
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing ferrierite-based catalysts with improved properties for skeletal isomerization of n-butene to isobutene
Haiqiang Hu,
Ming Ke *,
Ka Zhang,
Qiang Liu,
Pei Yu,
Yang Liu,
Changchun Li and
Wen Liu
*,
Ka Zhang,
Qiang Liu,
Pei Yu,
Yang Liu,
Changchun Li and
Wen Liu
State Key Laboratory of Heavy Oil Processing, China University of Petroleum, Beijing 102249, P. R. China. E-mail: keming@cup.edu.cn; Tel: +86 10 89733372
First published on 20th June 2017
Abstract
The crystal morphology and size of ferrierite catalysts were controlled by a suite of crystallization methods and structure-directing agents. In particular, nano-sized ferrierite could be obtained by using tetramethylammonium hydroxide and pyrrolidine as double structure-directing agents under dynamic crystallization. The catalysts were analysed by XRD, SEM, N2 adsorption–desorption, NMR, XRF, NH3-TPD, Py-IR, and TGA, and the characteristics were correlated with their catalytic performance in the skeletal isomerization of n-butene. The results clearly indicated that all the catalysts have a similar crystallinity, Si/Al ratio, and total number of acid sites, while a decrease in their crystal size leads to more pore mouth acid sites. The nano-sized ferrierite showed better stability and higher activity, which were related to their crystal morphology, size, and pore mouth acid. In addition, we further confirmed that high isobutene selectivity can be produced by monomolecular reaction.
1. Introduction
Skeletal isomerization of n-butene to isobutene is an important method to produce refinery-deficient isobutene, which is widely used in the synthesis of butyl rubber, polyisobutene, methyl tert-butyl ether, and methacrylate.1,2 Over the last few years, many 10 membered ring (MR) channel zeolites with shape-selective properties have been used extensively in industry and have been under intensive academic investigation.3–5 It appears that zeolites with ferrierite topology are much more effective than other zeolites in terms of activity, selectivity, and stability.Ferrierite is a medium-pore zeolite, containing a bi-dimensional pore system consisting of 10 MR channels (with dimensions of 0.43 × 0.55 nm) running parallel to the [001] direction and intersected by 8 MR channels (with dimensions of 0.35 × 0.48 nm) running parallel to the [010] direction. The 6 MR channels in the [001] direction and 8 MR channels intersect, which create [82686458] cavities, known as ferrierite cages.6,7 Although ferrierite is a natural zeolite, it has been synthesized through the hydrothermal synthesis method with many organic structure-directing agents (SDAs).8–12 However, reports on the synthesis of nano-sized ferrierite are very limited. Both Xue13 and Wuamprakhon14 successfully synthesized hierarchical ferrierite with a ball-shaped morphology comprising nanosheets. This hierarchical ferrierite showed greatly improved catalytic activities for the pyrolysis of low-density polyethylene and benzylation of toluene with benzyl chloride. Pinar,15 Lee,16 and Yang17,18 have all reported needle-like or rod-like ferrierite, and found that the ferrierite has a higher selectivity and better stability than the commercial ferrierite for the skeletal isomerization of n-butene. In addition, Hu19 obtained small-sized ferrierite through an aging step, and found that this ferrierite showed higher activity and isobutene yield than large-sized ferrierite. To the best of our knowledge, there is no report on the effect of morphology and crystal size on the initial activity and selectivity of the skeletal isomerization of n-butene.
There are at least three reaction mechanisms (bimolecular, monomolecular, and pseudo-monomolecular) on ferrierite that can be used to describe the skeletal isomerization of n-butene. In the bimolecular mechanism, two n-butenes dimerize to form C8, which undergoes isomerization and cracking, to form isobutene, n-butene, propene, and pentene.20,21 The monomolecular mechanism occurs on Brønsted acid sites through a protonated cyclopropyl cation, which can be broken down to a primary carbenium ion, and then rearrange to form isobutene.16,22,23 Even though the monomolecular mechanism can explain the high isobutene selectivity, some researchers do not support this view because this mechanism must go through a primary carbenium ion, which is highly energetically and thermodynamically unfavorable. Guisnet proposed a pseudo-monomolecular mechanism, in which formation of the primary carbenium ion is avoided. It is proposed with this mechanism that the active site is a tertiary carbenium ion, which reacts with n-butene to produce a secondary carbenium ion, and then a more stable tertiary carbenium ion is formed through rearrangement. The isobutene is produced through β-scission and the active site is regenerated.24,25
In this work, we successfully synthesized three ferrierite catalysts with different crystal morphologies and sizes. Nano-sized ferrierite was obtained through tetramethylammonium hydroxide (TMAOH) and pyrrolidine acting as SDAs under rotating crystallization. In addition, we studied how the crystal morphology and size influence the activity and selectivity for the skeletal isomerization of n-butene based on physicochemical properties of the ferrierite catalysts. We also displayed high isobutene selectivity over the fresh nano-sized ferrierite for the first time. Moreover, we also elucidated the deactivation mechanism in the reaction.
2. Experimental
2.1 Materials
The reagents used for the synthesis of ferrierite (FER) zeolite were sodium silicate (28.2 wt% SiO2 and 8.6 wt% Na2O, Beijing Red Star Guangsha Chemical Co., Ltd.), aluminum sulfate octadecahydrate (Analytical Reagent, Sinopharm Chemical Reagent Co., Ltd.), sulfuric acid (95–98%, Beijing Chemical Works), pyrrolidine (99+%, ACROS), tetramethylammonium hydroxide (25%, Sinopharm Chemical Reagent Co., Ltd.), ammonium chloride (Analytical Reagent), and deionized water.2.2 Catalyst preparation
Traditional ferrierite (T-FER) was synthesized following the conventional procedure reported in the literature26 and using pyrrolidine as SDA, crystallized from a gel with the molar composition: 20Na2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :37pyrrolidine:66SiO2:2Al2O3:6.3H2SO4:1600H2O. The gel was heated statically at 175 °C and under autogenous pressure for 55 h. Small ferrierite (S-FER) was obtained by rotation (60 rpm) at 175 °C and under autogenous pressure for 55 h with the same molar composition. The nano-sized ferrierite (N-FER) was synthesized with a slightly modified method where TMAOH was added into the gel together with pyrrolidine. The input gel molar composition was: 20Na2O:37pyrrolidine: 3TMAOH:66SiO2:2Al2O3:7.8H2SO4:1600H2O. Crystallization conditions were the same as for S-FER. The products were recovered by filtration, washed, and dried overnight. To obtain the proton form, all products were calcined in air at 550 °C for 6 h, and then ion-exchanged with 1 M NH4Cl solution at 90 °C for 4 h. After washing and drying, the products were calcined in air at 550 °C for 4 h.
:37pyrrolidine:66SiO2:2Al2O3:6.3H2SO4:1600H2O. The gel was heated statically at 175 °C and under autogenous pressure for 55 h. Small ferrierite (S-FER) was obtained by rotation (60 rpm) at 175 °C and under autogenous pressure for 55 h with the same molar composition. The nano-sized ferrierite (N-FER) was synthesized with a slightly modified method where TMAOH was added into the gel together with pyrrolidine. The input gel molar composition was: 20Na2O:37pyrrolidine: 3TMAOH:66SiO2:2Al2O3:7.8H2SO4:1600H2O. Crystallization conditions were the same as for S-FER. The products were recovered by filtration, washed, and dried overnight. To obtain the proton form, all products were calcined in air at 550 °C for 6 h, and then ion-exchanged with 1 M NH4Cl solution at 90 °C for 4 h. After washing and drying, the products were calcined in air at 550 °C for 4 h.
2.3 Catalyst characterization
X-ray diffraction (XRD) was used to measure the crystallization and phase of the catalysts performed with a PANalytical B. V. diffractometer apparatus with Cu Kα radiation (k = 0.15406 nm) at a voltage and current of 40 kV and 20 mA, respectively. The 2θ scanning range was from 5° to 50° with a scan rate of 4° min−1. The crystallinity of the self-made ferrierite catalysts was obtained by calculating the intensity of the peaks at 2θ = 9.3° and between 22° and 25.5°.26 The crystallinity of T-FER was set as 100%. Scanning electron microscopy (SEM) measurements were carried out to confirm the crystal morphology and size with a HITACHI SU8010 apparatus. The textural characterization of the ferrierite catalysts was performed by N2 adsorption–desorption at −196 °C with a US Micrometric TriStar II 3020 nitrogen adsorption analyzer. The ferrierite catalysts were degassed at 300 °C for 8 h under vacuum before the adsorption measurement. 13C and 27Al solid MAS NMR analyses were carried out on an AVANCE 300 NMR spectrometer. The 13C MAS NMR spectra were obtained at a frequency of 100.54 MHz with a 5 mm T3HX probe and spinning rate of 5 kHz. The pulse length and recycling delay were set as 4.5 μs and 3 s, respectively. The 27Al MAS NMR spectra were analyzed at a frequency of 78.16 MHz with a pulse of 1.8 μs and a recycling delay of 0.5 s, and by spinning the samples at a frequency of 10.0 MHz. X-ray fluorescence (XRF) was carried out on a Bruker S4 Pioneer to analyze the sodium content and Si/Al ratio. Ammonia temperature-programmed desorption (NH3-TPD) was used to study the acid properties of the catalysts with US Autosorb-1C-TCD-MS adsorption apparatus. Before NH3 adsorption, 100 mg of the samples was calcined in a flow of pure argon at 550 °C for 60 min and NH3 adsorption was performed at 100 °C for 40 min. Subsequently, purging was carried out with pure argon at 100 °C for 60 min to remove the physically-adsorbed NH3. Finally, the samples were heated from 100 to 650 °C using a temperature ramp of 10 °C min−1, and the quantity of NH3 desorbed was monitored using mass spectrometry. Fourier transform infrared (FT-IR) absorbance spectra were carried out on an US MAGNAIR-IR560 IR apparatus. Pyridine was used as the probe molecule to characterize the concentration and nature of acid sites located at or near the pore mouth. Approximately, samples with a quantity of 20 mg and diameter of 12 mm were obtained by a self-supporting wafer under a pressure of 10 MPa and pretreated at 400 °C for 30 min. The adsorption of pyridine was performed at 130 °C for 30 min, and subsequently, the physisorbed molecules were removed at 130 °C for 30 min. IR spectra were recorded at 200 and 350 °C. The weight loss of coke formed in the skeletal isomerization of n-butene was measured by thermogravimetric analysis (TGA). TGA was carried out on a Mettler-Toledo TGA/DSC-1 synchronous thermal analyzer. Approximately 20 mg of the coked ferrierite catalysts were used and heated from 100 to 800 °C at a heating rate of 10 °C min−1 under oxygen flow.2.4 Catalytic tests
The skeletal isomerization of n-butene to isobutene was carried out at 410 °C, under atmospheric pressure. For example, 0.3500 g of catalyst was placed in the fixed bed reactor with an inner diameter of 8 mm. The feed stream, a mixture of nitrogen and n-butene with a molar ratio of 3:1, was fed into the reactor to start the skeletal isomerization when heated to 410 °C. The products were analyzed online by gas chromatography (SHIMADZU 2010 plus) equipped with a flame ionization detector and a GS-Alumina 50 m × 0.53 mm capillary column. For every catalyst, the WSHV was set as 3.5, 10, 35, and 100 h−1 to obtain n-butene conversion over a large range.
Since equilibrium of 1-butene, trans-2-butene, and cis-2-butene was established rapidly through double-bond isomerization and all were transformed to isobutene. 1-Butene, trans-2-butene, and cis-2-butene were considered as the reactants.25,27
The n-butene conversion (C) was defined as:
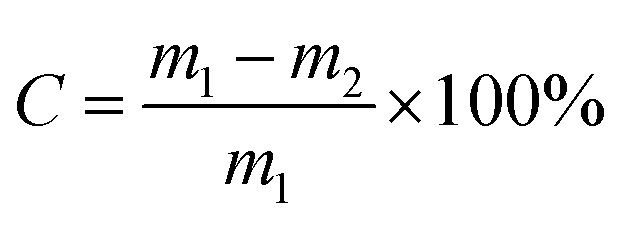 | (1) |
The selectivity of isobutene (Siso) was defined by:
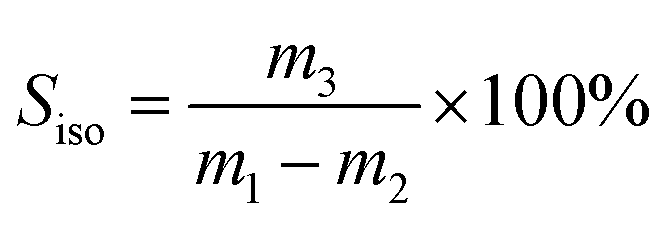 | (2) |
The yield of isobutene (Yiso) was calculated using:
 | (3) |
The yield of propene and pentene (Yc3+c5) was calculated by:
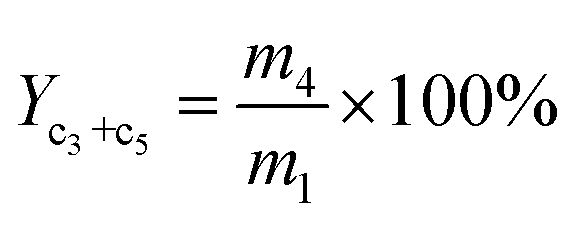 | (4) |
3. Results and discussion
3.1 Characterization of different catalysts
The crystal structure of the catalysts was analyzed by XRD. The XRD patterns of the catalysts synthesized using different methods all show the typical diffraction peaks of an FER-type structure without evidence of contamination by impure phase as shown in Fig. 1.26,28 The intensity of the X-ray diffraction lines for T-FER (Fig. 1a), S-FER (Fig. 1b), and N-FER (Fig. 1c) are similar, which means that all the catalysts have a similar crystallinity (as shown in Table 1). However, the XRD characterization also showed differences between the catalysts. That is, the catalysts synthesized in the presence of TMAOH had broader XRD peaks which are often seen with nanocrystalline materials.16,29 The nature of these catalysts was further evident in their SEM images. | ||
| Fig. 1 XRD patterns of the catalysts: (a) T-FER, (b) S-FER, (c) N-FER. | ||
| Sample | Si/Al | Na (%) | Crystallinity | SBET (m2 g−1) | Sext (m2 g−1) | Smic (m2 g−1) | Vt (cm3 g−1) | Vmic (cm3 g−1) | Vmes (cm3 g−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Si/Al ratio and sodium content: measured by XRF. Vmes = Vt − Vmic. | |||||||||
| T-FER (fresh) | 9.3 | <0.05 | 100 | 314 | 16 | 297 | 0.16 | 0.14 | 0.02 |
| T-FER (2000 min) | — | — | — | 7 | 2 | 5 | 0.02 | 0.02 | 0.00 |
| S-FER (fresh) | 10.2 | <0.05 | 103 | 315 | 23 | 292 | 0.18 | 0.14 | 0.04 |
| S-FER (2000 min) | — | — | — | 19 | 9 | 10 | 0.05 | 0.01 | 0.04 |
| N-FER (fresh) | 9.7 | <0.05 | 98 | 337 | 55 | 282 | 0.24 | 0.14 | 0.10 |
| N-FER (2000 min) | — | — | — | 61 | 33 | 28 | 0.11 | 0.03 | 0.08 |
Fig. 2 shows the SEM results of the different ferrierite catalysts. The crystal morphology and size were greatly influenced by the utilized crystallization method (rotating or static) and the utilized TMAOH. T-FER, synthesized without rotation and TMAOH, exhibited a large cuboid shape and a size of about 8 × 3 × 3 μm (Fig. 2a). This morphology was commonly observed.26,30,31 A different morphology consisting of small-sized crystallites (S-FER) was obtained when static crystallization was replaced by the rotating method. The S-FER possessed a plate-like shape, while the size was about 0.8 × 0.2 μm (Fig. 2b). A crystal sample consisting of nano-sized crystallites (N-FER) was produced when TMAOH was added under rotation. The N-FER as displayed in the SEM images (Fig. 2c and d) had a rectangular shape with a length of 120 nm and thickness of 30 nm. The rectangular crystallites were a crucial precursor, whereby the cuboid-shaped framework was formed by the stacking of these crystallites. The size of the crystal aggregates was about 0.7 μm, which was similar to that of the S-FER.
 | ||
| Fig. 2 SEM patterns of the catalysts: (a) T-FER, (b) S-FER, (c) and (d) N-FER. | ||
Nitrogen adsorption–desorption isotherms and pore size distribution (PSD) for the fresh and coked ferrierite catalysts are shown in Fig. 3. The isotherm of fresh T-FER and S-FER was similar to the type-I isotherm (Fig. 3A(d and e)), which is characteristic of micropore materials. It indicates that the T-FER and S-FER are comprised solely of micropores. The PSD, as shown in Fig. 3B(d and e), also indicates that few mesoporous structures existed within the crystals. However, the isotherm of fresh N-FER, as reveled in Fig. 3A(f), exhibited a combined characteristic of type I and IV with an obvious hysteresis loop at a relative pressure of 0.45, caused by capillary condensation of N2 in the mesopores. The PSD ranged from 1 to 70 nm, as shown in Fig. 3B(f), indicating the existence of a mesoporous structure among the stack of nano-sized crystallites.
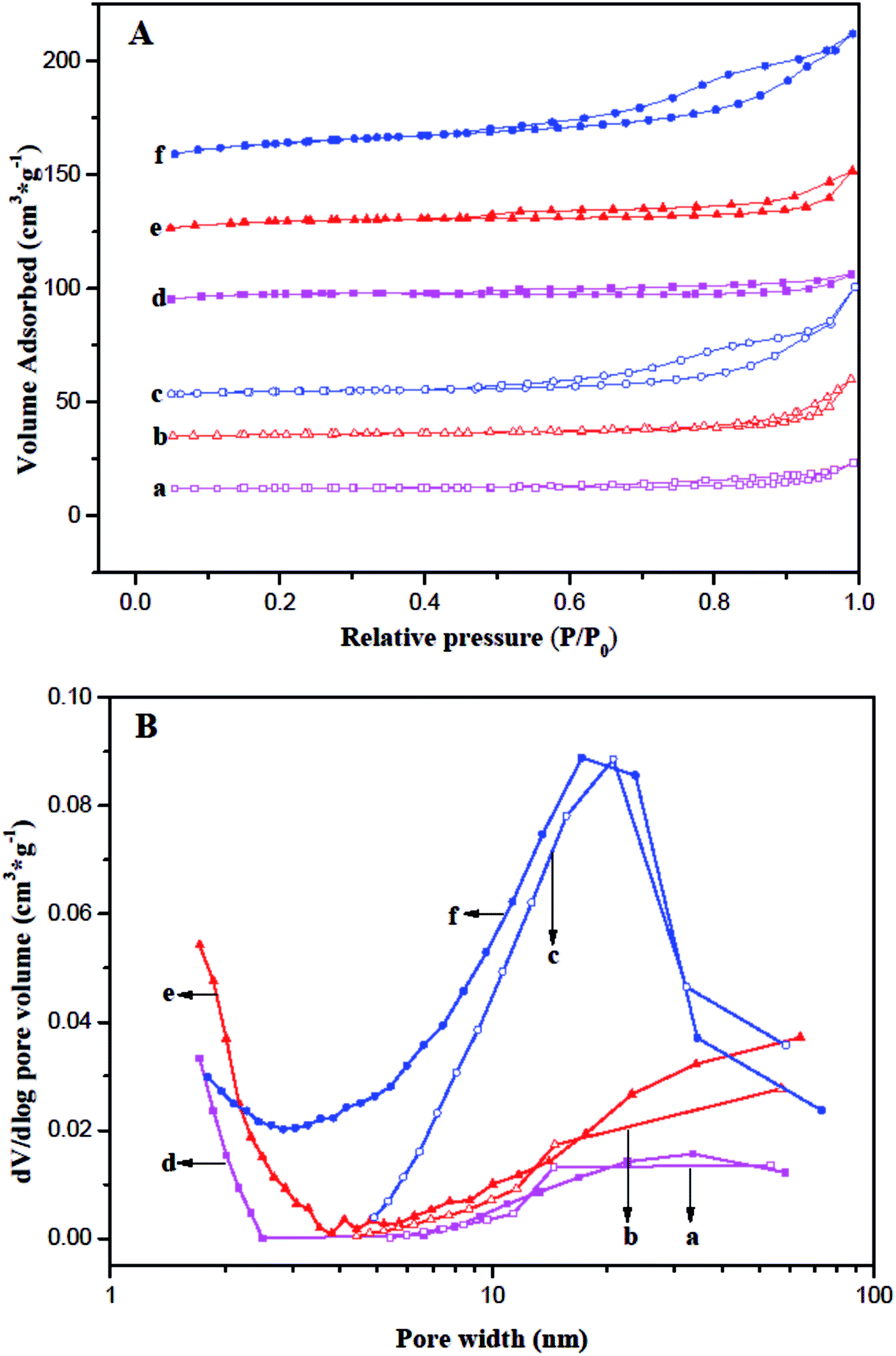 | ||
| Fig. 3 Comparison of (A) nitrogen adsorption–desorption isotherms (B) BJH pore size distribution: (a) T-FER (2000 min), (b) S-FER (2000 min), (c) N-FER (2000 min), (d) T-FER (fresh), (e) S-FER (fresh), and (f) N-FER (fresh). | ||
A summary of the physicochemical and textual properties is given in Table 1. It revealed the external surface area (Sext) and mesopore pore volume (Vmes) of the fresh N-FER were 55 m2 g−1 and 0.10 cm3 g−1, respectively, which were much larger than that of the fresh T-FER and S-FER. The BET surface area (SBET) and total pore volume (Vt) of the fresh N-FER were 337 m2 g−1 and 0.24 cm3 g−1, respectively, which were also larger than the data obtained for the fresh T-FER and S-FER. In contrast, the micropore surface area (Smic) of the fresh N-FER was a little lower than that of the fresh T-FER and S-FER, while all the fresh catalysts had the same micropore volume. Moreover, the coked ferrierite catalysts after 2000 min of stream at 410 °C, WSHV = 10 h−1 and under atmospheric pressure, labeled as T-FER (2000 min), S-FER (2000 min), and N-FER (2000 min), were also performed. The data obtained from the coked ferrierite catalysts indicates that the coking decreased the Smic and Vmic significantly, but decreased the Sext and Vmes only slightly. We can consider that the micropore was almost fully filled by coke, but the 10 MR pore mouth and mesopores are still accessible.
The solid-state MAS NMR spectra of the ferrierite catalysts are displayed in Fig. 4. The peaks at 23 and 50 ppm in the 13C MAS NMR spectrum (Fig. 4A) can be assigned to the –CH2 of pyrrolidine.32,33 This means that pyrrolidine exists in the three types of ferrierite. Moreover, the extra peak at 60 ppm in Fig. 4A(c) assigned to the methyl group of the TMA+ indicates that both TMA+ and pyrrolidine existed in the N-FER.13,15,34 Therefore, it suggests that the change in morphology and size should be caused by the interaction between pyrrolidine and TMA+ in the gel. The coordination of aluminum in the three ferrierite catalysts was also studied by 27Al MAS NMR and the results demonstrated that there was only one resonance at approximately 52 ppm assigned to the tetrahedral Al(OSi)4 species in the zeolite framework but no signal around 0 ppm (Fig. 4B), indicating that there was no extra framework of aluminum. Moreover, the sodium content and Si/Al ratio were also confirmed by XRF, as shown in Table 1. The sodium content was lower than 0.05% for all ferrierite catalysts, and the Si/Al ratio of C-FER, S-FER, and N-FER was 9.3, 10.2, and 9.7, respectively. By comparing the 27Al MAS NMR and XRF data, we can conclude that the three ferrierite catalysts had a similar framework Si/Al ratio.
 | ||
| Fig. 4 (A) 13C and (B) 27Al MAS NMR spectra of the catalysts: (a) T-FER, (b) S-FER, (c) N-FER. | ||
The strength and quantity of the acid sites in fresh and coked ferrierite catalysts were studied by the ammonia TPD method. The acid strength can be determined by the desorption temperature, and the quantity of acid sites can be obtained through the deconvolution of the desorption curve. As shown in Fig. 5A, all the fresh ferrierite catalysts showed two peaks at around 240 °C and 450 °C, which were assigned to the weak and strong acid of the catalysts, respectively. All the fresh ferrierite catalysts exhibited a similar quantity of total acid. The ratio of the strong to weak acid was about 1.248, 1.293, and 1.253 for T-FER, S-FER, and N-FER, respectively. The amount of total acid and the ratio of strong to weak acid, which were based on the limited data, did not appear to be influenced by the crystal morphology and size. It should be noted that the higher desorption temperature (the center of the second peak), corresponding to N-FER, was 454 °C, while the lower desorption temperature measured for T-FER and S-FER was 442 °C. The diameter of ammonia molecules is about 0.3 nm; therefore, it can be considered that the acid sites in the ferrierite channels are all accessible by these molecules.35,36 Therefore, NH3-TPD can determine all acid sites regardless of the channel or pore mouth. A similar framework Si/Al ratio seen in these fresh ferrierite catalysts evidenced by NMR and XRF may explain why these catalysts had a similar amount of total acid sites and strong to weak acid ratio. In addition, Fig. 5B shows the NH3-TPD results for the coked ferrierite catalysts. It was found that the amount of acid among these coked catalysts was in the order: N-FER (2000 min) > S-FER (2000 min) > T-FER (2000 min). Both the amount of strong and weak acid decreased compared to the fresh ferrierite, especially that of the strong acid. This may be caused by the coke blocking the ferrierite cage and micropore and causing modification of the pore mouth acid so that NH3 can only access part of the pore mouth acid.
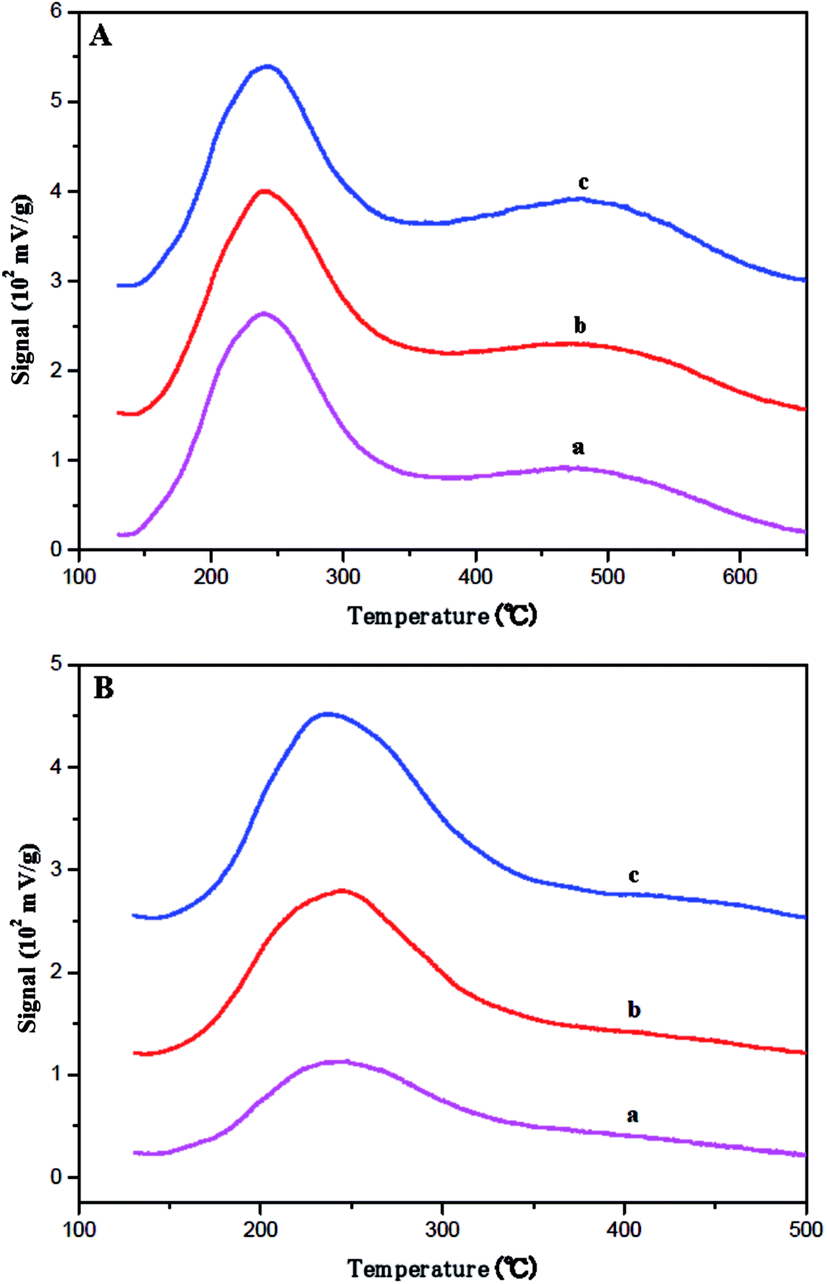 | ||
| Fig. 5 Ammonia TPD profiles for the (A) fresh catalysts: (a) T-FER, (b) S-FER, (c) N-FER and (B) aged catalysts: (a) T-FER (2000 min), (b) S-FER (2000 min), (c) N-FER (2000 min). | ||
According to the ref. 37 and 38, the kinetic diameter of pyridine is about 0.57 nm, which is bigger than the elliptical 10 MR. Some researchers37 reported that more than 40% of acid sites were not adsorbed by pyridine at 133 °C for 30 min. Therefore, pyridine (Py) was used as the probe molecule to measure the acid sites only located at or near the 10 MR pore mouths of fresh ferrierite at 130 °C for 30 min. Fig. 6 shows the IR spectra between 1600 and 1400 cm−1 with the presence of the key ring stretching peak of the adsorbed Py. All catalysts show Lewis acid and Brønsted acid. The Brønsted acid sites correspond to bridged hydroxyl groups (Si–OH–Al) and the Lewis acid sites can be generated by dehydrogenate of Brønsted acid sites.39 The bands at 1454 and 1545 cm−1 were attributed to the adsorption of pyridine on Lewis and Brønsted acid sites, respectively. The quantity of acid was calculated following the literature,40 in which the molar extinction coefficient of Lewis and Brønsted acid interacting with pyridine is given as 2.22 and 1.67 cm μmol−1, respectively. Table 2 lists the Lewis and Brønsted acidity determined by Py-IR characterization. It is noted that Brønsted and the total number of acidity sites decrease in the order: N-FER > S-FER > T-FER. Furthermore, N-FER and S-FER have a similar number of Lewis acid sites, while T-FER has less than these catalysts. This may be due to its smaller crystal size and larger external surface area, which leads to more pore mouths and makes more acid sites accessible to Py.
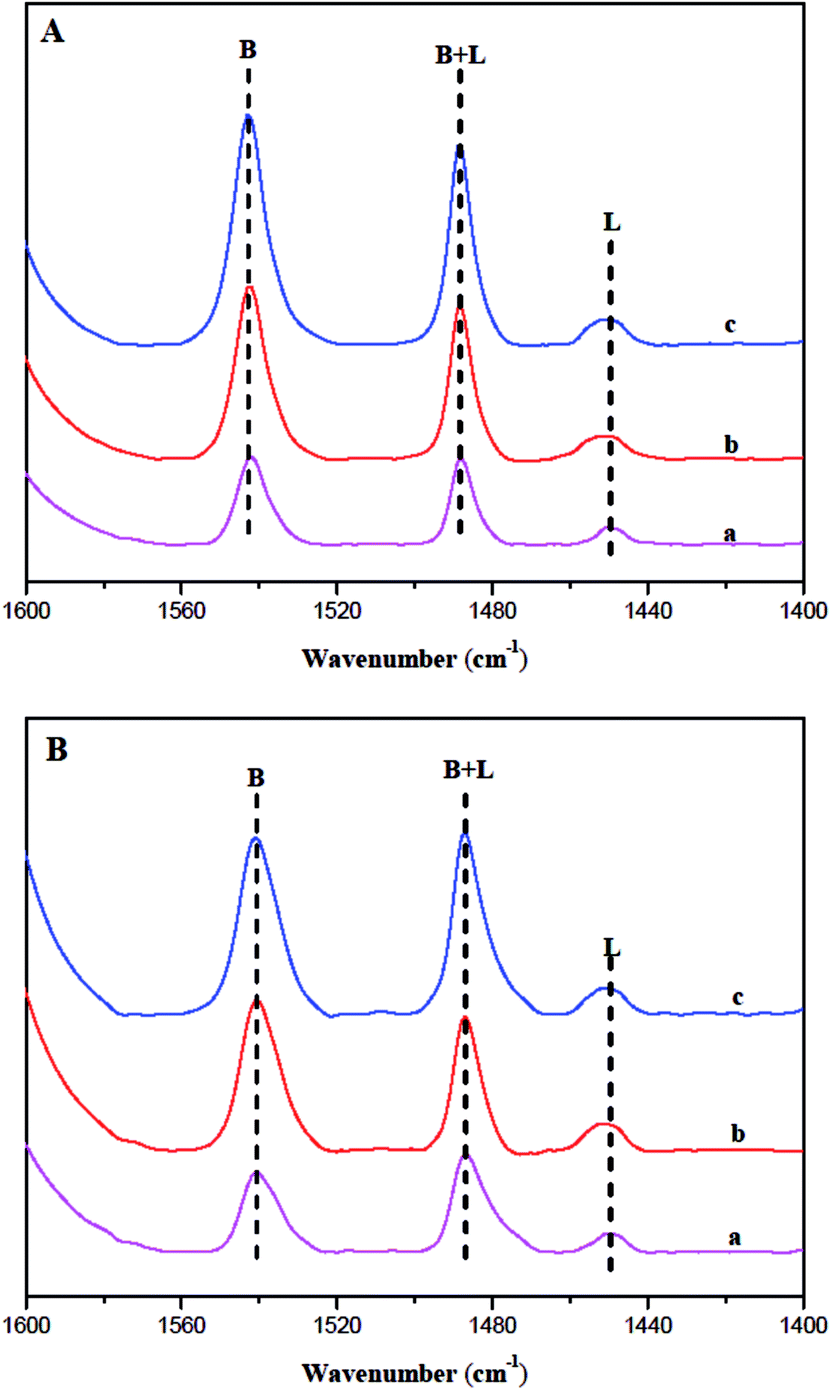 | ||
| Fig. 6 IR absorbance spectra of T-FER (a), S-FER (b), and N-FER (c) after adsorption of Py and desorption at (A) 200 °C and (B) 350 °C. | ||
| Sample | Acidity (μmol g−1) | ||||||
|---|---|---|---|---|---|---|---|
| WB | WL | SB | SL | TB | TL | TB+L | |
| T-FER | 11.1 | 1.2 | 67.5 | 10.2 | 78.6 | 11.4 | 90.0 |
| S-FER | 23.8 | 3.6 | 139.0 | 18.6 | 162.8 | 22.2 | 185.0 |
| N-FER | 62.8 | 3.0 | 169.2 | 18.0 | 231.9 | 21.00 | 252.9 |
The amount of carbonaceous depositions on ferrierite during skeletal isomerization of n-butene at 410 °C, WSHV = 10 h−1, and under atmospheric pressure for 2000 min, as determined by TGA is displayed in Fig. 7 and Table 3. According to the ref. 25 and 41, the temperature region can be divided into three parts. The temperature regions at 25–207 °C, 207–417 °C, and 417–717 °C correspond to the desorption of water, hydrogen-rich carbonaceous depositions (coke-type I), and highly condensed polyaromatic depositions (coke-type II), respectively. After the reaction run for TOS of 2000 min, there was a similar amount (8.0%) of carbonaceous depositions with a ratio of I/II = 0.054 formed on the three ferrierite catalysts. Jong demonstrated that carbon deposition was preferentially formed inside the ferrierite cage and channel system, followed by the outer surface of the zeolite in skeletal isomerization of n-butene.42,43 All the fresh catalysts had the same micropore volume, evidenced by nitrogen adsorption–desorption. Therefore, the coked catalysts should have a similar coke content when the micropore is almost filled.
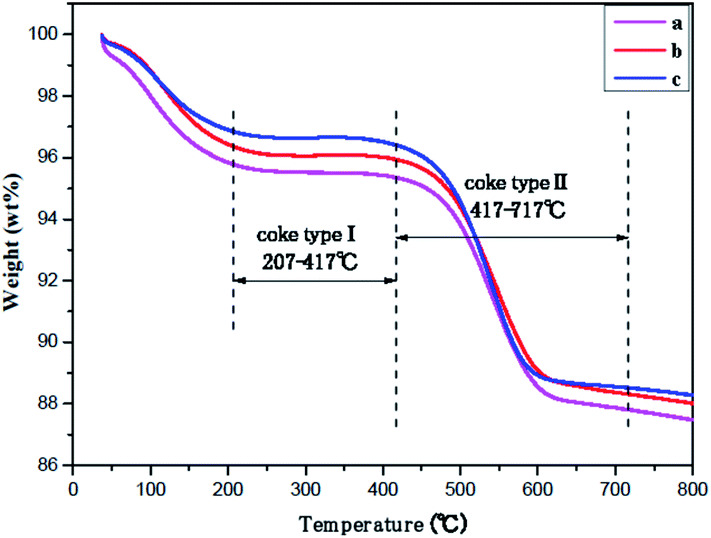 | ||
| Fig. 7 TGA curves over catalysts after 2000 min of stream under T = 410 °C, WHSV = 10 h−1, and atmospheric pressure: (a) T-FER (2000 min), (b) S-FER (2000 min), (c) N-FER (2000 min). | ||
| Sample | Coke I | Coke II | Coke I + II | Coke I/II |
|---|---|---|---|---|
| T-FER (2000 min) | 0.42% | 7.42% | 7.84% | 0.054 |
| S-FER (2000 min) | 0.41% | 7.62% | 8.03% | 0.054 |
| N-FER (2000 min) | 0.44% | 7.88% | 8.32% | 0.056 |
3.2 Catalytic properties
| Sample | WSHV (h−1) | C (%) | Siso (%) | Yiso (%) |
|---|---|---|---|---|
| T-FER | 3.5 | 83.8 | 17.1 | 14.4 |
| T-FER | 10 | 71.9 | 37.0 | 26.6 |
| T-FER | 35 | 52.7 | 60.8 | 32.1 |
| T-FER | 100 | 20.4 | 63.6 | 13.0 |
| S-FER | 3.5 | 84.4 | 16.3 | 13.7 |
| S-FER | 10 | 72.1 | 35.9 | 25.9 |
| S-FER | 35 | 52.7 | 69.9 | 36.9 |
| S-FER | 100 | 25.0 | 71.7 | 17.9 |
| N-FER | 3.5 | 86.0 | 15.3 | 13.1 |
| N-FER | 10 | 72.2 | 34.3 | 24.8 |
| N-FER | 35 | 52.9 | 79.3 | 41.8 |
| N-FER | 100 | 37.2 | 86.3 | 32.1 |
Despite the difference in morphology and size of the ferrierite catalysts, similar values of conversion and selectivity were obtained at lower WSHV (3.5 and 10 h−1). It suggests that all the acid sites in each of the independent channels were accessible at lower WSHV and that all the fresh ferrierite catalysts have a similar total acid amount and ratio of strong to weak acid, evidenced by NH3-TPD (Fig. 5A). In contrast, the higher the WSHV, the less n-butene that enters the 10-MR channels. Therefore, the pore mouth may dominate the reaction under higher WSHV. The conversion comparison among the three ferrierite catalysts at higher WSHV (100 h−1) shows that the highest conversion is obtained with N-FER, which presents the smallest crystal size but the highest number of pore mouth acid sites, as revealed by Py-IR. This is followed by S-FER, which also has a very small crystal size, but a lower number of pore mouth acid sites than N-FER. T-FER, with the smallest number of pore mouth acid sites among the three ferrierite catalysts, was the least active sample. In addition, it is interesting to note that the selectivity of the ferrierite catalysts was in the order: N-FER > S-FER > T-FER, which follows a similar order to the pore mouth Brønsted acid sites. According to the above results, it can be deduced that the formation of isobutene in ferrierite is related to the quantity of Brønsted acid sites present at or near the pore mouth of the 10-MR channels accessible to n-butene molecules. On the other hand, the presence of Lewis acid sites together with Brønsted acid sites would enhance the oligomerization44 and cracking.45 Therefore the non-selective bimolecular reaction is enhanced, which leads to a lower isobutene selectivity.
More importantly, we obtained a high isobutene selectivity (86.3%) with fresh N-FER. This indicates that high isobutene selectivity can be obtained without coking. According to the knowledge accumulated thus far, the monomolecular reaction mechanism proposes that the highly isobutene-selective active sites are the modified Brønsted acid sites,23,46 while the pseudo-monomolecular reaction mechanism considers that the highly isobutene-selective active sites are alkylaromatic tertiary carbenium ions on the coke molecules.24,47,48 In this study, we obtained high isobutene selectivity on fresh nano-sized ferrierite corresponding to the amount of Brønsted acid sites; therefore, a monomolecular reaction mechanism seems more likely.
 | ||
| Fig. 8 n-Butene conversion (A), isobutene selectivity (B), isobutene yield (C), and propene and pentene yield (D) for a long stream time on T-FER (a), S-FER (b), and N-FER (c). | ||
Although significant controversy remains on the reaction mechanism occurring over ferrierite, it is generally accepted that the nonselective bimolecular mechanism exists in fresh ferrierite.20,21,49,50 It can be considered that the monomolecular mechanism and bimolecular mechanism both existed in the fresh ferrierite, and the bimolecular reaction occurred more easily and faster.51 The carbonaceous deposits on the catalyst were produced rapidly in the first 100 min. Meanwhile, the non-selective acid sites were poisoned by coke,52,53 and the acid strength and density were also modified by coke54,55 on the coked ferrierite. Therefore, the non-selective bimolecular reaction was suppressed. This was confirmed by the decrease in propene and pentene. The monomolecular mechanism dominated the coked ferrierite and all the ferrierite catalysts exhibited a rapid decrease in conversion and a significant increase in selectivity. On the other hand, the nitrogen adsorption–desorption and TGA results show that Smic and Vmic of the coked ferrierite catalysts are almost fully blocked by carbonaceous depositions. The nitrogen adsorption–desorption and NH3-TPD results indicate that Sext and Vmes of the coked ferrierite catalysts are still accessible, and the coked N-FER (2000 min) has the largest values of Sext and Vmes (Sext = 33 m2 g−1 and Vmes = 0.08 cm3 g−1). The Sext (9 m2 g−1) and Vmes (0.04 cm3 g−1) of the coked S-FER (2000 min) is smaller, and those (Sext = 2 m2 g−1 and Vmes = 0.00 cm3 g−1) of the coked T-FER (2000 min) are the smallest. The amount of acid over these coked catalysts is in the order: N-FER (2000 min) > S-FER (2000 min) > T-FER (2000 min), which has the same sequence as that obtained with Sext and Vmes. The activity of the catalysts followed the same order with the amount of acid. Hence, we can conclude that the deactivation of ferrierite was caused by increased carbon deposition in the cage or channel of ferrierite, fully blocking the pore mouth and the external surface.46,47,56 Moreover, we found that both fresh and coked N-FER have bigger external surface area and mesopore volume, which leads to more pore mouth acid sites. This was evidenced by nitrogen adsorption–desorption, Py-IR and NH3-TPD. This explains why higher activity and better stability were obtained on N-FER after a longer stream time.
4. Conclusions
Ferrierite catalysts with different crystal morphologies and sizes were designed through hydrothermal synthesis in an alkali solution using different structure-directing agents and crystallization methods. All the catalysts obtained had a similar crystallinity and Si/Al ratio. The variation of crystal morphology and size did not change the total number of acid sites, but the decrease of crystal size increased the external surface area and mesopores, forming more pore mouth acid sites. In particular, nano-sized ferrierite, which showed more pore mouth acid sites and better stability and higher activity, was synthesized using pyrrolidine and TMAOH as double structure-directing agents under dynamic crystallization. Moreover, we observed that the initial activity and isobutene selectivity in skeletal isomerization of n-butene are related to the quantity and nature of acid and confirmed that the monomolecular reaction mechanism occurred on fresh ferrierite. Although high isobutene selectivity was obtained on fresh nano-sized ferrierite, better stability and higher selectivity required that carbonaceous materials modified the acid sites, suppressing the bimolecular reaction under a long stream time.Notes and references
- R. A. Sawichi, R. J. Pellet, D. G. Casey, R. V. Kessler, H. M. Huang, C. L. O'-Young and E. J. Kuhlmann, Hydrocarbon Eng., 1998, 3, 44–48 Search PubMed.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- M. Trombetta, G. Busca, S. Rossini, V. Piccoli, U. Cornaro, A. Guercio, R. Catani and R. J. Willey, J. Catal., 1998, 179, 581–596 CrossRef CAS.
- S. H. Lee, C. H. Shin and S. B. Hong, J. Catal., 2004, 223, 200–211 CrossRef CAS.
- L. Domokos, L. Lefferts, K. Seshan and J. A. Lercher, J. Catal., 2001, 203, 351–361 CrossRef CAS.
- P. A. Vaughan, Acta Crystallogr., 1966, 21, 983–990 CrossRef CAS.
- I. S. Kerr, Nature, 1966, 210, 294–295 CrossRef CAS.
- M. B. D. Santos, H. M. C. Andrade and A. J. S. Mascarenhas, Microporous Mesoporous Mater., 2016, 223, 105–113 CrossRef.
- M. Zhang, S. Xu, Y. Wei, J. Li, J. Chen, J. Wang, W. Zhang, S. Gao, X. Li, C. Wang and Z. Liu, RSC Adv., 2016, 6, 95855–95864 RSC.
- X. Chen, T. Todorova, A. Vimont, V. Ruaux, Z. Qin, J. P. Gilson and V. Valtchev, Microporous Mesoporous Mater., 2014, 200, 334–342 CrossRef CAS.
- P. Frontera, S. Candamano, A. Macario, F. Creab, L. A. Scarpino and P. L. Antonucci, Mater. Lett., 2013, 104, 72–75 CrossRef CAS.
- R. Arletti, G. Vezzalini, S. Quartieri, F. D. Renz and V. Dmitriev, Microporous Mesoporous Mater., 2014, 191, 17–27 CrossRef.
- T. Xue, H. P. Liu and Y. M. Wang, RSC Adv., 2015, 5, 12131–12138 RSC.
- P. Wuamprakhon, C. Wattanakit, C. Warakulwit, T. Yutthalekha, W. Wannapakdee, S. Ittisanronnachai and J. Limtrakul, Microporous Mesoporous Mater., 2016, 219, 1–9 CrossRef CAS.
- A. B. Pinar, L. Gomez-Hortiguela and J. Perez-Pariente, Chem. Mater., 2007, 19, 5617–5626 CrossRef.
- Y. Lee, M. B. Park, P. S. Kim, A. Vicente, C. Fernandez, I. S. Nam and S. B. Hong, ACS Catal., 2013, 3, 617–621 CrossRef CAS.
- B. Yang, J. G. Jiang, H. Xu, Y. Liu, H. Peng and P. Wu, Appl. Catal., A, 2013, 455, 107–113 CrossRef CAS.
- B. Yang, J. G. Jiang, H. Xu, Y. Liu, H. Peng and P. Wu, Microporous Mesoporous Mater., 2015, 203, 54–62 CrossRef CAS.
- Y. Hu, L. Liu, H. Zhang, L. Hu, C. Zhang and H. Zhang, React. Kinet., Mech. Catal., 2014, 112, 241–248 CrossRef CAS.
- J. Houžvička, O. Diefenbach and V. Ponec, J. Catal., 1996, 164, 288–300 CrossRef.
- M. Guisnet, P. Andy, N. S. Gnep, E. Benazzi and C. Travers, J. Catal., 1996, 158, 551–560 CrossRef CAS.
- D. M. Brouwer and H. Hogeveen, Prog. Phys. Org. Chem., 1972, 9, 179–240 CrossRef CAS.
- D. Jo, S. B. Hong and M. A. Camblor, ACS Catal., 2015, 5, 2270–2274 CrossRef CAS.
- P. Andy, N. S. Gnep, M. Guisnet, E. Benazzi and C. Traversy, J. Catal., 1998, 173, 322–332 CrossRef CAS.
- Y. P. Khitev, I. I. Ivanova, Y. G. Kolyagin and O. A. Ponomareva, Appl. Catal., A, 2012, 17, 124–135 CrossRef.
- R. Anand, R. B. Khomane, B. S. Rao and B. D. Kulkarni, Catal. Lett., 2002, 78, 189–194 CrossRef CAS.
- H. C. Lee, H. C. Woo, S. H. Chung, H. J. Kim, K. H. Lee and J. S. Lee, J. Catal., 2002, 211, 216–225 CrossRef CAS.
- X. Li, X. Liu, S. Liu, S. Xie, X. Zhu, F. Chen and L. Xu, RSC Adv., 2013, 3, 16549–16557 RSC.
- C. Liu, W. Gu, D. Kong and H. Guo, Microporous Mesoporous Mater., 2014, 183, 30–36 CrossRef CAS.
- N. L. Chauhan, Z. V. P. Murthy, J. Das and P. A. Parikh, Indian J. Chem. Technol., 2011, 18, 335–342 CAS.
- A. B. Pinar, P. A. Wright and L. Gómezhortigüela, Microporous Mesoporous Mater., 2010, 129, 164–172 CrossRef CAS.
- R. García, L. Gómezhortigüela, I. Díaz, E. Sastre and J. Pérezpariente, Chem. Mater., 2008, 20, 1099–1107 CrossRef.
- Y. R. Leshkov, M. Moliner and M. E. Davis, J. Phys. Chem. C, 2011, 115, 1096–1102 Search PubMed.
- R. García, L. Gómezhortigüela, T. Blascoc and J. Pérezparientea, Microporous Mesoporous Mater., 2010, 132, 375–383 CrossRef.
- L. Domokos, L. Lefferts, K. Seshan and J. A. Lercher, J. Mol. Catal. A: Chem., 2000, 162, 147–157 CrossRef CAS.
- P. Cañizares and A. Carrero, Appl. Catal., A, 2003, 248, 227–237 CrossRef.
- B. Wichterlová, Z. Tvarůžková, Z. Sobalík and P. Sarv, Microporous Mesoporous Mater., 1998, 24, 223–233 CrossRef.
- F. T. Starzyk, I. Stan, S. Abelló, A. Bonilla, K. Thomas, C. Fernandez, J. P. Gilson and J. P. Ramírez, J. Catal., 2009, 264, 11–14 CrossRef.
- J. B. Uytterhoeven, L. G. Christner and W. K. Hall, J. Phys. Chem., 1965, 69, 2117–2126 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- G. Elordi, M. Olazar, G. Lopez, P. Castano and J. Bilbao, Appl. Catal., B, 2011, 102, 224–231 CrossRef CAS.
- S. V. Donk, E. Bus, A. Broersma, J. H. Bitter and K. P. D. Jong, Appl. Catal., A, 2002, 237, 149–159 CrossRef.
- W. Q. Xu, Y. G. Yin, J. S. L. Suib and C. L. O'-Young, J. Phys. Chem., 1995, 99, 758–765 CrossRef CAS.
- S. Beran, P. Jiru and L. Kubelkova, J. Mol. Catal., 1982, 16, 299–304 CrossRef CAS.
- S. E. Tung and E. McIninch, J. Catal., 1968, 10, 166–174 CrossRef CAS.
- M. A. Asensi and A. MartíNez, Appl. Catal., A, 1999, 183, 155–165 CrossRef CAS.
- B. D. Ménorval, P. Ayrault, N. S. Gnep and M. Guisnet, J. Catal., 2005, 230, 38–51 CrossRef.
- B. D. Ménorval, P. Ayrault, N. S. Gnep and M. Guisnet, Appl. Catal., A, 2006, 1, 1–13 CrossRef.
- J. Houžvička and V. Ponec, Ind. Eng. Chem. Res., 1997, 36, 1424–1430 CrossRef.
- A. G. Stepanov, M. V. Luzgin, S. S. Arzumanov, H. Ernst and D. Freude, J. Catal., 2002, 211, 165–172 CAS.
- P. Mériaudeau, V. A. Tuan, N. H. Le and G. Szabo, J. Catal., 1997, 169, 397–399 CrossRef.
- W. Q. Xu, Y. G. Yin, S. L. Suib, J. C. Edwards and C. L. O. Young, J. Phys. Chem., 1995, 99, 9443–9451 CrossRef CAS.
- P. Mériaudeau, C. Naccache, L. N. Hung, V. A. Tuan and G. Szabo, J. Mol. Catal. A: Chem., 1997, 123, 1–4 CrossRef.
- W. Q. Xu, Y. G. Yin, S. L. Suib, J. C. Edwards and C. L. O. Young, J. Catal., 1996, 163, 232–244 CrossRef CAS.
- B. Wichterlova, N. Zilkova, E. Uvarova, J. Cejka, P. Sarv, C. Paganini and J. A. Lercher, Appl. Catal., A, 1999, 182, 297–308 CrossRef CAS.
- S. V. Donk, J. H. Bitter and K. P. D. Jong, Appl. Catal., A, 2001, 212, 97–116 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |