
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Temperature-responsive methacrylamide polyampholytes†
Lucy G. Weaver a,
Regine Stockmanna,
San H. Thangbc and
Almar Postma*b
a,
Regine Stockmanna,
San H. Thangbc and
Almar Postma*b
aCSIRO Agriculture and Food, Private Bag 16, Werribee, VIC 3030, Australia
bCSIRO Manufacturing, Bag 10, Clayton South, VIC 3169, Australia. E-mail: almar.postma@csiro.au
cMonash University School of Chemistry, Clayton Campus, VIC 3800, Australia
First published on 15th June 2017
Abstract
A series of methacrylamide-based polyampholytes were synthesised for the first time and their temperature responsive properties investigated. N-[3-(Dimethylamino)propyl]methacrylamide (DMAPMAm) and methacrylic acid (MAA) were copolymerised in equimolar amounts along with N-(tert-butyl)methacrylamide (tBMAm) via RAFT polymerisation, forming a series of polymers with varying tBMAm content. A variety of solvents were assessed to determine the optimal conditions for synthesis. The solution properties of the resultant polymers were then determined via turbidimetry and NMR. Results from this study show that tBMAm was an effective modulator of TCP events when it was present in levels of 16% or more of the total monomers in the polymer chain. Below this threshold, polymers displayed properties akin to traditional polyampholytes due to the antipolyelectrolyte effect. Above this level however, the polymers displayed properties analogous to other temperature-responsive polymer systems. Interestingly changes in both pH and addition of salt led to an increase in the polymer cloud points, and in some cases loss of thermoresponsivity entirely, despite the high level of anionic and cationic charges present in the side chains.
Introduction
Polymers that reversibly change their solution conformation in response to an applied external stimulus have found use in drug delivery, chromatography and tissue engineering, amongst other applications.1–4 Termed stimuli-responsive or smart polymers,5 the physical properties of these materials can be precisely controlled by modifying the external environment of the polymer.6–11 For example, the solution structures of poly(N-isopropylacrylamide) (PNIPAM)12–14 and poly(oligoethylene glycol)(meth)acrylate (POEG(M)A)15,16 copolymer systems in water both respond to changes in temperature. Upon heating above certain temperatures the polymers transition from a hydrophilic to a hydrophobic state, as evidenced by their structural transition from a random coil to a globule state and accompanying phase separation.17,18 This behaviour is characteristic of the lower critical solution temperature (LCST) phenomenon, an entropy driven process which is defined as the temperature at the minimum of the bimodal of the polymer phase diagram.19 At different polymer concentrations this transition is visually observed as a clouding of the otherwise transparent polymer–water solution at a particular temperature (cloud point temperature; TCP).20 As cloud temperatures are specific to each individual polymer, they can be modulated to higher or lower temperatures simply by changing the polymer composition to be more hydrophilic or hydrophobic, respectively.18 The cloud point of a polymer in solution can also be modified by including charged moieties that are inherently responsive to changes in pH or salt concentrations.21,22Our research group is focussing on the novel separation of analytes from aqueous food or food by-product streams.23,24 Therefore, we are developing new stimuli-responsive polymer systems with specifically engineered properties, such as mechanical strength and variant pH stability. Under challenging conditions, such as solution pH extremes, many of the common acrylate and methacrylate thermoresponsive copolymers with ester-linked side chains are immediately rendered non-compatible due to the propensity of the ester-linked side chain to be hydrolysed.25–29 N-Substituted (meth)acrylamide copolymers, however, present a viable alternative to their ester-linked counterparts, with their stability demonstrated.30 For our applications, polymers must also have a high level of anionic and cationic groups along the polymer chain to enable interaction with specific analytes. Polymers with these properties in a single chain are usually referred to as polyampholytes, and typically possess zwitterionic functionality. These polymers are distinct in both function and structure from polyelectrolytes, which only contain a single type of charged group (anionic or cationic) in the polymer chain. Charged groups in polyampholytes are usually distributed on separate side chain units, as distinct from polybetaines where the charges are combined on a single side chain unit. Whilst both polyampholytes and polybetaines are polyzwitterions, their difference in structure can lead to markedly different interactions of the polymers with target analytes.31–33
The solution properties of polyampholytes are governed by coulombic interactions between charged moieties in the polymer chains and they can display both polyelectrolyte and antipolyelectrolyte behaviour depending on the solution properties.31 The antipolyelectrolyte effect (APE) occurs when a polyampholyte is in the presence of low or no added salt and self-polymer interactions cause the polymer to be in a globule state, with extension to a random coil only achieved via the addition of a low molecular weight salt.31 In contrast to this, polymers that display the polyelectrolyte effect exhibit a random coil state and high solubility in pure water, with the cloud point of solutions usually decreasing as a the salt concentration is increased – typical of classic thermoresponsive polymers.19 Therefore, temperature-responsive polyampholytes present a fascinating dichotomy in solution properties that can be controlled by changes in pH and salt concentrations and copolymer composition. These properties will herein be investigated via novel polyampholyte copolymers containing weakly acidic and basic species in the one polymer chain.31
Experimental
Materials
N-[3-(Dimethylamino)propyl]methacrylamide (DMAPMAm), methacrylic acid (MAA) and 1,3,5-trioxane were purchased from Sigma-Aldrich. DMAPMAm and MAA were purified by running the monomer through a short column packed with hydroquinone monomethyl ether inhibitor removers (Sigma-Aldrich). N-(tert-Butyl)methacrylamide (tBMAm) was purchased from TCI Chemicals and was used as received. 4-((((2-Carboxyethyl)thio)carbonothioyl)thio)-4-cyanopentanoic acid (CETCPA) was obtained from Boron Molecular (BM1433) and was used as received. 4,4′-Azobis(4-cyanovaleric acid) (ACVA) was obtained from Sigma-Aldrich, and was recrystallised from methanol prior to use. All analytical grade solvents were purchased from Sigma-Aldrich or Merck and were used as received. NMR solvents were purchased from Cambridge Isotope Laboratories and were used as received.Instrumentation
Gel Permeation Chromatography (GPC) for all polymers was carried out on a Waters Alliance system equipped with an Alliance 2695 Separations Module (integrated quaternary solvent delivery, solvent degasser and autosampler system), a Waters column heater module, a Waters 2414 RDI refractive index detector, a Waters PDA 2996 photodiode array detector (210–400 nm at 1.2 nm) and 2 × Agilent PL-Aquagel columns (PL-Aquagel Mixed H (8 μm)), each 300 mm × 7.8 mm2, providing an effective molar mass range of 6000 to 6 × 107. The mobile phase was prepared with Milli-Q grade water with 200 mM sodium nitrate, 10 mM sodium phosphate and 200 ppm sodium azide at pH 8 and was filtered through a 0.45 μm membrane filter. The eluent had a flow rate of 1 mL min−1 at 30 °C. Number average (Mn) and weight average (Mw) molar masses were evaluated using Waters Empower-3 software. The GPC columns were calibrated with low dispersity polyethylene glycol/oxide (PEG/PEO) standards (Polymer Laboratories) ranging from 233 to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 016921 g mol−1, and molar masses are reported as PEG/PEO equivalents. A 3rd-order polynomial was used to fit the logMp vs. time calibration curve, which was near linear across the molar mass ranges. Polymer samples were freeze dried using a CHRIST Alpha 1-2 LDplus freeze dryer with Leybold Rotary Vane Vacuum Pump (Trivac D4B) attached and with the chamber cooled to −57 °C. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker BioSpin 400 MHz NMR spectrometer (Billerica, MA) at 298 K and using a 10 s delay between acquisitions. This was found to be a sufficient relaxation time to allow accurate analysis of vinyl peaks for monomer consumption and monomer conversion calculations for these polymers. Data was analysed using TopSpin software. Chemical shifts (δH) are reported in parts per million (ppm) and referenced to residual solvent signal (DMSO: δ 2.5 ppm) or internal standard TSP-d4: δ 0.0 ppm in D2O. In situ temperature ramp NMR experiments were also performed on a Bruker BioSpin 400 MHz NMR spectrometer. Samples in D2O (no TSP-d4) were loaded into the spectrometer at 298 K and heated to 348 K in 2.5° increments, with a 300 s equilibration time at each temperature before acquiring 1H (400.13 MHz) NMR spectra. All spectra were referenced to the residual H2O signal at 4.7 ppm. Cloud point (TCP) measurements were performed by heating a solution of the polymer in water, deuterated water (D2O) or buffer at different concentrations (1.0, 2.5, 5.0, 10.0 mg mL−1) from 15–100 °C, with a heating/cooling rate of 1.0 °C min−1, and 1 s equilibration time. Two heating/cooling cycles were conducted, with the reported TCP values calculated from absorbance measurements obtained in the first cycle. Absorbance (A) values were converted to percent transmittance (% T) by using this equation: % T = 100(10−A). Absorbance of the solutions during these cycles was recorded using a Shimadzu UV-1650 UV/Vis spectrometer with an 8-micro multi-cell temperature controlled cuvette chamber (path length 10 mm), operated using a Shimadzu temperature controller and Tm analysis software. Reported TCP values correspond to the temperature at half (50%) of the transmittance variation. Buffer solutions used were as follows: pH 3: citric acid–sodium phosphate dibasic, pH adjusted with orthophosphoric acid; pH 7: sodium phosphate–sodium phosphate dibasic, pH adjusted with orthophosphoric acid; pH 10.8: sodium bicarbonate–sodium carbonate buffer, pH adjusted with 1.0 M NaOH. The salt solutions used were either sodium bicarbonate (NaHCO3) or sodium chloride (NaCl) which was dissolved in Milli-Q grade water at a concentration of either 0.01, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, or 1.0 M.
016921 g mol−1, and molar masses are reported as PEG/PEO equivalents. A 3rd-order polynomial was used to fit the logMp vs. time calibration curve, which was near linear across the molar mass ranges. Polymer samples were freeze dried using a CHRIST Alpha 1-2 LDplus freeze dryer with Leybold Rotary Vane Vacuum Pump (Trivac D4B) attached and with the chamber cooled to −57 °C. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker BioSpin 400 MHz NMR spectrometer (Billerica, MA) at 298 K and using a 10 s delay between acquisitions. This was found to be a sufficient relaxation time to allow accurate analysis of vinyl peaks for monomer consumption and monomer conversion calculations for these polymers. Data was analysed using TopSpin software. Chemical shifts (δH) are reported in parts per million (ppm) and referenced to residual solvent signal (DMSO: δ 2.5 ppm) or internal standard TSP-d4: δ 0.0 ppm in D2O. In situ temperature ramp NMR experiments were also performed on a Bruker BioSpin 400 MHz NMR spectrometer. Samples in D2O (no TSP-d4) were loaded into the spectrometer at 298 K and heated to 348 K in 2.5° increments, with a 300 s equilibration time at each temperature before acquiring 1H (400.13 MHz) NMR spectra. All spectra were referenced to the residual H2O signal at 4.7 ppm. Cloud point (TCP) measurements were performed by heating a solution of the polymer in water, deuterated water (D2O) or buffer at different concentrations (1.0, 2.5, 5.0, 10.0 mg mL−1) from 15–100 °C, with a heating/cooling rate of 1.0 °C min−1, and 1 s equilibration time. Two heating/cooling cycles were conducted, with the reported TCP values calculated from absorbance measurements obtained in the first cycle. Absorbance (A) values were converted to percent transmittance (% T) by using this equation: % T = 100(10−A). Absorbance of the solutions during these cycles was recorded using a Shimadzu UV-1650 UV/Vis spectrometer with an 8-micro multi-cell temperature controlled cuvette chamber (path length 10 mm), operated using a Shimadzu temperature controller and Tm analysis software. Reported TCP values correspond to the temperature at half (50%) of the transmittance variation. Buffer solutions used were as follows: pH 3: citric acid–sodium phosphate dibasic, pH adjusted with orthophosphoric acid; pH 7: sodium phosphate–sodium phosphate dibasic, pH adjusted with orthophosphoric acid; pH 10.8: sodium bicarbonate–sodium carbonate buffer, pH adjusted with 1.0 M NaOH. The salt solutions used were either sodium bicarbonate (NaHCO3) or sodium chloride (NaCl) which was dissolved in Milli-Q grade water at a concentration of either 0.01, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, or 1.0 M.
General procedure for the preparation of statistical copolymers via RAFT
Weighed quantities of monomers (DMAPMAm, MAA and tBMAm), RAFT agent (CETCPA), initiator (ACVA) and the internal standard (1,3,5-trioxane) were combined into 25 mL screw-top vials and diluted with solvent (either Milli-Q grade water, 1,4-dioxane, or glacial acetic acid) to a set concentration (usually 2.0 M). Each mixture was agitated to solubilise all components and a small aliquot was taken from each vial for NMR analysis (t = 0 time point). A rubber septum was then fitted to the top of each vial and oxygen was removed via a nitrogen purge for 30 min. The vials were then placed in a temperature controlled oil bath that had been pre-heated to 70 °C. Aliquots were taken at various time points and analysed by NMR to follow the course of the polymerisations. At the conclusion of the reactions, each vial was cooled to room temperature and opened to oxygen to quench the polymerisation. The resultant polymers were isolated by dialysis (SpectraPor6 RC tubing, MWCO 3.5 kDa) and were dialysed against Milli-Q grade water. Water was replaced 6 times over the course of the dialysis. Cleaned samples were then transferred to round bottomed flasks, frozen via immersion in liquid nitrogen and then dried using a freeze dryer. All polymers were isolated as fluffy white solids. Monomer consumptions, overall polymer conversions and the ratio of monomers in the final polymers were calculated from 1H NMR spectra by comparison of the integrals of the vinylic protons at the beginning and the end of polymerisations against the integral of the internal standard at ∼5.2 ppm. Full experimental details of all polymerisations can be found in the ESI section.†Results and discussion
Polymer synthesis
Using a combination of N-[3-(dimethylamino)propyl]methacrylamide (DMAPMAm) and methacrylic acid (MAA) as the basic and acidic comonomers, respectively, we report a set of polyampholytes that have increasing amounts of N-(tert-butyl)methacrylamide (tBMAm) incorporated as the hydrophobic comonomer. All polymers were synthesised from a combination of DMAPMAm, MAA and tBMAm monomers, via RAFT polymerisation34 using 4-((((2-carboxyethyl)thio)carbonothioyl)thio)-4-cyanopentanoic acid (CETCPA) as the RAFT agent, as shown in Scheme 1.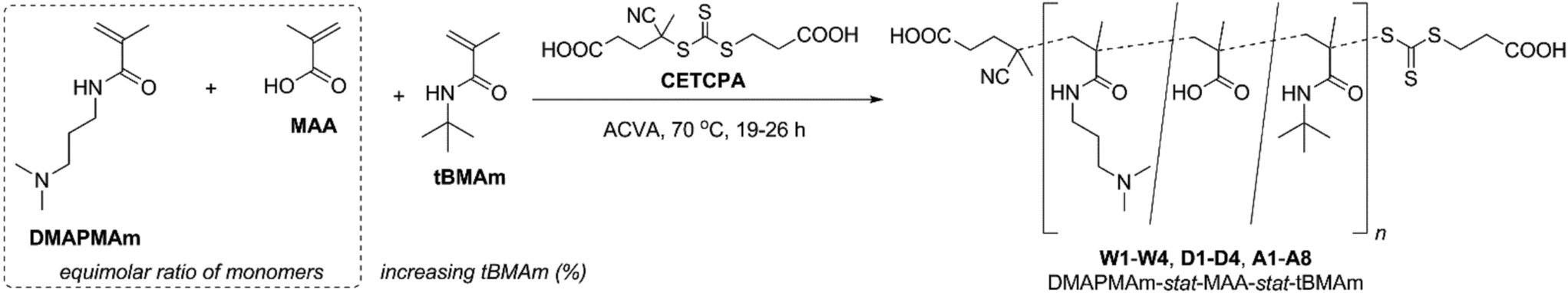 | ||
| Scheme 1 Synthesis of DMAPMAm-stat-MAA-stat-tBMAm copolymers W1–W4, D1–D4 and A1–A8 via RAFT polymerisation, using CETCPA, ACVA, and a range of different solvents (W = water; D = 1,4-dioxane; A = glacial acetic acid). | ||
In all cases the proportion of DMAPMAm to MAA was kept constant at a 1:1 ratio, and the content of tBMAm was varied from 0–50% (of total monomers), as shown in Table 1. The purpose of this was to try and incorporate equimolar amounts of charged monomers, whilst also reducing the overall hydrophilicity of the terpolymers through incorporation of the hydrophobic monomer tBMAm. We hypothesised that this would help modulate the cloud points of the final polymers in solution. The purpose of synthesising the polymers via RAFT polymerisations was to ensure that the polymer chain lengths were controlled, minimising any interference from chain lengths arising from differences in polymer chain composition.
| Polymer | Starting monomer ratios (%) | Reaction conditions | Experimental results | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DMAPMAm | MAA | tBMAm | Solvent | Mn,theo (g mol−1) | Conv.c (%) | Mn,NMR (g mol−1) | DPNMRd | % M1:M2:M3d |
Mne | Mwe | Đe | |
| a Trace amounts of EtOH was needed to solubilise some starting materials.b Precipitate formed after 1–2 h.c Percent (%) overall conversion of monomer to polymer as calculated from 1H NMR.d Calculated from the integrals of the vinylic peaks against the integral of the internal standard 1,3,5-trioxane.e Calculated from GPC (aqueous) against polyethylene glycol/oxide standards.f Could not be calculated accurately due to insolubility of components of the reaction mixture in 1,4-dioxane and deuterated 1H NMR solvents. | ||||||||||||
| W1 | 50 | 50 | 0 | H2Oa | 19200 |
74 | 18100 |
147 | 41:59:0 |
3200 | 4600 | 1.43 |
| W2 | 47.5 | 47.5 | 5 | H2Oa | 18300 |
70 | 17500 |
141 | 39:56:5 |
3400 | 5100 | 1.49 |
| W3 | 45 | 45 | 10 | H2Oa | 17600 |
67 | 16700 |
134 | 37:53:10 |
3900 | 7200 | 1.84 |
| W4 | 42.5 | 42.5 | 15 | H2Oa | 16300 |
62 | 15800 |
125 | 35:50:14 |
4200 | 9000 | 2.13 |
| D1 | 40 | 40 | 20 | 1,4-Dioxaneb | 18200 |
69f | 17900 |
138 | 50:50:0f |
7700 | 19400 |
2.50 |
| D2 | 35 | 35 | 30 | 1,4-Dioxaneb | 11700 |
44f | 11300 |
88 | 50:50:0f |
7800 | 20100 |
2.59 |
| D3 | 30 | 30 | 40 | 1,4-Dioxaneb | 16700 |
62f | 16500 |
124 | 34:36:30f |
6500 | 15800 |
2.42 |
| D4 | 25 | 25 | 50 | 1,4-Dioxaneb | 14900 |
55f | 14900 |
109 | 21:25:54f |
4800 | 11500 |
2.40 |
| A1 | 50 | 50 | 0 | AcOH | 17900 |
69 | 16600 |
137 | 39:61:0 |
14000 |
20700 |
1.48 |
| A2 | 47.5 | 47.5 | 5 | AcOH | 17500 |
67 | 16400 |
135 | 38:59:4 |
14100 |
20300 |
1.44 |
| A3 | 45 | 45 | 10 | AcOH | 18600 |
71 | 17600 |
143 | 37:55:7 |
11100 |
18200 |
1.64 |
| A4 | 42.5 | 42.5 | 15 | AcOH | 16100 |
61 | 14900 |
121 | 34:55:11 |
14600 |
20400 |
1.40 |
| A5 | 40 | 40 | 20 | AcOH | 18800 |
71 | 17800 |
142 | 34:51:16 |
10800 |
15800 |
1.47 |
| A6 | 35 | 35 | 30 | AcOH | 18200 |
68 | 17300 |
136 | 31:46:24 |
9800 | 14800 |
1.50 |
| A7 | 30 | 30 | 40 | AcOH | 16500 |
61 | 15700 |
122 | 26:42:32 |
8200 | 12600 |
1.54 |
| A8 | 25 | 25 | 50 | AcOH | 17400 |
64 | 16900 |
128 | 23:34:43 |
7100 | 10500 |
1.48 |
In determining the optimal conditions for the synthesis of these polymers, a variety of solvents were trialled (Table 1). Both DMF and absolute ethanol were initially trialled to synthesise 0–15% tBMAm copolymers (data not shown), however the contents of many of the reaction vials contained solid precipitates after only 1–2 hours of polymerisation, and low conversions were obtained as observed via NMR analysis. These same problems were also encountered when 1,4-dioxane was used (Table 1, forming polymers D1–D4), but were alleviated when Milli-Q grade water (Table 1, forming polymers W1–W4) or glacial acetic acid (Table 1, forming polymers A1–A8) were used. In the cases of both water and glacial acetic acid, all reaction components (and products) remained solubilised throughout the course of the polymerisation, which led to much higher monomer conversions. Following quenching of the polymerisation process, the polymers were purified using dialysis against Milli-Q grade water, and were freeze-dried to yield white fluffy solids. They were then analysed by 1H NMR and GPC, with the results shown in Table 1.
Total monomer conversions, molecular weights (Mn,NMR), the degree of polymerisation (DPNMR) and the percent monomer incorporation (% M1:M2:M3) were all determined from 1H NMR analysis of samples taken at relevant time points over the course of the polymerisation. By analysing the integrals of peaks corresponding to the vinylic moieties of the monomers against the peak corresponding to the internal standard 1,3,5-trioxane, monomer conversions and the relative ratio of monomers incorporated into each polymer was calculated.
GPC analysis of all polymers was also conducted, and compared with the data obtained from NMR. There was some agreement between the experimentally derived molecular weights determined from different experimental methodologies (Mn,GPC vs. Mn,NMR; Table 1), however in many cases large differences were observed due to differences in solvent (H2O vs. D2O vs. pH 8 buffer; see Experimental section for further details) and structural differences to the PEO used for calibration of the GPC. As the proportion of tBMAm relative to DMAPMAm and MAA is increased in the polymers however, these values become increasingly more disparate (A6–A8 in particular). This is most likely due to the increasing hydrophobicity of the polymer caused by altered interactions of the polymer side chains with the buffer solution at the 30 °C temperature used for GPC analysis.
GPC analysis showed the molar mass distributions to be in the range Đ = 1.40–2.59. For all polymers synthesised in acetic acid (A1–A8) the dispersity values were the lowest (Đ = 1.40–1.64, Fig. 1c) compared with polymers synthesised in water (Đ = 1.43–2.13; Fig. 1a) or 1,4-dioxane (Đ = 2.40–2.59; Fig. 1b). Despite using RAFT polymerisation conditions to synthesise these polymers, there was clearly a loss of livingness during the synthesis of all of these polymers as shown by the high polydispersity values. In order to obtain a set of polymers that had similar molecular weights, we extended the polymerisation times up to 26 hours, which must have led to increased termination and side reactions occurring. Furthermore, for polymers W1–W4, hydrolysis of the RAFT group is known to occur under prolonged water exposure during polymerisation and is the most likely reason for presence of the higher molecular weight tailing (Fig. 1a). In the case of 1,4-dioxane, the higher molecular weight shoulders presumably arose from the insolubility of the products in the solvent which was evidenced by precipitation of the products from the reaction after only 2 hours of heating. This would have caused a range of polymer chain lengths to be present at the conclusion of the reaction, and is reflected in the broad peaks obtained (Fig. 1b). Polymers A1–A8 (Fig. 1c) clearly display a narrower dispersity range, compared to polymers W1–W4 (Fig. 1a) and polymers D1–D4 (Fig. 1b), with minimal to no tailing effects observed in any of the acetic acid-synthesised polymers. This likely resulted from synthesis of these polymers under acidic conditions whereby hydrolysis of the RAFT end group was minimised.
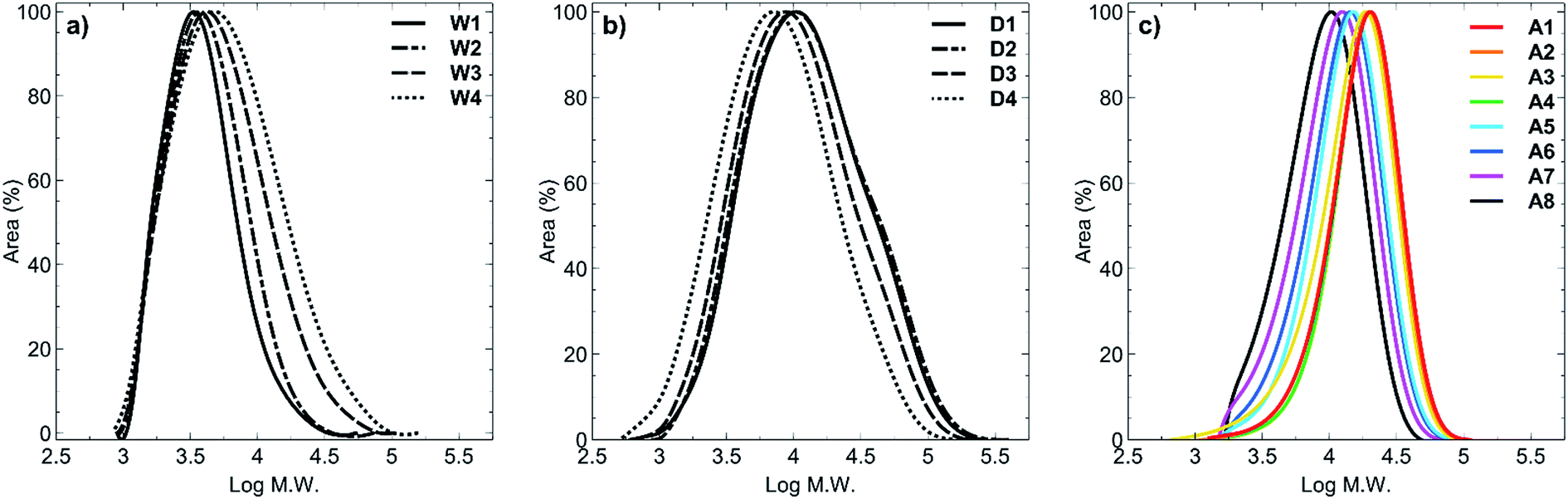 | ||
| Fig. 1 GPC traces of polymer sets (a) W1–W4, (b) D1–D4, and (c) A1–A8. Solvent: Milli-Q grade water containing 200 mM sodium nitrate, 10 mM sodium phosphate and 200 ppm sodium azide at pH 8; Mn are relative to polyethylene glycol/oxide (PEG/PEO) standards. | ||
The discrepancies in the above results are a clear indicator that the choice of solvent was crucial during the synthesis of these polymers, as previously highlighted. Solvent choice also impacted slightly on the monomer incorporation, particularly in the case of polymers W1–W4 and A1–A4. This was determined by 1H NMR analysis of aliquots of reaction solutions collected during the reaction. From this data, the mole fraction of each monomer at a particular time point was determined, and plotted against overall monomer conversion. One example is shown in Fig. 2 for polymers with a starting feed of 42.5:42.5:15 (DMAPMAm:MAA:tBMAm) synthesised in water (W4) versus the same polymer feed but synthesised in glacial acetic acid (A4). For a comparison of the remaining polymers, see Fig. S9–S15 in the ESI section.† Despite beginning with an initial 1:1 ratio of DMAPMAm to MAA, there is clearly greater incorporation of MAA into the polymer chain in both cases, as shown by the decrease in the mole fraction of MAA remaining over the course of the polymerisation (signifying its incorporation into the polymer chain). This is likely due to the different reactivity ratios of the two monomers, which have been reported as MAA 0.41 ± 0.08 and DMAPMAm 1.88 ± 0.15 for synthesis in water,35 compounded by the high self-association that has been reported for DMAPMAm.30 Whilst reactivity of this monomer pair might be expected to be vastly different in acetic acid rather than water, the incorporation of DMAPMAm does not significantly increase through this solvent change. There is, however, a much greater incorporation of tBMAm into the polymer chain when water is the solvent rather than acetic acid. Given that our research interests were focussed on observing and analysing the solution properties of these polymers, we did not estimate the reactivity ratio of these monomers during this study.
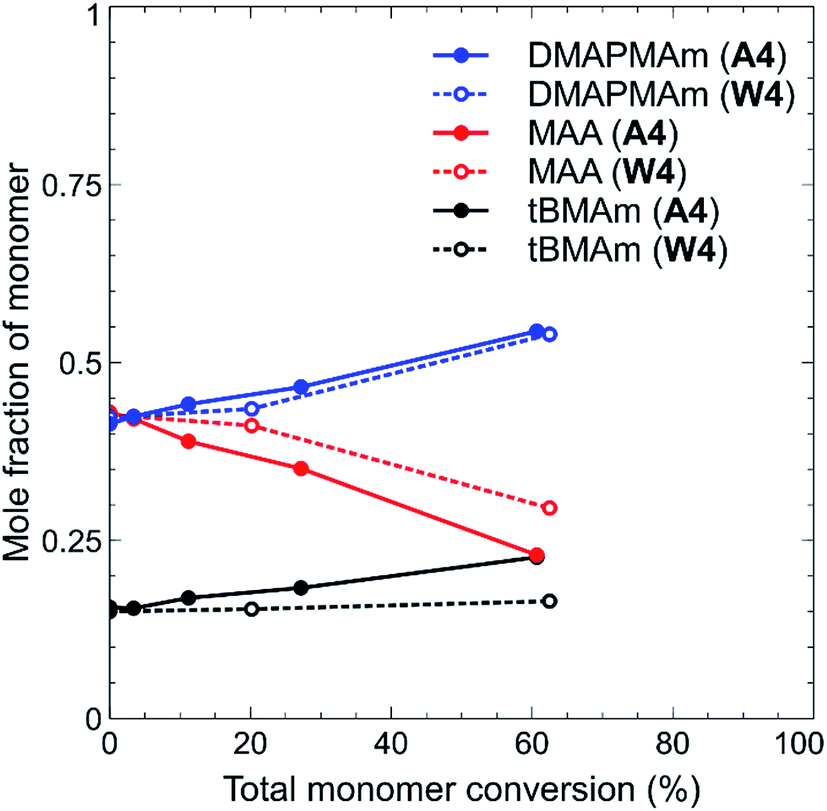 | ||
| Fig. 2 Mole fraction of monomer as a function of total monomer conversion (%) for polymers A4 and W4, as determined by 1H NMR analysis of reaction aliquots. Data points and lines are coloured according to each monomer: blue – DMAPMAm; red – MAA; black – tBMAm and are connected via solid lines (acetic acid-synthesised polymers) or dotted lines (water-synthesised polymers). | ||
In order to best conserve the starting monomer composition, water would appear at this point to be the optimal solvent choice for the synthesis of these polymers. However, as shown by the GPC data, the polymers that were obtained from synthesis in water were much smaller than theoretical values, and their overall monomer composition and broader distribution of polymer chain molecular weights was much more variable, as demonstrated by their relatively high Đ values. Therefore, the most reproducible and interesting polymers were obtained through synthesis in glacial acetic acid solvent. As such, only the stimuli-responsive properties of polymers A1–A8 will be discussed in the following sections.
Temperature-responsive properties of polymers A1–A8
The temperature-responsive properties of polymers A1–A8 at concentrations of 1.0, 2.5, 5.0 and 10.0 mg mL−1 in Milli-Q grade water were determined via UV-Vis spectroscopy (500 nm) at a heating/cooling rate of 1.0 °C min−1 over the temperature range of 15–100 °C. For each sample, the absorbance was measured and converted to the recommended percent transmittance (% T) values,19 and the cloud point temperature (TCP) values were determined as described in the Experimental section.The heating curves for polymers A1–A8 (1.0 mg mL−1) are shown in Fig. 3, with the corresponding curves for the remaining concentrations (2.5, 5.0 and 10.0 mg mL−1) shown in the ESI section (Fig. S16(a)–(c)†). In all cases, the polymer sets are ordered in increasing percent of tBMAm content, which ranges from 0%–43% A1–A8, respectively. This is shown in each graph through different data point colours and sizes, where the increasing size of the data point directly corresponds to the increasing tBMAm content in each polymer.
 | ||
| Fig. 3 Temperature (°C) dependence of the transmittance (%) of 1 mg mL−1 solutions of polymers A1–A8 in Milli-Q grade water. Data point sizes and colours are set according to the relevant incorporation of tBMAm into each polymer, with the calculated percent of tBMAm in each polymer listed in brackets beside the polymer code. | ||
In Fig. 3, it can clearly be seen that the phase transition of polymers A1–A4, which have a tBMAm content of 11% or lower, are not very sharp nor does the solution become significantly cloudy (solution transmittance is >85%). When the tBMAm content is 16% or higher however (A5–A8), the cloud point curves of the polymers become much sharper and the reduction in transmittance is much greater. This particular trend is graphically represented in Fig. 4, where polymers with a clearly defined TCP value (A5–A8) have been plotted against tBMAm content (%). By fitting a second order polynomial curve to this data, a very good correlation is observed between the cloud point temperatures and the hydrophobic content of the polymer chains. This analysis could potentially be used as a predictor of TCP based on tBMAm incorporation in this system, assuming the relative molar ratio and incorporation of DMAPMAm and MAA monomers into each polymer is kept constant. Similar results with good correlations are also observed at higher polymer concentrations, as shown in the ESI (Fig. S17(a)–(c)†).
 | ||
| Fig. 4 Cloud point temperatures (TCP, °C) vs. molar ratio of tBMAm content (%) in polymers A5–A8 at 1 mg mL−1. A clear correlation is observed between decreasing TCP and increasing tBMAm content. | ||
Polymer concentration effects on the cloud points were also studied, with TCP as a function of increasing polymer concentration plotted in Fig. 5(a) and (b). In all cases, the cloud point of each polymer decreased as the concentration was increased. This effect is caused by decreased solvation of the polymer in solution and is a commonly reported phenomenon for many different thermoresponsive polymers.19,36 What is most interesting from these results is that at a polymer concentration of 10 mg mL−1, the polymers containing little to no tBMAm content have similar cloud points to those containing a high percentage of tBMAm, despite them having very different cloud points at much lower polymer concentrations. For example, the TCP of A1 (0% tBMAm) at 10 mg mL−1 is 39 °C, whilst the TCP of A8 (43% tBMAm) at the same polymer concentration is 36 °C (a difference of 3 °C). At 1 mg mL−1 however, polymer A1 (0% tBMAm) doesn't display a measureable cloud point, whereas the cloud point for polymer A8 (43% tBMAm) is 45 °C; a difference of over 55 °C. These effects likely result from the increased self-association of polymers under these high concentrations, which leads to a decrease in hydrogen bonding capacity of the polymers with water and overall lowered TCP.
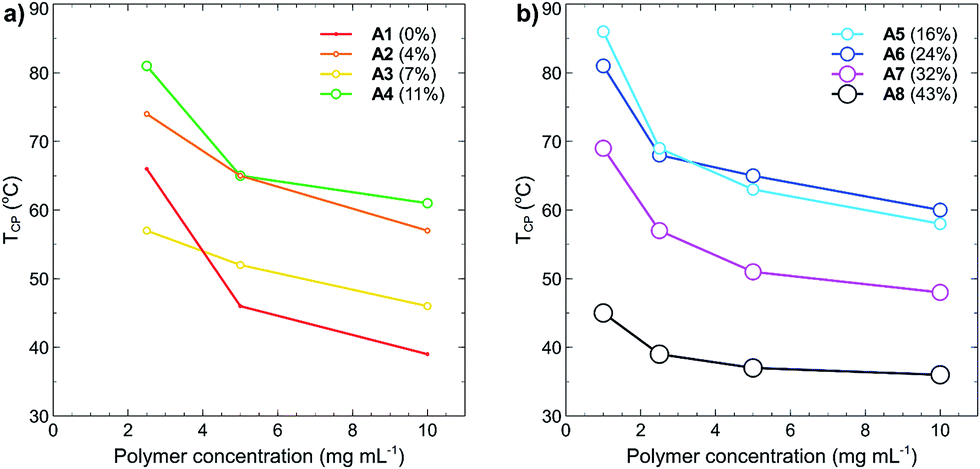 | ||
| Fig. 5 Cloud points (TCP, °C) of polymers (a) A1–A4 and (b) A5–A8 as function of polymer concentration (mg mL−1) in Milli-Q grade water. No TCP was recorded for polymers A1–A4 at 1 mg mL−1 (see Fig. 3). Data points are coloured and sized according to the increasing percent of tBMAm in each polymer. | ||
pH and salt effects on the polymer cloud points
By incorporating a balance of acidic and basic comonomers into the polymer chains, it was expected that these polymers would display additional responsiveness to changes in pH. To test this hypothesis, each polymer A1–A8 was dissolved at a concentration of 1 mg mL−1 in 10 mM buffer solutions that had been adjusted to pH 3, 7, and 10 (see Experimental section for buffer details). These values were selected as the pH extremities 3 and 10 were greater than the pKa's of each of the monomers in the chain, namely, 4.66 for MAA37 and 9.25 for DMAPMAm.38 The pH values of the polymer solutions were then measured using a pH probe to ensure that the final solution pH matched that of the buffer solution. In all cases no change in pH was observed from the measured pH of the buffer solution. Each polymer–buffer solution was then heated and analysed via turbidimetry (as previously outlined in the Experimental section) to determine the polymer cloud points. In all cases however, no decrease in transmittance was observed for any polymer solutions at any of the three pH's tested. This is most likely an effect of the polyzwitterionic nature of these polymers, which act as buffers themselves against changes in pH. At pH 3 and 10, the acid and base groups of the polymers were each neutralised, respectively, and so the polymers each contained a single charge at each of these pH's. This would have led to an increase in the solubility of the polymers in water akin to the behaviour of typical polyelectrolyte thermoresponsive polymers.The thermoresponsive properties of the polymers also changed from the addition of salt. Each of the polymers was dissolved at a concentration of 1 mg mL−1 in either NaHCO3 or NaCl solutions at salt concentrations ranging between 0.01–1.0 M. Again, the cloud points for all polymers increased drastically, with polymers A1–A5 displaying no TCP events at any of the salt concentrations tested. In the case of polymers A6–A8, no cloud points were observed in NaHCO3 salt solutions, whilst some TCP events were observed in NaCl solutions as shown in Fig. 6. Polymers A6, A7 and A8 all showed marked increases in their cloud points as salt was added, particularly when the concentration was increased from 0.01–0.1 M, with no cloud points observed for polymers A6 and A7 above this range. Whilst the cloud point of polymer A8 showed a steep increase in temperature at low salt concentrations, this increase plateaued off somewhat as the salt concentration was further increased up to 1.0 M.
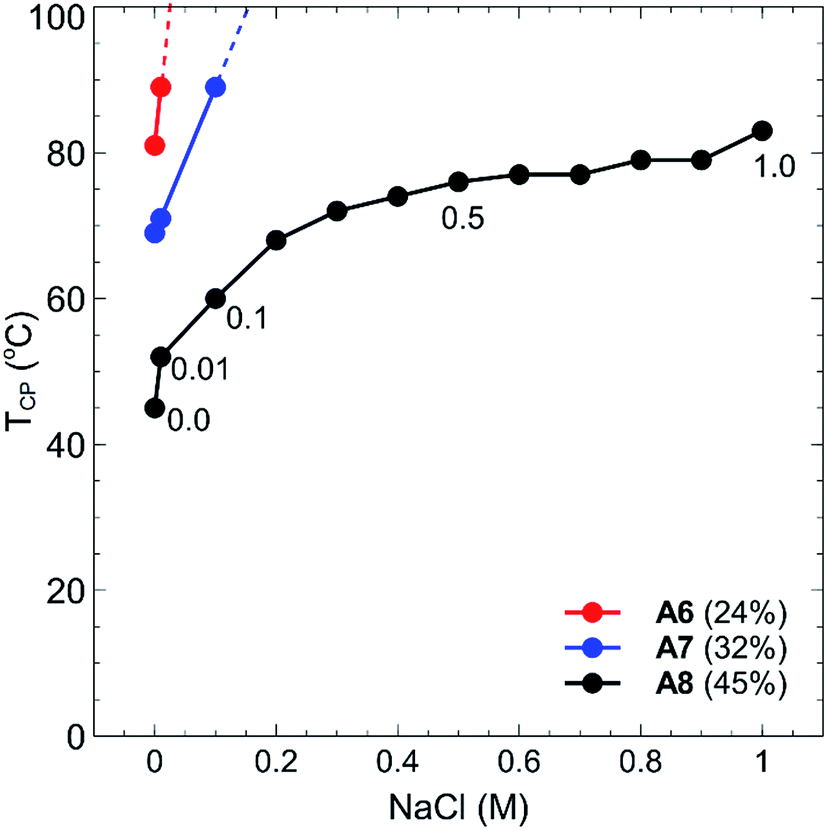 | ||
| Fig. 6 Cloud point temperatures (TCP, °C) vs. sodium chloride concentration (NaCl, M) for polymers A6 (red), A7 (blue) and A8 (black) at 1 mg mL−1 in Milli-Q grade water, with the tBMAm (%) in each polymer shown in brackets. | ||
As previously mentioned, polyelectrolytes and other non-charged thermoresponsive polymers generally display a decrease in their TCP as the salt concentration is increased. This is due to the fact that the addition of ions shields the polymer from water, which creates a more hydrophobic environment for the polymer and results in a decreased TCP value. Due to the presence of both anionic and cationic charged moieties in these polymers however, properties akin to traditional polyzwitterions are displayed, wherein polymer solubility is actually increased as a function of salt concentration due to the shielding of the electrostatic interactions of the polyampholytes.39
NMR studies of polymer A8
To examine these effects in more detail, NMR was employed to assess the structure of polymer A8 in the presence and absence of salt at temperatures above and below the determined cloud points. NMR is an excellent tool to study the thermoresponsive properties of polymers at the molecular level because decreased chain mobility in solution and the relative hydration states of specific moieties in a polymer chain can be visualised through broadening of polymer peaks above the polymer cloud point.19 In this study, 10 mg mL−1 solutions of polymer A8 both in the presence (0.01 M NaCl) and absence (0.0 M) of salt were heated in situ at 2.5 °C increments from 25 °C to 75 °C (300 s equilibration time at each temperature), with 1H NMR spectra acquired at the various temperature intervals. The arrayed NMR spectra of the temperature intervals (5 °C for simplicity) were then plotted and are shown in the ESI section (Fig. S18:† A8 in D2O (0 mM NaCl) and Fig. S19:† A8 in D2O (10 mM NaCl)). In both cases, all peaks exhibit broadening and downfield shifts as the temperature of the solution is increased. This indicates that there is loss of water surrounding the polymers as they transition from a hydrophilic state to a hydrophobic state. Furthermore, there is no observable difference here between the shape and/or broadening of each spectra from the addition of salt. This shows that salt does not appreciably affect the side chains in this polymer that might otherwise cause differences in hydration, which would be observed by sharpening or broadening of specific peaks corresponding to the polymer pendant groups.Data from these experiments was then collated to obtain further information about the temperature dependence of the polymer solubilities. Normalised integrals of the region between 0.5–4.0 ppm corresponding to peaks representing the side chain and backbone protons of the polymers were compared to the integral of residual H2O in each sample, and this data was plotted against temperature as shown in Fig. 7a. From this graph, it can clearly be seen that after 37.5 °C, the polymers start to aggregate and phase separate from the solution. Furthermore, there is no appreciable difference in the temperature at which this occurs despite the addition of salt. This can also be observed in the spectra (see ESI, Fig. S18 and S19†) where a decrease in the intensity of the peaks is noticeable as the temperature is increased. Shown alongside the NMR data in Fig. 7a is the equivalent data obtained for this polymer via turbidimetry (Fig. 7b). In this case, the onset of change in the polymer system occurs at slightly lower temperatures; 34 °C and 35 °C for the solution in pure water and D2O, respectively, and 36 °C for the 0.01 M NaCl solutions. This data is roughly in agreement with that observed during the temperature ramp experiments, with small changes most likely resulting from a combination of experimental error and slight differences in hydrogen vs. deuterium bonding interactions with the polymers. This data further demonstrates the polyzwitterionic properties of the polymers, where at high polymer concentrations they are able to accommodate small changes in salt concentrations.
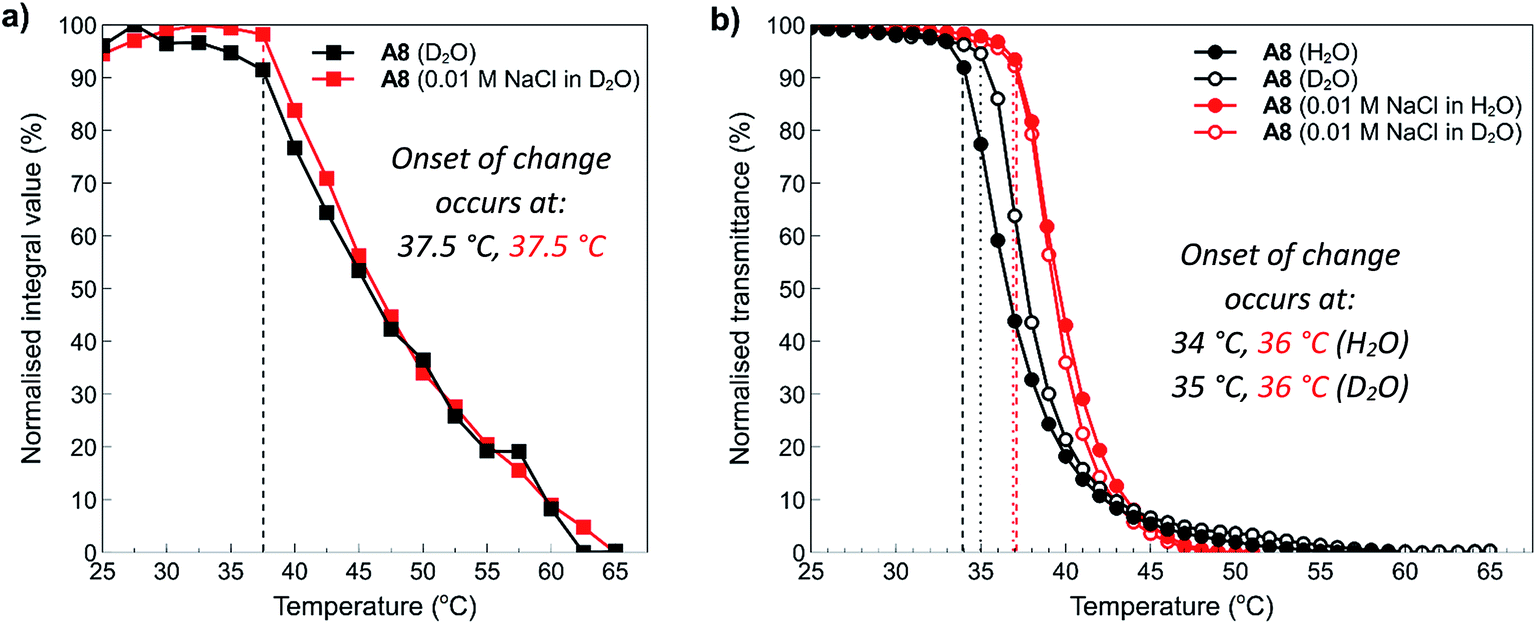 | ||
| Fig. 7 (a) Temperature dependence of the normalised integral of peaks corresponding to polymer side chain and backbone protons as measured by in situ temperature ramp 1H NMR experiments in D2O (black squares) and 10 mM NaCl in D2O (red squares). (b) Temperature dependence of the transmittance (%) of 10 mg mL−1 solutions of polymer A8 in Milli-Q grade water (filled black circles), D2O (open black circles), 0.01 M NaCl in Milli-Q grade water (filled red circles) and 0.01 M NaCl in D2O (open red circles). | ||
Conclusion
A series of N-[3-(dimethylamino)propyl]methacrylamide (DMAPMAm), methacrylic acid (MAA) and N-(tert-butyl)methacrylamide (tBMAm) terpolymers were synthesised via RAFT polymerisation. Polymer synthesis in a variety of solvents was trialled, and glacial acetic acid was determined to be the best solvent during this synthesis as the polymers obtained had a narrow molecular weight distribution that was close to theoretical values. Polymers A1–A8 with a range of tBMAm contents were then characterised for their response to changes in temperature, pH and salt concentrations. At all polymer concentrations ranging between 1–10 mg mL−1, polymer cloud points were successfully modulated by the inclusion of tBMAm, with the cloud points reduced as the tBMAm content was increased to 16% and above. Changes in pH and addition of salt both led to an increase in the polymer cloud points, and in many cases, rendered the polymers free of their thermoresponsive properties within the 15–100 °C temperature ranges investigated.Acknowledgements
The authors wish to acknowledge funding from the CSIRO's Office of the Chief Executive (OCE). Many thanks to Dr Roger Mulder for his help with the NMR experiments and subsequent analyses, and to Dr David Alexander for his help with the data analysis of the cloud point temperatures versus molar ratio of tBMAm content.References
- M. L. Wei, Y. F. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- M. Talelli, A. Duro-Castaño, G. Rodríguez-Escalona and M. J. Vicent, in Smart Polymers and their Applications, ed. M. R. Aguilar and J. S. Román, Woodhead Publishing, 2014, pp. 327–358, DOI:10.1533/9780857097026.2.327.
- C. A. Custódio, R. L. Reis, J. F. Mano and A. Del Campo, in Smart Polymers and their Applications, ed. M. R. Aguilar and J. S. Román, Woodhead Publishing, 2014, pp. 301–326, DOI:10.1533/9780857097026.2.301.
- M. A. Cohen Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef CAS PubMed.
- P. J. Roth and A. B. Lowe, Polym. Chem., 2017, 8, 10–11 RSC.
- A. S. Hoffman, Macromol. Symp., 1995, 98, 645–664 CrossRef CAS.
- G. Kocak, C. Tuncer and V. Bütün, Polym. Chem., 2017, 8, 144–176 RSC.
- J. Thevenot, H. Oliveira, O. Sandre and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 7099–7116 RSC.
- Z. Sekkat and W. Knoll, in Molecular Switches, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 423–515, DOI:10.1002/9783527634408.ch13.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- R. Hoogenboom, in Smart Polymers and their Applications, ed. M. R. Aguilar and J. S. Román, Woodhead Publishing, 2014, pp. 15–44, DOI:10.1533/9780857097026.1.15.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- R. Pelton, J. Colloid Interface Sci., 2010, 348, 673–674 CrossRef CAS PubMed.
- J. F. Lutz, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl. Chem., 1968, 2, 1441–1455 CrossRef CAS.
- R. Hoogenboom, in Complex Macromolecular Architectures, John Wiley & Sons (Asia) Pte Ltd, 2011, pp. 685–715, DOI:10.1002/9780470825150.ch22.
- Q. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Mater. Horiz., 2017, 4, 109–116 RSC.
- M. Constantin, M. Cristea, P. Ascenzi and G. Fundueanu, eXPRESS Polym. Lett., 2011, 5, 839–848 CrossRef CAS.
- R. Freitag and F. Garret-Flaudy, Langmuir, 2002, 18, 3434–3440 CrossRef CAS.
- A. Kumar, A. Srivastava, I. Y. Galaev and B. Mattiasson, Prog. Polym. Sci., 2007, 32, 1205–1237 CrossRef CAS.
- M. Liu, C. Taylor, B. Chong, L. Liu, A. Bilic, N. S. Terefe, R. Stockmann, S. Thang and K. de Silva, Eur. Polym. J., 2014, 56, 153–159 CrossRef.
- N. S. Terefe, O. Glagovskaia, K. De Silva and R. Stockmann, Food Bioprod. Process., 2014, 92, 208–225 CrossRef CAS.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- M. B. McCool and E. Senogles, Eur. Polym. J., 1989, 25, 857–860 CrossRef CAS.
- D. Gromadzki, P. Štĕpánek and R. Makuška, Mater. Chem. Phys., 2013, 137, 709–715 CrossRef CAS.
- K. Vallee-Rehel, V. Langlois and P. Guerin, J. Environ. Polym. Degrad., 1998, 6, 175–186 CrossRef CAS.
- P. van de Wetering, N. J. Zuidam, M. J. van Steenbergen, O. A. G. J. van der Houwen, W. J. M. Underberg and W. E. Hennink, Macromolecules, 1998, 31, 8063–8068 CrossRef CAS.
- D. M. Kamorin, M. Rumyantsev, O. A. Kazantsev, A. P. Sivokhin and S. I. Kamorina, J. Appl. Polym. Sci., 2017, 134, 44412 CrossRef.
- A. B. Lowe and C. L. McCormick, Chem. Rev., 2002, 102, 4177–4190 CrossRef CAS PubMed.
- M. J. Fevola, J. K. Bridges, M. G. Kellum, R. D. Hester and C. L. McCormick, J. Appl. Polym. Sci., 2004, 94, 24–39 CrossRef CAS.
- G. S. Georgiev, E. B. Kamenska, E. D. Vassileva, I. P. Kamenova, V. T. Georgieva, S. B. Iliev and I. A. Ivanov, Biomacromolecules, 2006, 7, 1329–1334 CrossRef CAS PubMed.
- G. Moad, Polym. Chem., 2017, 8, 177–219 RSC.
- R. A. Navolokina, V. A. Sinsokov, L. I. Abramova and S. M. Danov, Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Tekhnol., 1996, 39, 191–193 CAS.
- L. G. Weaver, R. Stockmann, A. Postma and S. H. Thang, RSC Adv., 2016, 6, 90923–90933 RSC.
- I. A. Efimenko, P. V. Ankudinova, L. G. Kuz'mina, A. V. Churakov, N. A. Ivanova, L. I. Demina and O. S. Erofeeva, Russ. J. Inorg. Chem., 2015, 60, 848–860 CrossRef CAS.
- O. A. Kazantsev, K. V. Shirshin, S. A. Kazakov and S. M. Danov, Russ. J. Inorg. Chem., 1996, 66, 2014–2018 CAS.
- J. Zhao, N. A. D. Burke and H. D. H. Stover, RSC Adv., 2016, 6, 41522–41531 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental section, 1H NMR spectra of polymers A1–A8, including the temperature ramp experiment, mole fraction of monomers vs. conversion of polymers A1–A3, A5–A8 and W1–W3, cloud point curves for polymers A1–A8 at polymer concentrations 2.5, 5.0 and 10.0 mg mL−1, and TCP (°C) vs. molar ratio of tBMAm (%) of polymers A5–A8 at 2.5, 5.0 and 10.0 mg mL−1. See DOI: 10.1039/c7ra04723a |
| This journal is © The Royal Society of Chemistry 2017 |