
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical and diffuse reflectance study on tetrahedral ε-Keggin-based metal–organic frameworks†
Zhong-Bin Nieac,
Min Zhangc,
Tan Su*bd,
Liang Zhaoc,
Yuan-Yuan Wangc,
Guang-Yan Sun *a and
Zhong-Min Suc
*a and
Zhong-Min Suc
aFaculty of Chemistry, Yanbian University, Yanji 133002, P. R. China. E-mail: gysun@ybu.edu.cn
bLaboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun 130021, P. R. China. E-mail: sutan_jlu@jlu.edu.cn
cInstitute of Functional Material Chemistry, National & Local United Engineering Lab for Power Battery, Department of Chemistry, Northeast Normal University, Changchun 130024, P. R. China
dState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China
First published on 20th June 2017
Abstract
Herein, two new polyoxometalate-based metal–organic frameworks consisting of Zn-ε-Keggin nodes and semi-rigid organic linkers, [TBA]3[PMo8VMo4VIO36(OH)4Zn4][CPBPC]2 (1) and [TBA]3[PMo8VMo4VIO37(OH)3Zn4][PTABAB][HPTABAB]·7H2O (2) (where CPBPC = 4′-(4-carboxylatophenoxy)-[1,1′-biphenyl]-4-carboxylate and PTABAB = 4,4′-((6-(phenylamino)-1,3,5-triazine-2,4-diyl)bis(azanediyl))dibenzoate), were successfully synthesized. In 1, Zn-ε-Keggin units were bridged by CPBPC fragments to form an eight-fold interpenetrating framework with diamond topology. In 2, the adjacent 2D layers are constructed from Zn-ε-Keggin units and PTABAB moieties interlocked with each other to generate a double layer, which was further packed in an AAA sequence. The electrochemical behaviours of these two compounds were studied, and they exhibit good electrocatalytic activities towards the reduction of nitrite ions. Moreover, their diffuse reflectance was also investigated to obtain their bandgaps of around 1.1 eV, suggesting that they are potential and promising semiconductor materials.
Introduction
Research on metal–organic frameworks (MOFs), also known as porous coordination polymers (PCPs), has developed rapidly driven largely by their underlying topologies in relation to designed synthesis1 and the upsurge of applications in gas storage and separation, sensing, catalysis, light harvesting and so on.2,3 As an emerging class of porous materials, MOFs assemble through coordination bonds between metal-based nodes (ions or clusters) and bridging organic linkers.4 Because of their structural and functional tunability, the study of MOFs has become one of the fastest growing topics in the fields of synthetic chemistry and materials science.5 In addition, MOF chemistry provides the potential to introduce desired properties and functionality prior to the assembly process by preselecting building blocks to contain desired structural and geometrical information that codes for a given underlying net. In this context, the molecular building block (MBB) approach, which permits access to MOFs with specific topologies, caught the researchers' eyes.6 However, the successful employment of MBB approach for the rational design and preparation of MOFs requires to meet the satisfaction of following factors: (i) the selection of an ideal topology that is exclusive for the assembly of its corresponding basic building units and (ii) the isolation of reaction conditions that consistently allow the in situ formation of the desired inorganic MBB, matching the basic building units of the targeted net.Polyoxometalate (POM) ions represent a well-defined library of inorganic clusters with oxygen-rich surfaces that can range in size from the nano- to micrometer scale.7,8 Interestingly, their properties such as solubility, stability, redox potential, and acidity can be finely-tuned by varying constituent elements.9 As a result, POMs offer an ideal platform to serve as secondary building units for the design and construction of POM-based MOF materials. However, it is still tremendously challenging to construct porous POM-based MOFs in which POM units acted as noded to directly connect with organic ligands.10
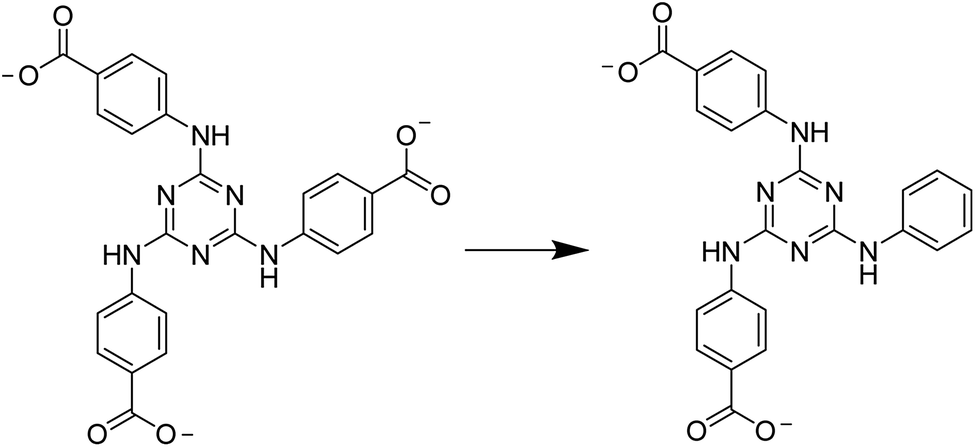
By taking into account the excellent features, ε-Keggin polymolybdate unit grafted by four Zn(II) ions (noted as Zn-ε-Keggin) was chosen for the following reasons: (i) Zn-ε-Keggin core can be formed in situ under mild conditions,11,12 (ii) the four exposed Zn(II) ions on the surface of Zn-ε-Keggin favours for the formation of tetrahedral arrangement,13 as those of O atoms in a tetrahedral SiO4 unit, and (iii) Zn-ε-Keggin unit possesses certain redox activity on the basis of Mo element. In this work, semi-rigid 4′-(4-carboxylatophenoxy)-[1,1′-biphenyl]-4-carboxylate (CPBPC) and 4,4′,4′′-((1,3,5-triazine-2,4,6-triyl)tris(azanediyl))tribenzoate (TATAB) were selected as bridging linkers to synthesize POM-based MOFs. Fortunately, [TBA]3[PMo8VMo4VIO36(OH)4Zn4][CPBPC]2 (1) and [TBA]3[PMo8VMo4VIO37(OH)3Zn4][PTABAB][HPTABAB]·7H2O (2) were isolated after a series of parallel experiments (PTABAB represents 4,4′-((6-(phenylamino)-1,3,5-triazine-2,4-diyl)bis(azanediyl))dibenzoate). Here, a decarboxylation reaction of TATAB was observed to generate PTABAB in situ under the reaction conditions. In these two compounds, Zn-ε-Keggin units were acted as metal nodes to directly connect with CPBPC or PTABAB fragments, respectively. Compound 1 possesses an eight-fold interpenetrating framework with diamond (dia) topology, whereas the adjacent 2D layers construct by Zn-ε-Keggin units and PTABAB moieties interlocked each other to generate a double layer in 2, which were further packed in an AAA sequence. The electrochemical behaviours of these two compounds and their electrocatalytic activity toward the reduction of nitrite ions were investigated. In addition, their diffuse reflectance performance was also measured and discussed.
Results and discussion
When semi-rigid H2CPBPC was introduced to the reaction system, crystals 1 was produced. Single-crystal X-ray diffraction analysis suggests that compound 1 crystallizes in the orthorhombic Pbcn space group (Table S1†). In the asymmetric unit of 1, there are half {ε-PMo8VMo4VIO36(OH)4Zn4} unit, one kind of CPBPC moiety (Fig. S1†) and 1.5 TBA+ ions. So far, only {ε-PMo12O40} core capped by four metal ions (La3+, Zn2+, or Ni2+) were documented in the literature.11–14 No naked ε-Keggin ions were isolated that presumably results from the high negative charge of naked ε-Keggin unit. In the {ε-PMo8VMo4VIO36(OH)4Zn4} unit, the Mo–Mo distances are around 2.6 and 3.2 Å, suggesting the presence of eight Mo(V) and four Mo(VI) species as reported. The valence bond calculations15 for compound 1 suggest that Mo(1), Mo(2), Mo(4) and Mo(6) atoms are in +5 oxidation state, while Mo(3) and Mo(5) centres are in +6 oxidation state (Table S2†). In addition, O(7) and O(19) sites are protonated according to the valence bond calculations. Moreover, the oxidation states of Mo element was evidenced by X-ray photoelectron spectroscopy (XPS) analysis (Fig. S2†). In compound 1, all four Zn2+ ions grafted on the ε-Keggin unit are four-coordinated by four oxygen atoms in a typical tetrahedral geometry (Fig. 1a), in which three oxygen atoms are provided by the polymolybdate cluster and another oxygen atom coming from the carboxylate group of CPBPC moiety. And the ε-Keggin units were extended by four CPBPC fragments to give rise to a 3D framework with dia topology (Fig. 1b). The large adamantanoid cage in a single dia net possesses a maximum dimension of 64.698 × 58.932 × 54.782 Å. The pore space of an individual net is sufficiently large to give rise to an eight-fold interpenetration in 1 (Fig. 1c and d and S3†).16 The interpenetration degree is up to ten found within diamondoid nets exclusively on the basis of coordinative bonds.17 The TBA+ cations are distributed orderly in the channel, acting as space filling and structural-templated species, as well as charge balance agents. Strikingly, even after interpenetration, the effective free volume of 1 reached 56.6% of the crystal volume (7386.9 Å3 out of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 054.5 Å3 unit cell volume) based on the PLATON calculations.18
054.5 Å3 unit cell volume) based on the PLATON calculations.18
 | ||
| Fig. 1 The structure of 1: (a) representation of the coordination mode of the Zn-ε-POM unit, (b) a single dia cage, (c) the 3D spacefill mode and (d) the schematic view of the [4 + 4] interpenetrated topology. | ||
Single-crystal X-ray diffraction analysis reveals that 2 crystallizes in the monoclinic space group P21/c. The framework of 2 was composed of one {ε-PMo8VMo4VIO37(OH)3Zn4} unit, one fully deprotonated PTABAB and one HPTABAB fragment. Similarly, the Mo oxidation states were also confirmed by valence bond calculations and XPS measurement (Table S2 and Fig. S4†), which is consistent with the result in compound 1. However, O(11) and O(25) sites are protonated, another H+ was delocalized to POM unit according to the valence bond calculations. The formula of 2 was determined as [TBA]3[PMo8VMo4VIO37(OH)3Zn4][PTABAB][HPTABAB]·7H2O by consideration of charge balance, squeeze result and TG analysis. It should be noted that the decarboxylation reaction of TATAB was happened and PTABAB was formed in situ under the hydrothermal conditions (Scheme 1). The hydrothermal decarboxylation reactions have recently been shown to occur in the presence of metal ions.19 In 2, each ε-Keggin unit was connected to four PTABAB fragments through four capped Zn(II) ions on the POM surface (Fig. 2a). The two types of PTABAB both play the role of bridging linkers with two sites (L1: two oxygen atoms from two different carboxylate groups, and L2: one oxygen atom from carboxylate group and one N atom from triazine ring, Fig. S5†). The Zn–O bond lengths were within the range of 1.89–1.96 Å, whilst the Zn–N bond length was 2.01 Å. Such connectivity leads to the formation of a 2D layer structure (Fig. 2b). Furthermore, two adjacent layers interlocked each other, resulting in a double layer (Fig. 2c and d and S6†). In addition, the adjacent double layers were packed in an AAA array (Fig. 2e and f). The disordered TBA+ countercations and water molecules were resided in the space formed between adjacent layers.
 | ||
| Scheme 1 In situ hydrothermal decarboxylation reaction from TATAB to PTABAB. | ||
 | ||
| Fig. 2 The structure of 2: (a) the coordination mode of the Zn-ε-POM unit, (b) the 2D layer formed by Zn-ε-Keggin cores and PTABAB fragments, the two-fold interpenetrated structure (c) and topology (d) between adjacent layers, (e) the AAA array and (f) topology of adjacent two-fold interpenetrated layers. | ||
The PXRD patterns of compounds 1 and 2 are presented in Fig. S7 and S8.† The diffraction peaks of both simulated and experimental patterns match well at the key positions, indicating the phase purities of these two compounds. Furthermore, compounds 1 and 2 are air-stable, maintaining their crystallinities for at least several months, and no efflorescence was observed. The IR spectra of these two compounds were presented in Fig. S9,† which confirms the presence of POM units, linkers and TBA+ ions. The TG measurements of the two compounds were carried out under N2 atmosphere from room temperature to 700 °C at a heating rate of 5 °C min−1 (Fig. S10†). Compound 1 shows a good thermal stability with decomposition starting at 325 °C. Subsequently, a continuous weight loss of ∼40% from 325 to 630 °C can be assigned to the release of all organic ligands and TBA+ ions. Compound 2 exhibits a continuous weight loss step from room temperate to 640 °C (∼45%), corresponding to the loss of all uncoordinated water molecules, organic ligands, and TBA+ ions.
The electrochemical behaviours of 1 and 2 on glassy carbon electrode (GCE) in 0.1 mol L−1 H2SO4 solution were carried out. The electrochemical behaviours of the two as-synthesized compounds are similar except for some slight potential shifts (Fig. 3 and S11†). As a result, we set 2 as an example to describe their electrochemical properties. As shown in Fig. 3a, the typical cyclic voltammograms (CVs) of 2 were studied at potentials covering the range from −0.4 to +0.6 V (vs. Ag/AgCl) at different scan rates (from 25 to 400 mV s−1). The half-wave potentials E1/2 = (Epa + Epc)/2 for 2 appeared at approximately −0.188 (I–I′), +0.079 (II–II′), and +0.183 V (III–III′) (scan rate: 50 mV s−1), respectively. These data are in agreement with those reported in the literature.20 When the scan rates were varied from 25 to 400 mV s−1, the peak potentials varied gradually: the cathodic peak potentials shift toward the negative direction and the corresponding anodic peak potentials to the positive direction with increasing scan rates. In addition, the approximate proportionality of the three redox peak currents to the scan rates from 25 to 400 mV s−1, indicating that the redox process is surface-controlled for 1- and 2-GCE (Fig. S12†).21 In addition, 1- and 2-GCE are highly stable, and the peak potentials change little with the increasing scan rates, which is especially useful for electrocatalytic study.
 | ||
| Fig. 3 The CVs of 2-GCE measured in 0.1 mol L−1 H2SO4 aqueous solution (a) at different scan rates (mV s−1) and (b) at the scan rate of 50 mV s−1 containing different concentrations of NaNO2, respectively. | ||
NaNO2 is present in processed and cured meats, which is used to preserve the meat. However, during the cooking or digestion process, NaNO2 can reacts with amine to form nitrosamines, which is very powerful carcinogen. In this sense, the detection and determination of NaNO2 is of great importance in biological denitrification or industrial companies related to food.22 Therefore, the electrocatalytic activities of these two compounds were investigated. Upon the addition of NaNO2, the reduction peak currents increase (Fig. 3b and S11b†), and the corresponding oxidation peak currents decrease, suggesting that nitrite ion is reduced.23 Both 1- and 2-GCE show better electrocatalytic activities towards reduction of NaNO2 (Fig. S13 and S14†) compared with other reported POM-based compounds.24
The UV-Vis diffuse reflectance spectra were monitored to achieve the optical bandgaps (Eg) of these two materials. Generally, the Eg was obtained as the intersection point between the energy axis and the line extrapolated from the linear portion of the adsorption edge in a plot of the Kubelka–Munk function F against E.25 As shown in Fig. 4, the optical absorption associated with Eg can be assessed at 1.10 and 1.06 eV for 1 and 2, suggesting the presence of an optical band gap. It should be noted that the Eg values of these two compounds are much lower than the reported POM-based materials.26 The results suggest that these two compounds are potential and promising semiconductor materials, and the extended Zn-ε-Keggin unit in the hybrid structures appears to be mainly responsible for their optical bandgaps.27
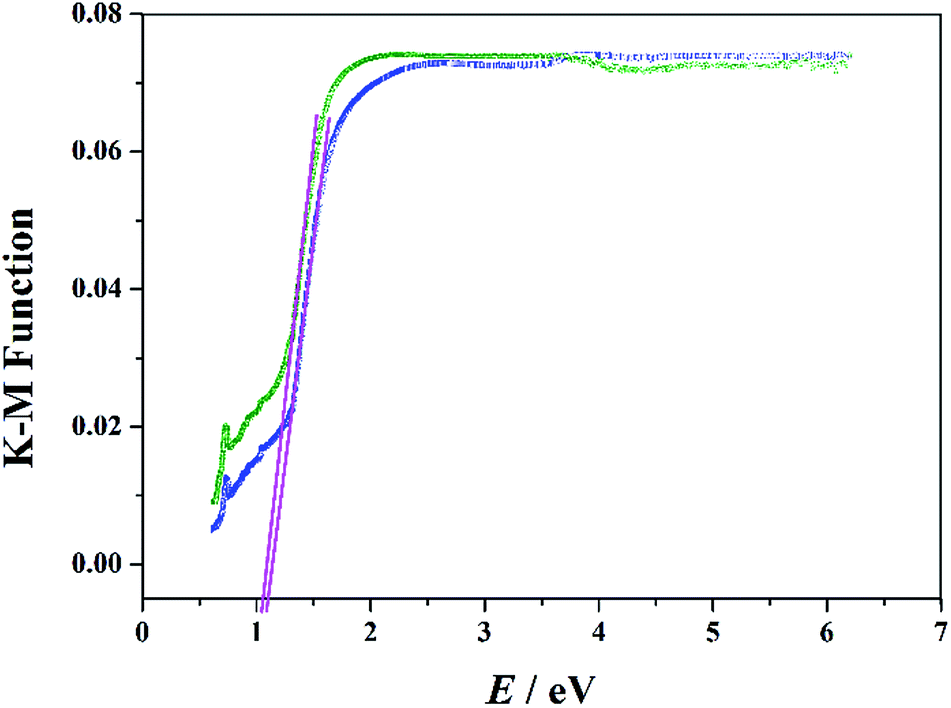 | ||
| Fig. 4 Diffuse reflectance UV-Vis spectra of Kubelka–Munk function versus energy for compounds 1 (blue line) and 2 (olive line). | ||
Conclusions
In conclusion, two novel POM-based MOFs were successful synthesized and characterized. They were both composed of Zn-ε-Keggin nodes and semi-rigid organic linkers. In compound 1, Zn-ε-Keggin cores were bridged by CPBPC moieties to generate a 3D dia framework with an eight-fold interpenetration, which can be described as two sets of normal fourfold nets. The adjacent 2D layers construct by Zn-ε-Keggin units and PTABAB fragments interlocked each other in compound 2 to generate a double layer, which was further stacked in an AAA sequence. In addition, these two compounds exhibit good electrocatalytic activities toward the reduction of nitrite ion, which is useful for the detection of NaNO2 in food chemistry. Furthermore, the title compounds possessing narrow bandgaps are potential semiconductor materials on the basis of the diffuse reflectance measurements. It is expected to design and prepare more novel Zn-ε-Keggin-based MOFs, and more investigation is ongoing in our lab.Acknowledgements
We thank the National Natural Science Foundation of China (Grants: 21401021 and 21501062), the Science and Technology Development Planning of Jilin Province (Grant: 20140203006GX) and the Open Project of the State Key Laboratory of Inorganic Synthesis and Preparative Chemistry (Grant: 2015-31) for financial support of this work.References
- (a) O. Delgado-Friedrichs and M. O'Keeffe, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, 351 CrossRef; (b) M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782 CrossRef PubMed.
- (a) J. Y. Lee, O. M. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC; (b) A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260 CrossRef CAS PubMed; (c) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC; (d) Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657 RSC; (e) M. Zhang, Z.-Y. Gu, M. Bosch, Z. Perry and H.-C. Zhou, Coord. Chem. Rev., 2015, 293–294, 327 CrossRef CAS.
- (a) H.-J. Son, S. Jin, S. Patwardhan, S. J. Wezenberg, N. C. Jeong, M. So, C. E. Wilmer, A. A. Sarjeant, G. C. Schatz, R. Q. Snurr, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 862 CrossRef CAS PubMed; (b) H. Sato, W. Kosaka, R. Matsuda, A. Hori, Y. Hijikata, R. V. Belosludov, S. Sakaki, M. Takata and S. Kitagawa, Science, 2014, 343, 167 CrossRef CAS PubMed; (c) B. Ugale, S. S. Dhankhar and C. M. Nagaraja, Inorg. Chem. Front., 2017, 4, 348 RSC.
- M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343 CrossRef CAS PubMed.
- (a) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed; (b) H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846 CrossRef CAS PubMed; (c) B. Tu, Q. Pang, E. Ning, W. Yan, Y. Qi, D. Wu and Q. Li, J. Am. Chem. Soc., 2015, 137, 13456 CrossRef CAS PubMed; (d) J.-S. Qin, D.-Y. Du, M. Li, X.-Z. Lian, L.-Z. Dong, M. Bosch, Z.-M. Su, Q. Zhang, S.-L. Li, Y.-Q. Lan, S. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 5299 CrossRef CAS PubMed; (e) S. Yuan, Y.-P. Chen, J.-S. Qin, W. Lu, L. Zou, Q. Zhang, X. Wang, X. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 8912 CrossRef CAS PubMed; (f) B. Zhou, Y. Hua, F. Cheng, J. Duan, L. Chen and W. Jin, Inorg. Chem. Front., 2017, 4, 234 RSC.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141 RSC.
- (a) H. N. Miras, J. Yan, D.-L. Long and L. Cronin, Chem. Soc. Rev., 2012, 41, 7403 RSC; (b) D.-Y. Du, L.-K. Yan, Z.-M. Su, S.-L. Li, Y. Q. Lan and E.-B. Wang, Coord. Chem. Rev., 2013, 257, 702 CrossRef CAS; (c) H. N. Miras, L. Vilà-Nadal and L. Cronin, Chem. Soc. Rev., 2014, 43, 5679 RSC.
- (a) J. Zhang, J. Hao, Y. Wei, F. Xiao, P. Yin and L. Wang, J. Am. Chem. Soc., 2010, 132, 14 CrossRef CAS PubMed; (b) X. Fang, P. Kögerler, Y. Furukawa, M. Speldrich and M. Luban, Angew. Chem., Int. Ed., 2011, 50, 5212 CrossRef CAS PubMed; (c) D.-Y. Du, J.-S. Qin, T.-T. Wang, S.-L. Li, Z.-M. Su, K.-Z. Shao, Y.-Q. Lan, X.-L. Wang and E.-B. Wang, Chem. Sci., 2012, 3, 705 RSC.
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag, Berlin, 1983 Search PubMed.
- D.-Y. Du, J.-S. Qin, S.-L. Li, Z.-M. Su and Y.-Q. Lan, Chem. Soc. Rev., 2014, 43, 4615 RSC.
- (a) A. Dolbecq, C. Mellot-Draznieks, P. Mialane, J. Marrot, G. Férey and F. Sécheresse, Eur. J. Inorg. Chem., 2005, 3009 CrossRef; (b) A. Dolbecq, P. Mialane, F. Sécheresse, B. Keita and L. Nadjo, Chem. Commun., 2012, 48, 8299 RSC.
- (a) L. M. R. Albelo, A. R. Ruiz-Salvador, D. W. Lewis, A. Gómez, P. Mialane, J. Marrot, A. Dolbecq, A. Sampierie and C. Mellot-Draznieks, Phys. Chem. Chem. Phys., 2010, 12, 8632 RSC; (b) G. Rousseau, L. M. Rodriguez-Albelo, W. Salomon, P. Mialane, J. Marrot, F. Doungmene, I. Mbomekallé, P. de Oliveira and A. Dolbecq, Cryst. Growth Des., 2015, 15, 449 CrossRef CAS.
- (a) L. M. Rodriguez-Albelo, A. R. Ruiz-Salvador, A. Sampieri, D. W. Lewis, A. Gómez, B. Nohra, P. Mialane, J. Marrot, F. Sécheresse, C. Mellot-Draznieks, R. N. Biboum, B. Keita, L. Nadjo and A. Dolbecq, J. Am. Chem. Soc., 2009, 131, 16078 CrossRef PubMed; (b) B. Nohra, H. E. Moll, L. M. R. Albelo, P. Mialane, J. Marrot, C. Mellot-Draznieks, M. O'Keeffe, R. N. Biboum, J. Lemaire, B. Keita, L. Nadjo and A. Dolbecq, J. Am. Chem. Soc., 2011, 133, 13363 CrossRef CAS PubMed.
- (a) P. Mialane, A. Dolbecq, L. Lisnard, A. Mallard, J. Marrot and F. Sécheresse, Angew. Chem., Int. Ed., 2002, 41, 2398 CrossRef CAS; (b) A. Dolbecq, P. Mialane, L. Lisnard, J. Marrot and F. Sécheresse, Chem.–Eur. J., 2003, 9, 2914 CrossRef CAS PubMed; (c) W. Wang, L. Xu, G. Gao, L. Liu and X. Liu, CrystEngComm, 2009, 11, 2488 RSC.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192 CrossRef.
- H.-L. Jiang, T. A. Makal and H.-C. Zhou, Coord. Chem. Rev., 2013, 257, 2232 CrossRef CAS.
- L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, Chem.–Eur. J., 2002, 8, 1519 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- (a) W. Chen, H. Yuan, J. Wang, Z. Liu, J. Xu, M. Yang and J. Chen, J. Am. Chem. Soc., 2003, 125, 9266 CrossRef CAS PubMed; (b) S.-T. Zheng, J. Zhang and G.-Y. Yang, Angew. Chem., Int. Ed., 2008, 47, 39093 Search PubMed.
- L. Qian and X. R. Yang, Electrochem. Commun., 2005, 7, 547 CrossRef CAS.
- L. Cheng, X. M. Zhang, X. D. Xi and S. J. Dong, J. Electroanal. Chem., 1996, 407, 97 CrossRef.
- (a) B. Keita, A. Belhouari, L. Nadjo and R. Contant, J. Electroanal. Chem., 1995, 381, 243 CrossRef; (b) M. J. Moorcroft, J. Davis and R. G. Compton, Talanta, 2001, 54, 785 CrossRef CAS PubMed.
- (a) D. Du, Y. Lan, X. Wang, K. Shao, H. Wang and Z. Su, Solid State Sci., 2010, 12, 128 CrossRef CAS; (b) J.-S. Qin, D.-Y. Du, W. Guan, X.-J. Bo, Y.-F. Li, L.-P. Guo, Z.-M. Su, Y.-Y. Wang, Y.-Q. Lan and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 7169 CrossRef CAS PubMed.
- (a) Y.-Q. Lan, S.-L. Li, K.-Z. Shao, X.-L. Wang and Z.-M. Su, Dalton Trans., 2008, 3824 RSC; (b) Y. Lan, S. Li, K. Shao, X. Wang, X. Hao and Z. Su, Dalton Trans., 2009, 940 RSC; (c) Z. Shi, Z. Zhang, J. Peng, X. Yu and X. Wang, CrystEngComm, 2013, 15, 7199 RSC.
- J. I. Pankove, Optical Processes in Semiconductors, Prentice-Hall, Englewood Cliffs, New Jersey, 1971, p. 34 Search PubMed.
- (a) Y. Xia, P.-F. Wu, Y.-G. Wei, Y. Wang and H.-Y. Guo, Cryst. Growth Des., 2006, 6, 253 CrossRef CAS; (b) H. Zang, Y. Lan, G. Yang, X. Wang, K. Shao, G. Xu and Z. Su, CrystEngComm, 2010, 12, 434 RSC; (c) D. Du, J. Qin, Y. Li, S. Li, Y. Lan, X. Wang, K. Shao, Z. Su and E. Wang, Chem. Commun., 2011, 47, 2832 RSC.
- Q.-G. Zhai, X.-Y. Wu, S.-M. Chen, Z.-G. Zhao and C.-Z. Lu, Inorg. Chem., 2007, 46, 5046 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic data and figures. CCDC 1538353 and 1538354. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra04771a |
| This journal is © The Royal Society of Chemistry 2017 |