acrylamide and N,N′-methylene-bisacrylamide (NNMBA) was successfully synthesized. It can be proved that the polysalen is an effective support for immobilizing palladium complex. The resulted polysalen–palladium catalyst exhibited high efficiency for the carbonylative Sonogashira coupling of aryl iodides with terminal alkynes in aqueous media without the addition of phosphine ligands, which is much higher than that of the homogeneous Pd–salen catalyst. The improvement in the activity might be attributed to the interactions between the polymeric supports with palladium that isolate the metal–salen complexes from each other. In addition, the catalyst offered practical advantages such as easy separation from the products and was reused without loss in the activity. The heterogeneous salen–Pd catalyzed carbonylation reaction provides a practical and environmentally friendly procedure for the synthesis of α,β-alkynyl ketone.Experimental
Materials and techniques
All chemicals were of reagent grade and used as purchased. All solvents were purified according to standard procedures before use. The products were purified by flash chromatography on silica gel and characterized by comparison of their spectra and physical data with authentic samples. The 1H and 13C NMR spectra were recorded on a Bruker Avance 400 MHz NMR spectrometer (400 and 100 MHz, respectively) in ppm with reference to tetramethylsilane (TMS) internal standard in CDCl3. GC-MS were measured on an Agilent 6890/5973 GC-MS. Elemental analyses were carried out on a Vario EL analyzer. Powder X-ray diffraction (XRD) measurements were performed using an X'Pert Pro multipurpose diffractometer (PANalytical, Inc.) with Ni-filtered Cu Kα radiation (0.15046 nm) at room temperature from 10.0° to 80.0° (wide angle). The ESI-MS was performed on Micro-QOF-II mass instrument. The X-ray photoelectron spectra (XPS) were obtained using a VG ESCALAB 210 spectrometer equipped with a Mg Kα X-ray radiation source (hν = 1253.6 eV) and charge referencing was measured against adventitious carbon (C 1s at 284.6 eV). High resolution transmission electron microscope (HRTEM) experiments were conducted in a JEM-2010 TEM with an accelerating voltage of 200 KV. The Fourier transform infrared spectra (FTIR) of the samples were recorded using an NEXUS 870 FTIR spectrometer by the KBr pellet method in the range of 400 to 4000 cm−1.Preparation of Pd–salen (salicylaldehydoethylenediamine)16b
To a stirred solution of 2-hydroxybenzaldehyde (1 mmol) in 5 mL of methanol, 1,2-ethylenediamine (0.5 mmol) was added dropwise and the reaction stirred for 20 min at room temperature. N,N′-Bis(salicylidene)-ethylenediamine was obtained by filtration. Dichloromethane (DCM) (5 mL) was added to dissolve the formed precipitate. To this solution Pd(OAc)2 (0.5 mmol) in DCM (5 mL) was added dropwise. The resulting reaction mixture was stirred for 18 to 20 h at room temperature. The product was filtered and washed with DCM and methanol to remove the unreacted ligand.Preparation of 5% NNMBA-crosslinked polyacrylamide (PAA)
N,N′-Methylene-bis-acrylamide (NNMBA) (1.14 g, 7.4 mmol) and acrylamide (10 g, 0.14 mol) were dissolved in methanol (250 mL). K2S2O8 (62 mg, 0.23 mmol) was added to the solution and stirred. The polymer began to precipitate within 30–40 min. The suspension was stirred at 70–75 °C for additional 5 h. The polymer formed was collected by filtration, washed several times with water and methanol and dried at 60 °C under reduced pressure over night.Preparation of crosslinked poly(acrylohydrazide) (PAH)
PAA (1 g) was added in small portions to an excess of well-stirred hydrazine hydrate (10 mL). The mixture was refluxed at 60–100 °C for 9 h. It was then poured slowly into a large volume of methanol (250 mL). The resin was filtered off and washed with NaCl solution until the filtrate was free from hydrazine. The gel was washed with distilled water and methanol and subsequently dried at 60 °C under reduced pressure over night.Preparation of crosslinked poly(acylohydrazide-imino-salicylaldehyde) (polysalen)
PAH (1 g, 2.6 mmol NH2) was added to salicyaldehyde (1.9 g, 15.6 mmol) in methanol (50 mL) stirred at 60 °C for 12 h. The resultant yellow product was filtered, washed with water, methanol and acetone and dried at 50 °C under reduced pressure.Preparation of polysalen–Pd complex
Crosslinked poly(acrylohydrazide-imino-salicylaldehyde) (1 g) was treated with a THF solution of Pd(OAc)2 (0.5 mmol, 112 mg). The mixture was heated to 50 °C for 5 h with a magnetic stirred and then allowed to proceed for additional 30 min at 100 °C. It should be noted that a decrease in the coloration of the solution, accompanied by a change in the colour of the beads from yellow to dark brown, indicated successful loading. In succession, the brown powder was obtained by filtration and extensive washed with water, ethanol and acetone. Respectively, then dried under vacuum at 50 °C for 24 h.General procedure for catalytic Sonogashira carbonylative coupling of iodobenzene and phenylaceylene in water
In a typical experiment, known quantities of iodobenzene (1.0 mmol), phenylacetylene (1.2 mmol), polysalen–Pd (30 mg, Pd 0.5 mol%), Et3N (0.4 mL, 2.4 mmol) and distilled water (5.0 mL) were charged into the reactor. The autoclave was closed, purged three times with CO, pressurized to 2.0 MPa with CO, and then stirred at 130 °C for 6 h. After cooling down to room temperature, the reaction mixture was analyzed by GC-MS and then worked up by removing water under vacuum and the residue was purified by chromatography on silica gel (eluting solvent hexane: ethyl acetate).General procedure for recycle reaction
After reaction, carbon monoxide was purged carefully and the catalyst was obtained by centrifugation separation. The catalyst could be directly reused for the next run after washed several times with ethanol and dried under vacuum, the base should be freshly added before the next carbonylation reaction.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21503243, 21403257) and the Independent Innovation Plan Foundations of Qingdao City of China (No. 16-5-1-36-jch).Notes and references
- R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783 CrossRef CAS PubMed.
- A. S. Karpow and T. J. J. Müller, Org. Lett., 2003, 5, 3451 CrossRef PubMed.
- T. R. Ward, B. J. Turunen, T. Haack, B. Neuenswander, W. Shadrick and G. I. Geory, Tetrahedron Lett., 2009, 50, 6494 CrossRef CAS PubMed.
- J. Kiji, T. Okano, H. Kimura and K. Saiki, J. Mol. Catal. A: Chem., 1998, 130, 95 CrossRef CAS.
- J. D. Kirkham, S. J. Edeson, S. Stokes and J. P. A. Harrity, Org. Lett., 2012, 14, 5354 CrossRef CAS PubMed.
- J. H. Shen, G. L. Cheng and X. L. Cui, Chem. Commun., 2013, 49, 10642 Search PubMed.
- S. Sakamoto, E. Honda, N. Ono and H. Uno, Tetrahedron Lett., 2000, 41, 1819 CrossRef.
- (a) J. F. Normant, Synthesis, 1972, 63 CrossRef CAS; (b) D. A. Alonso, C. Najera and M. C. Pacheco, J. Org. Chem., 2004, 69, 1619 CrossRef PubMed; (c) K. Y. Lee, M. J. Lee and J. N. Kim, Tetrahedron, 2005, 61, 8705 CrossRef CAS; (d) S. S. Palimkar, V. S. More and K. V. Srinivasan, Synth. Commun., 2008, 38, 1456 CrossRef CAS; (e) P. R. Likhar, M. S. Subhas, M. Roy, S. Roy and M. L. Kantam, Helv. Chim. Acta, 2008, 91, 259 CrossRef CAS; (f) I. R. Baxendale, S. C. Schou, J. Sedelmeier and S. V. Ley, Chem.–Eur. J., 2010, 16, 89 CrossRef CAS PubMed; (g) S. Atobe, H. Masuno, M. Sonoda, Y. Suzuki, H. Shinohara and S. Shibata, Tetrahedron Lett., 2012, 53, 1764 CrossRef CAS.
- J. W. Grate, T. Kobayashi and M. Tanaka, J. Chem. Soc., Chem. Commun., 1981, 333 Search PubMed.
- (a) D. V. Kadnikov and R. C. Larock, J. Organomet. Chem., 2003, 687, 425 CrossRef CAS; (b) E. Awuah and A. Capretta, Org. Lett., 2009, 11, 3210 CrossRef CAS PubMed; (c) X. Wu, J. Schranck, H. Neumann and M. Beller, Chem.–Eur. J., 2011, 17, 12246 CrossRef CAS PubMed; (d) X. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1 CrossRef CAS PubMed.
- (a) S. T. Gadge and B. M. Bhanage, RSC Adv., 2014, 4, 10367 RSC; (b) H. Zhao, M. Chen, J. Zhang and M. Cai, Green Chem., 2014, 16, 2515 RSC; (c) M. Genelot, V. Dufaud and L. Djakovitch, Adv. Synth. Catal., 2013, 355, 2604 CrossRef CAS.
- L. Xue, L. Shi, Y. Han, C. Xia, H. V. Huynh and F. Li, Dalton Trans., 2011, 40, 7632 RSC.
- (a) C. Zhang, J. Liu and C. Xia, Org. Biomol. Chem., 2014, 12, 9702 RSC; (b) S. P. Chavan, G. B. B. Varadwaj, K. Parida and B. Bhanage, Appl. Catal., A, 2015, 506, 237 CrossRef CAS.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed.
- P. Das and W. Linert, Coord. Chem. Rev., 2016, 311, 1 CrossRef CAS.
- (a) V. Ayala, A. Corma, M. Iglesias, J. A. Rincón and F. Sánchez, J. Catal., 2004, 224, 170 CrossRef CAS; (b) S. R. Borhade and S. B. Waghmode, Tetrahedron Lett., 2008, 49, 3423 CrossRef CAS.
- L. Ding, Z. Chu, L. Chen, X. Lü, B. Yan, J. Song, D. Fan and F. Bao, Inorg. Chem. Commun., 2011, 14, 573 CrossRef CAS.
- O. Bortolini and V. Conte, Mass Spectrom. Rev., 2006, 25, 724 CrossRef CAS PubMed.
- S. Syukri, W. Sun and F. E. Kühn, Tetrahedron Lett., 2007, 48, 1613 CrossRef CAS.
- (a) L. Lou, K. Yu, F. Ding, X. Peng, M. Dong, C. Zhang and S. Liu, J. Catal., 2007, 249, 102 CrossRef CAS; (b) G. T. Sfrazzetto, S. Millesi, A. Pappalardo, R. M. Toscano, F. P. Ballistreri, G. A. Tomaselli and A. Gulino, Catal. Sci. Technol., 2015, 5, 673 RSC; (c) S. M. Sadeghzadeh, RSC Adv., 2015, 5, 17319 RSC; (d) R. L. Oliveira, T. Nijholt, M. Shakeri, P. E. Jongh, R. J. M. K. Gebbink and K. P. Jong, Catal. Sci. Technol., 2016, 6, 5124 RSC; (e) X. Zheng, C. W. Jones and M. Weck, Chem.–Eur. J., 2006, 12, 576 CrossRef PubMed; (f) M. G. C. Kahn, J. H. Stenlid and M. Weck, Adv. Synth. Catal., 2012, 354, 3016 CrossRef CAS.
- B. Tamami and S. Ghasemi, Appl. Catal., A, 2011, 393, 242 CrossRef CAS.
- (a) Y. Wang, J. Liu and C. Xia, Tetrahedron Lett., 2011, 52, 1587 CrossRef CAS; (b) Y. Wang, J. Liu and C. Xia, Chin. J. Catal., 2011, 323, 1782 CrossRef.
- P. Subramanian, C. Clark, B. Wineher-Jensen, D. MacFarlane and L. Spiccia, Aust. J. Chem., 2009, 62, 133 CrossRef CAS.
- Y. Dong, X. Wu, X. Chen and Y. Wei, Carbohydr. Polym., 2017, 160, 106 CrossRef CAS PubMed.
- J. Lahaye, G. Nanse, A. Bagreev and V. Strelko, Carbon, 1999, 37, 585 CrossRef CAS.
- C. Ding, X. Qian and G. Yu, Cellulose, 2010, 17, 1067 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04910b |
| ‡ Y. Wang and X. Yang contributed equally to this work, both of them are the first authors. |
| This journal is © The Royal Society of Chemistry 2017 |