
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple and efficient Fmoc removal in ionic liquid†
M. L. Di Gioia *a,
P. Costanzod,
A. De Ninob,
L. Maiuolob,
M. Nardibc,
F. Olivitod and
A. Procopiod
*a,
P. Costanzod,
A. De Ninob,
L. Maiuolob,
M. Nardibc,
F. Olivitod and
A. Procopiod
aDipartimento di Farmacia e Scienze della Salute e della Nutrizione, Edificio Polifunzionale, Università della Calabria, 87030 Arcavacata di Rende, Cosenza, Italy. E-mail: ml.digioia@unical.it; Tel: +39 0984 493095
bDipartimento di Chimica, Università della Calabria, Cubo 12C, Arcavacata di Rende, CS, Italy
cDipartimento di Agraria, Università Telematica San Raffaele, Roma, Via di Val Cannuta, 247, 00166, Italy
dDipartimento di Scienze della Salute, Università Magna Graecia, Viale Europa, Germaneto, CZ, Italy
First published on 21st July 2017
Abstract
A mild method for an efficient removal of the fluorenylmethoxycarbonyl (Fmoc) group in ionic liquid was developed. The combination of a weak base such as triethylamine and [Bmim][BF4] makes the entire system more efficient for the cleavage at room temperature of various amines and amino acid methyl esters in short reaction times. The procedure works well even in the case of N-Fmoc amino acids bearing acid-sensitive protecting groups and of N-alkylated amino acid methyl esters. The solvent-free condition provides a complementary method for Fmoc deprotection in solution phase peptide synthesis and modern organic synthesis.
Introduction
Protective group practice represents an imperative strategy in multistep organic synthesis.1 Easy protection and deprotection methods are fundamental to enable the construction of chemical bonds and in the presence of pre-existing bonds.2 Hence, mild removal methods as well as finding new procedures for deprotection are highly desirable.The amine group is probably one of the most common functional groups found in biologically important molecules and the 9-fluorenylmethoxycarbonyl (Fmoc) group represents one of the most widely used protecting groups in the masking of amino functionality, finding particular utility in solution- and solid-phase peptide synthesis and chemistry.3
The classical means of Fmoc deprotection employ a large excess of a secondary amine, such as piperidine in DMF.4 However, piperidine is a controlled substance with usage in the synthesis of narcotic drugs and psychotropic substances, and leads to large amounts of toxic waste (20–50% solutions are required).5 Similarly, 1,8-diazabicyclo[5.4.0]undec-7-ene in DMF has found extensive usage for deprotection of difficult sequences6 but being a strong base (pKa 13.5) it promotes side reactions such as aspartimide formation.7 Furthermore, both agents are unfortunately not very suitable in the alternative liquid phase synthesis, nor the use of the conventional solvent DMF due to its low volatility.
Various bases with higher pKa values than piperidine can remove the Fmoc group with high efficacy though some of these bases induce side reactions and require enormous quantities of solvent. Consequently, there is an ongoing effort to search for reagents and solvents that are easily removable during mandatory intermediates isolation.8–10
With the increase in environmental concerns and governmental legislation, in order to meet the principles of green chemistry, the development of environmentally benign organic reactions has become a crucial and demanding research area in the modern chemical sciences.11 Thus, the development of an environmentally benign, efficient and simple methodology for a fundamental organic transformation is in great demand.
In this context, synthetic manipulations should be made to reduce the use of harmful compounds and volatile organic solvents in reactions, and their subsequent work-up.
Ionic liquids have attracted extensive research interest in recent years as an eco-friendly alternative to replace conventional organic solvents owing to their unique properties, such as non-inflammability, low volatility, negligible vapour pressure, reusability and high thermal stability. To date, many room-temperature ionic liquids (RTILs) have been used as solvents for several organic transformations.12
Among RTILs, ionic liquids containing imidazolium cations are typical since they are easily synthesized and as a powerful reaction media or catalyst show good performance in many reactions.13
Our recent interest has been in the development of new synthetic methods using ionic liquids as reaction media for the introduction and removal of different protecting groups.14
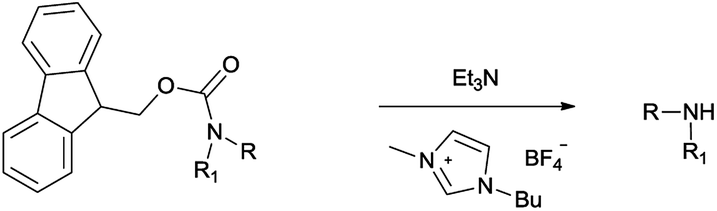
In this manuscript, we envisaged that the combination of a cheap, nontoxic and readily available amine such as triethylamine and an imidazolium-based IL could be appropriately adapted to fulfil the above requirements for the removal of the Fmoc protecting group.
Herein, we demonstrate a very simple method of cleavage by employing triethylamine in [Bmim][BF4] as recyclable reaction media, which will be a valuable addition to the existing Fmoc removal methods.
Results and discussion
In our initial experiments, we choose N-Fmoc aniline as a model substrate and the results for development and optimization of the deprotection studies are displayed in Table 1.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Temp. (°C) | Time (min) | Yieldb (%) |
| a Reaction conditions: Fmoc-aniline (1 mmol), Et3N (3 mmol) in 1 mL of solvent.b Isolated yield.c 1 mmol of Et3N.d 5 mL of DCM were used. | |||||
| 1 | [Bmim][BF4] | — | 25 | 300 | 0 |
| 2 | [Bmim][BF4] | Et3N | 25 | 5 | 90 |
| 3c | [Bmim][BF4] | Et3N | 25 | 300 | 62 |
| 4d | DCM | Et3N | 25 | 360 | 10 |
| 5 | [Bmim][Cl] | — | 80 | 240 | 30 |
| 6 | [Bmim][OH] | — | 80 | 240 | 45 |
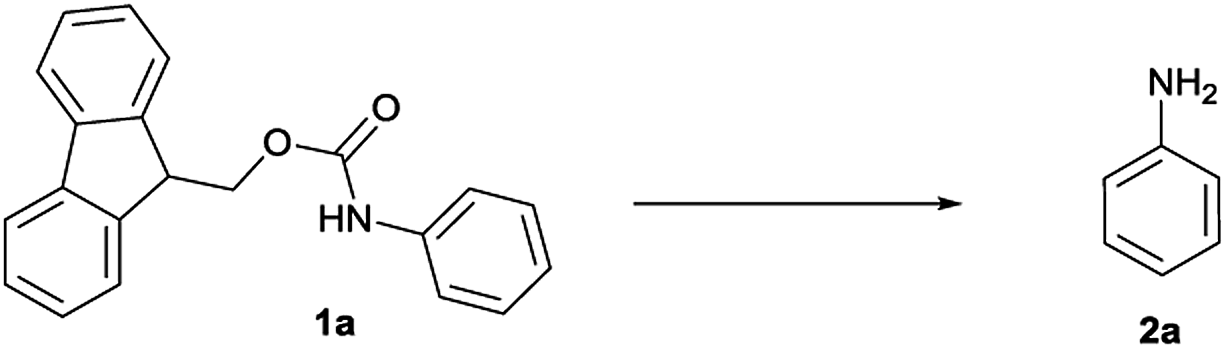
On treatment of the N-Fmoc aniline (1 mmol) with 1 mL of [Bmim][BF4] ionic liquid at room temperature, the deprotection did not take place (Table 1, entry 1). However, on adding 3 mmol of triethylamine, deprotection was smoothly accomplished after only 5 min in 90% yield (Table 1, entry 2). TLC analysis and ninhydrin assay confirmed the cleavage of the N-Fmoc group. Usage of less Et3N slowed down the reaction rate (5 h) and yield (Table 1, entry 3).
Changing the IL for a conventional solvent such as dichloromethane results in incomplete conversion after 6 h. The low yield (entries 4, Table 1) of the reaction conducted in DCM as the conventional solvent provides confirmation of the literature data,15 which reports that the cleavage of the Fmoc group by triethylamine in traditional organic solvents occurs very slowly (after 1080 min of reaction). This indicates that the reaction is probably strongly affected by the ionic liquid, which assists the process during the abstraction of the fluorenyl proton, enhancing the amine basicity.16 In addition, the ionic liquid has a stabilizing effect on the charged intermediates during the Fmoc removal.
Next, we tested other imidazolium based ionic liquids with basic properties for this transformation to avoid the use of Et3N in the reaction system. Employing 1-methyl-3-butyl imidazolium chloride ([Bmim][Cl]) or 1-methyl-3-butyl imidazolium hydroxide ([Bmim][OH]) as the solvent, catalyst and base in the deprotection reaction, the yields were low and the reaction times long (Table 1, entries 5 and 6). Furthermore, the use of these basic ILs required severe conditions of high temperature.
We found that the best results in terms of reaction rate (5 min) and product yield were the use 3 mmol Et3N in [Bmim][BF4] at room temperature (entry 2, Table 1).
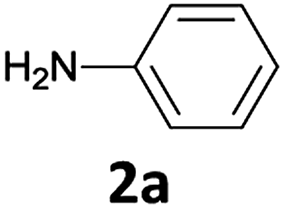
It is well known that during the deprotection of the Fmoc group, dibenzofulvene (DBF) is a by-product that needs to be efficiently removed. Particularly in peptide synthesis, a dibenzofulvene amine adduct can be responsible for side reactions.10 To prevent the formation of this undesired by-product, after the extraction of the free aniline from [Bmim][BF4] with diethyl ether, we added an aqueous 1 N HCl solution to protonate the amine. The undesired DBF was thus removed by diethyl ether extraction and the protonated aniline remained in the aqueous solution. The collected aqueous solution was then basified to neutral to give the native amine, which was finally recovered in ethereal solution.
Encouraged by the optimized solvent-free ionic liquid-catalyzed method for the deprotection of Fmoc-aniline, we applied the strategy to various Fmoc-protected aliphatic linear and cyclic amines (Table 2). The reactions carried out in [Bmim][BF4] at room temperature afforded the corresponding free amines in excellent yields and with short reaction times (4–8 min).
|
||||
|---|---|---|---|---|
| Entry | N-Fmoc-amine | Product | Time (min) | Yieldb (%) |
| a Reaction conditions: Fmoc-amine (1 mmol), Et3N (3 mmol), [Bmim][BF4] (1 mL) for 3–8 min at 25 °C. The free amine was obtained after a simple extraction procedure with diethyl ether from the ionic liquid and an appropriate work-up to remove the resultant dibenzofulvene.b Isolated yields after work-up.c Yield of the first of three runs using the same recovered IL. Yields of the subsequent runs were 88 and 89%, respectively. | ||||
| 1 | 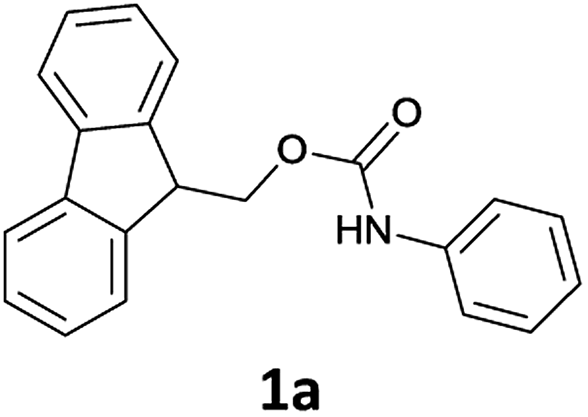 |
 |
5 | 90c |
| 2 | 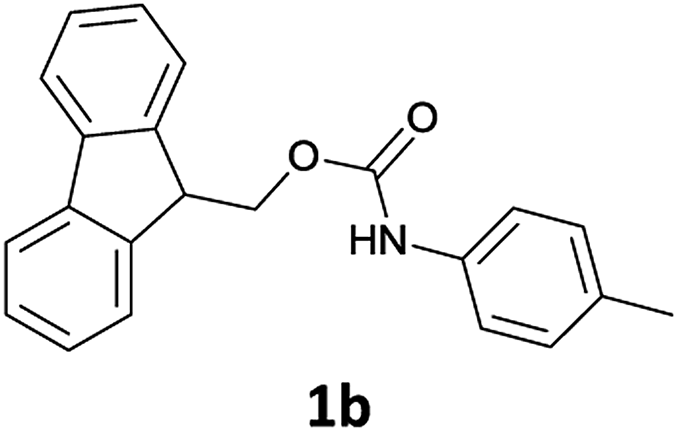 |
 |
6 | 91 |
| 3 | 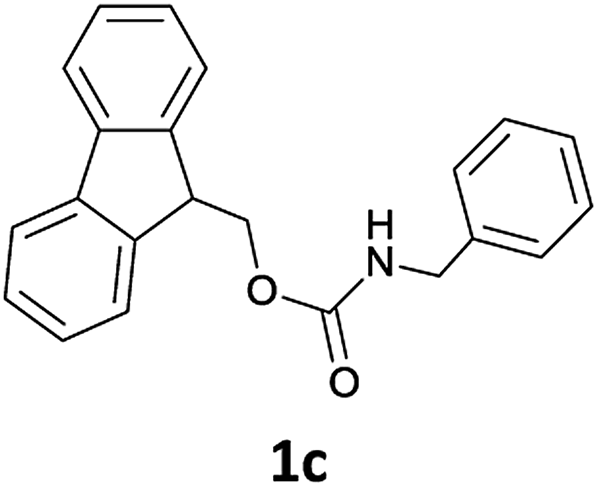 |
 |
5 | 85 |
| 5 | 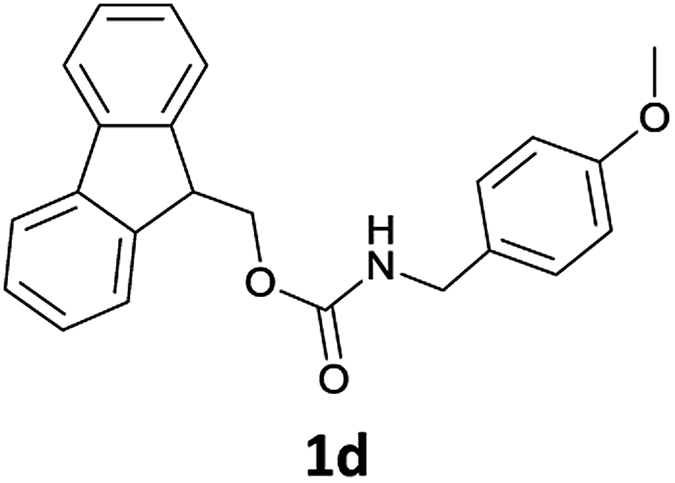 |
 |
8 | 80 |
| 6 | 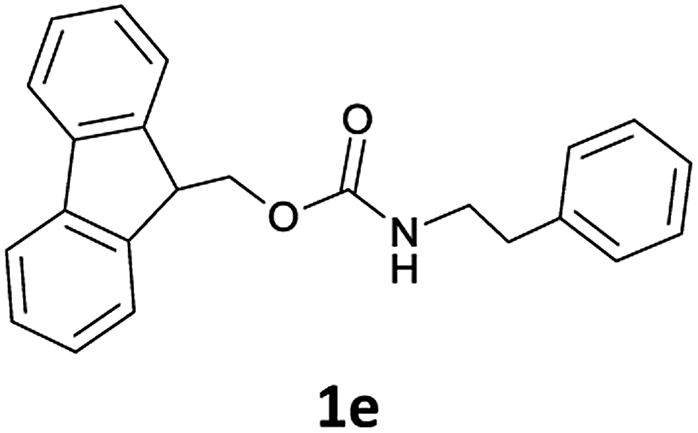 |
 |
7 | 82 |
| 7 | 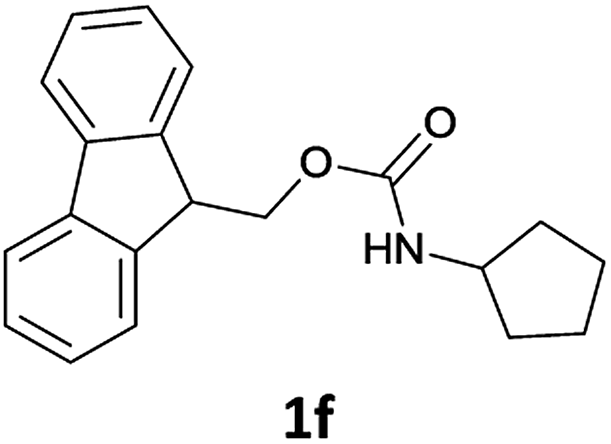 |
 |
7 | 92 |
| 8 | 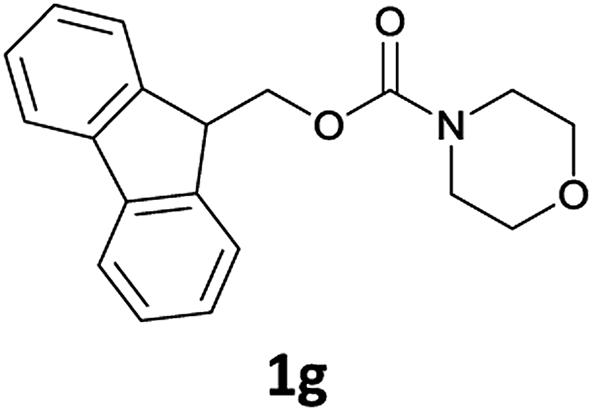 |
 |
4 | 93 |
| 9 | 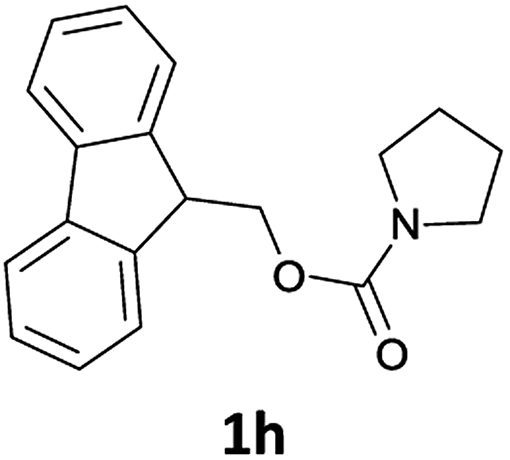 |
 |
4 | 93 |
| 10 |  |
 |
4 | 92 |
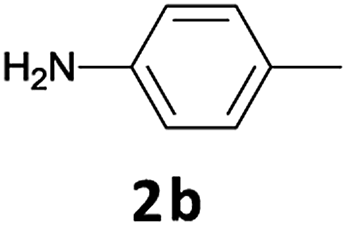
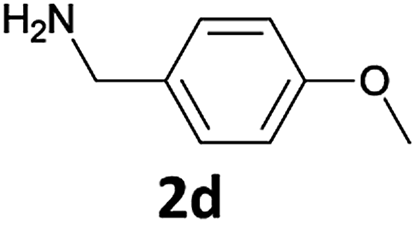
We observed that N-Fmoc-protected p-toluidine or derivatives of benzylamine easily undergo deprotection under the selected reaction conditions to afford the desired amines in excellent yields (Table 2).
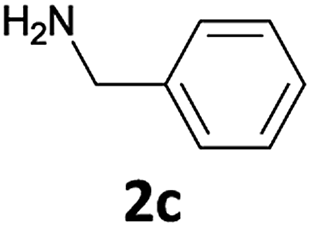
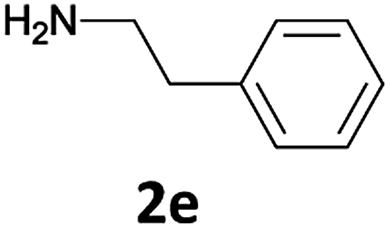
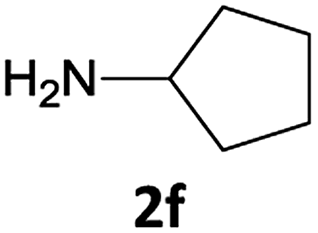
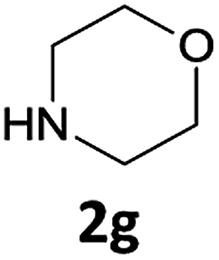
The method is also applicable for deprotection of N-Fmoc-protected aliphatic amines: the cleavage of N-Fmoc cyclopentylamine and 2-phenylethylamine (Table 2, entries 6 and 7) proceeded efficiently in high yields. The removal of the Fmoc group from secondary cyclic amines took a shorter reaction time and gave almost quantitative yields (Table 2, entries 8–9). It is evident that the resonance and inductive effects of the substituent groups on the tested amines have no influence on the Fmoc removal.
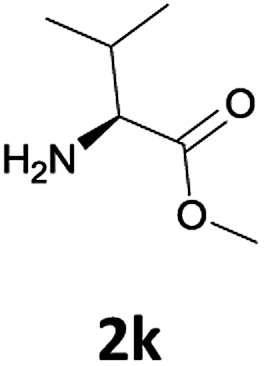
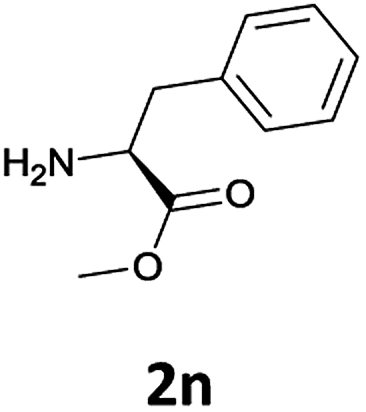
Considering the importance of the N-Fmoc protection strategy in peptide synthesis, we next examined the feasibility of N-Fmoc deprotection of amino acids. Therefore, the developed method was used in a series of lipophilic Fmoc-protected amino acid methyl esters (1j–1n, Table 3).
| Entry | Substrate | Product | Time (min) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: Fmoc-amine (1 mmol), Et3N (3 mmol), [Bmim][BF4] (1 mL) for 5–8 min at 25 °C. The free amino acid was obtained after a simple extraction procedure with diethyl ether from the ionic liquid and an appropriate work-up to remove the resultant dibenzofulvene. The free amino acids were characterized by GC/MS analysis after acetylation.b Isolated yields after work-up. | ||||
| 1 | 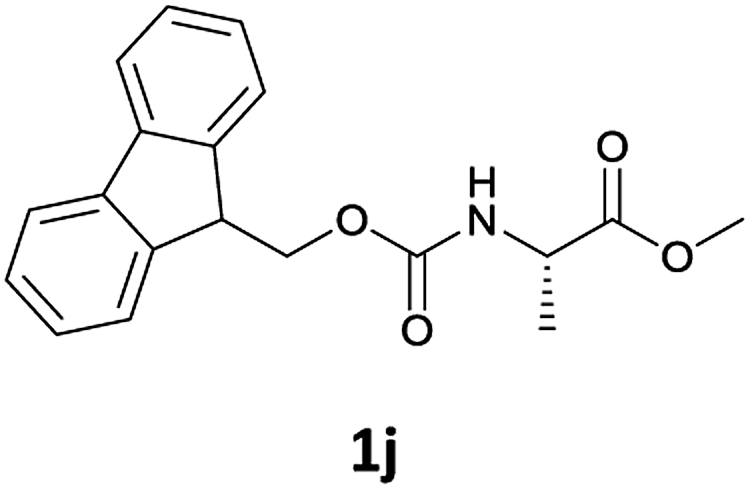 |
 |
8 | 88 |
| 2 | 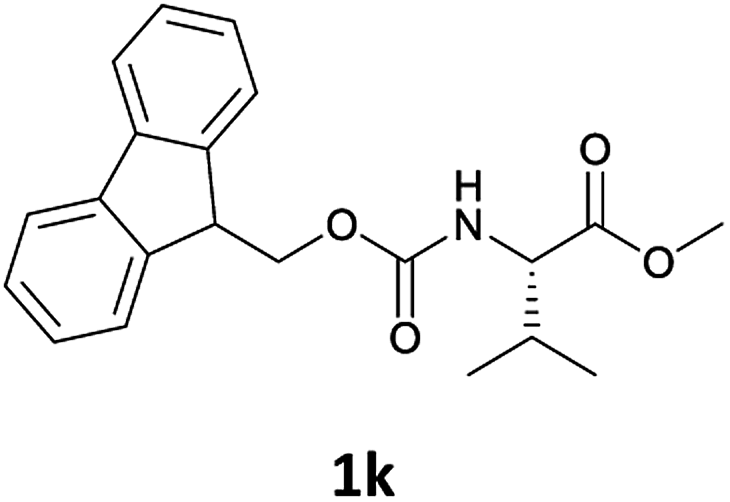 |
 |
10 | 86 |
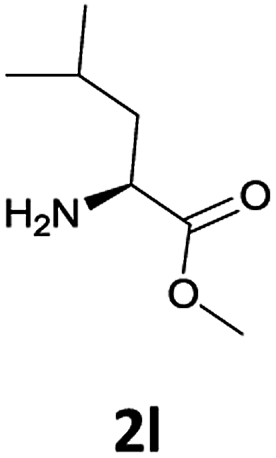
| 3 | 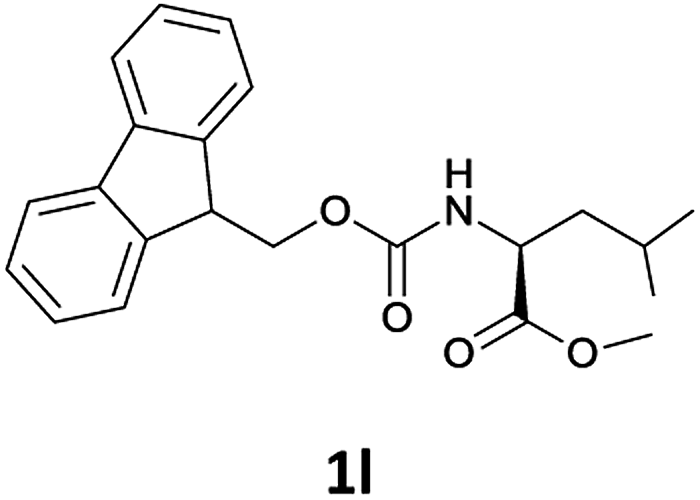 |
 |
10 | 81 |
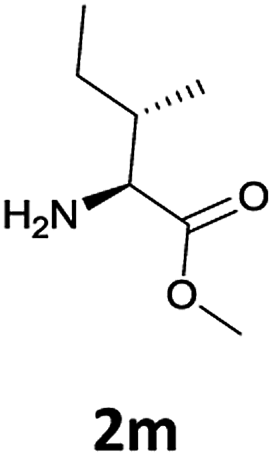
| 4 | 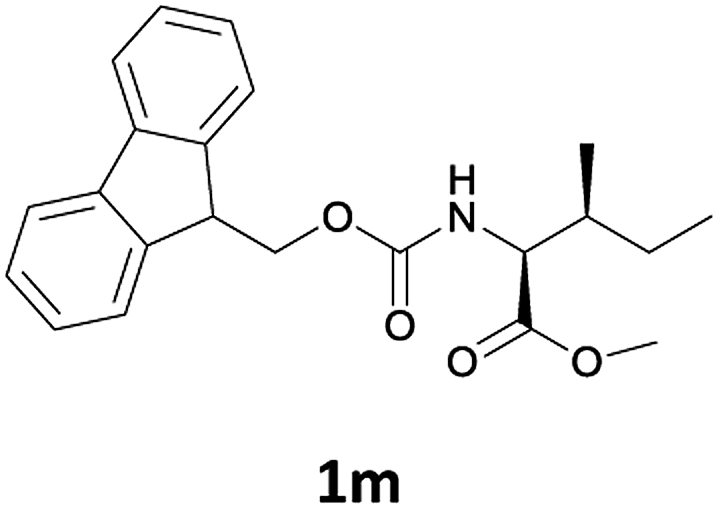 |
 |
10 | 79 |
| 5 | 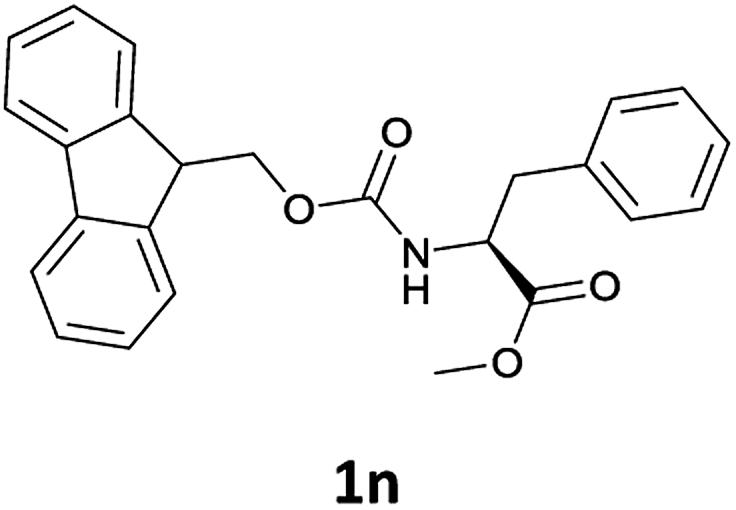 |
 |
10 | 82 |
| 6 | 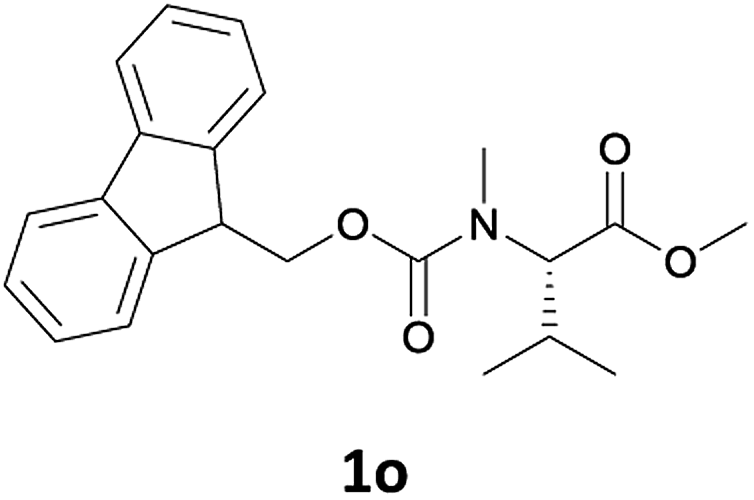 |
 |
12 | 75 |
| 7 | 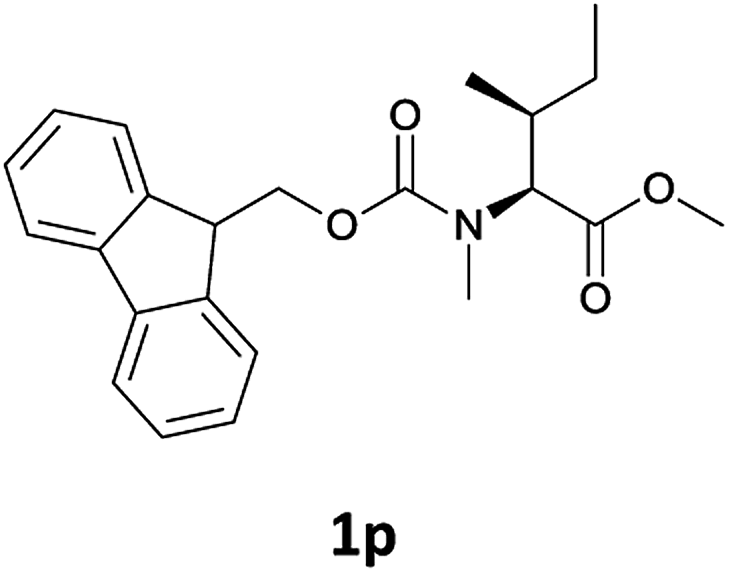 |
 |
15 | 83 |
| 8 |  |
 |
13 | 82 |
| 9 | 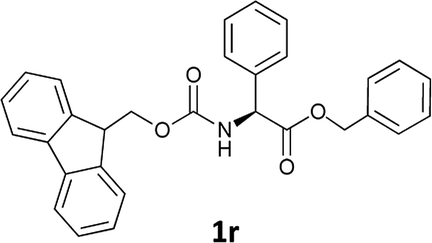 |
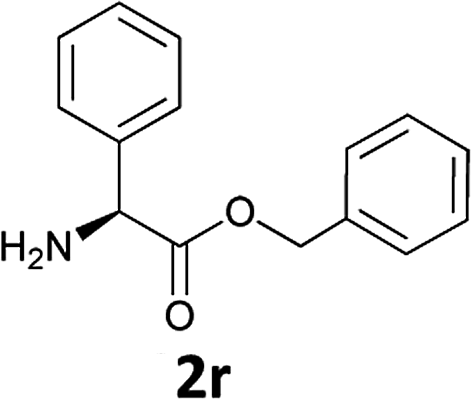 |
15 | 85 |
| 10 | 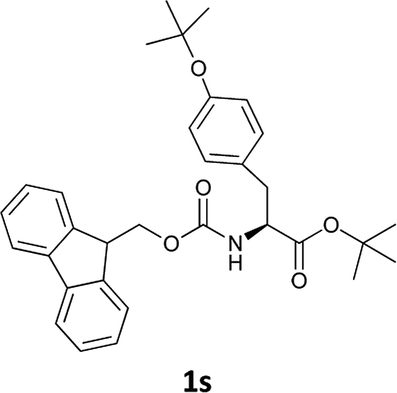 |
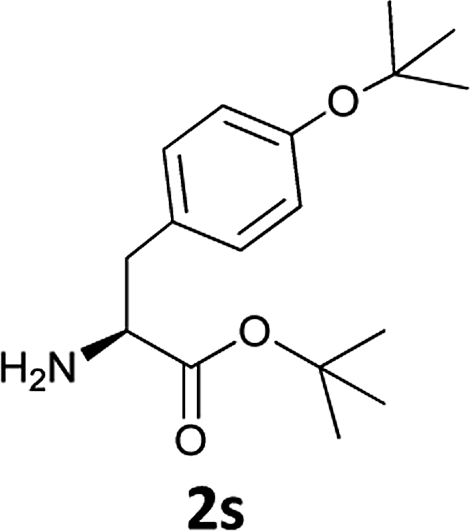 |
14 | 86 |
| 11 | 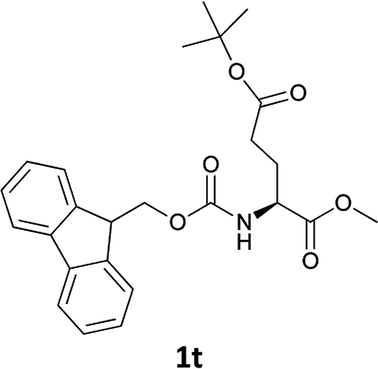 |
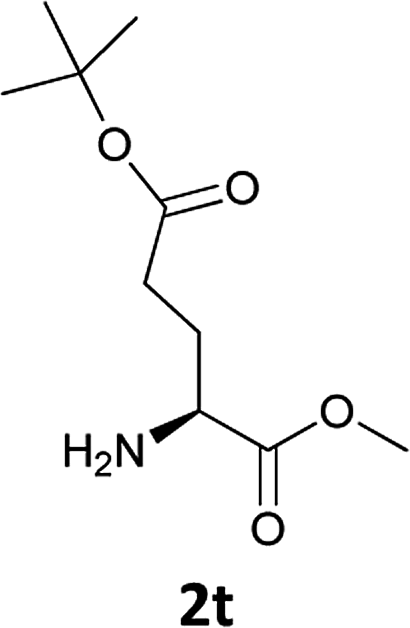 |
14 | 86 |
| 12 | 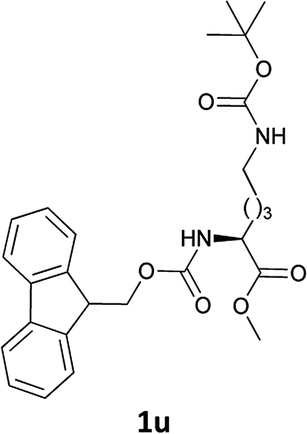 |
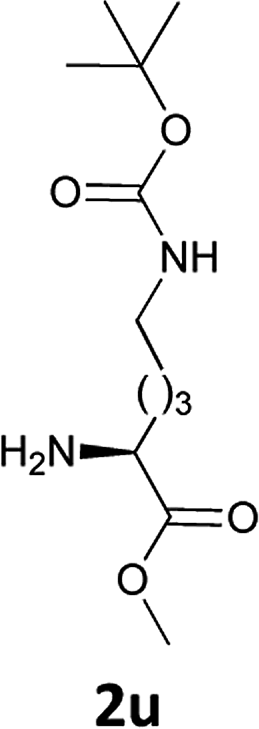 |
15 | 87 |
In order to demonstrate the successful removal of the Fmoc group in IL, compounds 1j–1n were characterized by GC/MS after their conversion into the corresponding N-acetyl derivatives by treatment with acetic anhydride under Schotten–Baumann conditions. Racemization at the α-carbon of the amino acid derivatives was not observed in any cases.
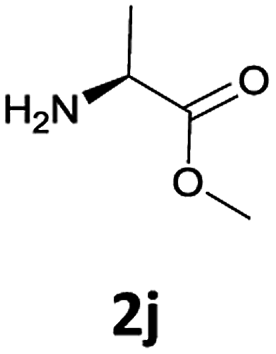
The stereochemical integrity was studied by chiral GC analysis. To this end, compound 2j (Table 3) obtained by IL-mediated Fmoc deprotection was acetylated to get Ac-L-Ala-OMe and compared with the methyl ester of DL-alanine opportunely acetylated. In Fig. 1 (panel a) two peaks were observed, which clearly show different retention times for the two enantiomers, Ac-D-Ala-OMe and Ac-L-AlaOMe. Instead, the chiral GC analysis of acetylated Ac-L-Ala-OMe revealed the presence of only one peak, thus strongly supporting the chiral integrity without undesired racemization (Fig. 1, panel b). Therefore, the treatment with Et3N in [Bmim][BF4] induced no racemization in the Fmoc deprotection process of amino acids.
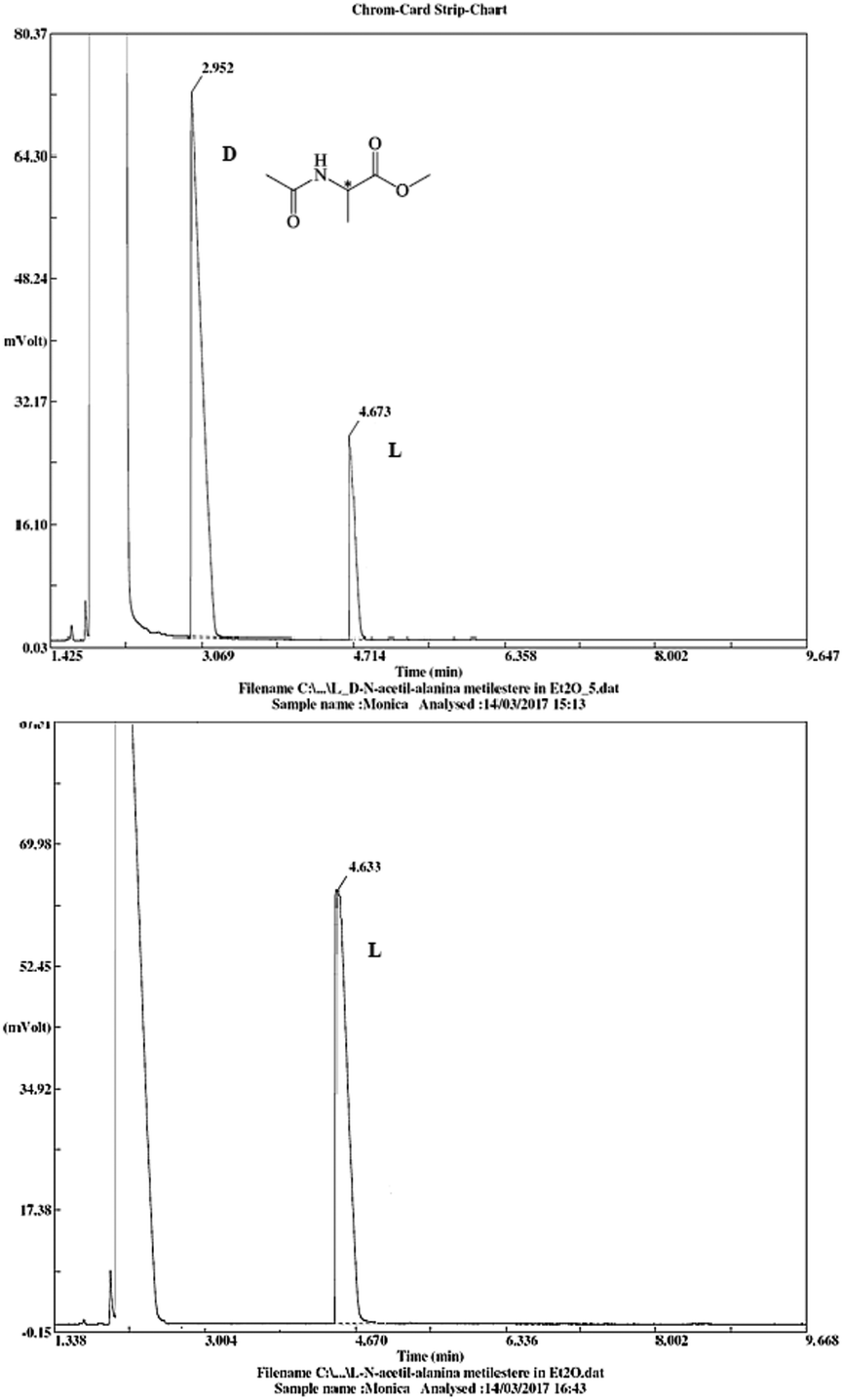 | ||
| Fig. 1 Chiral GC data for Ac-DL-Ala-OMe and Ac-L-Ala-OMe (25 m × 0.25 mm, diethyl tert-butyldimethylisilyl-β-cyclodextrin chiral capillary column). | ||
The applicability of this method was then tested for a set of N-Fmoc-amino acids containing functionalized side chains with acid-labile protecting groups (e.g., t-Bu, Boc, benzyl) to make the adopted procedure useful for peptide synthesis.
In particular, we selected the masking groups commonly used during peptide synthesis that are orthogonal to the Fmoc group (see Table 3, entries 8–12). In addition, in this case, the Fmoc-amino acids we checked gave high yields of the corresponding derivatives unmasked on the α-amino function (Table 3, entries 8–12) and the acid-labile protective groups of all the products obtained remained intact at the end of the process.
To avoid undesired removal of the acid-sensitive side chain protecting groups, the work-up of the ethereal extract from IL was performed with a 5% solution of citric acid.
The times required for the cleavage were short and no chromatographic purification was necessary.
The methodology is also successful when applied to N-Fmoc amino acid benzyl and tert-butyl esters (Table 3, entries 8–10).
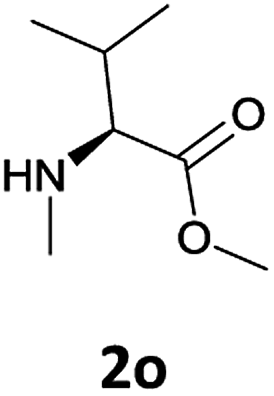
Finally, we tested our procedure for the N-deprotection of N-Fmoc-N-alkyl amino acid derivatives.
N-Methyl-α-amino acids are very important building blocks in the synthesis of peptidomimetics, covering an important role in medicinal chemistry.17 Many efforts have been directed at optimizing strategies for their synthesis and use in peptide chemistry.18
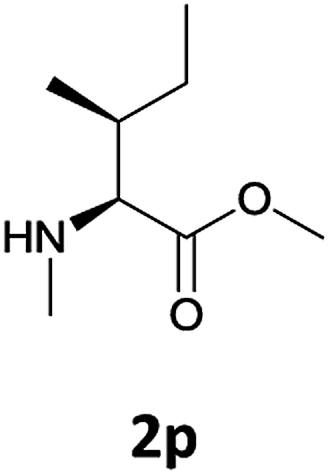
We were delighted to observe that the N-amino group was easily deprotected when two different N-Fmoc-N-alkylated-amino acid methyl esters (1o and 1p, Table 3) were treated with triethylamine in [Bmim][BF4] ionic liquid at room temperature. As already described above, a conventional acid–base work-up was employed for these analogues to remove the side product (i.e., DBF) formed during the process. The N-deprotected-N-alkylated products were obtained in good yields (entries 6 and 7, Table 3). The chemical yields of 2o (entry 6 in Table 3) were not so high (75%) compared to the other products. The possible explanation for this low yield compared to the other products lies in the steric bulk of N-alkyl amino acids, which reduces the reaction rate and leads to undesirable byproducts.
The Fmoc protecting group is widely used in solid phase peptide synthesis and its removal is generally accomplished by treatment with piperidine (20–50%) in DMF.4 Although highly successful and readily automated, solid-phase synthesis suffers a series of problems due to the heterogeneous reaction conditions, such as the huge amount of solvent required during preparation or purification steps. Thus, alternative, more environmentally friendly liquid phase methodologies are particularly desirable to restore homogeneous reaction conditions and the ionic liquids could be particularly suitable for amino acid and peptide synthesis or transformations since the polarity of ionic solvents is ideal for such species.
In this context, our research report proposes the use of ionic liquids as alternative green solvents in peptide synthesis according to the Fmoc-strategy in homogeneous conditions.19 Important advantages of our method are the absence of organic solvents, easy work-up, and the possibility to recycle the ionic liquid. Additionally, the solid support bearing the growing peptide chain would turn out to be inseparable from the ionic liquid phase. Thus, the proposed method should be considered as an alternative to the solid-phase synthesis of peptides.
Indeed, in other ways, almost simultaneously with the demonstration that ionic liquids were particularly convenient solvents for peptide coupling,20 a number of recent reports propose new insights into supported peptide synthesis where strategies in which peptide elongation could be performed on immobilized species using ionic liquid moieties as the soluble support are proposed. In that case, the IL-supported species can be dissolved in a solvent, and the reaction can be conducted in a homogeneous solution as an alternative to the solid-phase peptide synthesis.21
In view of green chemistry, one of the important features of the reactions conducted in ionic medium is the possibility of reuse the catalytic system for the subsequent runs with only a gradual decrease in activity and with no production of waste.11
We investigated the recycling capability of [Bmim][BF4] in the deprotection of the Fmoc group. After extraction with Et2O to separate the products from the reaction mixture, the residual ionic liquid was directly reused for the next deprotection. The results are displayed in Table 2 and show a slight decrease in the yield after the third recycling, indicating no significant loss of efficiency of the ionic liquid. The results of the presented protocol show that Et3N/[Bmim][BF4] is an efficient reagent system to remove the Fmoc group.
Conclusions
The use of greener and more efficient methodologies for organic transformations is fundamental for the development of more sustainable processes.We have reported a simple and convenient protocol based on the use of [Bmim][BF4] and the easily accessible triethylamine for the deprotection of Fmoc amines and amino acid methyl esters.
Important advantages of this method are: absence of organic solvents, use of a weak, inexpensive and readily available base such as Et3N, short reaction times, high yields, easy work-up, and finally the possibility to recycle the ionic liquid.
The procedure is also advantageous in that it avoids any hazardous chemicals. The protocol is an improvement over the existing methodologies for the removal of the Fmoc protecting group in solution phase.
Experimental section
General
Commercially available reagents were purchased from Sigma-Aldrich Chemical Co. (Milano, Italy) and used as supplied unless stated otherwise. All syntheses were carried out in atmospheric conditions. 1H NMR spectra were recorded at 300 MHz, while 13C NMR spectra were measured at 75 MHz. Spectral analysis was performed at 293 K on diluted solutions of each compound by using CDCl3 as the solvent. Chemical shifts (δ) are reported in ppm and referenced to CDCl3 (7.25 ppm for 1H and 77.0 ppm for 13C spectra). Coupling constants (J) are reported in hertz (Hz). Reaction mixtures were monitored by thin layer chromatography (TLC) using Merck Silica gel 60-F254 precoated glass plates, and UV light (254 nm) or 0.2% ninhydrin in ethanol and charring reagent as the visualizing agent. Evaporation of solvents was performed at reduced pressure using a rotary vacuum evaporator. Chiral GC analyses were carried out using a Thermo Gas Chromatograph instrument. Chiral GC analyses of enantiomeric compounds Ac-DL-AlaOMe and Ac-L-AlaOMe were performed using a 25 m × 0.25 mm, diethyl tert-butyldimethylisilyl-β-cyclodextrin chiral capillary column. The GC-MS Shimadzu workstation constituted a GC 2010 (equipped with a 30 m-QUADREX 007-5MS capillary column, operating in “split” mode, 1 mL min−1 flow of He as carrier gas) and a 2010 quadrupole mass-detector. LC-MS analyses were carried out using an Agilent 6540 UHD Accurate-Mass Q-TOF LC-MS (Agilent, Santa Clara, CA) fitted with an electrospray ionisation source (Dual AJS ESI) operating in positive ion mode. Chromatographic separation was achieved using a C18 RP analytical column (Poroshell 120, SB-C18, 50 × 2.1 mm, 2.7 μm) at 30 °C with an elution gradient from 5% to 95% of B over 13 min, A being H2O (0.1% FA) and B CH3CN (0.1% FA). The flow rate was 0.4 mL min−1.N-Fmoc amines 1a–i and N-Fmoc α-amino acid methyl esters 1j–u were prepared according to a previously published protocol.14b Spectral data of 1a–p agreed with those already reported for the same compounds prepared previously.14b
General procedure for the N-Fmoc removal of amines 1a–i in [Bmim][BF4]
To a magnetically stirred mixture of N-Fmoc-protected amines 1a–i (1 mmol) and [Bmim][BF4] (1 mL), Et3N (3 mmol) was added and the mixture was stirred at ambient temperature for 4–12 min. TLC was used to monitor the reaction. Diethyl ether was added after the completion of reaction and the IL settled at the bottom. The supernatant was decanted off and the IL was washed with Et2O (3 × 2 mL). The combined Et2O extracts were acidified with an aqueous solution of 1 N HCl and separated. The aqueous phase was then basified with sat. aq NaHCO3 and finally extracted with diethyl ether. The organic phase was dried over Na2SO4 and filtered. The products were isolated after evaporation of the diethyl ether to yield the free amines 2a–i in 80–93% yields. Spectroscopic data were compared to those of the pure products (see ESI†).General procedure for the N-Fmoc removal of amino acid methyl esters 1j–q in [Bmim][BF4]
To a magnetically stirred mixture of N-Fmoc amino acid methyl esters 1j–p (1 mmol) and [Bmim][BF4] (1 mL), Et3N (3 mmol) was added and the mixture was stirred at ambient temperature for 12 min. TLC was used to monitor the reaction. Diethyl ether was added after the completion of the reaction and the IL settled at the bottom. The supernatant was decanted off and the IL was washed with Et2O (3 × 2 mL). The combined Et2O extracts were acidified with an aqueous solution of 1 N HCl and separated. The aqueous phase was then basified with sat. aq NaHCO3 and finally extracted with diethyl ether. The organic phase was dried over Na2SO4 and filtered. The products were isolated after evaporation of the diethyl ether to yield the free amino acid methyl ester 2j–p in 75–88% yields. Compounds 2j–p were acetylated in order to perform GC/MS analysis. N-Acetylation was achieved dissolving 2j–p in DCM (5 mL) and adding acetic anhydride (1 mL) and a 9% aqueous solution of NaHCO3 (5 mL). The mixture was maintained under magnetic stirring at room temperature for 4 h. The organic layer was separated and the aqueous phase was extracted with three additional portions of DCM (3 × 10 mL). The combined organic layers were washed with a 9% aqueous solution of NaHCO3, twice with aqueous HCl 1 N, and once with brine, and then finally dried (Na2SO4). The solvent was evaporated under reduced pressure to afford the corresponding N-acetyl derivatives 3j–p as colourless oils in quantitative yield. Spectroscopic data for 3j–n were comparable to those reported in the literature.2aRecycling of [Bmim][BF4] in the synthesis of 2a
At the end of the first experiment and after the recovery of the free aniline, the IL was treated with 5% aqueous HCl (2 mL) and subjected to consecutive extractions with ethyl acetate and diethyl ether. The ionic liquid was recovered by removing water under vacuum. The IL was finally dried at 100 °C for 1 h and reused for the next run without any significant loss in its efficiency (88 and 89% yields).Conflict of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by grants from MIUR (MinisteroItaliano dell'Università e della Ricerca, ex-60% funds).Notes and references
- P. J. Kocienski, Protecting Groups, Georg Thieme Verlag, New York, 3rd edn, 2005 Search PubMed.
- (a) R. De Marco, M. L. Di Gioia, A. Leggio, A. Liguori and M. C. Viscomi, Eur. J. Org. Chem., 2009, 3795–3800 CrossRef CAS; (b) A. Procopio, G. Cravotto, M. Oliverio, P. Costanzo, M. Nardi and R. Paonessa, Green Chem., 2011, 13, 436–443 RSC; (c) M. Nardi, A. Cozza, L. Maiuolo, M. Oliverio and A. Procopio, Tetrahedron Lett., 2011, 52, 4827–4834 CrossRef CAS; (d) M. Nardi, A. Cozza, A. De Nino, M. Oliverio and A. Procopio, Synthesis, 2012, 44, 800–804 CrossRef CAS; (e) M. Oliverio, P. Costanzo, R. Paonessa, M. Nardi and A. Procopio, RSC Adv., 2013, 3, 2548–2552 RSC; (f) A. De Nino, L. Maiuolo, M. Nardi, R. Pasceri, A. Procopio and B. Russo, Arabian J. Chem., 2016, 9, 32–37 CrossRef CAS; (g) M. Nardi, N. H. Cano, A. De Nino, M. L. Di Gioia, L. Maiuolo, M. Oliverio, A. Santiago, D. Sorrentino and A. Procopio, Tetrahedron Lett., 2017, 58, 1721–1726 CrossRef CAS.
- (a) A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS PubMed; (b) M. Nardi, N. H. Cano, P. Costanzo, M. Oliverio, G. Sindona and A. Procopio, RSC Adv., 2015, 5(24), 18751–18760 RSC.
- (a) M. Bayermann, M. Bienert, H. Niedrich, L. A. Carpino and D. Sadat-Aalaee, J. Org. Chem., 1990, 55, 721–728 CrossRef; (b) L. A. Carpino, D. Sadat-Aalaee and M. Beyermann, J. Org. Chem., 1990, 55, 1673–1675 CrossRef CAS; (c) F. Alberho, N. Kneib-Cordonier, S. Biancalana, L. Gera, R. I. Masada, D. Hudson and G. Barany, J. Org. Chem., 1990, 55, 3730–3743 CrossRef; (d) E. Atherton, H. Fox, D. Harktss, C. J. Logan, R. C. Sheppard and B. J. Wilhams, Chem. Commun., 1978, 537–539 RSC; (e) E. Atherton, H. Fox, D. Harkiss and R. C. Sheppard, Chem. Commun., 1978, 539–540 RSC; (f) C.-D. Chang, A. M. Felix, M. H. Jimenez and J. Meienhofer, Int. J. Pept. Protein Res., 1980, 15, 485–494 CrossRef CAS PubMed.
- (a) N. Zinieris, L. Leondiadis and N. Ferderigos, J. Comb. Chem., 2005, 7, 4–6 CrossRef CAS PubMed; (b) C. F. Vergel Galeano, Z. J. Rivera Monroy, J. E. Rosas Pérez and J. E. García Castañeda, J. Mex. Chem. Soc., 2014, 58(4), 386–392 Search PubMed.
- Proceedings of the 31st European Peptide Symposium, ed. M. Lebl, M. Meldal, K. J. Jensen and T. Hoeg-Jensen, European Peptide Society, 2010 Search PubMed.
- K. Ralhan, V. G. KrishnaKumara and S. Gupta, RSC Adv., 2015, 5, 104417–104425 RSC.
- L. A. Carpino, Acc. Chem. Res., 1987, 20, 401–407 CrossRef CAS.
- N. Zinieris, L. Leondiadis and N. Ferderigos, J. Comb. Chem., 2005, 7, 4–6 CrossRef CAS PubMed.
- (a) D. Orain, J. Ellard and M. Bradley, J. Comb. Chem., 2002, 4, 1–16 CrossRef CAS PubMed; (b) J. Hachmann, J. Comb. Chem., 2006, 8(2), 149 CrossRef CAS PubMed; (c) C. Chen, B. Rajagopal, X. Yu Liu, K. Lin Chen, Y. C. Tyan, F. Lin and P. C. Lin, Amino Acids, 2014, 46, 367–374 CrossRef CAS PubMed.
- (a) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998, p. 30 Search PubMed; (b) P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed; (c) M. Nardi, N. Herrera Cano, P. Costanzo, M. Oliverio, G. Sindona and A. Procopio, RSC Adv., 2015, 5, 18751–18760 RSC; (d) N. Herrera Cano, J. G. Uranga, M. Nardi, A. Procopio, D. A. Wunderlin and A. N. Santiago, Beilstein J. Org. Chem., 2016, 12, 2410–2419 CrossRef PubMed.
- (a) M. J. Earle, P. B. McCormac and K. R. Seddon, Chem. Commun., 1998, 2245–2246 RSC; (b) F. Liu, M. B. Abrams, R. T. Baker and W. Tumas, Chem. Commun., 2001, 433–434 RSC; (c) E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2001, 124, 926–927 CrossRef; (d) J. M. Xu, Q. Wu, Q. Y. Zhang, F. Zhang and X. F. Lin, Eur. J. Org. Chem., 2007, 1798–1802 CrossRef CAS; (e) B. C. Ranu, A. Saha and R. Dey, Curr. Opin. Drug Discovery Dev., 2010, 13, 658–668 CAS; (f) A. K. Chakraborti and S. Raha Roy, J. Am. Chem. Soc., 2009, 131, 6902–6903 CrossRef CAS PubMed; (g) A. De Nino, L. Maiuolo, P. Merino, M. Nardi, A. Procopio, D. Roca-Lopez, B. Russo and V. Algieri, ChemCatChem, 2015, 7, 830–835 CrossRef CAS.
- (a) P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, 2004 Search PubMed; (b) M. A. P. Martins, C. P. Frizzo, D. N. Moreira, N. Zanatta and H. G. Bonacorso, Chem. Rev., 2008, 108, 2015–2050 CrossRef CAS PubMed; (c) J. Dupont and J. Spencer, Angew. Chem., Int. Ed., 2004, 43, 5296–5297 CrossRef CAS PubMed; (d) J. Ranke, S. Stolte, R. Storman, J. Arning and B. Jastor, Chem. Rev., 2007, 107, 2183–2206 CrossRef CAS PubMed; (e) K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC; (f) Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC; (g) P. Dominguez de Maria, Angew. Chem., Int. Ed., 2008, 47, 6960–6968 CrossRef CAS PubMed; (h) D. Coleman and N. Gathergood, Chem. Soc. Rev., 2010, 39, 600–637 RSC; (i) S. Majumdar, J. De, A. Chakraborty, D. Roy and D. K. Maiti, RSC Adv., 2015, 5, 3200–3205 RSC.
- (a) M. L. Di Gioia, A. Barattucci, P. Bonaccorsi, A. Leggio, L. Minuti, E. Romio, A. Temperini and C. Siciliano, RSC Adv., 2014, 4, 2678–2686 RSC; (b) M. L. Di Gioia, A. Gagliardi, A. Leggio, V. Leotta, E. Romio and A. Liguori, RSC Adv., 2015, 5, 63407–63420 RSC.
- (a) C.-D. Chang, M. Waki, M. Ahmad, J. Meienhofer, E. O. Lundell and J. D. Haug, Int. J. Pept. Protein Res., 1980, 15, 59–66 CrossRef CAS PubMed; (b) G. B. Fields, Methods Mol. Biol., 1994, 35, 17–27 CAS.
- D. Millán, M. Rojas, J. G. Santos, J. Morales, M. Isaacs, C. Diaz and P. Pavez, J. Phys. Chem. B, 2014, 118, 4412–4418 CrossRef PubMed.
- (a) M. L. Di Gioia, A. Leggio, A. Liguori, F. Perri, C. Siciliano and M. C. Viscomi, Amino Acids, 2010, 38, 133–143 CrossRef CAS PubMed; (b) E. Belsito, R. De Marco, M. L. Di Gioia, A. Liguori, F. Perri and M. C. Viscomi, Eur. J. Org. Chem., 2010, 4245–4252 CrossRef CAS; (c) R. De Marco, M. L. Di Gioia, A. Liguori, F. Perri, C. Siciliano and M. Spinella, Tetrahedron, 2011, 67, 9708–9714 CrossRef CAS; (d) C. Siciliano, A. Barattucci, P. Bonaccorsi, M. L. Di Gioia, A. Leggio, L. Minuti, E. Romio and A. Temperini, J. Org. Chem., 2014, 79(11), 5320–5326 CrossRef CAS PubMed; (e) M. L. Di Gioia, A. Leggio, F. Malagrinò, E. Romio, C. Siciliano and A. Liguori, Mini-Rev. Med. Chem., 2016, 16, 683–690 CrossRef CAS PubMed.
- (a) R. De Marco, M. L. Di Gioia, A. Leggio, A. Liguori, F. Perri, C. Siciliano and M. C. Viscomi, Amino Acids, 2010, 38, 691–700 CrossRef PubMed; (b) A. Leggio, D. Alò, E. L. Belsito, M. L. Di Gioia, E. Romio, C. Siciliano and A. Liguori, J. Pept. Sci., 2015, 21(8), 644–650 CrossRef CAS PubMed; (c) M. L. Di Gioia, E. L. Belsito, A. Leggio, V. Leotta, E. Romio, C. Siciliano and A. Liguori, Tetrahedron Lett., 2015, 56, 2062–2066 CrossRef CAS.
- (a) Y. E. Jad, T. Govender, H. G. Kruger, A. El-Faham, B. G. de la Torre and F. Albericio, Org. Process Res. Dev., 2017, 21, 365–369 CrossRef CAS; (b) Y. Jad, G. Acosta, S. Khattab, B. Torre, T. Govender, H. Kruger, A. El-Faham and F. Albericio, Amino Acids, 2016, 48, 419–426 CrossRef CAS PubMed; (c) Y. E. Jad, G. A. Acosta, T. Govender, H. G. Kruger, A. El-Faham, B. de la Torre and F. Albericio, ACS Sustainable Chem. Eng., 2016, 4, 6809–6814 CrossRef CAS.
- J. C. Plaquevent, J. Levillain, F. Guillen, C. Malhiac and A. C. Gaumont, Chem. Rev., 2008, 108, 5035–5060 CrossRef CAS PubMed.
- (a) W. Miao and T. H. Chan, Acc. Chem. Res., 2006, 39, 897–908 CrossRef CAS PubMed; (b) W. Miao and T. H. Chan, J. Org. Chem., 2005, 70(8), 3251–3255 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copy of 1H and 13C NMR spectra for compound N-acetyl N-methyl valine methyl ester. Copy of 1H spectra for N-acetyl N-methyl isoleucine methyl ester. See DOI: 10.1039/c7ra04425a |
| This journal is © The Royal Society of Chemistry 2017 |