
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-dimensional architecture of Ag/CeO2 nanorod composites prepared by dealloying and their electrocatalytic performance†
Xiaolong Zhangab,
Guijing Lib,
Xiaoping Songa,
Sen Yang *a and
Zhanbo Sun*a
*a and
Zhanbo Sun*a
aSchool of Science, MOE Key Laboratory for Non-Equilibrium Synthesis and Modulation of Condensed Matter, Key Laboratory of Shaanxi for Advanced Functional Materials and Mesoscopic Physics, State Key Laboratory for Mechanical Behavior of Materials, Xi'an Jiaotong University, Xi'an 710049, P. R. China. E-mail: szb@mail.xjtu.edu.cn; yangsen@mail.xjtu.edu.cn; Fax: +86 29 82665995; Tel: +86 29 82665995
bDepartment of Engineering Mechanics, Shijiazhuang Tiedao University, Shijiazhuang 050043, P. R. China
First published on 23rd June 2017
Abstract
A novel three-dimensional (3D) architecture of Ag/CeO2 nanorods with high electrocatalytic activity was prepared by dealloying melt-spun Al–Ag–Ce alloys in NaOH aqueous solutions for NaBH4 electro-oxidation. The nanorod composite was composed of CeO2 nanorods and large-size Ag nanoparticles. After undergoing calcination at 573 K in air, many highly dispersed small Ag nanoparticles were generated and deposited on the surface of the CeO2 nanorod framework. Well-defined Ag/CeO2 interfaces were created via the anchoring of small Ag nanoparticles on the CeO2 nanorods, and the nanorods were connected by larger conductive Ag nanoparticles. Electrochemical measurements showed that the mass specific current of the Ag/CeO2 composites was 2.5 times higher than that of pure Ag for BH4− oxidation. The Ag/CeO2 composites exhibited enhanced catalytic activity owing to the 3D nanorod architecture, strong interfacial interactions between small Ag nanoparticles and CeO2 nanorods, and high concentrations of surface oxygen species.
1. Introduction
The direct borohydride fuel cell (DBFC) is considered a promising power source for the direct conversion of chemical fuels to electricity due to its high specific capacity and cell voltage.1–4 The electrocatalytic oxidation of borohydride is a crucial reaction at DBFC anodes. Noble metal-based nanostructured anode electrocatalysts, such as Pt, Pd and Au, have exhibited high catalytic activities for the electro-oxidation of borohydride.1,2,4–6 However, the high cost and low reserves of noble metals have restricted their application, and thus have stimulated extensive studies to develop low-cost electrocatalysts with high efficiencies. To efficiently utilize noble metals, one can tune their structures to control particle size or combine them with metal oxides to form composites.7,8 The construction of a 3D noble metal/oxide architecture could take full advantage of optimized structures and maximize metal/oxide interface area.9–11Ceria (CeO2) is known to be an excellent additive to promote various catalytic reactions because of its ability to store and release oxygen with a reversible Ce4+/Ce3+ transition, high oxygen carrying capacity and lower price.12–16 CeO2/noble metal composites can often give rise to significant improvements in their activities due to the synergistic interface interaction between them, which are widely exploited in the field of fuel cells,17,18 methane oxidation,19 CO oxidation11,20 etc. Among noble metals, Ag is less expensive and more abundant than other noble metals. Ag–CeO2 composites have been demonstrated to be very competent at catalyzing many important oxidation reactions.21–25 However, Ag nanoparticles wrapped by a CeO2 shell might not be suitable for electrocatalytic applications due to the low electron conductivity of CeO2. Consequently, it is necessary to enhance the structural design of catalysts in order to reduce the negative effects associated with low electron conductivity and the lack of attachment between Ag and CeO2. To improve the conductivities of CeO2-containing catalysts, several strategies that focus on the optimization of morphologies or structures have been adopted, one of which involves supporting the catalysts on electrically conductive materials such as carbon, carbon nanotubes, or graphene.14,17,18,26 However, the many synthetic steps, high calcination temperatures and harsh conditions of the process are difficult to handle.
Recently, dealloying has been recognized as an effective method for the fabrication of nanoporous noble metals and their alloys, which have good conductivities.27,28 In our previous work, we designed a novel nanoporous Ag@CeO2 structure in which CeO2 nanoparticles were loaded on the surface of Ag ligaments via dealloying, and their electrocatalytic activity was effectively enhanced.29 There was only a small amount of CeO2 added in the composite, which still requires a considerable amount of Ag as the conductive material. In addition, annealing temperature and time also strongly influence on the performance of the hybrid Ag-containing nanocomposites.29–31 Therefore, it is of great importance to fully utilize noble metal/oxide interfaces to generate stable Ag/CeO2 electrocatalysts with low Ag contents and high performances.
In this work, we fabricated well-defined Ag/CeO2 interface nanostructures and assembled a monolithic Ag/CeO2 composite for electrocatalysis via a facile dealloying method. A 3D Ag/CeO2 architecture composed of CeO2 nanorods as the framework and Ag particles connecting them was directly constructed by dealloyed Al–Ce–Ag precursor alloys. After calcination, the ultra-small Ag nanoparticles were loaded in situ on the CeO2 architecture, resulting in the formation of bimodal Ag. The electrocatalytic performance of this 3D Ag/CeO2 architecture was investigated and exhibited an enhanced activity and good stability for the electrooxidation of sodium borohydride. The formation mechanism of this unique structure was also discussed.
2. Experimental
2.1 Preparation of the Ag/CeO2 nanorod architecture
Al–Ag–Ce alloys with nominal compositions of Al81Ag15Ce4, Al79Ag15Ce6, Al84Ag10Ce6, Al85Ag15 and Al90Ce10 alloys were prepared from pure Al (99.90 wt%), pure Ag (99.99 wt%) and pure Ce (99.90 wt%) by arc-melting using high-purity Ar as the protective atmosphere. Subsequently, the pre-alloyed ingots were re-melted in a quartz tube and heated by high frequency induction melting. Then, the metal solution was blown onto the surface of a single roller melt spinning at the speed of 33 m s−1. The melt-spun resulting ribbons were typically 20–60 μm in thickness and 2–4 mm in width.The melt-spun ribbons were dealloyed in a 5 wt% NaOH aqueous solution at 333 K for 12 h. The as-dealloyed samples were rinsed several times with reverse osmosis water and then with dehydrated alcohol. Subsequently, the dealloyed Al–Ag–Ce and Al90Ce10 ribbons were calcined at 573 K for 2 h (hereafter, Al–Ag–Ce ribbons-573 K).
2.2 Characterization methods
The phase constitutions of the prepared samples were analyzed using a Bruker D8 Advance X-ray diffractometer (XRD). The semiquantitative analysis for the dealloyed Al–Ag–Ce ribbons-573 K was carried out by Jade 6.5 software using reference intensity ratio (RIR) method from the ratio of XRD peak integrated intensity (peak area). The chemical analysis was determined by X-ray fluorescence spectrometry (XRF) using an S8 Tiger Bruker spectrometer. The microstructures and compositions were characterized using a JEM-2100 high-resolution Transmission Electron Microscope (TEM) and a JSM-7000F field emission Scanning Electron Microscope (SEM) equipped with an INCA X-Sight Oxford Energy Dispersive X-ray Spectrometer (EDX). All the SEM photos were acquired in the secondary electron imaging (SEI) mode. The compositions of the samples were determined by EDX.X-ray photoelectron spectroscopy (XPS) studies were performed using an Axis Ultra Kratos (UK) multifunctional spectrometer using monochromatic Al Kα radiation. Smoothing, background removal and peak fitting were carried out with a CasaXPS analysis software. N2 adsorption measurements were performed on a Micromeritics ASAP 2020 instrument at −77 K, and the samples were outgassed for 2 h in vacuum on the degassing port of the analyzer at 523 K prior to the Brunauer–Emmett–Teller (BET) surface area measurement. The surface areas of the samples were determined by the BET method, and the pore size distribution was determined by the desorption branch of the isotherm using the Barrett–Joyner–Halenda (BJH) algorithm.
2.3 Electrochemical measurements
Electrochemical measurements were performed on a Versa STAT MC workstation in a standard three-electrode cell with a Pt net serving as the counter electrode and a saturated Ag/AgCl, KCl electrode used as the reference. The working electrodes were prepared as follows: 5.0 mg of the as-prepared composite, 1.0 mg of acetylene black, 200.0 μL of isopropanol, and 200.0 μL of Nafion solution (DuPont: 0.5 wt%) were ultrasonically mixed. Four microliters (μL) of the catalyst ink was placed on a polished 3 mm diameter glassy carbon working electrode. The performances of the catalysts towards NaBH4 electro-oxidation was evaluated by cyclic voltammetry (CV) and chronoamperometric (CA) curves, which were collected in 0.5 M KOH + 0.01 M NaBH4 solution at room temperature under the protection of high-purity N2. The 0.5 M KOH electrolyte was deaerated with high purity nitrogen for thirty minutes before the electrochemical measurements. The current density was normalized by the geometric area of the glassy carbon working electrode and by the mass of Ag. The catalytic performance of the nanoporous Ag without CeO2 prepared from dealloyed Al85Ag15 alloy was also carried out under the same experimental conditions. Electrochemical impedance spectroscopy (EIS) was obtained in the frequency range of 1000 kHz to 1 Hz at −0.8 V using a CHI-660E electrochemical workstation (Shanghai, China). The results were represented in Nyquist plots.3. Results and discussion
3.1 Microstructure of the Ag/CeO2 nanorod architecture
Fig. 1 shows the XRD patterns of precursor alloys before and after dealloying, and the dealloyed ribbons calcined in air. The as-quenched alloy ribbons, as shown in Fig. 1a, consisted of an α-Al (Ag, Ce) solid solution, Ag7.32Al9.86Ce1.61 and an unidentified Al–Ce intermetallic compound (denoted AlxCey). After the ribbons were dealloyed, the diffraction peaks of the α-Al, AlxCey and Ag7.32Al9.86Ce1.61 intermetallic compounds disappeared, and only that of fcc-Ag could be detected (Fig. 1b). This result suggests that the precursor alloys are decomposed during dealloying. The diffraction peaks related to Ce could not be detected in the as-dealloyed ribbons. However, when the dealloyed samples were calcined at 573 K, the diffraction peak of CeO2 could be detected, indicating that Ce existed in another form for the as-dealloyed samples. | ||
| Fig. 1 XRD patterns of the melt-spun Al–Ag–Ce ribbons before and after dealloying and the dealloyed ribbons calcined at 573 K. | ||
The relative amounts of Ag and CeO2 in the Ag/CeO2 composites were determined from XRD data (Fig. S1†) using RIR analysis and the results are shown in Table S1.† The chemical composition of the Ag/CeO2 samples by XRF is also presented in Table S1.† The amount of CeO2 in the calcined samples increased with increasing Ce content in the precursor alloys. The EDX results of the melt-spun Al–Ag–Ce ribbons and the dealloyed ribbons-573 K are provided in Table S2.† The results of the melt-spun Al–Ag–Ce ribbons indicated that the ribbons contained Al, Ag and Ce, with a near nominal composition. According to the Ag/Ce ratio for the dealloyed ribbons-573 K, the nominal Ag amounts (wt%) in Ag/CeO2 prepared from the dealloyed Al81Ag15Ce4, Al79Ag15Ce6, and Al84Ag10Ce6 ribbons-573 K were 70.68%, 59.58%, and 49.73%, respectively; all of which were close to their actual contents as confirmed by XRF. The results demonstrated that the Ag/Ce atomic and weight ratios were in good agreement with those of the precursor alloys.
Fig. 2 shows the microstructures of the dealloyed Al–Ag–Ce samples-573 K. The surfaces of the dealloyed Al–Ag–Ce samples-573 K exhibited a nanocomposite structure composed of a nanoporous matrix and with a needle-like a phase morphology embedded in the matrix. The average particle size was approximately 50 nm and the length of the needle-like phase was approximately 100–200 nm. For the as-dealloyed Al81Ag15Ce4-573 K sample, as shown in Fig. 2a, its surface exhibited an open, particle-channel structure, and the corresponding cross-section image showed a similar morphology (Fig. 2b). When the Ce content in the precursory alloy increased to 6% (Al79Ag15Ce6), as shown in Fig. 2c and d, the needle-like morphology became clearer, and the fraction of the phase increased drastically, while the larger nanoparticles could still be observed. For the dealloyed Al84Ag10Ce6 ribbons-573 K, the surface was similar, but the width of the needle-like phase increased and a few large particles could be observed, especially in the cross-view, as shown in Fig. 2d. The insets of Fig. 2b, d and f show the section-view of low-magnification SEM images. It can be seen that sections of the monolithic samples have been dealloyed through whole ribbons, and the average ribbon thickness is about 23.0 (±3.0) μm. Therefore, the structure of the dealloyed Al–Ag–Ce ribbons-573 K is a 3D Ag/CeO2 architecture.
 | ||
| Fig. 2 Plan-view SEM images of the dealloyed Al81Ag15Ce4 (a), Al79Ag15Ce6 (c) and Al84Ag10Ce6 (e) ribbons calcined at 573 K. (b), (d) and (f) showing the corresponding section-view. Insets are low magnification section-view images with the substrate. | ||
TEM and HRTEM images of the Ag/CeO2 architecture are presented in Fig. 3. The dealloyed Al79Ag15Ce6 samples, as shown in Fig. 3a, consisted of larger particles with a size of 20–70 nm and nanorods with the width of 7–16 nm. After the samples were calcined, some small nanoparticles could be observed on the surfaces of the nanorods (Fig. 3b) and the corresponding selected area electron diffraction (SAED) pattern is presented in Fig. 3c. And the diffraction ring patterns were identified as Ag(111) and the (111), (200) and (311) reflections of CeO2. The high magnification TEM image in Fig. 3d shows that the Ag/CeO2 architecture from the dealloyed Al79Ag15Ce6-573 K clearly contained many ultra-small nanoparticles that were highly dispersed and loaded on the surfaces of the CeO2 nanorods. As shown in Fig. 3e, a typical HRTEM image focusing on a specific part of an individual nanorod (circled area, Fig. 3d), reveals that the d-spacings were 0.24 and 0.31 nm, corresponding to the (111) plane of Ag and (111) plane of CeO2, respectively. According to these results, it can be concluded that the larger particles were fcc-Ag while the needle-like phase was CeO2 nanorods.
 | ||
| Fig. 3 TEM images of the Ag/CeO2 nanorod architecture prepared from the dealloyed Al79Ag15Ce6 ribbons (a), the dealloyed sample (b and d) and the dealloyed Al84Ag10Ce6 sample (f) calcined at 573 K. SAED patterns (c) corresponding to (b) and an HRTEM image (e) of the dealloyed Al79Ag15Ce6 ribbon-573 K. TEM (g) and HRTEM (h) images of CeO2 without Ag prepared from dealloyed Al90Ce10 ribbon-573 K. A TEM images (i) of nanoporous Ag from the dealloyed Al81Ag15 ribbon. | ||
The ultra-small spherical Ag nanoparticles with a diameter of approximately 8 nm were found to be anchored and uniformly distributed on the surface of the CeO2 nanorods with a width of 18 nm. And the interface was created in the Ag/CeO2 nanorod architecture from the dealloyed Al79Ag15Ce6 ribbons-573 K. The Ag particle size distribution histograms have been given in Fig. S2.† The result showed that the Ag nanoparticles in the dealloyed Al79Ag15Ce6 ribbons-573 K exhibited a bimodal size distribution: one was around 5.91 nm, and the other was centered at 37.80 nm. When the Ag content decreased to 10 at% in the precursor alloys, the number of larger particles obviously decreased while the CeO2 nanorods dramatically increased. The TEM/HRTEM observation confirmed that the small nanoparticles supported on the CeO2 nanorods were indeed ultra-small Ag nanoparticles. The TEM image of a CeO2 sample (Fig. 3g) also displayed a nanorod structure, and the d-space of the HRTEM image confirmed that the nanorods were CeO2 (Fig. 3h). For comparison, the TEM image of Ag was also shown in Fig. 3i and it exhibited a typical nanoporous structure. The aforementioned results demonstrate that the structure of the 3D Ag/CeO2 nanorod architecture was constructed from the CeO2 nanorods, the uniformly distributed large-sized Ag nanoparticles, and the ultra-small Ag nanoparticles that were anchored on the CeO2 nanorods.
N2 adsorption–desorption isotherms and pore size distributions of the Ag/CeO2 nanorod architecture are shown in Fig. 4. All the isotherms were of type IV and exhibited H3 hysteretic loops of the mesoporous structure, which may be a result of nanorod stacking.32 The BET specific surface areas and other physicochemical parameters of Ag/CeO2 and nanoporous Ag are listed in Table 1. It is evident that the specific surface area increased along with the Ce content of the precursor alloys, resulting from a rapid increase in the amount of CeO2 nanorods. Meanwhile, the BET surface areas of the Ag/CeO2 sample prepared from the Al79Ag15Ce6 ribbons-573 K could reach up to 35 m2 g−1, which was much larger than that of the nanoporous Ag (8 m2 g−1). In addition, it had the smallest calculated pore size, narrowest pore size distribution, and largest pore volume. These results indicated that the Ce content in the precursor alloys plays multiple important roles in determining the surface properties of the Ag/CeO2 nanorod architecture.
 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherms (a) and corresponding pore size distributions (b) of dealloyed Al81Ag15Ce4, Al79Ag15Ce6 and Al84Ag10Ce6 ribbons calcined at 573 K. | ||
| Samples | SBET (m2 g−1) | DP (nm) | VP (cm3 g−1) | Osur/(Olat + Osur + Obon)% | Ce3+/(Ce3+ + Ce4+)% |
|---|---|---|---|---|---|
| Al81Ag15Ce4 | 29 | 13 | 0.1779 | 40.54 | 15.91 |
| Al79Ag15Ce6 | 35 | 10 | 0.1990 | 42.20 | 20.98 |
| Al84Ag10Ce6 | 37 | 11 | 0.1879 | 39.08 | 23.89 |
| Al85Ag15 | 8 | 13 | 0.0283 | N.A | N.A |
XPS spectra of Ce 3d and O 1s peaks of the Ag/CeO2 nanorod architecture are shown in Fig. 5. The spectra of Ce 3d exhibited multiple peaks and could be fitted with ten components with the assignments defined in Fig. 5a. The peaks labeled u and v, u′′ and v′′, u′′′ and v′′′ were characteristics of Ce4+, while the other two double labeled u0 and v0 and v′ and u′ were related to Ce3+.33,34 The Ce 3d spectrum was a mixture of both Ce3+ and Ce4+ oxidation state. The concentration of Ce3+ was reflected by the integrated areas of the corresponding peaks, which increased with increasing Ce content in the precursor alloys. The surface Ce3+ concentrations of total Ce on the Ag/CeO2 nanorod architecture obtained from different Al–Ag–Ce alloys were 15.91%, 20.98% and 23.89%, respectively (Table 1). The broad O 1s peak (Fig. 5b) at about 529, 532 and 533 eV for Ag/CeO2 were assigned to lattice oxygen (Olat), surface adsorbed oxygen (Osur) and weakly bonded oxygen species (Obon), respectively.20,34 The highly mobile Osur oxygen species is generally more important for oxidation reactions compared to the Olat and Obon species.35,36 The relative concentration ratios of Osur/(Olat + Osur + Obon) for each sample are estimated from the areas of the Olat, Osur and Obon peaks (Table 1). The highest relative content of Osur was obtained on the surface of the Ag/CeO2 sample from the dealloyed Al79Ag15Ce6 alloy (42.20%). The results indicate that the ratio of Ag/Ce in the precursor alloys has a significant influence on the concentration of Ce3+ and the active surface oxygen species.
 | ||
| Fig. 5 XPS spectra of Ce 3d (a) and O 1s (b) of the Ag/CeO2 nanorod architecture prepared from the dealloyed Al–Ag–Ce ribbons calcined at 573 K. | ||
Fig. S3† shows the Ag 3d XPS spectra of Ag/CeO2 nanorods and nanoporous Ag. For the dealloyed Al85Ag15 ribbons, the two peaks of Ag 3d5/2 and Ag 3d3/2 at 368.2 and 374.2 eV could be assigned to metallic silver. After the addition of Ce into the Al–Ag alloy, the binding energy of the Ag/CeO2 nanorods slightly shifted to a lower value of 368.0 eV, indicating the occurrence of a partial charge transfer between Ag and CeO2,23 as well as the formation of an oxidation state for Ag nanoparticles anchored on the surface of the composite.25
The dealloying of the melt-spun Al–Ag–Ce ribbons combined with calcination led to the formation of bimodal Ag nanoparticles supported on the 3D CeO2 nanorod architecture. The formation mechanism can be depicted as follows. As the removal of Al from alloys in the NaOH aqueous solution, the Ag and Ce atoms are released and form a mixture of Ag and Ce. Some fast diffused Ag adatoms are hindered by Ce and gradually accumulate, resulting in the formation of Ag nanoparticles with a size of 20–70 nm. Ce atoms at the surface of the Ce-rich Ag–Ce mixture can quickly react with hot water to generate nuclei of cerium hydroxide. The formation process of CeO2 nanorod has been discussed in our previous work.37 During the formation of CeO2, nanorods from different nuclei bond with each other to form the 3D architecture. These processes mainly occur on the surface scale of the Ag–Ce mixture with an outer layer of Ce oxide, and only the CeO2 surface scale is formed. Some Ag atoms are also enriched in the mixture. When undergoing further oxidation, especially calcination, the Ce atoms would diffuse from the Ag–Ce mixture toward the mixture-oxide interface, while Ag diffuses in the opposite direction according to published reports.38–40 The mixture–oxide interface is stable until the Ce is completely oxidized to CeO2, thus forming a highly rugged alloy–oxide interface.38–40 As a consequence, the aggregated Ag atoms or clusters in other sides formed Ag nanoparticles on the surface of the CeO2 nanorods, where they are partly embedded and immobilized. After the Al–Ag alloys were dealloyed, nanoporous Ag exhibited an interpenetrating porous structure with some bicontinuous Ag ligaments (Fig. 3i). When 6 at% Ce was added into the Al–Ag alloy and dealloyed, bicontinuous Ag ligaments became small isolated Ag particles and they did not form bicontinuous Ag ligaments (Fig. 3a). After calcination, CeO2 nanorod-based frameworks build the 3D architecture while small isolated Ag particles grew and aggregated to form larger particles, which then serve as the current collector for transferring the generated electrons at the contacted interface. The calcination process essentially promotes a close contact between the Ag nanoparticles and the CeO2 nanorods.
3.2 Electrocatalytic oxidation of sodium borohydride on Ag/CeO2 electrodes
In order to optimize the heat treatment temperature of the prepared composites, the dealloyed Al79Ag15Ce6 ribbons were calcined at between 373 and 673 K. Fig. S4† shows the CV curves of BH4− oxidation on the Ag/CeO2 electrodes. According to the literature,41,42 the oxidation peaks at around −0.3 V in the CV curves of the prepared electrodes corresponded to the direct oxidation of BH4−. The current density peaks of all the calcined Ag/CeO2 ribbons for BH4− oxidation obviously increased with increased calcination temperatures up to 573 K. When the temperature further increased to 673 K, the oxidation peak showed a slight decrease, which may result from growth of Ag nanoparticles and the slight coarsened structure of Ag/CeO2 composite. These results indicated that the calcination temperature had an effect on the catalytic activities toward the NaBH4 oxidation and the optimized treatment temperature is 573 K in the investigated temperatures.Catalytic activity is closely related to the structure and surface composition of the electrode. Fig. 6 shows the cyclic voltammetry recorded for the Ag/CeO2 nanorod and Ag electrodes with 0.01 M NaBH4 + 0.5 M KOH solution at a scan rate of 100 mV s−1. The current densities of the CV plots in Fig. 6a and b were calculated using surface specific current and mass specific current, respectively. The current densities of the anode peaks for BH4− oxidation on Ag and that of Ag/CeO2 from Al81Ag15Ce4, Al79Ag15Ce6 and Al84Ag10Ce6 electrodes were 16.89, 26.61, 25.65 and 16.24 mA cm−2, respectively (Fig. 6a). Furthermore, an accurate estimation of the intrinsic electrocatalytic activities of the catalysts could be made if the current density is calculated from the unit mass of Ag (as estimated in Table S2†). The current densities of the CV curves in Fig. 6b were calculated based on the Ag mass. The highest specific mass current density of the oxidation peak, 2.97 mA mg−1, was obtained from an Ag/CeO2 electrode prepared from the Al79Ag15Ce6 alloy-573 K, which was about 2.5 times higher than that from the nanoporous Ag electrode prepared from Al85Ag15 under the same conditions. These results reveal that the introduction of low-cost CeO2 not only effectively reduces the required amount of Ag, but also results in an initial increase of catalytic activity for BH4− oxidation in comparison with pure Ag counterparts.
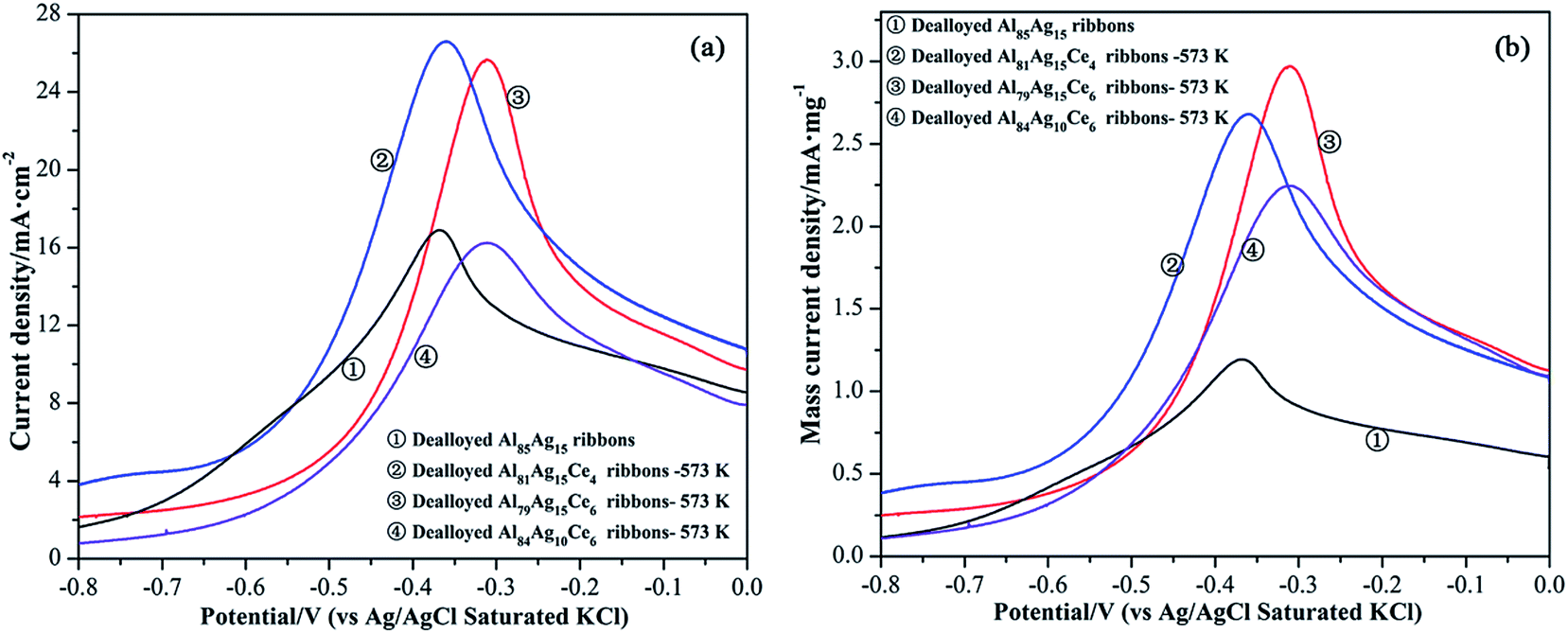 | ||
| Fig. 6 CV curves of BH4− oxidation on the Ag/CeO2 nanorods and Ag electrodes in 0.01 M NaBH4 + 0.5 M KOH at a scan rate of 100 mV s−1. Ag/CeO2 was prepared through the dealloyed Al–Ag–Ce alloys calcined at 573 K. Current density was given on the basis of surface specific current (a) and mass specific current (b). | ||
Fig. 7a shows the effect of scan rate on the current densities of the Ag/CeO2 electrodes obtained from the Al79Ag15Ce6 alloy-573 K for NaBH4 electrooxidation in 0.01 M NaBH4 + 0.5 M KOH. The peak current density increased and shifted to more positive potentials with increasing scan rate and no corresponding reduction peak occurred in the negative scan, which demonstrates that the electrochemical reaction of NaBH4 was irreversible.31,42 The peak current density varied linearly with the square root of the scan rate, as shown in Fig. 7b, which proved that the electrochemical reaction process was controlled by diffusion and mass transport43 within the investigated scan rates range on the Ag/CeO2 electrodes.
 | ||
| Fig. 7 CV curves of 0.01 M NaBH4 in a 0.5 M KOH solution on the Ag/CeO2 nanorod electrodes at different scan rates (a) and variation plot of the peak current density vs. the square root of the voltage scan rate (b). The electrode was prepared from the dealloyed Al79Ag15Ce6 ribbons calcined at 573 K. | ||
Fig. 8 shows the chronoamperometric response of Ag and Ag/CeO2 electrodes in 0.01 M NaBH4 + 0.5 M KOH solution. The current density of the catalysts electrodes declined over time during BH4− electrooxidation. After approximately 75 s, the current density became stable and only slightly declined. The current density stabilized at approximately 2.67, 2.42, 1.49 and 2.21 mA cm−2 for the Ag/CeO2 and Ag electrodes, respectively, after reaction for 480 s. The Ag/CeO2 catalysts prepared from the Al79Ag15Ce6 and Al81Ag14Ce4 ribbons-573 K exhibited higher catalytic activity for BH4− electrooxidation than Ag catalysts without CeO2, which was consistent with the CV measurements.
 | ||
| Fig. 8 CA curves (at −0.4 V vs. Ag/AgCl, KCl) for Ag/CeO2 nanorods and Ag electrode in 0.01 M NaBH4 + 0.5 M KOH. | ||
It is very important for an electrocatalyst to maintain increased stability against degradation. Electrocatalytic stability of the Ag/CeO2 electrode was studied by numerous cycling in a 0.01 M NaBH4 + 0.5 M KOH solution between −1.0 and 0.6 V. Fig. 9 shows a set of continuous CVs up to 81 scanning cycles for the Ag/CeO2 electrode at 20 mV s−1. It can be seen that the peak current density on the prepared electrode at around −0.4 V decreased with potential cycling with respect to the BH4− oxidation reaction. This indicated that the catalyst was not sufficiently stable during long-term cycling. Degradation of the catalyst may be related to the fast decay of electrochemical reaction due to the low concentration of NaBH4,44 which is similar to the reported nano-Au(Co)/Ti and graphene–cobalt–nickel nano-ensemble electrocatalysts.44,45
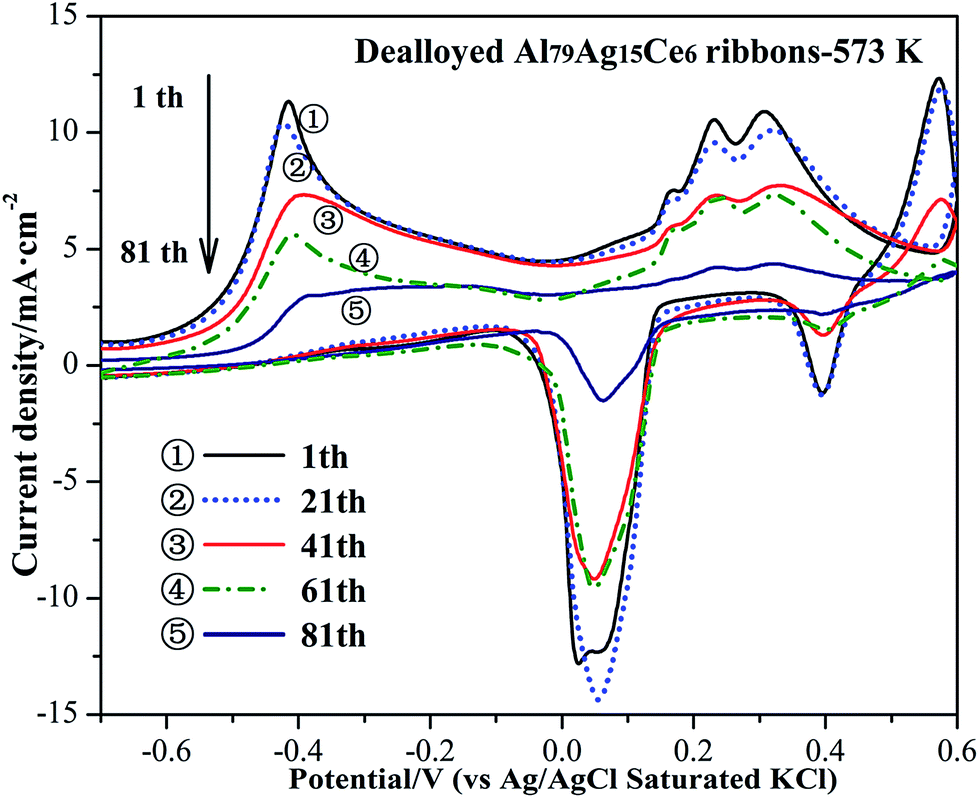 | ||
| Fig. 9 Long-term CVs of Ag/CeO2 catalyst recorded in 0.01 M NaBH4 + 0.5 M KOH at 20 mV s−1. The catalyst was prepared from the dealloyed Al79Ag15Ce6 ribbons calcined at 573 K. | ||
EIS can be used to determine the charge transfer resistance of the electrode surface during an electrochemical reaction. Fig. 10 shows the impedance plots of the Ag and Ag/CeO2 electrodes. The spectra display a semicircle in the high frequency region owing to the electron transfer limited process, while the straight line in the low frequency region can be attributed to the diffusion process.46 The electron transfer resistance (Rct) at the electrode surface is proportional to the semicircle diameter of the Nyquist plots. Compared with the Ag electrodes, the diameters of the Nyquist circle for Ag/CeO2 obtained from Al81Ag15Ce4 and Al79Ag15Ce6 alloys showed only a slight increase, revealing that the ability for charge transfer at the Ag/CeO2 electrode surface was similar to that at the Ag electrode surface. Although the Ag content and conductivity of the Ag/CeO2 electrodes were less than those of pure Ag, the Rcts of Ag/CeO2 did not increase. This could be attributed to the electronic inductive effect of the interfacial interaction. With further increases in the Ce content, the diameter of the semicircle visibly increased, indicating that the excess CeO2 blocked electron transfer from the electrolyte to the electrode surface.
 | ||
| Fig. 10 Nyquist diagrams of the Ag and Ag/CeO2 electrodes in 0.01 M NaBH4 + 0.5 M KOH. | ||
Our results show that the catalytic activity of the Ag/CeO2 nanorod electrodes was higher than that of the pure Ag electrodes and that the mass current density of the Ag/CeO2 catalyst was about 2.5 times as high as that of pure Ag. The relatively high electrocatalytic activity of the Ag/CeO2 with different Ag/Ce ratios can be attributed to the high concentration oxygen vacancies and the strong interfacial interaction between ultra-small Ag nanoparticles and the CeO2 nanorods that resulted from the unique structure.
It has been demonstrated that Ag and Ag-based nanocomposites are excellent catalytic materials as anode materials for BOR and the reaction on the Ag electrodes mainly occur on the Ag2O surface.29,41,47,48 The surface oxidation of Ag electrodes has a considerable catalytic effect on the BOR for Ag-based catalysts.14,15 Shimizu et al. have reported that the active oxygen species on the Ag–Ce catalyst has a similar activity to that of the lattice oxygen of Ag2O.49 On one hand, the XPS results suggested that the Ce3+ concentration on the surface of the samples increased with increasing Ce content in the precursor alloys and evidenced a corresponding high amount of oxygen vacancies, which can significantly improve intrinsic electrocatalytic activity (Fig. 6b). The oxygen vacancies absorb oxygen species (i.e., OHads) and can generate adsorbed atomic oxygen and hydroxyls, which greatly influences the redox properties of the supported metal-species.50 The abundant oxygen vacancies provide absorption sites for surface oxygen species to form and transform the active oxygen and electron, which are transferred to Ag nanoparticles through their interface.29,51,52 On the other hand, a robust interaction exists between CeO2 nanorods and the Ag nanoparticles, which can greatly enhance their electrocatalytic performance for BH4− oxidation. After adding Ce to the precursor alloy, the XPS peaks of Ag 3d displayed a slightly negative shift to lower values due to the electron transfer (Fig. S3†). The occurrence of electron transfer between Ag and CeO2, associated with strengthened interfacial interactions, was responsible for facilitating the uptake of electrons by reactant molecules, as reported elsewhere for Ag@CeO2 catalyst.23,53 According to Luches et al.,15 the increase in Ce3+ concentration along with the amount of small Ag nanoparticles deposited also supported the postulate that part of CeO2 is reduced by electron transfer. These evidence indicated that the addition of Ce to the precursor alloy modifies the electronic structure of Ag and produces an electronic interaction between Ag and CeO2. In addition, the lower binding energy can decrease the chemisorption ability of the adsorbate, and allow the efficient removal of the adsorbed reaction intermediate during electrochemical reactions, thus resulting in an improvement of activity. Therefore, the enhanced catalytic activity for BOR should be attributed to the high fraction of the active oxygen species and the strengthened interactions between Ag and CeO2.
The unique 3D Ag/CeO2 nanorod architecture provides a high specific surface area and improved access to the Ag species. Firstly, CeO2 nanorods and Ag particles produce interconnected porous channels that could provide a larger reaction space for catalytic reactions, which can effectively facilitate conductive ion transfer and reactant molecules transport for the electrocatalysis.31,54 A suitable amount of CeO2 in the Ag/CeO2 samples not only decreased the pore size but also narrowed the pore size distribution (Fig. 4), leading to higher catalytic activity (Fig. 6) and electrochemical stability (Fig. 8) of the Ag/CeO2 electrode. Secondly, highly conductive Ag particles can serve as a bridge to promote the transfer of electrons. After calcination, the in situ growth of ultra-small Ag nanoparticles supported on the CeO2 nanorods facilitated the formation of well-defined interfaces, and provided a high interfacial area. These resulted in enhanced interfacial interaction between Ag and CeO2, effectively reducing any additional contact resistance. EIS results suggested that the reinforced interfacial electronic interaction between Ag and CeO2 can overcome the adverse impact of low electron conductivity of CeO2 nanorods.17,29 Such a unique structure can provide more active sites and efficiently improve the electrochemical performance toward BH4− oxidation.
4. Conclusions
A 3D Ag/CeO2 nanorod architecture was prepared by a facile dealloying method, and bimodal Ag nanoparticles supported on the 3D CeO2 nanorod architecture formed after calcination. Ag nanoparticles with an average size of approximately 8 nm were loaded on the CeO2 nanorod framework, generating nanoscale interfaces. The highly conductive large-sized Ag particles with diameter of 20–70 nm connected the nanostructures as bridges for electron transport. This novel structure can fully utilize Ag to improve the conductivity of the synthesized composites for electrocatalysis. The interfaces between the Ag nanoparticles and the CeO2 nanorods can promote the generation of oxygen vacancies and active oxygen species. The 3D Ag/CeO2 architecture exhibits enhanced catalytic activity for BH4− oxidation, which can be attributed to the unique structures and the nanoscale Ag/CeO2 interfacial interaction. This work provides a simple strategy for the fabrication of 3D rare earth oxide/noble metal nanostructures to amplify noble metal/oxide interactions.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51371135).References
- E. Gyenge, Electrochim. Acta, 2004, 49, 965–978 CrossRef CAS.
- K. Cheng, D. Cao, F. Yang, D. Zhang, P. Yan, J. Yin and G. Wang, J. Power Sources, 2013, 242, 141–147 CrossRef CAS.
- P.-Y. Olu, A. Bonnefont, M. Rouhet, S. Bozdech, N. Job, M. Chatenet and E. Savinova, Electrochim. Acta, 2015, 179, 637–646 CrossRef CAS.
- R. Valiollahi, R. Ojani and J.-B. Raoof, Electrochim. Acta, 2016, 191, 230–236 CrossRef CAS.
- I. Merino-Jimenez, M. Janik, C. P. de Leon and F. Walsh, J. Power Sources, 2014, 269, 498–508 CrossRef CAS.
- P.-Y. Olu, B. Gilles, N. Job and M. Chatenet, Electrochem. Commun., 2014, 43, 47–50 CrossRef CAS.
- H.-J. Qiu, X. Shen, J. Wang, A. Hirata, T. Fujita, Y. Wang and M. Chen, ACS Catal., 2015, 5, 3779–3785 CrossRef CAS.
- X. Wang, D. Liu, S. Song and H. Zhang, J. Am. Chem. Soc., 2013, 135, 15864–15872 CrossRef CAS PubMed.
- G. Xi, J. Ye, Q. Ma, N. Su, H. Bai and C. Wang, J. Am. Chem. Soc., 2012, 134, 6508–6511 CrossRef CAS PubMed.
- B. Wu and N. Zheng, Nano Today, 2013, 8, 168–197 CrossRef CAS.
- H.-P. Zhou, H.-S. Wu, J. Shen, A.-X. Yin, L.-D. Sun and C.-H. Yan, J. Am. Chem. Soc., 2010, 132, 4998–4999 CrossRef CAS PubMed.
- S. Hosokawa, M. Taniguchi, K. Utani, H. Kanai and S. Imamura, Appl. Catal., A, 2005, 289, 115–120 CrossRef CAS.
- D. Valechha, S. Lokhande, M. Klementova, J. Subrt, S. Rayalu and N. Labhsetwar, J. Mater. Chem., 2011, 21, 3718–3725 RSC.
- Y. Zhou, Y. Gao, Y. Liu and J. Liu, J. Power Sources, 2010, 195, 1605–1609 CrossRef CAS.
- P. Luches, F. Pagliuca, S. Valeri, F. Illas, G. Preda and G. Pacchioni, J. Phys. Chem. C, 2011, 116, 1122–1132 Search PubMed.
- A. Younis, D. Chu and S. Li, J. Mater. Chem. A, 2015, 3, 13970–13977 CAS.
- Y. Y. Chu, Z. B. Wang, Z. Z. Jiang, D. M. Gu and G. P. Yin, Adv. Mater., 2011, 23, 3100–3104 CrossRef CAS PubMed.
- M. A. Scibioh, S.-K. Kim, E. A. Cho, T.-H. Lim, S.-A. Hong and H. Y. Ha, Appl. Catal., B, 2008, 84, 773–782 CrossRef CAS.
- A. Gomathi, S. M. Vickers, R. Gholami, M. Alyani, R. W. Man, M. J. MacLachlan, K. J. Smith and M. O. Wolf, ACS Appl. Mater. Interfaces, 2015, 7, 19268–19273 CAS.
- E. Varga, P. Pusztai, A. Oszkó, K. Baan, A. Erdőhelyi, Z. Kónya and J. Kiss, Langmuir, 2016, 32, 2761–2770 CrossRef CAS PubMed.
- D. Duan, H. Liu, X. You, H. Wei and S. Liu, J. Power Sources, 2015, 293, 292–300 CrossRef CAS.
- T. Mitsudome, Y. Mikami, M. Matoba, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2012, 51, 136–139 CrossRef CAS PubMed.
- J. Zhang, L. Li, X. Huang and G. Li, J. Mater. Chem., 2012, 22, 10480–10487 RSC.
- S. Chang, M. Li, Q. Hua, L. Zhang, Y. Ma, B. Ye and W. Huang, J. Catal., 2012, 293, 195–204 CrossRef CAS.
- L. Ma, D. Wang, J. Li, B. Bai, L. Fu and Y. Li, Appl. Catal., B, 2014, 148, 36–43 Search PubMed.
- H. Chen, J. Duan, X. Zhang, Y. Zhang, C. Guo, L. Nie and X. Liu, Mater. Lett., 2014, 126, 9–12 CrossRef CAS.
- Z. Zhang, Y. Wang, Z. Qi, W. Zhang, J. Qin and J. Frenzel, J. Phys. Chem. C, 2009, 113, 12629–12636 CAS.
- C. Xu, R. Wang, M. Chen, Y. Zhang and Y. Ding, Phys. Chem. Chem. Phys., 2010, 12, 239–246 RSC.
- G. Li, F. Lu, X. Wei, X. Song, Z. Sun, Z. Yang and S. Yang, J. Mater. Chem. A, 2013, 1, 4974–4981 CAS.
- A. Manshina, A. Povolotskiy, A. Povolotckaia, A. Kireev, Y. Petrov and S. Tunik, Appl. Surf. Sci., 2015, 353, 11–16 CrossRef CAS.
- X. Zhang, C. Wei, Y. Song, X. Song and Z. Sun, Int. J. Hydrogen Energy, 2014, 39, 15646–15655 CrossRef CAS.
- L. Wang, J. Ding, Y. Chai, Q. Liu, J. Ren, X. Liu and W.-L. Dai, Dalton Trans., 2015, 44, 11223–11234 RSC.
- X. Yao, Y. Xiong, W. Zou, L. Zhang, S. Wu, X. Dong, F. Gao, Y. Deng, C. Tang and Z. Chen, Appl. Catal., B, 2014, 144, 152–165 CrossRef CAS.
- D. Devaiah, T. Tsuzuki, T. Boningari, P. G. Smirniotis and B. M. Reddy, RSC Adv., 2015, 5, 30275–30285 RSC.
- Y. R. Zheng, M. R. Gao, Q. Gao, H. H. Li, J. Xu, Z. Y. Wu and S. H. Yu, Small, 2015, 11, 182–188 CrossRef CAS PubMed.
- S. Cai, D. Zhang, L. Zhang, L. Huang, H. Li, R. Gao, L. Shi and J. Zhang, Catal. Sci. Technol., 2014, 4, 93–101 CAS.
- X. Zhang, K. Li, W. Shi, C. Wei, X. Song, S. Yang and Z. Sun, Nanotechnology, 2016, 28, 045602 CrossRef PubMed.
- C. Wagner, J. Electrochem. Soc., 1956, 103, 571–580 CrossRef CAS.
- C. Wagner, J. Electrochem. Soc., 1952, 99, 369–380 CrossRef CAS.
- B. D. Lichter and C. Wagner, J. Electrochem. Soc., 1960, 107, 168–180 CrossRef CAS.
- E. Sanlı, H. Çelikkan, B. Z. Uysal and M. L. Aksu, Int. J. Hydrogen Energy, 2006, 31, 1920–1924 CrossRef.
- H. Çelikkan, M. Şahin, M. L. Aksu and T. N. Veziroğlu, Int. J. Hydrogen Energy, 2007, 32, 588–593 CrossRef.
- M. S. Ahmed, D. Park and S. Jeon, J. Power Sources, 2016, 308, 180–188 CrossRef CAS.
- S. Saha, S. Ganguly, D. Banerjee and K. Kargupta, Int. J. Hydrogen Energy, 2015, 40, 1760–1773 CrossRef CAS.
- L. Tamašauskaitė-Tamašiūnaitė, A. Jagminienė, A. Balčiūnaitė, A. Zabielaitė, A. Žielienė, L. Naruškevičius, J. Vaičiūnienė, A. Selskis, R. Juškėnas and E. Norkus, Int. J. Hydrogen Energy, 2013, 38, 14232–14241 CrossRef.
- X. Kang, J. Wang, H. Wu, I. A. Aksay, J. Liu and Y. Lin, Biosens. Bioelectron., 2009, 25, 901–905 CrossRef CAS PubMed.
- E. Sanli, B. Z. Uysal and M. L. Aksu, Int. J. Hydrogen Energy, 2008, 33, 2097–2104 CrossRef CAS.
- D. Duan, Q. Wang, H. Liu, X. You, S. Liu and Y. Wang, J. Solid State Electrochem., 2016, 1–13 Search PubMed.
- K. I. Shimizu, H. Kawachi and A. Satsuma, Appl. Catal., B, 2010, 96, 169–175 CrossRef CAS.
- H. Tan, J. Wang, S. Yu and K. Zhou, Environ. Sci. Technol., 2015, 49, 8675–8682 CrossRef CAS PubMed.
- X. Liu, K. Zhou, L. Wang, B. Wang and Y. Li, J. Am. Chem. Soc., 2009, 131, 3140–3141 CrossRef CAS PubMed.
- S. Hu, Y. Wang, W. Wang, Y. Han, Q. Fan, X. Feng, Q. Xu and J. Zhu, J. Phys. Chem. C, 2015, 119, 3579–3588 CAS.
- Y.-Y. Wang, Y. Shu, J. Xu and H. Pang, CrystEngComm, 2017, 19, 684–689 RSC.
- C. Dong, H. Zhong, T. Kou, J. Frenzel, G. Eggeler and Z. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 20215–20223 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04651k |
| This journal is © The Royal Society of Chemistry 2017 |