
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA DFT analysis on the radical scavenging activity of oxygenated terpenoids present in the extract of the buds of Cleistocalyx operculatus†
Thi Chinh
Ngo
a,
Duy Quang
Dao
 *a,
Minh Thong
Nguyen
b and
Pham Cam
Nam
c
*a,
Minh Thong
Nguyen
b and
Pham Cam
Nam
c
aInstitute of Research and Development, Duy Tan University, 03 Quang Trung, Danang, Vietnam. E-mail: daoduyquang@gmail.com
bThe University of Danang, Campus in Kon Tum, 704 Phan Dinh Phung, Kon Tum, Vietnam
cDepartment of Chemistry, The University of Da Nang – University of Science and Technology, 54 Nguyen Luong Bang, Lien Chieu, Danang, Vietnam
First published on 14th August 2017
Abstract
The antioxidant capacity of twenty-one oxygenated monoterpene and oxygenated desquiterpene compounds in the extract from Cleistocalyx operculatus has been computationally evaluated. Calculated by (RO)B3LYP/6-311++G(2df,2p)//B3LYP/6-311G(d,p) model chemistries, the thermochemical parameters, namely BDE, IE, PDE, PA and ETE in the gas phase, water and ethanol were determined. In addition, quantum descriptors, which allow the evaluation of the reactivity and stability of the resulting radicals like chemical potential (µ), hardness (η) and global electrophilicity (ω) were also computed. Potential energy surfaces of the reactions between CH3OO˙ and HO˙ radicals with falcarinol and α-vetivone, two typical and potential antioxidant molecules, were established to give more insight into the antioxidant mechanism. The obtained results underline falcarinol as the most effective antioxidant with the lowest BDE of 66.5 kcal mol−1 and PA of 341.3 kcal mol−1 in the gas phase. Among the reactions on falcarinol, the H-abstraction at C3–H position by both CH3OO˙ and HO˙ radicals are favorable with the energy barriers of −18.7 and −54.7 kcal mol−1, respectively. Moreover, NBO analysis helps to clarify the mechanism of antioxidant action which shows that the third lone pair of electrons on O1-atom of CH3OO˙ radical is donated to an unoccupied σ*-antibonding orbital on C3–H, and on the C4![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C5, C6C7 triple bonds. Similarly, attack of CH3OO˙ and HO˙ radicals on α-vetivone demonstrates that H-abstraction reactions are also more feasible than the addition ones with ΔH values of −7.3 and −41.9 kcal mol−1, respectively. For all considered reactions, the antioxidant molecules preferentially interact with HO˙ radical.
C5, C6C7 triple bonds. Similarly, attack of CH3OO˙ and HO˙ radicals on α-vetivone demonstrates that H-abstraction reactions are also more feasible than the addition ones with ΔH values of −7.3 and −41.9 kcal mol−1, respectively. For all considered reactions, the antioxidant molecules preferentially interact with HO˙ radical.
1. Introduction
Essential oils, aromatic and volatile liquids, are principally extracted from various parts (flowers, seed, leaves, fruits…) of numerous natural plants (tea, pepper, ginger, thyme…).1–5 They have been attractive subjects for applications in industry (for flavoring food, perfumes, cosmetics, etc.) as well as in pharmaceutical and medicinal domains.6–9 The essential oils are generally rich in terpenoid compounds, mainly mono-, sesqui- and desquiterpenes which are hydrocarbon or oxygenated compounds.7,10 These interesting oils have already been well-known in a number of applications such as antimicrobial, anticancer, antifungal, antiviral and especially antioxidant.4,11–17 For example, the results obtained by 2,2-diphenyl-1-picrylhydrazyl (DPPH) and nitric oxide scavenging assays clearly indicate that the essential oils isolated from fresh rhizomes of garlic (Allium Sativum) of the family Alliaceae are effective in scavenging free radicals, so they potentially have a powerful antioxidant role.16 In the work of Bozin et al.17 the antioxidant activities of the essential oils of Lamiaceae species including Ocimum basilicum L., Origanum vulgare L., and Thymus vulgaris L. were evaluated to play a role in a free radical scavenging capacity (RSC), together with effects on lipid peroxidation (LP). The investigated essential oils exhibit high RSC against DPPH radical and inhibit a very strong lipid peroxidation induced by both Fe2+/ascorbate and Fe2+/H2O2. Otherwise, the antioxidant properties of the essential oils extracted from Cleistocalyx operculatus are also well-known.18 A variety of organic compounds including monoterpenes, oxygenated monoterpenes, sesquiterpenes, oxygenated desquiterpenes and diterpenes were experimentally identified and measured antioxidant properties on the basis of the scavenging activities of the stable 2,2-diphenyl-1-picrylhydrazyl (DPPH) free radical.18 Additionally, the antioxidant mechanism of these three hydrocarbon terpenoid classes including monoterpenes, sesquiterpenes and diterpenes has theoretically been clarified in our previous study.19 Some of them (α-terpinene, γ-terpinene, cembrene and abieta-7,13-diene) have promising antioxidant properties. The ability to scavenge reactive oxygen species (ROS), one of essential requirements for antioxidant role, of selected α-terpinene was consequently ascertained via its interaction with hydroperoxyl radicals (HOO˙).19Moreover, several experimental works in literature20–23 have been discussed that oxygenated compounds available in Cleistocalyx operculatus like falcarinol, α-vetivone and so on, represent also as potential health promoting compounds. In fact, falcarinol, one of aliphatic C17-polyacetylenes which can be found in common food plants such as carrot, celeriac, parsnip and parsley, shows a number of interesting bioactivities such as antibacterial, antimycobacterial, and antifungal activity, anti-inflammatory, anti-platelet-aggregatory, as well as neuritogenic and serotonergic effects.20,21,23 α-vetivone, one of the most important constituents isolated and identified from crude vetiver (Vetiveria zizanioides L.) oil, has strong antioxidant activities via DPPH free radical scavenging assay and the Fe2+-metal chelating assay.24
Hence, in this paper, our interest is set on two classes namely oxygenated monoterpenes and oxygenated desquiterpenes, whose structures contain oxygen atom. For more detail, the insight into the theoretical mechanism of antioxidant activity of these compounds will be clarified via three mechanisms of antioxidant action including hydrogen atom transfer (HAT), single electron transfer followed by-proton transfer (SET-PT) and sequential proton loss electron transfer (SPLET). First and foremost, the calculations of various thermochemical parameters including bond dissociation enthalpy (BDE), proton affinity (PA), electron transfer enthalpy (ETE) and ionization energy (IE) are performed using the density functional theory (DFT) method at the model chemistry of (RO)B3LYP/6-311++G(2df,2p)//B3LYP/6-311G(d,p) in the gas phase, ethanol and water which is a typical polar solvent (ε = 78.36).
In the next step, the global descriptive parameters including chemical potential (µ), chemical hardness (η) and global electrophilicity (ω) of neutral compounds will be calculated to elucidate their reactivity and stability. Finally, potential energy surface (PES) of all possible addition reactions as well as H-abstraction ones between the most potential antioxidant compounds with CH3OO˙ and HO˙ radical will be established to give more insights into free radical scavenging mechanism. In parallel, singly-occupied molecular orbital (SOMO), atomic spin density (ASP) and natural bond orbital (NBO) analysis of the optimized transition states will also be taken into account in order to explain clearly the mechanism of reactions.
2. Computational method
All calculations were performed using the Gaussian 09 software.25 The geometry optimization and the vibrational frequency calculation of each compound and the corresponding radical, cationic radical and anion were primarily performed at the B3LYP/6-311G(d,p) level of theory. Their single point electronic energies were then calculated at the restricted-open shell (RO)B3LYP/6-311++G(2df,2p) level of theory. Generally, three common mechanisms of antioxidant action leading the same final products have been proposed and widely accepted as follows:19,26• Hydrogen atom transfer (HAT):
| R–H → R˙ + H˙ (BDE) |
• Single electron transfer followed by proton transfer (SET-PT)
| R–H → RH+˙ + e− (IE) |
| RH+˙ → R˙ + H+ (PDE) |
• Sequential proton loss electron transfer (SPLET)
| R–H → R− + H+ (PA) |
| R− → R˙ + e− (ETE) |
The reaction enthalpies of an antioxidant in gas phase at 298.15 K and 1 atm are calculated as follows:
| BDE = H(R˙) + H(H˙) − H(R–H) | (1) |
| IE = H(RH˙+) + H(e−) − H(R–H) | (2) |
| PDE = H(R˙) + H(H+) − H(RH ˙+) | (3) |
| PA = H(R−) + H(H+) − H(R–H) | (4) |
| ETE = H(R˙) + H(e−) − H(R−) | (5) |
The Htrans, Hrot, and Hvib are the translational, rotational and vibrational contributions to the enthalpy, respectively. E0 is the total energy at 0 K and ZPE is the zero-point vibrational energy. The enthalpies of H-atom (H˙), proton (H+), and electron (e−) are taken from literature.27 Vibrational frequencies obtained at the B3LYP/6-311G(d,p) level of theory were scaled by a factor of 0.9669.28 The calculation in solvents was based on integral equation formalism of polarizable continuum model (IEF-PCM) at the same level of theory as in the gas phase.29,30 The influence of water and ethanol on the thermodynamic properties was discussed.
To evaluate reactivity and stability of the studied compounds we used global theoretical descriptors that were calculated at B3LYP/6-311++G(2df,2p)//B3LYP/6-311G(d,p). Finite differences method proposed by Pearson and Parr is used to approximately determine chemical potential (µ),31,32 chemical hardness (η)33 and global electrophilicity (ω)34 on the basis of vertical electron affinity (EA) and ionization energy (IE) of chemical species as follows:
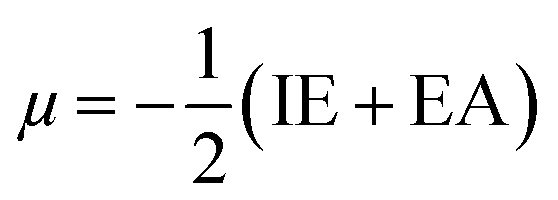
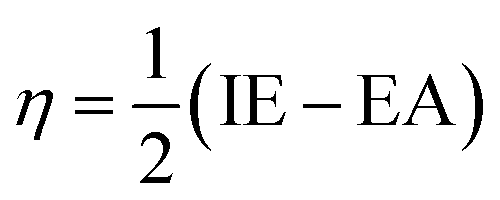
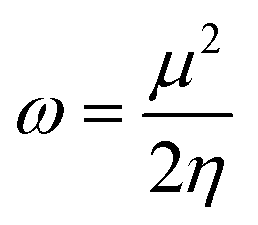
Potential energy surface (PES) of the most potential antioxidant compound was also established. The calculation of geometric optimization and vibrational frequencies respectively of transition states, of reactant complexes and of product complexes were investigated at B3LYP/6-311G(d,p) level of theory for all possible adduction reactions as well as H-abstraction ones.
3. Results and discussion
3.1. Thermochemical parameters characterizing antioxidant capacity
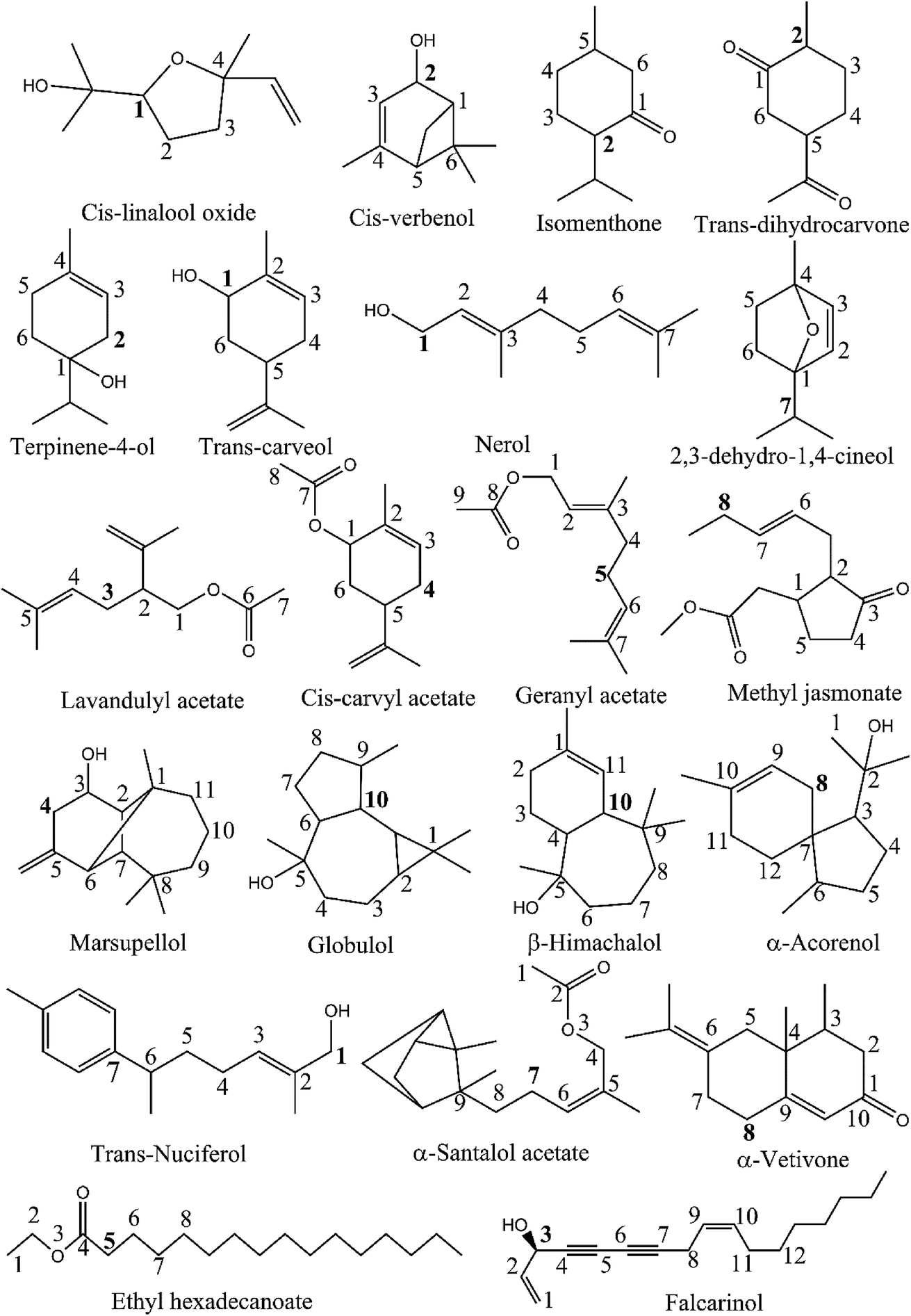 | ||
| Fig. 1 Chemical structure of 21 studied compounds. | ||
As observed in the previous study,19 the C–H bonds neighboring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds are detected to be the easiest breaking bonds in comparing to other ones due to the reason that the electron-withdrawing inductive effect (−I) of the π-bond induces an electron-releasing phenomenon from the carbon atoms, and consequently increases the polarization of the C–H bonds. Therefore, these bonds were reasonably selected to compute their strength at the higher level of theory of (RO)B3LYP/6-311++G(2df,2p)//B3LYP/6-311G(d,p). The BDEs of weakest bonds are present in Table 1. Cartesian coordinates and enthalpies of all parent molecules and resulted radicals, anions optimized at B3LYP/6-311G(d,p) level of theory in the gas phase, water and ethanol are presented in Table S1 ESI.†
C double bonds are detected to be the easiest breaking bonds in comparing to other ones due to the reason that the electron-withdrawing inductive effect (−I) of the π-bond induces an electron-releasing phenomenon from the carbon atoms, and consequently increases the polarization of the C–H bonds. Therefore, these bonds were reasonably selected to compute their strength at the higher level of theory of (RO)B3LYP/6-311++G(2df,2p)//B3LYP/6-311G(d,p). The BDEs of weakest bonds are present in Table 1. Cartesian coordinates and enthalpies of all parent molecules and resulted radicals, anions optimized at B3LYP/6-311G(d,p) level of theory in the gas phase, water and ethanol are presented in Table S1 ESI.†
| Compounds | Bonds | BDEs (kcal mol−1) | PDEs (kcal mol−1) | |||
|---|---|---|---|---|---|---|
| Gas phase | Water | Ethanol | Gas phase | |||
| Oxygenated monoterpenes | cis-Linalool oxide | C1–H | 91.9 | 93.2 | 92.5 | 217.9 |
| cis-Verbenol | C2–H | 78.5 | 80.2 | 79.7 | 205.6 | |
| Isomenthone | C2–H | 81.0 | 81.9 | 81.3 | 199.1 | |
| trans-Dihydrocarvone | C2–H | 84.8 | 85.7 | 85.1 | 201.6 | |
| Terpinen-4-ol | C2–H | 83.5 | 84.9 | 84.2 | 206.0 | |
| trans-Carveol | C1–H | 76.9 | 78.9 | 78.2 | 202.7 | |
| Nerol | C1–H | 79.2 | 81.0 | 80.4 | 213.8 | |
| 2,3-Dehydro-1,4-cineol | C7–H | 95.2 | 96.8 | 96.1 | 212.1 | |
| Lavandulyl acetate | C3–H | 82.7 | 83.7 | 83.0 | 211.2 | |
| cis-Carvyl acetate | C4–H | 83.6 | 85.1 | 84.5 | 209.8 | |
| Geranyl acetate | C5–H | 83.1 | 84.3 | 83.7 | 217.5 | |
| Methyl jasmonate | C8–H | 83.3 | 84.8 | 84.1 | 206.4 | |
| Oxygenated desquiterpene | Marsupellol | C4–H | 82.6 | 84.4 | 83.7 | 213.3 |
| Globulol | C10–H | 92.8 | 94.2 | 93.6 | 234.4 | |
| β-Himachalol | C10–H | 82.3 | 84.0 | 83.3 | 217.0 | |
| α-Acorenol | C8–H | 81.2 | 82.8 | 82.1 | 216.6 | |
| trans-Nuciferol | C1–H | 82.4 | 84.1 | 83.5 | 215.5 | |
| α-Santalol acetate | C7–H | 82.2 | 83.6 | 83.0 | 218.7 | |
| α-Vetivone | C8–H | 78.1 | 79.2 | 78.6 | 210.0 | |
| Ethyl hexadecanoate | C5–H | 92.1 | 93.6 | 92.9 | 206.8 | |
| Falcarinol | C3–H | 66.5 | 69.3 | 67.9 | 194.4 | |
Table 1 demonstrates that the BDEs (C–H) of studied compounds vary from 66.5 to 95.2 kcal mol−1. The lowest BDE (C–H) of 66.5 kcal mol−1 is indicated for falcarinol which contains two conjugated triple bonds and two double bonds in its structure. It means that the number of CC double and CC triple bonds present in molecular system has a significant effect on the BDEs. In fact, the lower BDE (C–H) (i.e. 78.1 kcal mol−1) of α-vetivone compared to the others is also related to the presence of two conjugated π-bonds nearby that C position (as can be seen in Fig. 1). As the number of conjugated double/triple bonds increases, the electrons associated with conjugated systems have more space to delocalize and require less energy to change states. This results are coherent with the previous study.19
In addition, the presence of OH group also contributes to the changing of the BDE values. In fact, the C–H bonds located nearby both OH group and unsaturated bonds possess lower BDE values compared to other ones. For example, the BDEs of cis-verbenol, trans-carveol and nerol are, in principle, lower than 80 kcal mol−1, being 79.2, 76.9 and 78.6 kcal mol−1, respectively.
Regarding the effect of solvents on BDEs, there is occurred no remarkable difference between the results computed in the gas phase and two solvents (water and ethanol). For example, the BDEs calculated in the gas phase, water and ethanol of falcarinol are 66.5, 69.3 and 67.9 kcal mol−1, respectively. Similarly, the calculated values in these three phases for trans-carveol are 76.9, 78.9 and 78.2 kcal mol−1, respectively. This leads to conclude that hydrogen atom transfer is favored in nonpolar environments.
| Compounds | IE | EA | µ | η | ω |
|---|---|---|---|---|---|
| cis-Linalool oxide | 8.76 | −0.42 | −4.17 | 4.59 | 1.89 |
| cis-Verbenol | 8.40 | −0.44 | −3.98 | 4.42 | 1.79 |
| Isomenthone | 8.69 | −0.38 | −4.15 | 4.53 | 1.91 |
| trans-Dihydrocarvone | 8.69 | −0.25 | −4.22 | 4.47 | 1.99 |
| Terpinen-4-ol | 8.65 | −0.42 | −4.12 | 4.54 | 1.87 |
| trans-Carveol | 8.52 | −0.39 | −4.07 | 4.45 | 1.86 |
| Nerol | 8.13 | −0.51 | −3.81 | 4.32 | 1.68 |
| 2,3-Dehydro-1,4-cineol | 8.95 | −0.50 | −4.23 | 4.72 | 1.89 |
| Lavandulyl acetate | 8.27 | −0.49 | −3.89 | 4.38 | 1.73 |
| cis-Carvyl acetate | 8.47 | −0.39 | −4.04 | 4.43 | 1.84 |
| Geranyl acetate | 8.15 | −0.45 | −3.85 | 4.30 | 1.72 |
| Methyl jasmonate | 8.42 | −0.29 | −4.06 | 4.36 | 1.90 |
| Marsupellol | 8.49 | −0.55 | −3.97 | 4.52 | 1.75 |
| Globulol | 8.14 | −0.39 | −3.88 | 4.27 | 1.76 |
| β-Himachalol | 8.21 | −0.38 | −3.92 | 4.29 | 1.79 |
| α-Acorenol | 8.09 | −0.67 | −3.71 | 4.38 | 1.57 |
| trans-Nuciferol | 8.02 | −0.35 | −3.83 | 4.19 | 1.76 |
| α-Santalol acetate | 8.27 | −0.34 | −3.97 | 4.31 | 1.83 |
| α-Vetivone | 8.06 | 0.02 | −4.04 | 4.02 | 2.03 |
| Ethyl hexadecanoate | 9.09 | −0.32 | −4.38 | 4.71 | 2.04 |
| Falcarinol | 8.35 | −0.27 | −4.04 | 4.31 | 1.89 |
In parallel with IE values, the global reactivity descriptors including chemical potential (µ), chemical hardness (η) and global electrophilicity (ω) calculated based on IE and EA values give more information about the tendency of selectivity and chemical reactivity of parent compounds and their radical formed via easiest C–H breaking bonds (data present in Table 2). For example, chemical hardness (η), which is defined as the resistance of cloud polarization or deformation of chemical species, is used to predict the reactivity of molecules. The lowest value of hardness means the highest reactivity. Regarding the hardness values present in Table 2, α-vetivone is seen to be the most reactive in comparison with the hardness of the other compounds noted 4.02 eV. In terms of global electrophilicity (ω) representing the capacity of a system to acquire an electron, a higher ω value demonstrates the more reactive molecule. Among these studied compounds, α-vetivone and ethyl hexadecanoate show as the most reactive ones with the ω value of 2.03 and 2.04 eV, respectively.
The following step of electron transfer via SET-PT mechanism is proton transfer process from C–H group of the formed cationic radical that is characterized by proton dissociation enthalpy (PDE). The PDE values for deprotonation of all cationic radical species are displayed in Table 1 and indicates that the easiest deprotonation generally found at the C–H position with the lowest BDE. As a result, the lowest PDEs are 194.4 and 199.1 kcal mol−1 corresponding to the cationic radical formed from falcarinol and isomenthone, respectively. It is noted that falcarinol demonstrates both the easiest homolytic and heterolytic H-dissociation.
| Compounds | Bonds | PA (kcal mol−1) | ETE (kcal mol−1) | ||||
|---|---|---|---|---|---|---|---|
| Gas phase | Water | Ethanol | Gas phase | Water | Ethanol | ||
| cis-Linalool oxide | O–H | 369.1 | 57.2 | 74.9 | 51.0 | 70.7 | 89.2 |
| cis-Verbenol | O–H | 369.9 | 54.1 | 71.9 | 46.3 | 69.7 | 88.2 |
| Isomenthone | C2–H | 363.2 | 50.5 | 67.9 | 32.3 | 52.2 | 71.1 |
| trans-Dihydrocarvone | C2–H | 356.5 | 50.7 | 68.1 | 43.9 | 57.9 | 76.4 |
| Terpinen-4-ol | O–H | 369.6 | 56.2 | 74.0 | 47.5 | 68.4 | 86.9 |
| trans-Carveol | O–H | 370.3 | 53.6 | 71.4 | 45.9 | 70.8 | 89.2 |
| Nerol | C4–H | 377.3 | 70.0 | 87.1 | 23.7 | 38.2 | 57.4 |
| 2,3-Dehydro-1,4-cineol | C6–H | 400.7 | 90.1 | 107.7 | 13.6 | 31.9 | 50.6 |
| Lavandulyl acetate | C7–H | 369.2 | 56.4 | 74.0 | 43.6 | 64.2 | 82.8 |
| cis-Carvyl acetate | C8–H | 367.6 | 56.1 | 73.6 | 45.1 | 64.5 | 83.2 |
| Geranyl acetate | C9–H | 370.0 | 56.7 | 74.3 | 42.7 | 63.9 | 82.5 |
| Methyl jasmonate | C2–H | 356.2 | 49.7 | 67.1 | 44.5 | 58.1 | 77.0 |
| Marsupellol | O–H | 366.3 | 53.7 | 71.4 | 48.6 | 69.2 | 87.7 |
| Globulol | O–H | 365.2 | 55.9 | 73.5 | 49.2 | 66.2 | 84.8 |
| β-Himachalol | O–H | 367.2 | 56.8 | 74.4 | 48.3 | 66.5 | 85.1 |
| α-Acorenol | O–H | 366.2 | 55.9 | 73.5 | 49.1 | 67.1 | 85.7 |
| trans-Nuciferol | O–H | 372.7 | 53.1 | 70.9 | 44.4 | 72.1 | 90.5 |
| α-Santalol acetate | C1–H | 369.0 | 56.9 | 74.5 | 43.8 | 63.7 | 82.3 |
| α-Vetivone | C8–H | 351.0 | 44.1 | 61.4 | 41.7 | 55.9 | 74.8 |
| Ethyl hexadecanoate | C5–H | 369.1 | 58.7 | 76.2 | 37.6 | 55.8 | 74.4 |
| Falcarinol | C8–H | 341.3 | 41.6 | 58.7 | 46.3 | 51.8 | 70.9 |
It is generally observed that heterolytic cleavage is favored at O–H and C–H position located nearby CC double bonds. The best antioxidant based on SPLET mechanism is falcarinol with the PAs computed in the gas phase of 341.3 kcal mol−1. In the meantime, when calculating in polar solvents, one observed a dramatic decrease in PAs by comparison with the values measured in the gas phase. In fact, the PA of cis-linalool oxide in the gas phase is 369.1 kcal mol−1 while its values in water and ethanol are 57.2 and 74.9 kcal mol−1, respectively. Similarly, the PAs of falcarinol are 341.3, 41.6 and 58.7 kcal mol−1 assigned to the calculation in the gas phase, water and ethanol, respectively. This observation can be explained by the higher solvation enthalpy of proton in water and ethanol compared to that in the gas phase, and also it is in a good agreement with the results obtained in previous studies.19,26,35 Indeed, Thong et al. showed the PAs of α-mangostin are 326.0 and 23.6 kcal mol−1 at B3LYP/6-31+G(d,p) level of theory in the gas phase and water, respectively.26 The PAs obtained for a hydroxylchalcone by Wang and coworkers are 354.6 kcal mol−1 in the gas phase and 58.5 kcal mol−1 in water at B3LYP/6-311++G(2d,2p) level of theory.35 In conclusion, the deprotonation process of an antioxidant is favored in polar environments.
Regarding the electron transfer enthalpy (ETE) which represents the electron donating ability of anion formed in the first step of the SPLET mechanism, one observed that the ETEs in the gas phase (as seen in Table 3) are relatively lower than IEs (data in Table 2). For example, ETE of cis-linalool oxide in the gas phase is 51.0 kcal mol−1 while IE is 206.1 kcal mol−1 (or 8.94 eV). It means that the electron transfer from anionic form is more favorable than that from neutral one. This is coherent with the results reported in previous studies.19,26,36,37
Among studied compounds, falcarinol seems to be the most efficient antioxidant molecule via both HAT and SPLET mechanisms. And α-vetivone is also a promising antioxidant compound. Therefore, in the following section, falcarinol and α-vetivone are considered as a model molecule to investigate their interaction with CH3OO˙ and HO˙ radicals which are chosen as representative free radical model for ROO˙ and RO˙ one.
3.2. Interaction of typical free radicals with potential antioxidant molecules
Falcarinol which contains two conjugated triple bonds and two double bonds shows as the easiest H-donating compound with the BDE (C3–H) in the gas phase of 66.5 kcal mol−1. It can play a role in scavenging free radical via either H-abstraction at C3–H or addition at the unsaturated bonds. Reactions between falcarinol and CH3OO˙ radical will be discussed in turn the H-abstraction channel at C3–H and the addition channel at C1C2, C9C10 double and C4C5, C6C7 triple bonds (see Fig. 1). The potential energy surfaces (PES) of these reactions are established and displayed in Fig. 2. All optimized structures and the corresponding energies of the species related to the mentioned reaction were computed at B3LYP/6-311G(d,p) level. IRC plots for all transition states related to reactions of CH3OO˙ and HO˙ radicals with falcarinol are presented in Fig. S2 of ESI.†
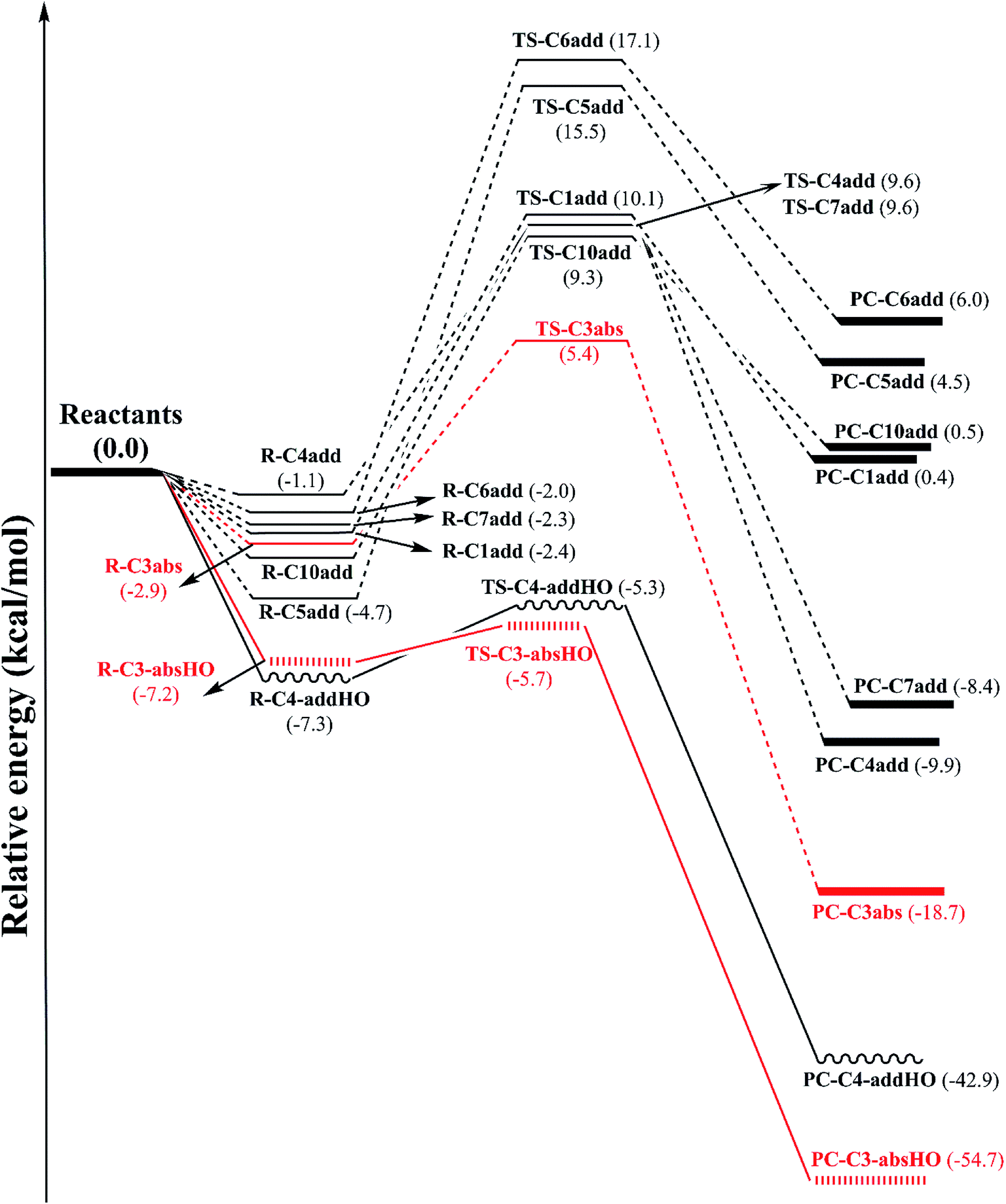 | ||
| Fig. 2 Potential energy surface (PES) at B3LYP/6-311G (d,p) level of theory of H-abstraction reaction at easiest breaking bond and addition reaction at unsaturated bond positions between CH3OO˙ and HO˙ radicals and falcarinol. (The abbreviation containing HO indicates for the reactions with HO˙, the rest is of the ones with CH3OO˙). | ||
| CH3OO˙/HO˙ + FALC–H → FALC˙ + CH3OOH/H2O |
PES of H-abstraction and radical addition to unsaturated bonds reactions of falcarinol with CH3OO˙ and HO˙ radical is displayed in Fig. 2. All the optimized geometries of transition state (TS) and product complex (PC) are shown in Fig. 3 and 4. TS structure of H-abstraction reaction between falcarinol with DPPH radical is also presented in Fig. S4 of ESI.†
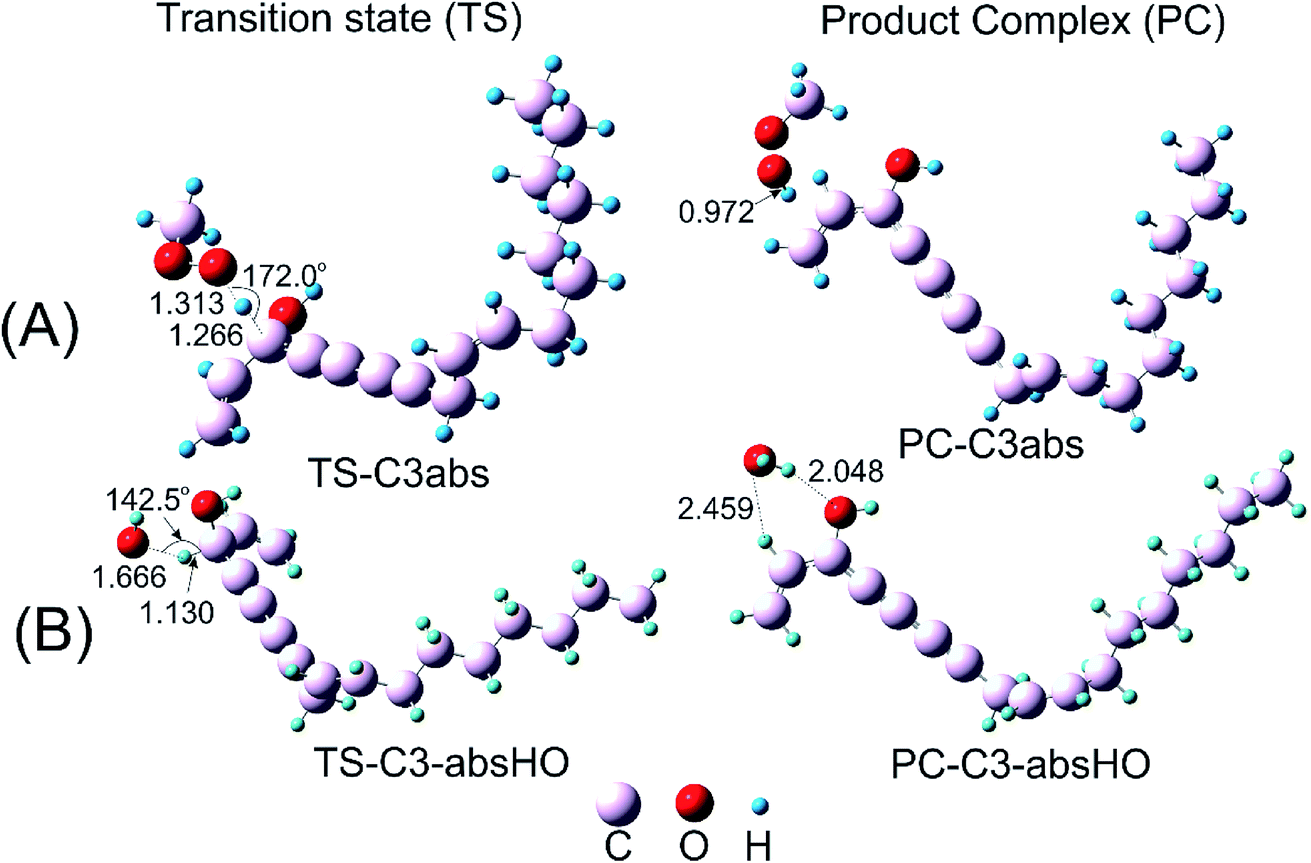 | ||
| Fig. 3 Optimized geometries of transition state (TS) and product complex (PC) species for the H-atom abstraction between falcarinol and (A) CH3OO˙ and (B) HO˙ radical at the B3LYP/6-311G(d,p) level of theory, (distances are given in angstroms). | ||
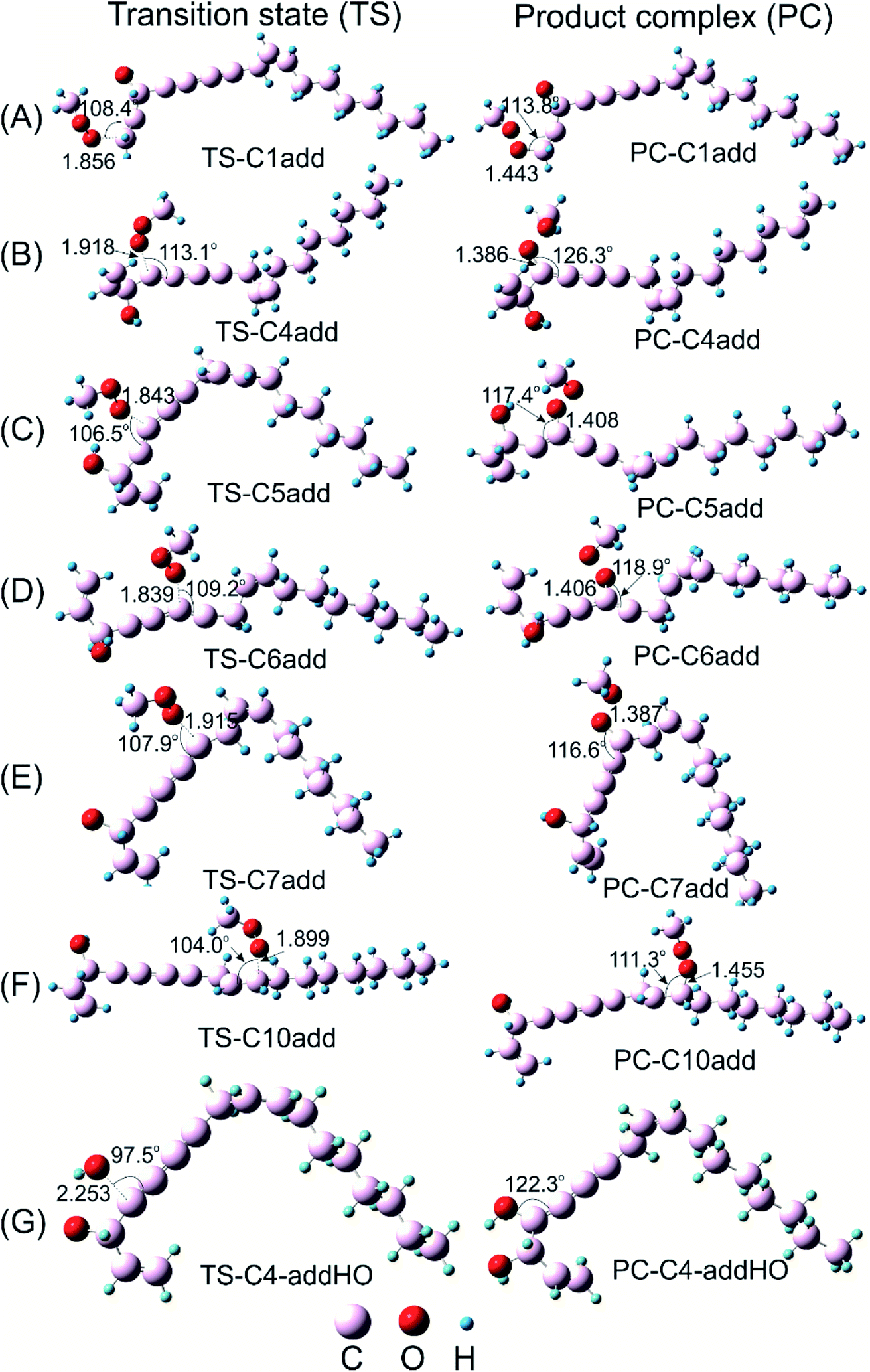 | ||
| Fig. 4 Optimized geometries of transition state (TS) and product complex (PC) of addition reaction at unsaturated bond positions between falcarinol and CH3OO˙ radical (A–F) and HO˙ radical (G). All calculations were performed at B3LYP/6-311G(d,p) level of theory. | ||
The H-atom transfer from C3–H bond of falcarinol to the CH3OO˙ radical is occurred in initiation step and demonstrated as an important one in interrupting the chain reactions.38 In the H-abstraction reaction, the CH3OO˙ radical and falcarinol reactants can form a hydrogen-bonded reactant complex (R–C3) at energy lying below the separated reactants by −2.3 kcal mol−1. In this state, a hydrogen bond is generated between O-atom of CH3OO˙ and H-atom at C3–H of falcarinol, and CH3OO⋯H–C3 distance is 2.447 Å (as seen in Fig. 3). Then, the H-atom of falcarinol tends to form a chemical bond with the O-atom of the radical via a transition state (TS-C3abs) lying at 5.4 kcal mol−1 above the reactants where the CH3OO⋯H–C3 distance is shorter. The H-atom is situated in the middle of O and C3 atoms and the CH3OO⋯H and H–C3 distance are 1.313 and 1.266 Å, respectively. The C3–H–O angle is relatively bent, being 172° (Fig. 3). After passing these states, the reaction pathway reaches the product complex (PC-C3abs) containing FALC˙ radical and CH3OOH at −18.7 kcal mol−1 below the reactants, in which a covalent chemical bond of 0.972 Å is formed between O and H-atom. It is worth noting that the H-abstraction reaction at the easiest breaking bond requires lower reaction barriers compared to the addition ones (see Fig. 2). In addition, the Gibbs energy (ΔG) presented in Table 4 shows that the H-abstraction reaction by CH3OO˙ radical is exergonic with a negative ΔG value of −10.0 kcal mol−1.
| Reactions | ΔH, kcal mol−1 | ΔG, kcal mol−1 |
|---|---|---|
| FALC–C3H + CH3OO˙ | −18.7 | −10.0 |
| FALC–C10 + CH3OO˙ | 0.5 | 12.6 |
| FALC–C7 + CH3OO˙ | −8.4 | 3.1 |
| FALC–C6 + CH3OO˙ | 6.0 | 18.0 |
| FALC–C5 + CH3OO˙ | 4.5 | 16.6 |
| FALC–C4 + CH3OO˙ | −9.9 | 1.7 |
| FALC–C1 + CH3OO˙ | 0.4 | 12.4 |
| FALC–C3H + HO˙ | −54.7 | −47.1 |
| FALC–C4 + HO˙ | −42.9 | −32.2 |
| VET–C8H + CH3OO˙ | −7.3 | 1.5 |
| VET–C8H + HO˙ | −41.9 | −36.2 |
| VET–C9 + CH3OO˙ | 4.8 | 18.0 |
| VET–C9 + HO˙ | −25.3 | −14.3 |
Additionally, the radical scavenging ability of falcarinol has been studied via another reactive radical such as HO˙. H-atom abstraction at C3–H position and addition reactions at C4C5 triple bond that showed as the most feasible ones in the reaction with CH3OO˙ were taken into account. These reactions are chosen because they are shown as the most feasible ones in terms of energy. These reactions pass transition states, called TS-C3-absHO and TS-C4-addHO, at −5.7 and −5.3 kcal mol−1 lying under reactants that are followed by product complexes, namely PC-C3-absHO and PC-C4-addHO, at considerably negative energy of −54.7 and −42.9 kcal mol−1, respectively. It is noted that these reactions are exergonic, showing negative ΔG values of −47.1 and −32.2 kcal mol−1, in sequence (Table 4). In comparison with the reactions with CH3OO˙ radical, it is observed that the ones with HO˙ are more feasible and higher exergonic.
Since α-vetivone appears as a promising antioxidant via HAT and SET-PT mechanism, attack of CH3OO˙ and HO˙ radicals on this molecule was similarly investigated. The reactions were performed at easiest C8–H breaking bond and C9C10 double bonds (as shown in Fig. 5). IRC plots for all transition states related to reaction of CH3OO˙ and HO˙ radicals with α-vetivone are presented in Fig. S3 of ESI.†
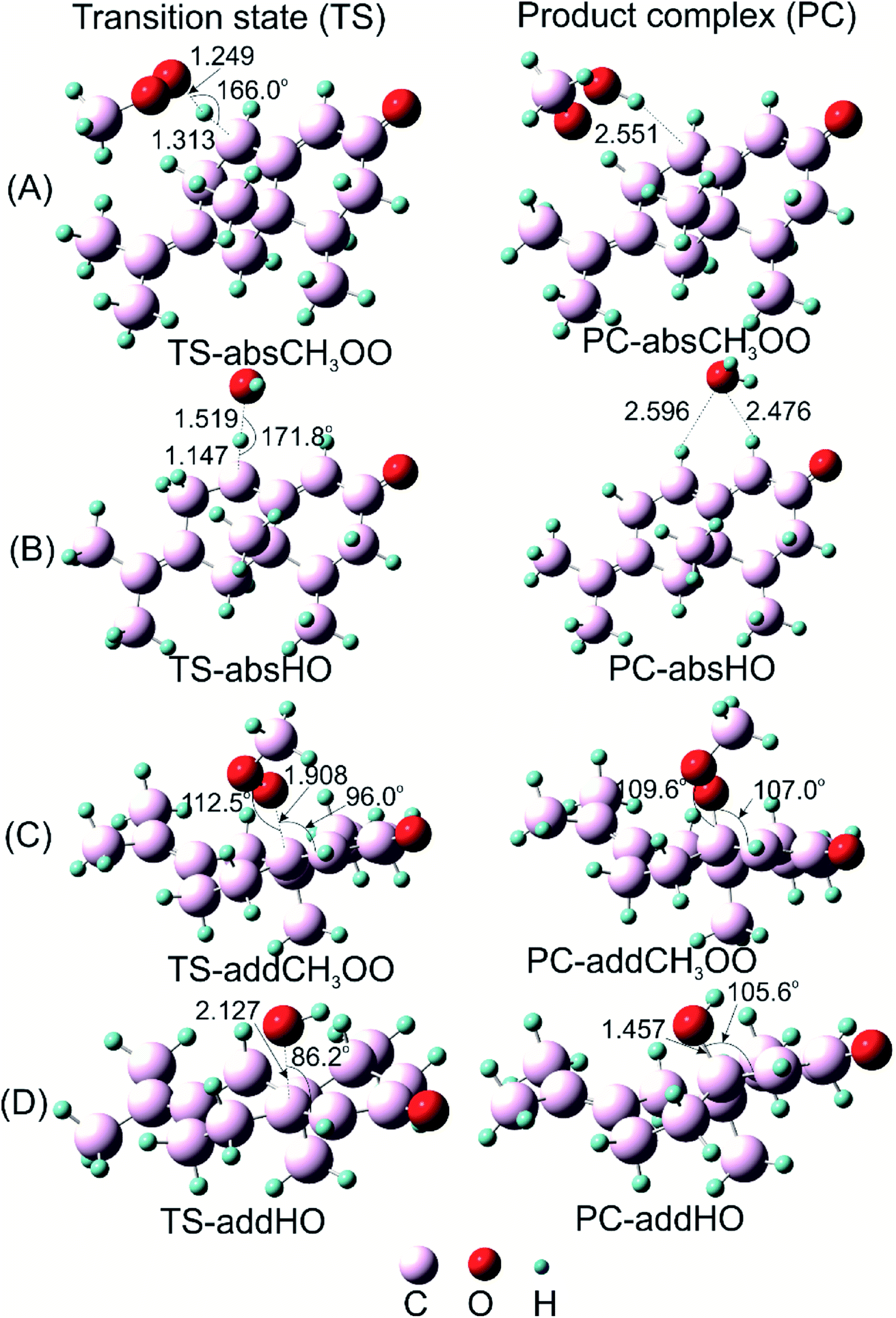 | ||
| Fig. 5 Optimized geometries of transition states (TS) and product complexes (PC) of H-atom abstraction reactions of α-vetivone with (A) CH3OO˙ radical, (B) HO˙ radical at C8–H position; and of addition reactions to double bonds for (C) CH3OO˙ radical and (D) HO˙ radical at C9 position. | ||
Before attaining product complex (PC), all reactions firstly pass transition state (TS) with energy of 12.3, 9.3, −3.6 and −4.7 kcal mol−1 corresponding to TS-addCH3OO, TS-absCH3OO, TS-absHO and TS-addHO, in turn (as seen in Fig. 6). The results underline that the H-atom abstractions by both radicals are more prominent than the addition ones. Indeed, PC-absCH3OO and PC-absHO are found at −7.8 and −41.9 kcal mol−1 while PC-addCH3OO and PC-addHO are determined at 0.8 and −25.3 kcal mol−1, respectively (Fig. 6). As energy of product complex of the reaction with HO˙ are much lower than the one with CH3OO˙ radical, the scavenging of HO˙ is indicated to be more favorable. Moreover, the results show that the H-abstraction reaction with CH3OO˙ is slightly endergonic with low positive ΔG value of 1.5 kcal mol−1 (as shown in Table 4), while the addition one is more strongly endergonic with notably positive ΔG value of 18.0 kcal mol−1. Oppositely, the reactions with HO˙ are more exergonic with meaningfully negative ΔG values of −36.2 and −14.3 kcal mol−1 corresponding to H-abstraction and addition reactions, respectively (Table 4). This observation follows the same tendency as in the case of falcarinol. Note that the antioxidant molecules preferentially react with reactive HO˙ radical.
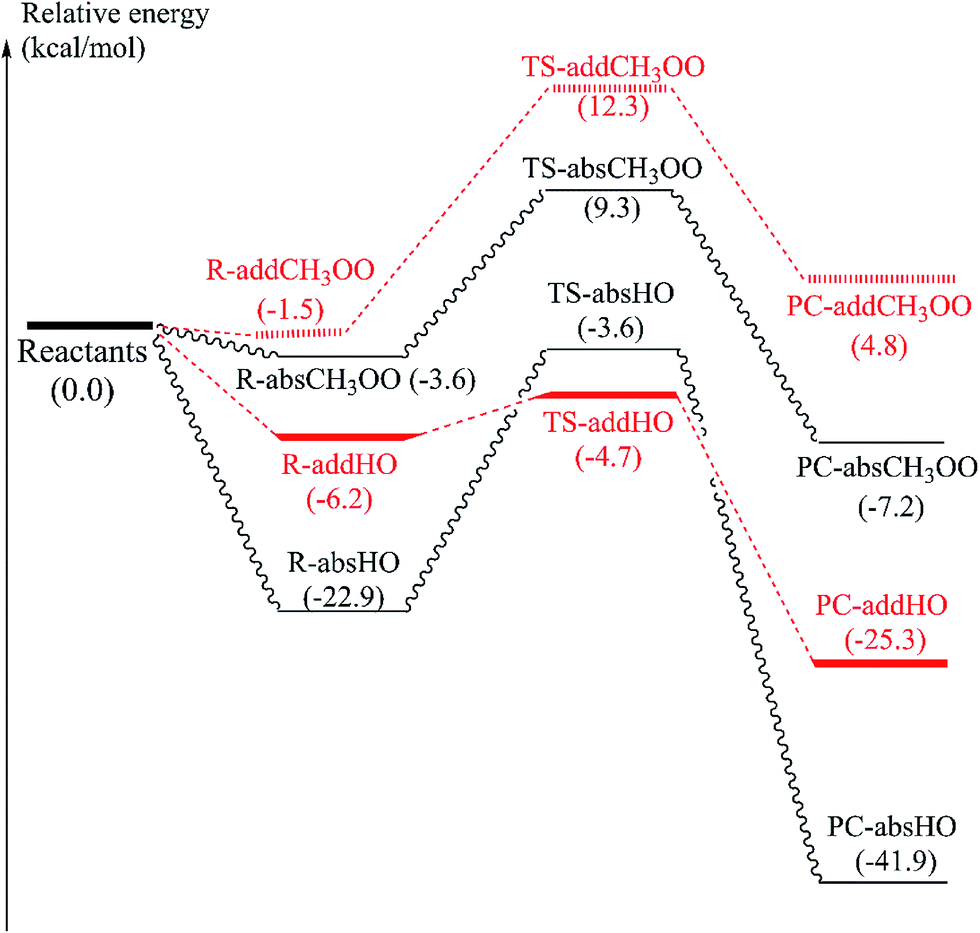 | ||
| Fig. 6 PES at B3LYP/6-311G(d,p) level of theory of H-atom abstraction reaction at easiest C8–H breaking bond and addition reaction at C9 positions for α-vetivone and CH3OO˙ and HO˙ radicals. | ||
Concerning H-atom abstraction reaction at C3, it is widely accepted that there are two mechanisms which generate the same products as HAT: sequential electron proton transfer (SEPT) and proton coupled electron transfer (PCET). They correspond to single electron transfer followed by or occurred simultaneously with proton transfer, respectively.39 In order to distinguish which kinds of mechanism falcarinol undergoes, natural bond population (NPA) charge, atomic spin density (ASP) and singly-occupied molecular orbitals (SOMO) analyses are systematically investigated for transition state of HAT reaction (Fig. 7A). As a result, the NPA charge indicates that O1-atom of the CH3OO˙ radical carries a negative charge, i.e. −0.3771e, while H3-atom which split from C3 atom on falcarinol, shows a positive value of 0.1712e. So the CH3OO˙ free radical possesses high nucleophile character which favors an attack to the H3–C3 electrophile active sites. As observed on ASP distribution in Fig. 7A, ASP distribution which consists in coefficient of the natural orbital carrying unpaired electron, of the transition state concentrates on two heavy atoms which undergo the H atom exchange, i.e. C3 and O1 atoms. This is corresponding to a HAT process.40 Indeed, Mulliken APS analyses show that O and C3 atoms carry high positive spin density of 0.4366 and 0.2273, respectively, while APS of H atom is slightly negative, i.e. −0.0247. Furthermore, the SOMO has a significant atomic orbital density oriented along the transition vector, essentially localized on the C3⋯H3⋯O1 vector, with a node plane located at the migrating H3 transferring atom. The NBO analysis indicates that the third lone pair of electrons on O1 atom, LP(3)O1, is donated to an unoccupied σ* antibonding orbital on C3–H3, σ*(1)C3–H3, in the HAT process with stabilization energy equal to 44.2 kcal mol−1 in forming a new σ-bond O1–H3 on CH3OOH (Table 5).
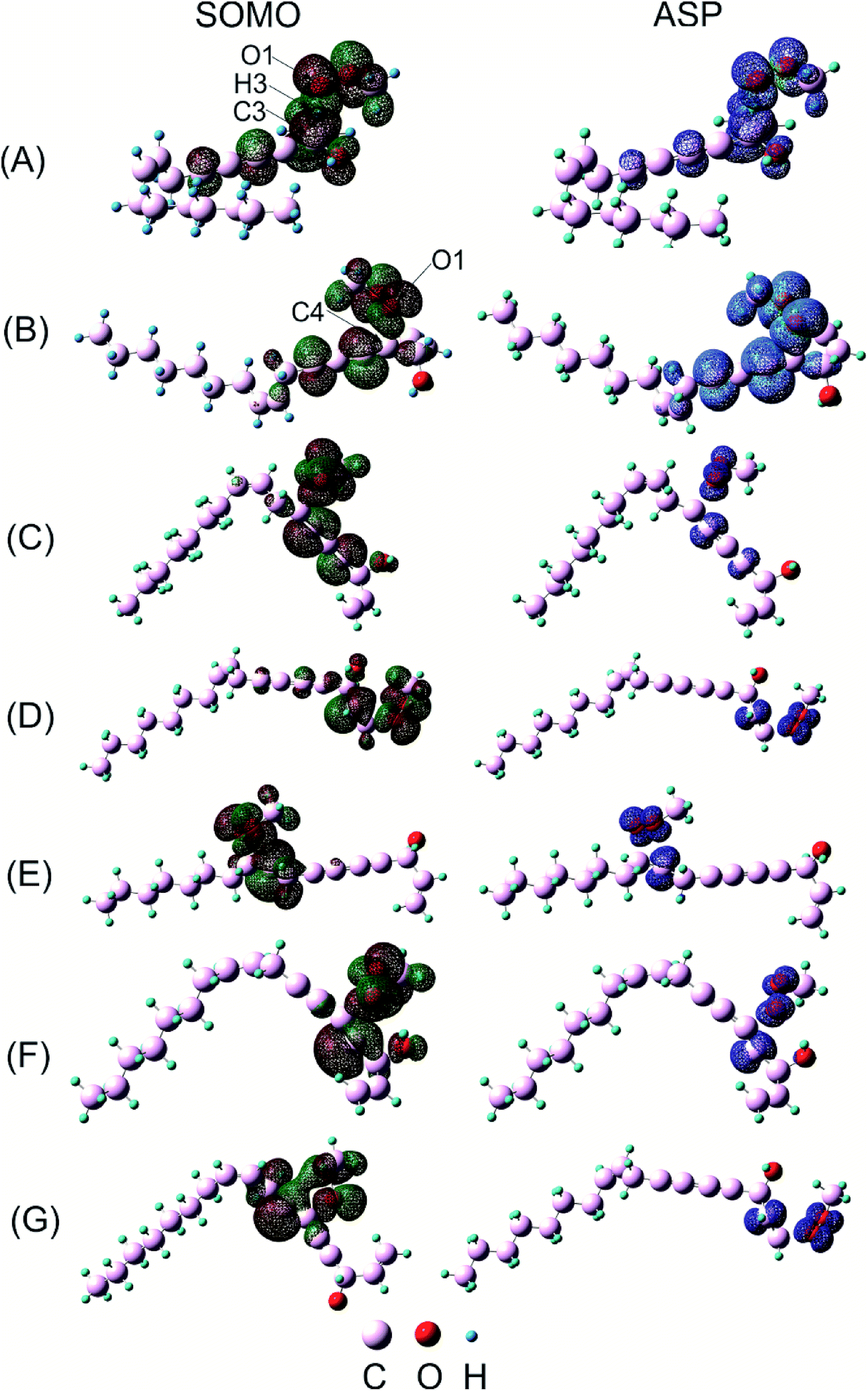 | ||
| Fig. 7 SOMO densities surface and atomic spin density (ASP) of (A) H-abstraction at C3 and addition reactions at (B) C4; (C) C7; (D) C1; (E) C10; (F) C5and (G) C6 between CH3OO˙ radical and falcarinol. (Orbital density is visualized at isovalue of 0.02 and spin density is mapped from −5 × 10−4 to 5 × 10−4 at isovalue of 0.004). | ||
| Donor NBO (i) | Acceptor NBO (j) | E(2), kcal mol−1 | |
|---|---|---|---|
| a π denotes π bonding orbital. σ* denotes sigma antibonding orbital. LP symbolizes a lone pair of electrons. LP(n) symbolizes the nth lone pair of electrons. | |||
| FALC–C3H + CH3OO˙ | LP(3) O1 | σ*(1) C1–H3 | 44.2 |
| FALC–C10 + CH3OO˙ | LP(3) O1 | σ*(2) C9–C10 | 34.6 |
| FALC–C7 + CH3OO˙ | LP(3) O1 | σ*(3) C6–C7 | 28.9 |
| π(3) C6–C7 | σ*(2) C4–C5 | 16.8 | |
| σ*(2) C6–C7 | σ*(3) C4–C5 | 58.1 | |
| σ*(3) C6–C7 | σ*(2) C4–C5 | 56.2 | |
| FALC–C6 + CH3OO˙ | LP(3) O1 | σ*(2) C6–C7 | 40.1 |
| σ*(3) C6–C7 | σ*(3) C4–C5 | 15.7 | |
| σ*(2) C6–C7 | σ*(2) C4–C5 | 13.1 | |
| FALC–C5 + CH3OO˙ | LP(3) O1 | σ*(2) C4–C5 | 38.6 |
| FALC–C4 + CH3OO˙ | LP(3) O1 | σ*(3) C4–C5 | 28.9 |
| π(3) C4–C5 | σ*(2) C6–C7 | 15.7 | |
| σ*(2) C4–C5 | σ*(3) C6–C7 | 59.5 | |
| σ*(3) C4–C5 | σ*(2) C6–C7 | 53.8 | |
| FALC–C1 + CH3OO˙ | LP(3) O1 | σ*(2) C1–C2 | 39.4 |
For the radical addition reaction to C4C5 bond (Fig. 7B), the NPA charge calculation shows that O1-atom of the free radical carries high negative charge, i.e. −0.3639e, in acting as a nucleophile. Meanwhile, the C4 atom where the free radical attacks, carries small positive charge, i.e. 0.1411e, in showing as an electrophile. The Mulliken spin density distribution shows high spin density concentrated on O1 atom which has a tendency to approach to C4 carbon atom (Fig. 7B). Meanwhile, the SOMO shows that p orbital on O1-atom has high tendency to overlap with π-orbital on the C4C5 triple bond. The NBO analysis confirms that the third lone pair of electron on O1 atom, LP(3)O1 is transferred to the third unoccupied σ* antibonding orbital of the triple bond, σ*(3)C4–C5, with stabilization energy equal to 28.9 kcal mol−1. This process tends to form σ-bond C4–O1, and results in loss of one π-bond between C4 and C5 atoms which changes it to a double bond. The observation for the other addition reactions is quite similar (Fig. 7C–G) (Table 5). The electron densities will then be shifted from bonding and antibonding orbitals of C4C5 bond like π(3) C4C5, σ*(2) C4–C5 and σ*(3) C4–C5 to the others vacant antibonding orbitals such as σ*(2) C6–C7, σ*(3) C6–C7 with the stabilization energy equal to 15.7, 59.5 and 53.8 kcal mol−1, respectively. The observation for the other addition reactions is quite similar (Fig. 7C–G).
4. Conclusions
The thermochemical parameters of twenty-one oxygenated monoterpene and desquiterpene compounds extracted from Cleistocalyx operculatus have been studied in detail. Falcarinol is determined as the most effective antioxidant on the basis of both HAT and SPLET mechanisms. In fact, the BDE and PA in the gas phase of falcarinol are 66.5 kcal mol−1 and 341.3 kcal mol−1. The effective property of falcarinol is a result of the presence of both OH group and unsaturated double/triple bonds in this molecule. Regarding the effect of solvent, it is observed that the hydrogen atom transfer is favorable in nonpolar environment while deprotonation process is supported by polar one where protons show higher solvation enthalpy. Calculated quantum chemical parameters underline that α-vetivone represent as the most reactive compounds with the chemical hardness of 4.02 eV and electrophilicity being 2.03 eV. This molecule also displays as one of the easiest electron removal with IE value of 8.06 eV.From the potential energy surface (PES) point of view, the interaction between CH3OO˙ radical and falcarinol were clarified. The H-abstraction reaction at C3–H position is determined as the most favored with the energy barriers of −18.7 kcal mol−1. Among the addition reactions, the ones at C4 position of C4C5 and C7 of C6C7 triple bonds are the most favored with the energy barriers of −9.9 and −8.4 kcal mol−1, respectively. And it is shown that the H-abstraction reaction is exergonic with a negative ΔG value of −10.0 kcal mol−1, while all addition reactions are endergonic with positive ΔG value from 1.7 to 16.6 kcal mol−1.
Interaction between falcarinol and reactive HO˙ radical was also considered. The PES for H-atom abstraction reaction at the easiest C3–H breaking bond and the addition reaction at C4 position were investigated. In comparing to the reactions with CH3OO˙, it is observed that falcarinol reacts more strongly with HO˙ radical with significantly lower ΔH and ΔG values, i.e. −54.7 and −47.1 kcal mol−1, respectively for H-abstraction reaction, and −42.9 and −32.2 kcal mol−1, respectively for addition one.
Singly-occupied molecular orbital (SOMO), natural population atomic (NPA) charge, atomic spin density (ASP) and natural bond orbital (NBO) analysis of the optimized transition states of the reactions between falcarinol and CH3OO˙ radical have been performed, as an example, to clarify the mechanism of these reactions. The mechanism is determined by the transfer of the third lone pair of electrons on O1-atom of CH3OO˙ radical to an unoccupied σ* antibonding orbital on C3–H3, and on the triple C4C5, C6C7 bonds.
In the case of α-vetivone, attack of CH3OO˙ and HO˙ at easiest C8–H breaking bond and at C9C10 double bonds was similarly studied. The results show that the H-atom abstraction reactions by both radicals are more prominent than the addition ones. Indeed, the energy of product complex for H-abstraction by CH3OO˙ and HO˙ radicals are found −7.2 and −41.9 kcal mol−1 while the ones of addition reaction are determined at 4.8 and −25.3 kcal mol−1, respectively. The reactions with HO˙ are more exergonic than the ones with CH3OO˙ radical with meaningfully negative ΔG values of −36.2 and −14.3 kcal mol−1 corresponding to H-abstraction and addition reactions, respectively.
According to the results obtained for falcarinol, it could conclude that the antioxidant molecules preferably react with reactive HO˙ radical.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.06-2015.09.References
- C. Cabrera, R. Gimenez and M. C. Lopez, J. Agric. Food Chem., 2003, 51, 4427–4435 CrossRef CAS PubMed.
- G. Singh, S. Maurya, C. Catalan and M. P. De Lampasona, J. Agric. Food Chem., 2004, 52, 3292–3296 CrossRef CAS PubMed.
- A. I. Hussain, F. Anwar, S. T. Hussain Sherazi and R. Przybylski, Food Chem., 2008, 108, 986–995 CrossRef CAS PubMed.
- K. A. Youdim, S. G. Deans and H. J. Finlayson, J. Essent. Oil Res., 2002, 14, 210–215 CrossRef CAS.
- M. Hyldgaard, T. Mygind and R. L. Meyer, Front. Microbiol., 2012, 3, 12 Search PubMed.
- B. Bozin, N. Mimica-Dukic, N. Simin and G. Anackov, J. Agric. Food Chem., 2006, 54, 1822–1828 CrossRef CAS PubMed.
- V. R. Preedy, Essential Oils in Food Preservation, Flavor and Safety, Academic Press, 2015 Search PubMed.
- H. Robert, in Handbook of Essential Oils, CRC Press, 2015, pp. 381–431 Search PubMed.
- L.-B. Maria, in Handbook of Essential Oils, CRC Press, 2015, pp. 619–653 Search PubMed.
- K. H. Baser and B. Gerhard, in Handbook of Essential Oils, CRC Press, 2015, pp. 1–3 Search PubMed.
- M. K. Fasseas, K. C. Mountzouris, P. A. Tarantilis, M. Polissiou and G. Zervas, Food Chem., 2008, 106, 1188–1194 CrossRef CAS.
- S. Chatterjee, Z. Niaz, S. Gautam, S. Adhikari, P. S. Variyar and A. Sharma, Food Chem., 2007, 101, 515–523 CrossRef CAS.
- H.-Y. Chen, Y.-C. Lin and C.-L. Hsieh, Food Chem., 2007, 104, 1418–1424 CrossRef CAS.
- C. Turek and F. C. Stintzing, Compr. Rev. Food Sci. Food Saf., 2013, 12, 40–53 CrossRef CAS.
- J. G. Lopez-Reyes, D. Spadaro, A. Prelle, A. Garibaldi and M. L. Gullino, J. Food Prot., 2013, 76, 631–639 CrossRef PubMed.
- R. Lawrence and K. Lawrence, Asian Pac. J. Trop. Biomed., 2011, 1, S51–S54 CrossRef.
- B. Bozin, N. Mimica-Dukic, N. Simin and G. Anackov, J. Agric. Food Chem., 2006, 54, 1822–1828 CrossRef CAS PubMed.
- N. T. Dung, J. M. Kim and S. C. Kang, Food Chem. Toxicol., 2008, 46, 3632–3639 CrossRef CAS PubMed.
- T. C. Ngo, D. Q. Dao, N. M. Thong and P. C. Nam, RSC Adv., 2016, 6, 30824–30834 RSC.
- S. Purup, E. Larsen and L. P. Christensen, J. Agric. Food Chem., 2009, 57, 8290–8296 CrossRef CAS PubMed.
- J. F. Young, L. P. Christensen, P. K. Theil and N. Oksbjerg, Dose-Response, 2008, 6, 239–251 CrossRef CAS PubMed.
- I.-Y. Choi, J. H. Lim, S. Hwang, J.-C. Lee, G.-S. Cho and W.-K. Kim, Free Radical Res., 2010, 44, 541–551 CrossRef CAS PubMed.
- P. C. Lars, Recent Pat. Food, Nutr. Agric., 2011, 3, 64–77 CrossRef.
- H.-J. Kim, F. Chen, X. Wang, H. Y. Chung and Z. Jin, J. Agric. Food Chem., 2005, 53, 7691–7695 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- N. M. Thong, D. T. Quang, N. H. T. Bui, D. Q. Dao and P. C. Nam, Chem. Phys. Lett., 2015, 625, 30–35 CrossRef.
- J. E. Bartmess, J. Phys. Chem., 1994, 98, 6420–6424 CrossRef CAS.
- K. K. Irikura, R. D. Johnson and R. N. Kacker, J. Phys. Chem. A, 2005, 109, 8430–8437 CrossRef CAS PubMed.
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- J. Tomasi, B. Mennucci and E. Cancès, J. Mol. Struct.: THEOCHEM, 1999, 464, 211–226 CrossRef CAS.
- I. B. Obot, D. D. Macdonald and Z. M. Gasem, Corros. Sci., 2015, 99, 1–30 CrossRef CAS.
- H. Chermette, J. Comput. Chem., 1999, 20, 129–154 CrossRef CAS.
- R. P. Iczkowski and J. L. Margrave, J. Am. Chem. Soc., 1961, 83, 3547–3551 CrossRef CAS.
- R. G. Parr, L. v. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- G. Wang, Y. Xue, L. An, Y. Zheng, Y. Dou, L. Zhang and Y. Liu, Food Chem., 2015, 171, 89–97 CrossRef CAS PubMed.
- M. Li, W. Liu, C. Peng, Q. Ren, W. Lu and W. Deng, Int. J. Quantum Chem., 2013, 113, 966–974 CrossRef CAS.
- A. Pérez-González and A. Galano, Int. J. Quantum Chem., 2012, 112, 3441–3448 CrossRef.
- M. C. Foti, J. Agric. Food Chem., 2015, 63, 8765–8776 CrossRef CAS PubMed.
- S. Olivella, J. M. Anglada, A. Solé and J. M. Bofill, Chem.–Eur. J., 2004, 10, 3404–3410 CrossRef CAS PubMed.
- J. M. Mayer, D. A. Hrovat, J. L. Thomas and W. T. Borden, J. Am. Chem. Soc., 2002, 124, 11142–11147 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04798c |
| This journal is © The Royal Society of Chemistry 2017 |