
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePaclitaxel-loaded pluronic F127/P123 silica nanocapsules with surface conjugated rhTRAIL for targeted cancer therapy
Xin Liu a,
Yuebin Lib,
Jian Heb,
Tingting Zhaob,
Changmei Chena,
Haoshuang Gub and
Xingguo Wang*a
a,
Yuebin Lib,
Jian Heb,
Tingting Zhaob,
Changmei Chena,
Haoshuang Gub and
Xingguo Wang*a
aHubei Collaborative Innovation Center for Green Transformation of Bioresources, The Faculty of Life Science, Hubei University, Wuhan 430062, China. E-mail: xgwang@hubu.edu.cn
bHubei Collaborative Innovation Center for Advanced Organic Chemical Materials, Hubei Key Laboratory of Ferro & Piezoelectric Materials and Devices, Faculty of Physics & Electronic Sciences, Hubei University, Wuhan 430062, China
First published on 12th June 2017
Abstract
A novel anticancer drug PFPSNT, paclitaxel (PTX)-loaded polymeric F127/P123 silica nanocapsules conjugated with TRAIL (tumor necrosis factor (TNF)-related apoptosis-inducing ligand), was designed and synthesized. Transmission electron microscopy (TEM), dynamic light scattering (DLS), UV-vis absorption and particle size analysis revealed that the synthesized PFPSNT was a spherical particle with a size of 24 nm and excellent colloidal stability. Drug loading efficiency (DL%) and encapsulation ratio (ER%) were 0.39% and 26.5%, and its cumulative release profile was linear. In vitro study showed that PFPSNT had marked cytotoxic and apoptotic activities towards HepG2 (liver cancer cell line) and CaSki (cervical cell line) cells with the IC50 values of 921.07 ng mL−1 and 236.24 ng mL−1, respectively. The antitumor efficacy of PFPSNT was evaluated by using a HepG2-xenografted BALB/c nu/nu mice model. In vivo study showed PFPSNT markedly inhibited HepG2 hepatocellular carcinoma, and its antitumor efficacy was more superior than those of TRAIL, PTX or PTX/F127/P123 silica nanocapsules. Our studies provided a novel strategy to synthesize anticancer nanodrugs with mutiple functions, and demonstrated that PFPSNT was a very effective anticancer drug. This novel nanodrug should be a promising drug for targeted cancer therapy to treat complex cancers.
1. Introduction
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) has been reported as a target with great potential for several anti-cancer therapeutics since the mid-1990s.1 TRAIL, a homotrimeric type 2 transmembrane protein, functions as a ligand and selectively triggers cell death in transformed cells without toxicity towards normal cells.2 When TRAIL binds to the death receptors DR4 (TRAIL-RI) and DR5 (TRAIL-RII), it initiates the process of the caspase-8-dependent apoptosis to induce cell apoptosis via an extrinsic apoptotic pathway.2,3 The recombinant TRAIL and agonistic antibodies, as drugs targeting the TRAIL pathway, have been shown robust anticancer activity in a number of preclinical studies4–6 Nevertheless, some cancer cells display strong resistance against TRAIL-induced apoptosis due to dysfunction of death receptors, defects in caspase protein or overexpression of prosurvival proteins (e.g. IAP and Bcl-2 family members).7 TRAIL resistance can be alleviated or reversed by combination therapy.8–11 Ray S et al.12 have reported that the combination of chemotherapy drugs and TRAIL can significantly enhance the apoptosis of tumor cells and reduce the dose of chemotherapy drugs. TRAIL combined with doxorubicin indeed displayed strong inhibitions for a wide variety of tumors.13,14 Paclitaxel (PTX), a cytoskeletal drug targeting tubulin, is commonly used in clinical therapy. It is usually used as an inhibitor to inhibit tumor cell proliferation through stabilizing microtubule network and inhibiting microtubule dynamics.15 It has been reported that PTX can be used as a sensitizing agent towards TRAIL.16–19 However, PTX, as a chemotherapeutic drug, has poor aqueous solubility and low therapeutic index.20 In clinical therapy, PTX is used as a mixture solution composed of 50% (v/v) dehydrated alcohol and 50% (v/v) cremophor EL (polyoxyl 35 castor oil) with 5–20-fold dilution with saline or dextrose solution before administration.21 Like other chemotherapeutic drugs, its usage causes several side effects, such as hair loss, muscle and joint pains, and diarrhea.22 Improving PTX solubility and reduce its side effects are certainly required. Recently, Xin H. et al.23 has found that the embedment of PTX in nanocapsules can significantly improve PTX solubility.Nanocapsules are made up of a nontoxic polymeric membrane and an inner liquid core at the nanoscale level.24 Nanocapsules used as vesicular systems for cancer treatment have a myriad of benefits, including the increase of drug dissolution rate, drug delivery, controllable release and precise targeting.25,26 Up to today, several amphiphilic organic molecules including lipids, block copolymers and polypeptides27–29 have been used to assemble micelles to encapsulate hydrophobic anticancer drugs, such as paclitaxel, doxorubicin and cisplatin.30–32 Silica nanocapsules (SNCs) combine the merits of silica (e.g. mechanical robustness, biocompatibility and great versatility) and capsular nanostructure (e.g. large inner cavity, low density and favourable colloidal properties). SNCs attract people's great attention due to its small size with good colloidal stability, silica shell providing a pathway for chemical drug molecules to diffuse out of the cavity and its onion-like structure favoring loading capacity and release rate of biomedical drugs.33 To make nanodrugs more specific for targeting cancer therapy, linking nanocapsules with a delivery target for cancer therapy becomes a hotspot of current research.34
More recently, the biodistribution experiment has demonstrated the preferential accumulation of 125I-rhTRAIL in CasKi-xenografted tumors, indicating that TRAIL can also be used as a delivery target vector.35 Thus, it is possible for us to link chemotherapeutic drugs, silica nanocapsules and TRAIL together to generate a novel anticancer drug with mutifunctions. The synthesized drugs are predicted to increase dissolution rate, controllable release, co-delivery of chemotherapeutic drugs with TRAIL, precise targeting and robust anticancer activity. In this study, we developed a novel strategy to construct an anticancer targeting drug by using rhTRAIL as a delivery target vector. At first, PTX was embedded inside the pluronic F127/P123 silica nanocapsules, and the then PTX-loaded nanocapsules were conjugated with rhTRAIL to form the PTX/F127/P123 silica nanocapsules conjugated with rhTRAIL (PFPSNT). The size and morphology of the PFPSNT, loading parameters for PTX embedment in the nanocapsules and PTX release from the nanocapsules were also examined. Finally, cytotoxic and apoptotic activities as well as the efficacy of inhibiting tumor growth of the PFPSNT were respectively evaluated by using two cancer cell lines HepG2 and CaSki and the HepG2-xenografted nu/nu mouse. Our studies show that the PFPSNT indeed offers a promise of a novel anti-cancer drug with strong tumor-killing efficacy and sustained release of PTX.
2. Materials and methods
2.1 Materials and reagents
Pluronic F127 (EO106PO70EO106, MW = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 g mol−1) and P123 (EO20PO70EO20, MW = 5800 g mol−1) were purchased from Sigma Aldrich Co. LLC. The rhTRAIL protein (ab157018) was purchased from Abcam Trading (Shanghai) Company Ltd. Paclitaxel (PTX), N-hydroxysuccinimide (NHS), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), acetonitrile, fluorescein diacetate (FDA), tetramethoxysilane (TMOS), pyrene were purchased from Aladdin Industrial Corporation. Chloroform, tetrahydrofuran (THF), methanol, ethanol, xylene, hydrogen peroxide were purchased from Sinopharm Chemical Reagent Co. Ltd. Dimethyl sulfoxide (DMSO) was purchased from Biosharp. The purity of all reagents purchased was ≥98%. Phosphate-buffered saline (PBS) and Dulbecco's modified Eagle's medium (DMEM) were purchased from Hyclone. Trypsin–EDTA (0.25%) was purchased from Life Technologies. Fetal bovine serum (FBS) was purchased from Zhejiang Tianhang Biological Technology Co., Ltd. DAPI dihydrochloride and penicillin–streptomycin (100×) was purchased from Beyotime Technologies. Poly formaldehyde (4%) was purchased from Boster Biological Technology. HepG2 cell line was obtained as a gift from College of Life Sciences, Wuhan University, and CaSki cell line were purchased from FineTest Biotech Co., Ltd.
600 g mol−1) and P123 (EO20PO70EO20, MW = 5800 g mol−1) were purchased from Sigma Aldrich Co. LLC. The rhTRAIL protein (ab157018) was purchased from Abcam Trading (Shanghai) Company Ltd. Paclitaxel (PTX), N-hydroxysuccinimide (NHS), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), acetonitrile, fluorescein diacetate (FDA), tetramethoxysilane (TMOS), pyrene were purchased from Aladdin Industrial Corporation. Chloroform, tetrahydrofuran (THF), methanol, ethanol, xylene, hydrogen peroxide were purchased from Sinopharm Chemical Reagent Co. Ltd. Dimethyl sulfoxide (DMSO) was purchased from Biosharp. The purity of all reagents purchased was ≥98%. Phosphate-buffered saline (PBS) and Dulbecco's modified Eagle's medium (DMEM) were purchased from Hyclone. Trypsin–EDTA (0.25%) was purchased from Life Technologies. Fetal bovine serum (FBS) was purchased from Zhejiang Tianhang Biological Technology Co., Ltd. DAPI dihydrochloride and penicillin–streptomycin (100×) was purchased from Beyotime Technologies. Poly formaldehyde (4%) was purchased from Boster Biological Technology. HepG2 cell line was obtained as a gift from College of Life Sciences, Wuhan University, and CaSki cell line were purchased from FineTest Biotech Co., Ltd.
2.2 Carboxylation of pluronic F127
Based on the protocol reported previously,36 a modified method was used to carboxylate pluronic F127. Briefly, 3.15 g pluronic F127 was dissolved in the mixture of 13 mL anhydrous toluene and 0.7 mL pyridine with heating and stirring in a container filled with nitroge. The final concentration of F127 was 0.25 mM. When the temperature approached to 70 °C, 75 mg succinic anhydride (SA) or 73.5 mg maleic anhydride (MA) was added. The mixture was continuously heated and stirred. Then, the reflux condensation of the mixture was performed at 90 °C for 1–5 h. Finally, the distillated residue was washed with chilly ether, and then the synthesized product F127-COOH was dried by vacuuming at 40 °C for 24 h. Succinic anhydride was chosen to be used for carboxylation of pluronic F127 in the following experiments.2.3 Preparation of PTX/F127/P123 silica nanocapsules
PTX/F127/P123 silica nanocapsules (PTX-NCs) were prepared by using a modified method based on the thin-film hydration37 and the addition of tetramethoxysilane (TMOS)38;39. Briefly, the carboxylated F127 (90 mg) and pluronic P123 (180 mg) were mixed and dissolved in 1 mL chloroform, and then sonicated for 15 minutes at room temperature. 4 mg of PTX was added into the F127/P123 solution, followed by sonication for another 15 minutes to form a highly transparent solution. The PTX/F127/P123 solution was evaporated in a drying oven at 75 °C under the vacuum state. Then, the resultant thin film was hydrated with 4 mL water by sonication to obtain a micelle solution. 35 μL TMOS, dissolved in 0.5 mL THF, was taken out and dropped into the micelle solution with stirring at 1000 rpm, and then the mixed solution was continuously stirred for 3 days so that THF was evaporation and TMOS was completely hydrolyzed to form the silica shell. Finally, the prepared PTX-NCs were filtrated through 0.22 μm filter membrane to remove those unincorporated drug aggregate. All experimental steps were performed at room temperature unless otherwise stated. F127/P123 silica nanocapsules (NCs) without PTX and PTX loaded F127/P123 nanomicelle (PTX-NMs) without addition of TMOS were also made respectively under the similar conditions.2.4 Characterization of PTX-NCs
The morphology of the PTX-NCs was observed by transmission electron microscope (TEM, Tecnai G2 20, 200 kV). The size and size distribution of PTX-NCs and PTX-NMs were measured by dynamic light scattering (DLS) (Malvern Zetasizer Nano-S). All measurements were conducted in a glass cuvette and used water as the dispersant. Colloidal stability was also determined by DLS. The aqueous suspension of the PTX-NCs and PTX-NMs was diluted for 100–1000 times with distilled water and stirred at room temperature for 4 hours before DLS measurements. UV/visible absorption spectra were recorded on a UV-3600 spectrophotometer (Shimadzu).2.5 Loading efficiency and release of PTX
The loading parameters for PTX were measured at 229 nm on an UV-visible spectrophotometer. Drug loaded efficient (DL%) and encapsulation ratio (ER%) were calculated according to the following formula as reported by Zhang et al.40| DL (%) = (weight of the PTX embedded in nanocapsules)/(total weight of the PTX plus nanocapsules) × 100 | (1) |
| ER (%) = (weight of the PTX embedded in nanocapsules)/(weight of the feeding PTX) × 100 | (2) |
PTX release behaviors of PTX-NCs, PTX-NMs and PSPFNT were measured by HPLC (Dionex UltiMate 3000 PumP) using the dialysis method as reported by Andersen et al.41 Briefly, 3 mL PTX-NCs was loaded into a dialysis tubing with the molecular-weight cutoff (MWCO) of 8000 g mol−1, and then dialysized in 30 mL PBS containing 0.1% Tween 80 (pH = 7.4).42 An aliquot of 0.2 mL was taken out at 0.5, 1, 2, 4, 6, 8, 10 and 12 h. Meanwhile, the equal amount of release medium (0.2 mL) was supplemented at each time point to keep total volume unchanged. The content of PTX was estimated by HPLC. Meanwhile, PTX release from stock solution was also conducted under the same condition as a positive control.
2.6 Surface conjugation of PTX-NCs with TRAIL
The carboxylic groups of the carboxylated F127 on the surface of PTX-NCs were conjugated with the amino groups of TRAIL proteins to form PFPSNT under the catalysis of EDC and NHS.43 Briefly, EDC (5 mg), NHS (6 mg) and 1 mL PTX-NCs were mixed in 3 mL of PBS (pH = 6.4), followed by shaking for 1 h to active the carboxylic group of the F127. The reaction mixture were dialysized in 2 L PBS (pH = 6.4) for 12 h to remove the superfluous EDC and NHS in case self ligation occurred between carboxylic groups and amino groups of TRAIL proteins. Then, 1 mL of the TRAIL with a his-tag at its C-terminal (25 μg mL−1) was added, and the mixture was shaken for 24 h at 4 °C. The Ni-affinity chromatography was used to collected the PFPSNT and remove those uncrosslinked nanocapsules. In this experiment, a fluorescent dye pyrene, instead of PTX, was embedded into the F127/P123 silica nanocapsules to estimate conjugation efficiency of the nanocapsules with TRAIL. After Ni-affinity chromatography, the unbound and bound nanocapsules were measured at 338 nm which is the characteristic absorbance for pyrene. Binding capacity was calculated by the eqn (3).| Binding capacity (%) = bound nanocapsules/(bound plus unbound nanocapsules) × 100 | (3) |
2.7 Cytotoxicity assessment and cellular uptakes of TRAIL-pyrene-NCs
Cytotoxicity assay was performed by using Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Inc) to assess the cytotoxicity of TRAIL, TRAIL-NCs, PTX, PFPSNT and PTX-NCs, in which HepG2 and CaSki cell lines were used respectively. Both HepG2 and CaSki cells were cultured in the DMEM medium containing 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin/streptomycin at 37 °C with 5% CO2. Briefly, cells were inoculated into a 96 well plates (LAB-TEK, Chambered Coverglass System) at a density of 8000 cells per well. After pre-incubation overnight, the media were replaced with the fresh DMEM, and then TRAIL, TRAIL-NCs, PTX, PTX-NMs, PTX-NCs and PFPSNT were respectively added to the well at final concentrations of 0–100 μg mL−1. After 24 hour incubation, 10 μL CCK-8 solution was added to each well, followed by incubating another 4 h. The optical absorbance for each well was measured at 450 nm on a Benchmark Plus microplate spectrophotometer (Bio-RAD). The absorbance of each experimental group was calculated by using the value measured in the experimental group minus that of the blank control containing only DMEM medium. All results were again normalized with the positive control group in which cells and the medium were used without additing any nanocapsules or drugs. The date presented in this study was the mean calculated from three independent experiments.Cellular uptakes of TRAIL-pyrene-NCs by HepG2 cells were also examined using fluorescence microscopy. Briefly, HepG2 cells were plated onto the sterile coverslip in a 6-well plate (LAB-TEK, Chambered Coverglass System). Cell adensity was 1 × 106 cells per well. After incubation for 8 h, the medium was replaced with fresh medium containing TRAIL-pyrene-NCs or pyrene-NCs at final concentrations of 2 μg mL−1, and cells were continually incubated for 24 h. All cells in each well were stained by FDA for 15 minutes, and then washed with PBS three times. The coverslip were taken out and cells were observed under a fluorescence microscope (Olympus Fluoview 1000). FDA fluorescent image was acquired at λex = 494 nm and λem = 520 nm, while the pyrene fluorescent image was acquired at λex = 336 nm and λem = 384 nm.
2.8 PFPSNT therapy of HepG2 xenografts
All animal studies were carried out under the guidelines of the laboratory animal center in Tongji Medical College, Huazhong University of Science and Technology. 4 week-old female athymic BALB/c nu/nu nude mice were purchased from Beijing HFK Bioscience Co., Ltd. The average weight of nude mouse was 15 g. HepG2 cells were harvested and mixed with 80% matrigel, and then injected into the right flank of each mouse. Inoculum dose for each mouse was 3 × 106 cells by injecting a total volume of 100 μL. When tumor diameter approached to 0.4–0.6 cm, the nude mice bearing HepG2 xenograft were randomized into five groups (5 mice in each group). 0.35 mg kg−1 TRAIL (7 μg per mouse), 3.5 mg kg−1 PTX (70 μg per mouse), PTX-NCs (70 μg PTX per mouse), 100 μL PFPSNT (7 μg TRAIL and 70 μg PTX per mouse); or 100 μL PBS (as control) were respectively used to treat the nude mice via intraperitoneal (IP) injection. The tumor size was routinely measured, and the tumor volume was calculated according to the eqn (4). The relative tumor volumes were calculated as V/V0 (V: the tumor volume after treatment V0: the tumor volume before treatment). After 14 day treatment, all mice were sacrificed.| Tumor volume (V) = [(tumor length) × (tumor width)2]/2 | (4) |
The anti-tumor efficiency was also evaluated by histological examination. The tumors were taken out from the mice and fixed in 4% paraformaldehyde solution. The fixed tumors were embedded with paraffin and sectioned into thin pieces, and then stained with Hematoxylin and Eosin staining (H&E staining),44 DAPI (4′,6-diamidino-2-phenylindole) staining45 and TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling)46 were respectively performed by using the in situ cell death detection kit (Roche, Applied Science). Alterations of tumor tissues and cells at histopathological and apoptosis levels were observed under a light microscope and a confocal laser scanning microscope (Olympus, CX-21). Major organs including heart, liver, spleen, lung and kidney in each group were also collected and examined.
2.9 Statistical analysis
Date are presented as mean ± SDs. Statistical significances were determined using the two way ANOVA.47 P-values of <0.05 were considered statistically significant.3. Results and discussion
3.1 Characterization of PTX-NCs
It was reported that PTX/F127/P123 nanomicelles were able to be fabricated by using the thin-film hydration method if pluronic F127 and P123 were mixed with PTX.48 The authors found that F127, P123 and PTX was completely dissolvable in a common water-miscible organic solvent (e.g. chloroform), and pluronic F127/P123 in the molten state was an excellent solvent for water-insoluble PTX. However, the resultant thin film was easily hydrated with water by sonication. Recently, another research group found that the hydrophobic PPO (polypropylene oxide) segments and hydrophilic PEO (polyethylene oxide) segments of PPO–PEO block copolymers could rapidly form a core and shell of the micelles. When TMOS was added, it entered the juncture of PEO and PPO, and then hydrolyzed to form silicon dioxide (SiO2). Finally, SiO2 deposited between the core and shell of the micelles to form a thin silica layer. This novel type of nanocapsules with a silica layer was more stabile and soluble in aqueous phase.38,49 To obtain multifunctional anti-cancer drug with low toxicity, controllable release and precise targeting, pluronic F127 and P123 were used to make the PTX-loaded F127/P123 silica nanocapsules by combining two methods as mentioned above. The reason for choosing F127 and P123 was that the hydrophilic PEO segment of F127 was longer than that of P123. A long hydrophilic PEO segment might enhance the solubility of the synthesized nanocapsules, whereas a short PEO segment of P123 might benefit PTX-loading capacity in the nanocapsule.In order to crosslink TRAIL with the PTX-NCs, pluronic F127 was initially carboxylated, and then the F127-COOH was used to replace F127 during the preparation of PTX-NCs. Fig. 1B shows the FTIR spectra of F127 and carboxylated F127. The stretching vibration of hydroxyl group of F127 at 1630 cm−1 disappeared in the carboxylated F127, and the latter emerged a novel peak at 1735 cm−1 attributing to its carbonyl group. The analysis of FTIR spectra indicated that the pluronic F127 indeed was carboxylated successfully to form the F127-COOH. Its carboxylation efficiency, determined by conductometric titration, reached up to 96.5% or 92.8%, when succinic anhydride or maleic anhydride was used.
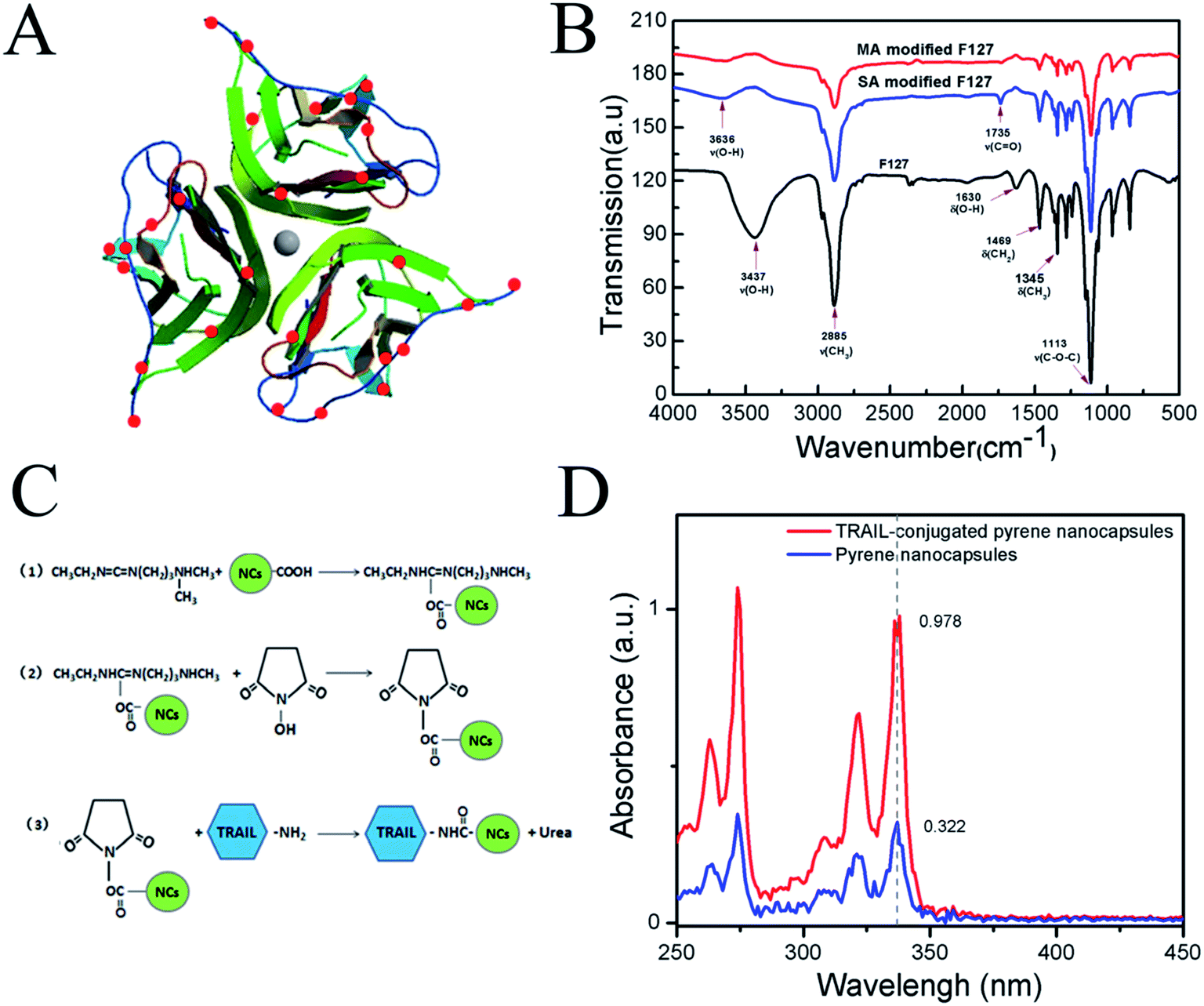 | ||
| Fig. 1 (A) A molecular model presenting amino groups on the surface of TRAIL analyzed by PyMol images; (B) FTIR spectra of carboxylated-F127 block copolymers; (C) a schematic describing the reaction process of the conjugation of PTX-NCs with TRAIL; (D) wavelength scanning of TRAIL-conjugated pyrene/F127/P123 silica nanocapsules and pyrene/F127/P123 silica nanocapsules. | ||
The morphology and size of the PTX-NCs were examined. The synthesized PTX-NCs were observed by transmission electron microscopy (TEM), and their morphology and sizes were shown in Fig. 2A. As shown in Fig. 2A, the PTX-NCs indeed exhibited a typical structure of silica nanocapsules. The dark silica shell displayed a significantly high electron density, and its thickness measured by the software nanomeasure was 5.28 nm. The bright core was composed of PPO blocks of F127 and P123 containing PTX. The particle size measured by the TEM images was 12.98 nm. As shown in Fig. 2A, the nanocapsules appeared to link each other. This might perhaps result from the short PEO chains of P123 unable to provide enough steric hindrances to keep the silica nanocapsules from agglomerating together. Pluronic P123/F127 mixed polymeric nanocapsules appeared aggregation was reported by Hsu B. Y. W. et al.50 In addition, the nanocapsules also showed obvious Tyndell effects when a laser was used to illuminate them (see the inset in Fig. 2A).
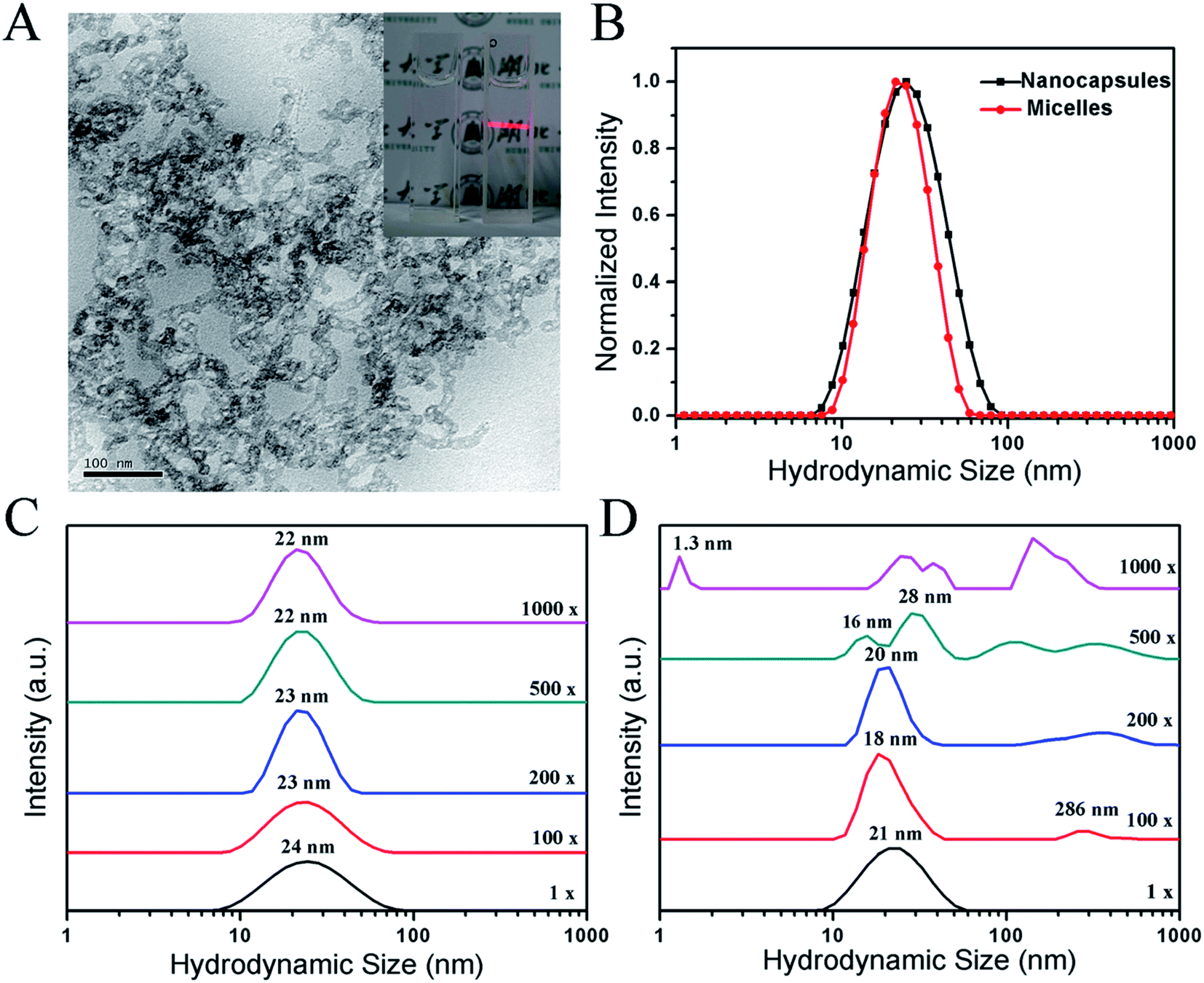 | ||
| Fig. 2 Characterization of PTX-NCs and PTX-NMs. (A) TEM images of PTX-NCs; (B) the size determination of PTX-NCs and PTX-NMs by DLS; DLS tests of colloidal stability for PTX-NCs (C) and PTX-NMs (D). | ||
The size of the PTX-NCs was determined by using DLS, and the results were summarized in Fig. 2B. The PTX-NCs showed a relatively narrow peak of size distribution with a low polydispersity index (PDI), similar to that for the PTX-NMs. However, the average diameter for the PTX-NCs was close to 24 nm with an acceptable PDI of 0.217. By contrast, the average diameter for the PTX-NMs was 21 nm with a PDI value of 0.122. The size of 24 nm obtained from the DLS measurement was larger than that from the TEM image. The explanation for size discrepancy could be that the particle size measured by DLS includes the extension of free PEO chains into water, but TEM images do not.
Colloidal stability is another important index of nanocapsules. Polymeric micelles will gradual disintegrate when they are diluted below the critical micelle concentration (CMC).51 By contrast, silica nanocapsules would avoid of disintegration due to their silica-shell formation. The colloidal stability of the PTX-NCs and the PTX-NMs were compared. After dilutition of 100–1000 folds with dH2O, DLS assays were performed, and the results were summarized in Fig. 2C and D. As shown in Fig. 2C, the size distributions of PTX-NCs were almost unchanged even if it was diluted to 1000 folds. Different from the PTX-NCs, the PTX-NMs displayed a marked change in its size distributions when its dilution was in excess of 200 folds. When the dilution approached to 500 or 1000 folds, several new peaks appeared. In addition, the hydrodynamic sizes of the PTX-NMs also showed a gradual increase at 100–200 fold dilutions. The appearance of several new peaks implied that the PTX-NMs were easily disintegrated under the dilution conditions, and the embedded PTX might easily be exposed to water solvent. These results in Fig. 2C and D clearly demonstrated that the PTX-NCs were more stable than the PTX-NMs.
3.2 Loading capacity and release of PTX in PTX-NCs
The PTX-loading parameters for both PTX-NCs and PTX-NMs were examined by measuring the PTX absorption at 229 nm. The results were summarized in Table 1. The effective concentrations of PTX in the PTX-NCs and the PTX-NMs were 265.4 μg mL−1 and 852.3 μg mL−1, which were much larger than 0.3 μg mL−1 for PTX intrinsic water solubility.22 More than 800-fold increase of PTX-loading concentration in either nanocapsules or micelles illustrated that the microencapsulation of PTX markedly improved its aqueous solubility. As shown in Table 1, DL and ER values were given 0.39% and 26.5% for the PTX-NCs and 1.26% and 85.2% for the PTX-NMs. Low DL and ER values for the PTX-NCs might result from the formation of silica shell when TMOS entered into the core of micelles. Such physical alteration might affect the package of PTX in silica nanocapsules. Although DL and ER values of the PTX-NCs were less than those for the PTX-NMs, the silica shell depositing on the interface between hydrophobic PPO core and hydrophilic PEO chains could prevent drug release directly from the hydrophobic core.| DL (%) | ER (%) | PTX concentration in nanoparticles (μg mL−1) | |
|---|---|---|---|
| a DL: drug loaded efficient; ER: encapsulation ratio. | |||
| PTX-NMs | 1.26 | 85.2% | 852.3 |
| PTX-NCs | 0.39 | 26.5% | 265.4 |
The in vitro cumulative release profiles of PTX for the PTX-NMs, PTX-NCs and PFPSNT were shown in Fig. 3. PTX release from stock solution was also investigated as a positive control. It was found that more than 70% PTX in the stock solution were released in the first 2 h. This suggested that PTX could freely diffuse through the dialysis membrane. As shown in Fig. 3, a hyperbolic curve for PTX cumulative release profile indicated that PTX release from the PTX-NMs was quite rapid at early stage. More than 30% of PTX in the PTX-NMs was released at 30 min, and nearly 56% PTX was released at 2 h. By contrast, the PTX-NCs released only 2.3% and 10.4% of PTX at the same time points. Even at 12 hours, the amount of PTX released from PTX-NCs was less than 62%. More importantly, the cumulative release profile of PTX was almost linear in this case, suggesting that PTX release was slow and steady during the testing process. These results clearly demonstrated that the PTX-NCs indeed sustained the release of PTX. Such a gradual release of PTX could benefit anticancer therapy because the sustained release of PTX from silica nanocapsules would decrease PTX concentration in blood. Less PTX in blood reduced the nonspecific toxicity of PTX to normal cells. In addition, both PFPSNT and PTX-NCs showed similar profiles of PTX cumulative release. No significant difference between release profiles of PFPSNT and PTX-NCs indicated that TRAIL conjugation did not affected the release behavior of PTX from silica nanocapsules.
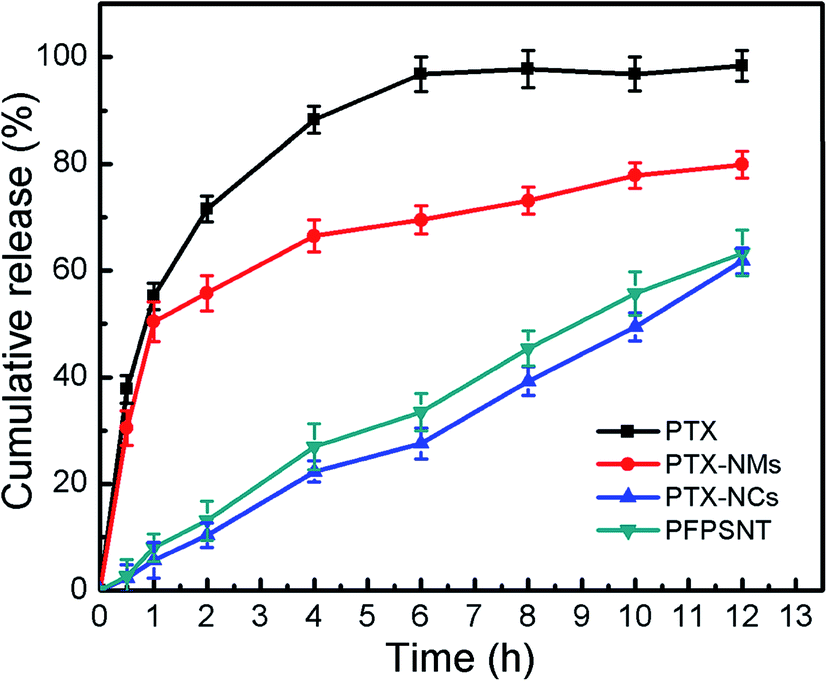 | ||
| Fig. 3 Cumulative release of PTX from PTX-NMs, PTX-NCs and PFPSNT. | ||
3.3 Conjugation efficiency of PTX-NCs and TRAIL
The carboxylic groups of the carboxylated F127 on the surface of PTX-NCs were conjugated with the amino groups of TRAIL to form a PTX/F127/P123 silica nanocapsule-TRAIL (PFPSNT). 3D image of the surface amino groups of TRAIL was analysed by using PyMol software52 and shown in Fig. 1A. The TRAIL protein, used in this study, contains 168 amino acids, in which only 10 lysine and arginine residues with a free amino group exist. The analysis by PyMol software found that only 9 amino groups were exposed on the surface of each TRAIL subunit. Thus, a trimeric TRAIL, acting as an active form, contains 27 amino groups on its surface. Fig. 1C illustrated the reaction scheme of TRAIL conjugation with PTX-NCs. Firstly, EDC activated the carboxylic group of the nanocapsules to form an o-acylisoureaester intermediate. Then NHS reacted with the o-acylisoureaester intermediate to form N-hydroxysuccinimide-ester. N-Hydroxysuccinimide-ester of nanocapsules was stable, and actively reacted with the amino group of TRAIL. Finally, TRAIL-conjugated silica nanocapsules were obtained.To determine the conjugation efficiency, a fluorescent dye, pyrene, was loaded into the F127/P123 silica nanocapsules instead of PTX, and then TRAIL was conjugated with the pyrene/F127/P123 silica nanocapsules. The reason for using pyrene was that its absorbances at 338 nm was proportional to its content. The TRAIL-conjugated pyrene/F127/P123 silica nanocapsules and the pyrene/F127/P123 silica nanocapsules without TRAIL were separated by using Ni-chelating affinity chromatography, and then measured at 338 nm specific for pyrene. Fig. 1D showed that the absorbances at 338 nm were 0.978 for TRAIL-conjugated pyrene/F127/P123 silica nanocapsules and 0.322 for unconjugated nanocapsules. It was calculated that 75.2% nanocapsules were successfully coupled with TRAIL protien.
3.4 In vitro cytotoxicity and targeting capability of PFPSNT
The cytotoxicity of the nanocapsules (NCs) without PTX and TRAIL and anti-tumor cell capacity of PTX, PTX-NCs, TRAIL, TRAIL-NCs and PFPSNT were examined using CCK-8 assays. HepG2 and CaSki cells were respectively used in these assays. As shown in Fig. 4A and B, cell viabilities of HepG2 and CaSki cells were almost 100% when they were incubated with NCs for 24 h, suggesting that the blank NCs did not show obvious cytotoxicity towards both cancer cells. The relative viabilities of CaSki cells treated respectively with PTX and PTX-NCs showed a comparable decrease with the increase of PTX concentration. In the case of HepG2, trearments by PTX or PTX-NCs also showed a similar result. No significant difference between PTX and PTX-NCs treatments indicated again that the NCs did not have observable cytotoxicity. The decrease of relative viabilities might mainly come from the cytotoxicity of PTX. This conclusion was also supported by the half maximal inhibitory concentrations (IC50) values of PTX (671.4 μg mL−1 for HepG2, 194.4 μg mL−1 for CaSki) and PTX-NCs (626.5 μg mL−1 for HepG2, 172.6 μg mL−1 for CaSki). IC50 values of TRAIL were 5.514 μg mL−1 for HepG2 and 1.78 μg mL−1 for CaSki. Similarly, IC50 values of TRAIL-NCs were 4.883 μg mL−1 for HepG2 and 2.092 μg mL−1 for CaSki. No significant difference between IC50 values of TRAIL and TRAIL-NCs for both two cell lines, indicating that the bioactivity of TRAIL was not influenced after it was conjugated to silica nanocapsules. In addition, PFPSNT showed the lowest cell viability with the IC50 values of 921.1 ng mL−1 for HepG2 and 236.2 ng mL−1 for CaSki. The PFPSNT had a superior anticancer effect as compared to the PTX-NCs and TRAIL. Those results obtained from in vitro studies demonstrated that the surface conjugation of the PTX-NCs with TRAIL was indeed able to enhance the efficacy of killing cancer cells.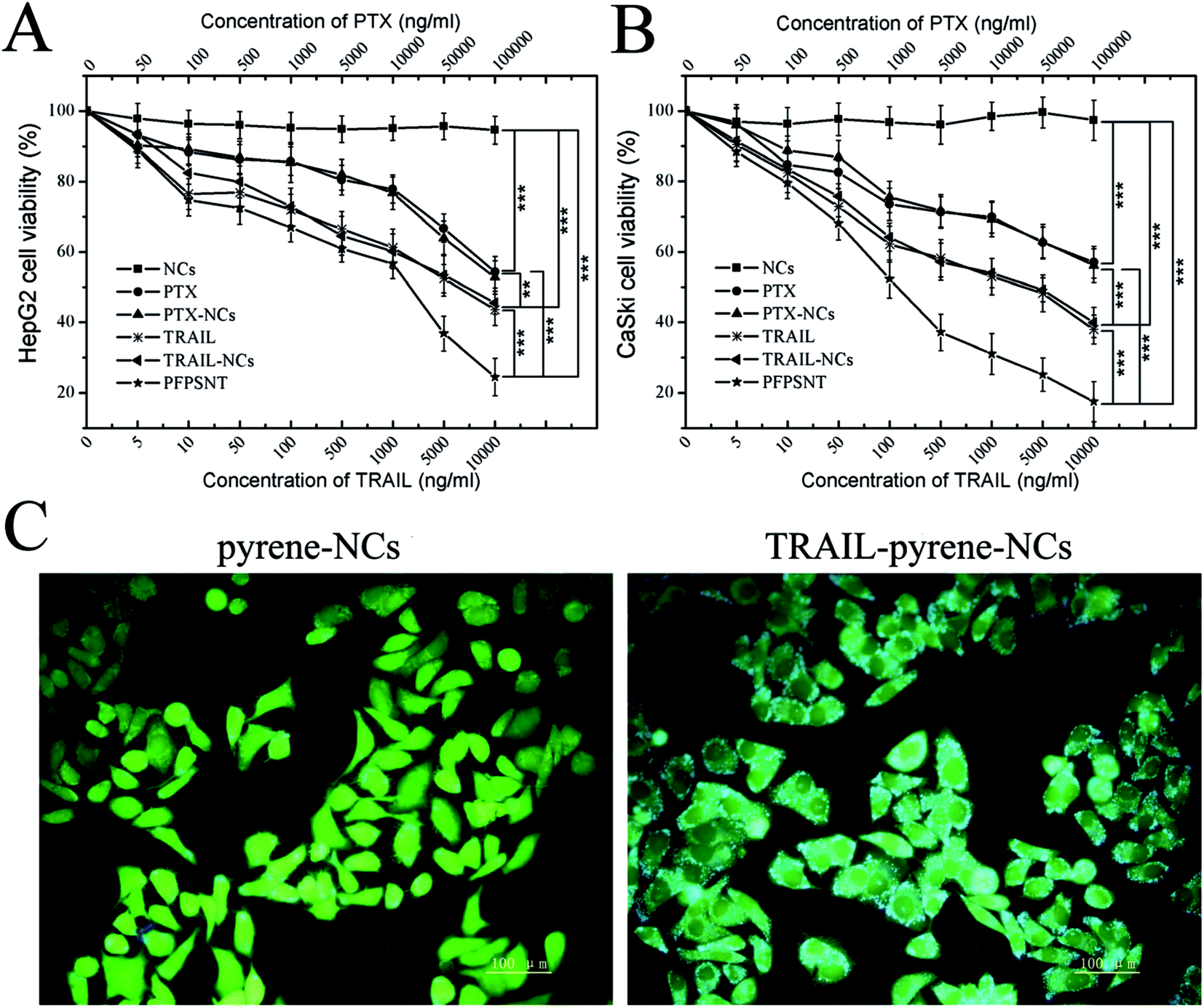 | ||
| Fig. 4 Synergistic cytotoxicity evaluations. Viabilities of CaSki cells (A) and HepG2 cells (B) treated with NCs, PTX, PTX-NCs, TRAIL, TRAIL-NCs and PFPSNTs; (C) cellular uptake of TRAIL-pyrene-NCs in HepG2. The results from statistic analysis were marked in (A) and (B).*** Means P values of <0.01;** means P values of <0.05. | ||
To test the targeting capability of the PFPSNT, a fluorescent dye pyrene, instead of PTX, was encapsulated into the nanocapsules. Pyrene gives blue emission fluorescence at the 338 nm excitation. When HepG2 cells were cultured with TRAIL-pyrene-NCs or pyrene-NCs without TRAIL conjugation, the cells treated with TRAIL-pyrene-NCs showed a significant increase in the intensity of blue fluorescence compared to those cells treated with pyrene-NCs (Fig. 4C). The cells treated with pyrene-NCs without TRAIL conjugation mainly displayed green fluorescence which came from FDA staining. This result demonstrated that TRAIL indeed had a greatly targeting capacity.
3.5 Anticancer efficacy of PFPSNT on HepG2 xenografts
Anticancer efficacy of PFPSNT was investigated in vivo by using the nude mice bearing HepG2 xenografts. The body weights and tumor volumes were measured every two days and summarized in Fig. 5. The mean body weights of mice in all five groups increased slowly with a similar manner (see Fig. 5A). However, the tumor volumes in each group were significantly different (see Fig. 5B). In the control group, five tumors growed rapidly, and the mean volume increased approximately 13 fold in two weeks. The tumor volumes in other three groups treated with PTX, PTX-NCs or TRAIL showed somewhat increase, but much slowly than that for control group. This result indicated that PTX or TRAIL alone was able to suppress tumor growth as expected. In the group of PFPSNT treatment, the PFPSNT showed marked tumor inhibition. After 14 day treatments, the tumors disappeared from two mice in this group, and other three tumors stoped their growth. | ||
| Fig. 5 The body weights (A) and the relative tumor volumes (B) of HepG2 xenografted nu/nu mice treated with PBS, PTX, PTX-NCs, TRAIL and PFPSNT, respectively. (C) Photographs of excised tumors and the mice after 14 day treatments. (D) Mean weights of five tumors in each group after 14 day treatments.*** Presented significant difference (p < 0.01), and** means P values of <0.05. | ||
After 14 day treatments, the mice were sacrificed and tumors were collected. The photographs of all tumors obtained from five groups were shown in Fig. 5C. The tumor sizes in the control group were significantly larger than those for other four groups. In the PFPSNT-treated group, the sizes of tumors were the smallest, and two tumors disappeared post treatment. Each tumor was weighted, and average weights for each group were shown in Fig. 5D. Average tumor weights were 478 ± 129 mg for the control group, 172 ± 56 mg for the PTX-treated group, 116 ± 47 mg for PTX-NCs-treated group, 88 ± 25 mg for TRAIL-treated group and 16 ± 15 mg for the PFPSNT-treated group. These results obtained from in vivo study clearly show that PFPSNT indeed displays high anticancer activity.
3.6 Histological evaluations of HepG2 xenografts and major organs of HepG2 xenografted mice
Tumor necrosis and apoptosis were visualized by staining with H&E, TUNEL and DAPI. As shown in Fig. 6A, there were obvious tumor cells with little red cytoplasm and big purple nucleus, while vascular in tumor also could be found. Tumor necrosis area of H&E stained tumor tissues were statistical analysis by Image-pro Plus and SPSS. The mean tumor necrosis area of PFPSNT, TRAIL, PTX-NCs, PTX and control groups were 38.8%, 31.1%, 23.9%, 17.2% and 9.4%. The tumor tissues from five groups were stained with TUNEL and observed under fluorescence microscope. In this experiment, apoptotic cells displayed green fluorescence. As shown in Fig. 6A, the tumor tissue treated with PFPSNT showed much stronger green fluorescence as compared to other four groups, indicating that the combination of TRAIL and PTX indeed induced cell apoptosis, which was more than those treated with single therapeutic approach alone. DAPI staining showed a smaller amount of nucleus presented in the tumor tissues of the PFPSNT-treated group, indicating that more cell necrosis happened in this group. Tumor tissues photos viewed in the bright field and merged picture were also shown in Fig. 6A.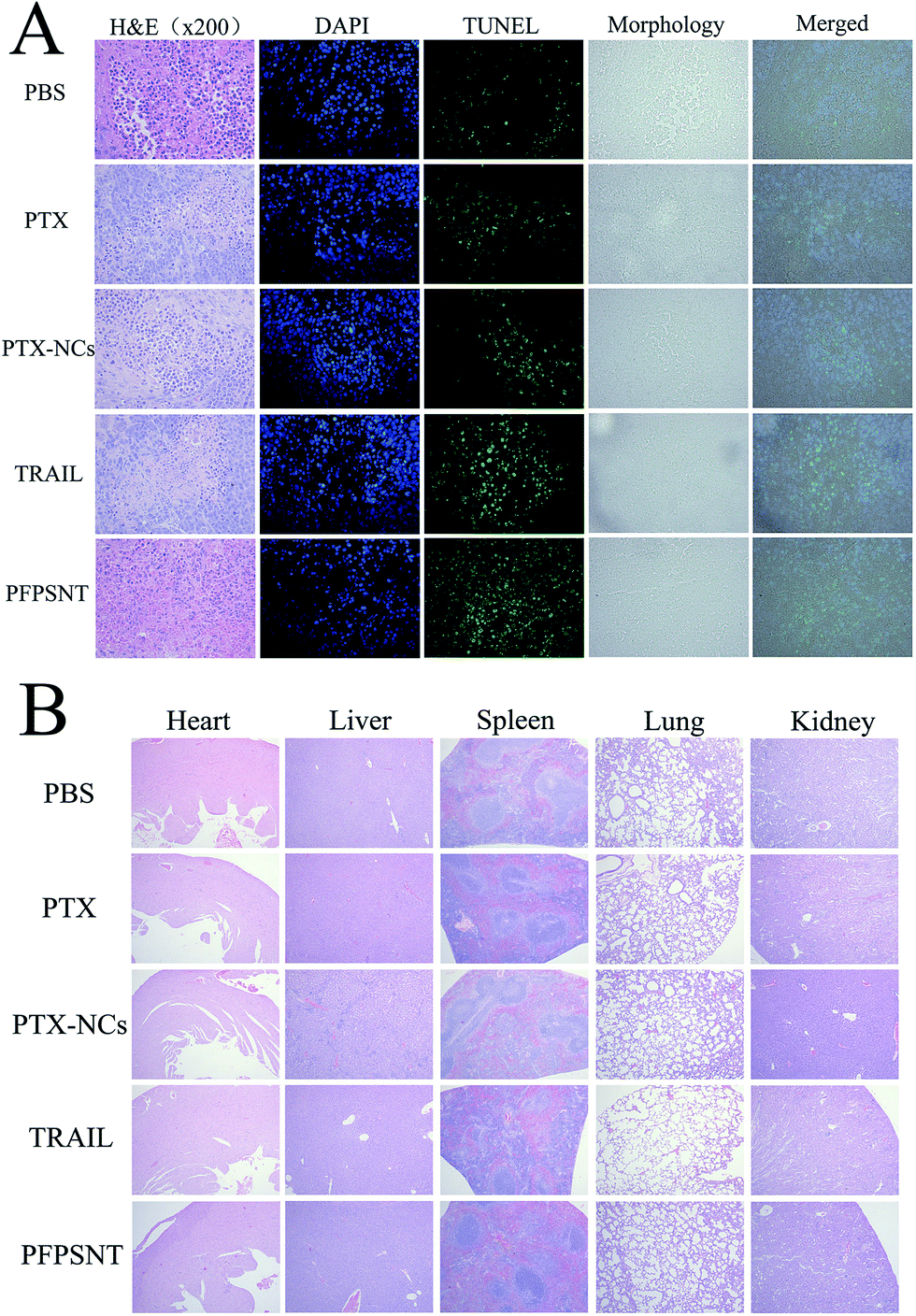 | ||
| Fig. 6 Histological observations of HepG2 xenografts and major organs of HepG2-xenografted nu/nu mice treated with PTX, PTX-NCs, TRAIL or PFPSNT. (A) The tumor tissues were stained with H&E, TUNEL and DAPI respectively, and (B) all tissues of mouse organs were stained by H&E staining. | ||
In vivo toxicity was also examined via H&E staining of major organs of the mice bearing HepG2 xenografts used in this study, including heart, liver, spleen, lung and kidney. As shown in Fig. 6B, no obvious lesion or inflammation was found in all histological samples collected from five groups. These results showed that the nano material and TRAIL protein used in this study had a good biocompatibility, and could be used as drug carriers for further clinical treatment.
4. Conclusion
In this study, we describe a novel strategy to develop an anticancer drug for targeting cancer therapy, in which PTX-NCs was made firstly and then conjugated with TRAIL to form a novel nano-drug PFPSNT. We expected that TRAIL linked to the carboxylic groups of the F127 on the surface of PFPSNT acted as a direct anticancer reagent and a delivery vector targeting DR4 and DR5 death receptors of cancer cells. Meanwhile, PTX embedded in pluronic F127/P123 silica nanocapsules acted as another anticancer reagent and performed a sustained release in order to reduce its side effects during cancer therapy. Fig. 7 presented the technical route for synthesizing PFPSNT and a possible anticancer mechanism. At first, PFPSNT targets cancer cells via interactions of TRAIL with death receptors DR4 and DR5, and then TRAIL induces apoptosis of tumor cells. Meanwhile, PTX released from PFPSNT goes directly into tumor cells to cause cell necrosis. As expected, the pluronic F127/P123 silica nanocapsules prepared in this study were spherical nanoparticles with a size of 24 nm, and more stabile than the F127/P123 nanomicelles. Polymeric F127/P123 silica nanocapsule indeed enhanced PTX solubility, and sustained PTX release. In both in vitro and in vivo studies, the PFPSNT made in this study showed a strong capability for killing HepG2 and CaSki cells and restraining the growth of HepG2 xenografts without impairment of host organs. As a result, PFPSNT could be considered as a promising drug to treat complex tumors in targeting cancer therapy. | ||
| Fig. 7 A schematic presenting the synthetical route of the novel nano-drug PFPSNT and its possible anticancer mechnism. | ||
Ethical statement
Animals, used in this study, were maintained in accordance with the guidelines of the national standard of China for laboratory animal—requirements of environment and housing facilities (GB 14925-2010), and approved by the institutional committee of the Laboratory Animal Center, Huazhong University of Science and Technology.Acknowledgements
This work was supported by the grant (51303046) founded by National Natural Science Foundation of China, and the science and technology project of Finance Department of Hubei Province. The authors also would like to thank the Laboratory Animal Center, Huazhong University of Science and Technology for its kind assistances and guidelines of animal experiments.References
- G. Pan, K. O'Rourke and A. M. Chinnaiyan, et al., The receptor for the cytotoxic ligand TRAIL, Science, 1997, 276, 111–113 CrossRef CAS PubMed.
- R. M. Pitti, S. A. Marsters and S. Ruppert, et al., Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family, J. Biol. Chem., 1996, 271, 12687–12690 CrossRef CAS PubMed.
- J. J. Song and Y. J. Lee, Differential cleavage of Mst1 by caspase-7/-3 is responsible for TRAIL-induced activation of the MAPK superfamily, Cell. Signalling, 2008, 20, 892–906 CrossRef CAS PubMed.
- R. S. Herbst, S. G. Eckhardt, R. Kurzrock, S. Ebbinghaus, P. J. O'Dwyer, M. S. Gordon, W. Novotny, M. A. Goldwasser, T. M. Tohnya and B. L. Lum, et al., Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer, J. Clin. Oncol., 2010, 28, 2839–2846 CrossRef CAS PubMed.
- J. C. Soria, E. Smit, D. Khayat, B. Besse, X. Yang, C. P. Hsu, D. Reese, J. Wiezorek and F. Blackhall, Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in combination with paclitaxel, carboplatin, and bevacizumab in patients with advanced non-squamous non-small-cell lung cancer, J. Clin. Oncol., 2010, 28, 1527–1533 CrossRef CAS PubMed.
- J. Wiezorek, P. Holland and J. Graves, Death receptor agonists as a targeted therapy for cancer, Clin. Cancer Res., 2010, 16, 1701–1708 CrossRef CAS PubMed.
- L. Zhang and B. Fang, Mechanisms of resistance to TRAIL-induced apoptosis in cancer, Cancer Gene Ther., 2005, 12, 228–237 CrossRef CAS PubMed.
- C. S. Mitsiades, S. P. Treon and N. Mitsiades, et al., TRAIL/Apo2L ligand selectively induces apoptosis and overcomes drug resistance in multiple myeloma: therapeutic applications, Blood, 2001, 98, 795–804 CrossRef CAS PubMed.
- S. Fulda, W. Wick and M. Weller, et al., Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo, Nat. Med., 2002, 8, 808–815 CAS.
- M. Garofalo, G. D. Leva and G. Romano, et al., miR-221&222 Regulate TRAIL Resistance and Enhance Tumorigenicity through PTEN and TIMP3 Downregulation, Cancer Cell, 2009, 16, 498–509 CrossRef CAS PubMed.
- U. M. Nazim, J. K. Jeong, J. W. Seol, J. Hur, S. K. Eo, J. H. Lee and S. Y. Park, Inhibition of the autophagy flux by gingerol enhances TRAIL-induced tumor cell death, Oncol. Rep., 2015, 33, 2331–2336 CAS.
- S. Ray and A. Almasan, Apoptosis Induction in Prostate Cancer Cells and Xenografts by Combined Treatment with Apo2 Ligand/Tumor Necrosis Factor-related Apoptosis-inducing Ligand and CPT-11, Cancer Res., 2003, 63, 4713–4723 CAS.
- I. Kim, H. J. Byeon and T. H. Kim, et al., Doxorubicin-loaded porous PLGA microparticles with surface attached TRAIL for the inhalation treatment of metastatic lung cancer, Biomaterials, 2013, 34, 6444–6453 CrossRef CAS PubMed.
- S. Bae, K. Ma and T. H. Kim, et al., Doxorubicin-loaded human serum albumin nanoparticles surface-modified with TNF-related apoptosis-inducing ligand and transferrin for targeting multiple tumor types, Biomaterials, 2012, 33, 1536–1546 CrossRef CAS PubMed.
- E. Pasquier, M. Carré, B. Pourroy, L. Camoin, O. Rebaï, C. Briand and D. Braguer, Antiangiogenic activity of paclitaxel is associated with its cytostatic effect, mediated by the initiation but not completion of a mitochondrial apoptotic signaling pathway, Mol. Cancer Ther., 2004, 3, 1301–1310 CAS.
- L. Li, X. Wen, Z. Bu, X. Cheng, X. Xing, X. Wang, L. Zhang, T. Guo, H. Du, B. Fan and J. Ji, Paclitaxel enhances tumoricidal potential of TRAIL via inhibition of MAPK in resistant gastric cancer cells, Oncol. Rep., 2016, 35, 3009–3027 CAS.
- J. Gong, D. Yang, S. Kohanim, R. Humphreys, L. Broemeling and R. Kurzrock, Novel in vivo imaging shows up-regulation of death receptors by paclitaxel and correlates with enhanced antitumor effects of receptor agonist antibodies, Mol. Cancer Ther., 2006, 5, 2991–3000 CrossRef CAS PubMed.
- T. B. Hunter, N. J. Manimala, K. A. Luddy, T. Catlin and S. J. Antonia, Paclitaxel and TRAIL synergize to kill paclitaxel-resistant small cell lung cancer cells through a caspase-independent mechanism mediated through AIF, Anticancer Res., 2011, 31, 3193–3204 CAS.
- R. Nimmanapalli, C. L. Perkins, M. Orlando, E. O'Bryan, D. Nguyen and K. N. Bhalla, Pretreatment with paclitaxel enhances apo-2 ligand/tumor necrosis factor-related apoptosis-inducing ligand induced apoptosis of prostate cancer cells by inducing death receptors 4 and 5 protein levels, Cancer Res., 2001, 61, 759–763 CAS.
- E. K. Rowinsky and R. C. Donehower, Paclitaxel (taxol), N. Engl. J. Med., 1995, 332, 1004–1014 CrossRef CAS PubMed.
- J. D. Adams, K. P. Flora and B. R. Goldspiel, et al., Taxol: a history of pharmaceutical development and current pharmaceutical concerns, JNCI Monogr., 1993, 15, 141–147 Search PubMed.
- H. Gelderblom, J. Verweij and K. Nooter, et al., Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation, Eur. J. Cancer, 2001, 37, 1590–1598 CrossRef CAS PubMed.
- H. Xin, L. Chen and J. Gu, et al., Enhanced anti-glioblastoma efficacy by PTX-loaded PEGylated poly(ε-caprolactone) nanoparticles: in vitro and in vivo evaluation, Int. J. Pharm., 2010, 402, 238–247 CrossRef CAS PubMed.
- Y. Saito, Nanoparticles and filled nanocapsules, Carbon, 1995, 33, 979–988 CrossRef CAS.
- C. E. Mora-Huertas, H. Fessi and A. Elaissari, Polymer-based nanocapsules for drug delivery, Int. J. Pharm., 2010, 385, 113–142 CrossRef CAS PubMed.
- P. Couvreur, G. Barratt and E. Fattal, et al., Nanocapsules Technology: A review, Crit. Rev. Ther. Drug Carrier Syst., 2002, 19, 99–134 CrossRef CAS PubMed.
- A. L. Savian, D. Rodrigues and J. Weber, et al., Dithranol-loaded lipid-core nanocapsules improve the photostability and reduce the in vitro irritation potential of this drug, Mater. Sci. Eng., C, 2015, 46, 69–76 CrossRef CAS PubMed.
- R. H. Staff, M. Gallei and M. Mazurowski, et al., Patchy nanocapsules of poly(vinylferrocene)-based block copolymers for redox-responsive release, ACS Nano, 2012, 6, 9042–9049 CrossRef CAS PubMed.
- F. Chécot, J. Rodríguez-Hernández and Y. Gnanou, et al., pH-responsive micelles and vesicles nanocapsules based on polypeptide diblock copolymers, Biomol. Eng., 2007, 24, 81–85 CrossRef PubMed.
- S. Peltier, J. M. Oger and F. Lagarce, et al., Enhanced Oral Paclitaxel Bioavailability After Administration of Paclitaxel-Loaded Lipid Nanocapsules, Pharm. Res., 2006, 23, 1243–1250 CrossRef CAS PubMed.
- D. Ren, F. Kratz and S. W. Wang, Protein nanocapsules containing doxorubicin as a pH-responsive delivery system, Small, 2011, 7, 1051–1060 CrossRef CAS PubMed.
- V. Chupin, A. I. P. M. De Kroon and B. De Kuijff, Lipid organization in cisplatin nanocapsules: lipid-coated aggregates of cisplatin with high cytotoxicity, Cell. Mol. Biol. Lett., 2005, 10, 81–84 Search PubMed.
- Y. Zhang, B. Y. Hsu, C. Ren, X. Li and J. Wang, Silica-based nanocapsules: synthesis, structure control and biomedical applications, Chem. Soc. Rev., 2015, 44, 315 RSC.
- K. Tatematsu, M. Iijima and N. Yoshimoto, et al., Bio-nanocapsules displaying various immunoglobulins as an active targeting-based drug delivery system, Acta Biomater., 2016, 35, 238–247 CrossRef CAS PubMed.
- X. G. Wang, T. Jandl and E. Dadachova, et al., Effect of naive and radiolabeled rhTRAIL on the cervical cancer xenografts in mice, Ther. Delivery, 2014, 5, 139–147 CrossRef CAS PubMed.
- S. J. Huang, S. L. Sun and T. H. Feng, et al., Folate-mediated chondroitin sulfate-Pluronic 127 nanogels as a drug carrier, Eur. J. Pharm. Sci., 2009, 38, 64–73 CrossRef CAS PubMed.
- F. Szoka Jr and D. Papahadjopoulos, Comparative Properties and Methods of Preparation of Lipid Vesicles (Liposomes), Annu. Rev. Biophys. Bioeng., 1980, 9, 467–508 CrossRef CAS PubMed.
- Y. Zhang, M. Wang and Y. Zheng, et al., PEOlated Micelle/Silica as Dual-Layer Protection of Quantum Dots for Stable and Targeted Bioimaging, Chem. Mater., 2013, 25, 2976–2985 CrossRef CAS.
- T. Zhao, X. Liu and Y. Li, et al., Fluorescence and drug loading properties of ZnSe:Mn/ZnS-Paclitaxel/SiO2 nanocapsules templated by F127 micelles, J. Colloid Interface Sci., 2016, 490, 436–443 CrossRef PubMed.
- W. Zhang, Y. Shi and Y. Chen, et al., Multifunctional Pluronic P123/F127 mixed polymeric micelles loaded with paclitaxel for the treatment of multidrug resistant tumors, Biomaterials, 2011, 32, 2894–2906 CrossRef CAS PubMed.
- A. Andersen, D. J. Warren and P. F. Brunsvig, et al., High sensitivity assays for docetaxel and paclitaxel in plasma using solid-phase extraction and high-performance liquid chromatography with UV detection, BMC Clin. Pharmacol., 2006, 6, 1–10 Search PubMed.
- M. Tsai, Z. Lu and M. G. Wientjes, et al., Paclitaxel-loaded polymeric microparticles: Quantitative relationships between in vitro, drug release rate and in vivo, pharmacodynamics, J. Controlled Release, 2013, 172, 737–744 CrossRef CAS PubMed.
- J. Hong, D. Xu and P. Gong, et al., Covalent binding of α-chymotrypsin on the magnetic nanogels covered by amino groups, J. Mol. Catal. B: Enzym., 2007, 45, 84–90 CrossRef CAS.
- P. Demaerel, H&E Staining, Encyclopedia of Diagnostic Imaging, Springer, Berlin, Heidelberg, 2008 Search PubMed.
- F. Otto, DAPI staining of fixed cells for high-resolution flow cytometry of nuclear DNA, Methods Cell Biol., 1990, 33, 105–110 CrossRef CAS PubMed.
- S. W. Perry, L. G. Epstein and H. A. Gelbard, Simultaneous in situ detection of apoptosis and necrosis in monolayer cultures by TUNEL and trypan blue staining, BioTechniques, 1997, 22, 1102–1106 CAS.
- R. R. Wilcox, Two-way ANOVA, Applying Contemporary Statistical Techniques, 2003 Search PubMed.
- W. Zhang, J. Hao and Y. Shi, et al., Paclitaxel-loaded Pluronic P123/F127 mixed polymeric micelles: formulation, optimization and in vitro characterization, Int. J. Pharm., 2009, 376, 176–185 CrossRef PubMed.
- H. Tan, N. S. Liu and B. He, et al., Facile synthesis of hybrid silica nanocapsules by interfacial templating condensation and their application in fluorescence imaging, Chem. Commun., 2009, 6240–6242 RSC.
- B. Y. W. Hsu, C. Teh and H. Tan, et al., PEO surface-decorated silica nanocapsules and their application in in vivo imaging of zebrafish, RSC Adv., 2012, 2, 12392–12399 RSC.
- N. Rapoport, Stabilization and activation of Pluronic micelles for tumor-targeted drug delivery, Colloids Surf., B, 1999, 16, 93–111 CrossRef CAS.
- R. P. Shringi, PyMol Software for 3D Visualization of Aligned Molecules, Biomaterials, 2005, 26, 63–72 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |