
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Shedding light on the effective fluorophore structure of high fluorescence quantum yield carbon nanodots†
W. Wanga,
B. Wangc,
H. Embrechtsab,
C. Damma,
A. Cadranel c,
V. Straussc,
M. Distasoab,
V. Hinterbergerab,
D. M. Guldic and
Wolfgang Peukert*ab
c,
V. Straussc,
M. Distasoab,
V. Hinterbergerab,
D. M. Guldic and
Wolfgang Peukert*ab
aInstitute of Particle Technology (LFG), Friedrich-Alexander-Universität Erlangen-Nürnberg, Cauerstraße 4, 91058 Erlangen, Germany. E-mail: Wolfgang.peukert@fau.de; Fax: +49 9131 85 29402; Tel: +49 9131 85 29400
bInterdisciplinary Center of Functional Particle Systems, Friedrich-Alexander-Universität Erlangen-Nürnberg, Haberstraße 9a, 91058 Erlangen, Germany
cDepartment of Chemistry and Pharmacy, Interdisciplinary Center for Molecular Materials (ICMM), Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstr. 3, 91058 Erlangen, Germany
First published on 9th May 2017
Abstract
Carbon nanodots (CD) have great potential for imaging and sensing applications, due to their unique luminescence properties in combination with their low toxicity. Although CDs are currently the subject of intensive research activity, the exact structure of their effective fluorophore species has to be clarified because it can depend on the starting materials and reaction conditions. Most importantly, the limited photostability of some CDs presents an impediment to the much-needed breakthroughs required in the field. In this work, the photobleaching of CDs synthesized hydrothermally from citric acid and urea under various conditions is studied in detail. By visualizing changes in the absorption and fluorescence properties of CDs over time upon exposure to UV-light by means of in situ spectroscopic probes, we demonstrate a trade-off between high initial quantum yield and photostability. We found that the CDs consist of low-molecular weight fluorophores bound to π-conjugated domains (carbon core). In the case of CDs synthesized at higher reaction temperatures and/or for longer reaction times, lower initial quantum yields and higher photostability originate from partial conversion of the low-molecular weight fluorophores to π-conjugated domains during synthesis. Thus, the remaining surface bound fluorophores are bound to larger π-conjugated domains, which, in turn, enable dissipation of absorbed UV-energy and protection against photochemical damage. Reference experiments were performed with citrazinic acid – a pyridone-based structure – using ultrafast time-resolved spectroscopy, steady-state spectroscopy, and theoretical DFT simulations. We conclude that pyridone-like structures are most likely responsible for both the high quantum yields and the photobleaching of CDs.
Introduction
In recent years, the use of carbon-based compounds for applications requiring high fluorescence quantum yields, low-cost, biocompatibility, and environmentally friendly device production has attracted immense attention.1,2 In particular, nano-sized carbon particles are promising candidates for display and lighting applications due to their superior fluorescent properties and low toxicity. Although different structures, depending on starting materials and reaction conditions, are discussed3–6 as the fluorescence origin, the immense potential of these so-called carbon dots (CD) has already been successfully proven in several fields such as sensing, optoelectronics, photocatalysis, drug delivery et cetera.7–12 Importantly, the properties of CDs are strongly dependent on their synthesis conditions and reaction precursors. “Bottom-up” fabrication methods from molecular precursors are widely employed, because they allow for high-yield reactions from ecologically friendly starting materials and because the CD products have ultra-high emission quantum yields, low toxicity, and excellent biocompatibility.13,14 “Bottom-up” CD synthesis is relatively simple and inexpensive. However, the resulting CDs have a complex structure and fluorescence can origin from different structure elements.In contrast, “top-down” CDs, as, for example, produced from larger carbon materials, have a well-defined aromatic backbone and the photoluminescence is commonly assigned to quantum confinement effects.15
High fluorescence quantum yields (QY) have been reported for CDs in recent years.3–6,16,17 Since the QYs of “bottom-up” synthesized CDs are much higher than those of “top-down” produced CDs, these CDs are preferred for optoelectronic applications. Understanding of the exact nature of the fluorescent centers in “bottom-up” produced CDs is important, but challenging. Therefore only few research groups presented results of structural investigations.3–6 In this context, the 1 nm size of “bottom-up” synthesized CDs and their environmentally-sensitive structure are the biggest obstacles to developing efficient purification procedures and direct characterization techniques. Notably, intense laser irradiation and/or electrical currents applied during characterization can easily destroy the CD structure, leading to false structural information. Correlating the origin of fluorescence with the basic structure is, nevertheless, of outmost importance to realizing applications based on “bottom-up” synthesized CDs.
QY and photostability are two very important performance metrics to be kept in mind when developing CD applications. The limited photostability of CDs is a large obstacle that must be overcome. Recent studies have revealed that photobleaching and photoblinking compromise the photostability of CDs. Photobleaching relates to permanent photochemical degradation, while photoblinking refers to temporary transformations between dark and emissive states. Both phenomena are commonly encountered in CDs. For example, Khan et al. confirmed the reversible photoswitching of red emissive CDs, enabling promising CD applications for reversible and saturable optical linear fluorescence transition (RESOLFT).18 Moreover, Chizhik et al. correlated their experimental findings to the surface charge influenced photoswitching of CDs and demonstrated the potential of stochastic optical switching in super resolution microscopy.19 The fluorescence fluctuation, which is a “disadvantage” at first glance, turns into a significant advantage in super-resolution imaging, especially in optical fluctuation imaging applications.
In this paper, we use well-detectable changes in optical properties occurring during the photobleaching of blue fluorescent CDs as a tool to elucidate their structure and origin of fluorescence. To this end, strongly emitting CDs were synthesized via hydrothermal treatment of a mixture of citric acid and urea. In situ UV/Vis-absorption and fluorescence-probes were employed to track changes in CD absorption and luminescence during photobleaching induced by monochromatic 365 nm light. In addition to characterization of the CDs by conventional steady-state spectroscopic tools, we also investigated the excited state dynamics by ultrafast absorption and emission spectroscopy. We found that upon UV light exposure, carbonization initiates effects, which impact the fluorescence. Finally, from comparative spectroscopic investigations and theoretical simulations on citrazinic acid, a model pyridone structure, we conclude that a pyridone-based structure attached to the CD π-conjugated domains is responsible for the high fluorescence QYs and also for the photobleaching observed in citric acid derived CDs.
Results and discussion
Changes in the optical properties of aqueous CD samples upon UV exposure were recorded as a function of CD synthesis conditions, that is, reaction temperatures and times, using two in situ optical probes. The UV/Vis-absorption and fluorescence spectra from a typical sample are shown in Fig. 1, while results for samples synthesized under different conditions are shown in Fig. S1 in the ESI.† All of the UV/Vis-absorption spectra displayed peaks at around 220 and 340 nm. These two peaks are ascribed to the π–π* (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/CN bonds) and n–π* transitions, respectively.20 As shown in Fig. S3 in the ESI,† the CD absorption increases upon photobleaching throughout the entire spectral region – the only notable exception being the peak at 340 nm. These changes are particularly pronounced in the region around 270 nm. From the latter, we infer the formation of aromatic π-conjugated structures, stemming most likely either from a carbonization process, a realignment of CD surface structures, or a loss of surface functional groups during photobleaching.21 Notably, the increase in absorption at 270 nm is most evident in CDs synthesized at lower reaction temperatures and shorter reaction times: CDs synthesized at 180 °C for 3 h (abbr. CD180,3) were subject to pronounced photobleaching, while CDs synthesized at 210 °C for 17 h (abbr. CD210,17) were comparatively stable during UV exposure.
C/CN bonds) and n–π* transitions, respectively.20 As shown in Fig. S3 in the ESI,† the CD absorption increases upon photobleaching throughout the entire spectral region – the only notable exception being the peak at 340 nm. These changes are particularly pronounced in the region around 270 nm. From the latter, we infer the formation of aromatic π-conjugated structures, stemming most likely either from a carbonization process, a realignment of CD surface structures, or a loss of surface functional groups during photobleaching.21 Notably, the increase in absorption at 270 nm is most evident in CDs synthesized at lower reaction temperatures and shorter reaction times: CDs synthesized at 180 °C for 3 h (abbr. CD180,3) were subject to pronounced photobleaching, while CDs synthesized at 210 °C for 17 h (abbr. CD210,17) were comparatively stable during UV exposure.
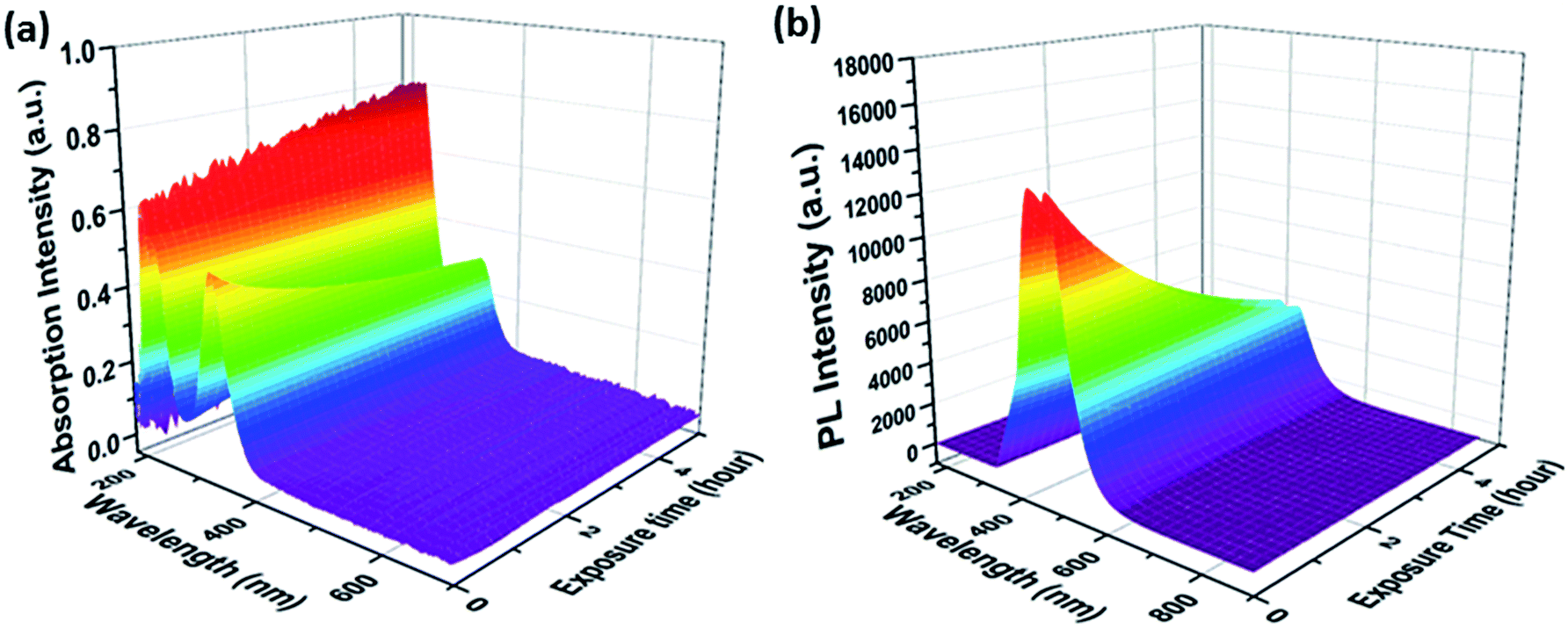 | ||
| Fig. 1 (a) UV/Vis-absorption and (b) fluorescence spectra of an aqueous CD210,3 suspension as a function of UV light exposure time. | ||
In all cases, the intensity of the fluorescence at 440 nm decreased during UV exposure. In this study, the mildest reaction conditions for synthesizing citric acid-based CDs, that is, 180 °C for 3 h, led to the highest QYs. CDs synthesized under such mild conditions are, however, susceptible towards UV light-induced photobleaching, as documented in Fig. 2. In sharp contrast, CDs synthesized at higher reaction temperatures and/or longer reaction times are more resistant towards photobleaching, but show lower starting QYs. In other words, the temporal evolution of the absorption at 270 nm under UV light exposure correlates inversely with that of the fluorescence intensity (Fig. S4 in the ESI†). The aforementioned steady-state absorption analysis demonstrates the destructive effects of UV-induced carbonization in terms of fluorescence. A dependence of the fluorescence QY on the temperature during hydrothermal CD synthesis from citric acid and urea was also found by the group of Okuyama.3–5 The highest QY was found for CDs synthesized at 150–170 °C, at higher temperatures the QY is reduced due to graphitization.3–5 In contrast, the CDs synthesized by Okuyama's group are photostable indicating a different structure of the fluorophore although the starting materials are the same and only their molar ratio is different from this study.
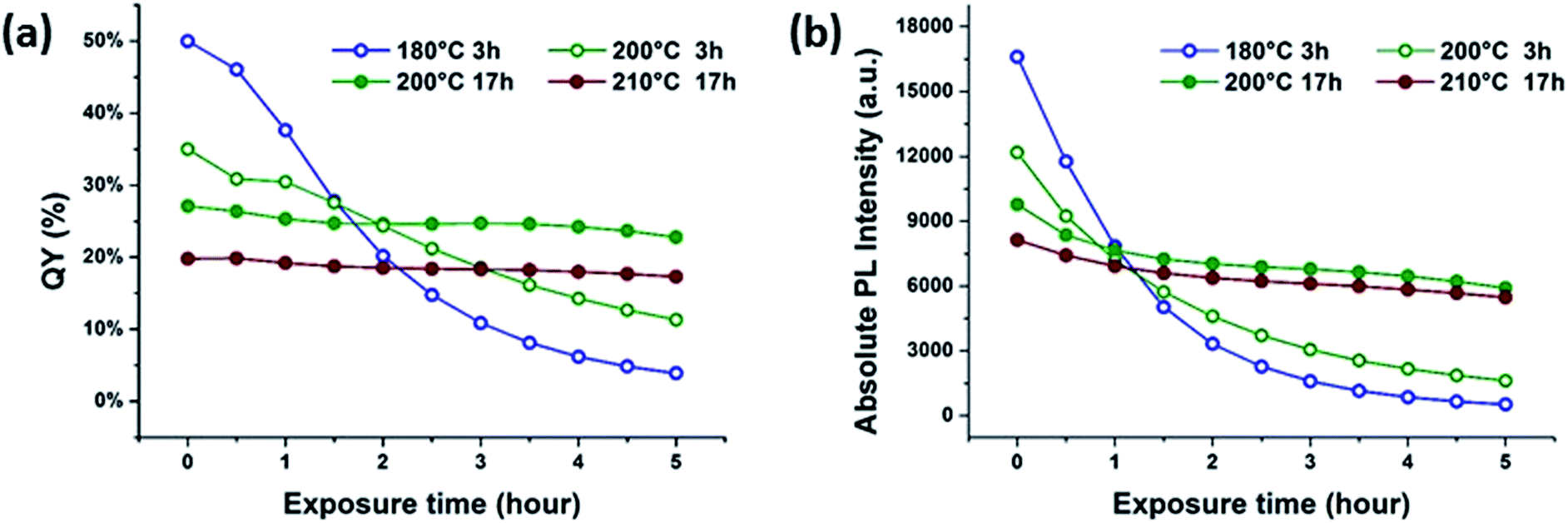 | ||
| Fig. 2 Correlations of (a) QY and (b) absolute fluorescence degradation for aqueous CD suspensions under UV exposure. | ||
A similar cross-correlation between initial QY and photostability was reported for CDs synthesized by reaction of citric acid with ethylene diamine, L-cysteine or cysteamine.6,16 A rationale was proposed, suggesting that small fluorophores are formed in the initial steps of CD synthesis. Low reaction temperatures play hereby a significant role. As the reaction comes closer to reaching completion, due to longer heating times or higher reaction temperatures, more carbogenic π-conjugated domains are formed. Overall, a dehydration of the precursors or the low-molecular weight fluorophores results in the partial consumption of the fluorophores.
The basic skeleton of the π-conjugated domains serves as an anchor for unconsumed fluorophores. Peripheral binding of the latter to π-conjugated domains increases the fluorescence of CDs. Notably, the number of π-conjugated domains increases as a function of reaction temperature and reaction time. As an immediate consequence, more fluorophores are bound. These peripheral bound fluorophores are expected to dissipate energy from the π-conjugated domains, preventing photochemical damage.
This would explain the higher photobleaching resistance of CDs fabricated at longer reaction times and higher reaction temperatures. Energy dissipation due to the integration of fluorophores into a larger carbon network results in overall lower QYs. The sum of these effects is hypothesized to be responsible for the trade-off between high initial QY and photostability of citric acid derived “bottom-up” synthesized CDs.
The QYs of CDs synthesized under harsher reaction conditions (Fig. 2a), including those synthesized at 200 and 210 °C for 17 h, remain nearly constant during UV exposure. Due to the definition of QY, which is the ratio of fluorescence relative to the corresponding absorption, a constant QY does not necessarily entail a completely stable fluorescence. Upon UV exposure, both the 365 nm absorption and the fluorescence intensities decrease as a function of time, leading to the false interpretation that CDs synthesized under extreme conditions are perfectly photostable. A closer look at the absolute fluorescence intensity plots, however, reveals that all samples are susceptible to photobleaching upon UV exposure, as shown in Fig. 2b. For example, the fluorescence intensity plots demonstrate that even CDs prepared at high reaction temperatures and/or long reaction times exhibit photobleaching upon exposure to UV. The degree of photobleaching is, however, much lower in comparison with CDs synthesized under milder conditions.
In contrast to semiconductor and “top-down” graphene-based quantum dots, the fluorescence of “bottom-up” synthesized CDs relates to surface and carbogenic structures rather than to quantum confinement effects.3–6,22 As such, the fluorescence of “bottom-up” CDs depends mainly on the molecular-like surface states and the π-conjugated domains, but not on the particle size. To validate this statement, a dialysis membrane with 1 kDa pores was used to separate unbound fluorophores from the CDs. Only small molecules diffuse out of the cellulose dialysis bag. According to Fig. S5 in the ESI,† the QY of the dialyzed solution featuring only the unbound fluorophores is much higher than that of the purified CD suspension. At the same time, a much faster photobleaching is detected for the unbound fluorophores. The behavior of the fluorescent molecules in solution is closer to that of the unpurified synthesized mixtures, especially those prepared at lower reaction temperature and/or shorter reaction time. Our observation supports the notion that the fluorescent molecules and their derivatives play a dominant role in the observed high QYs and photobleaching properties of CDs.
Until now, the presence of peripherally bound fluorophores – also known as peripheral effective fluorophores – has only been discussed in a few studies.3–6,23,24 The working hypothesis implies that CD fluorescence is caused by fluorescing carbonyl and carboxyl groups functionalized edges,25 disordered edges,26 polycyclic aromatic hydrocarbons,27 pyridinic nitrogen3–6 or carbonyl groups.28
To characterize the peripheral effective fluorophore structure and to propose a mechanism for CD formation, the fluorescence properties of products resulting from closely related carboxylic acid and amino containing precursors were compared.
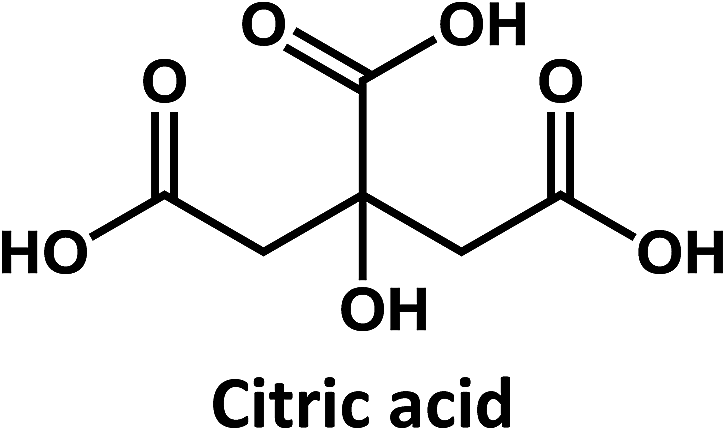
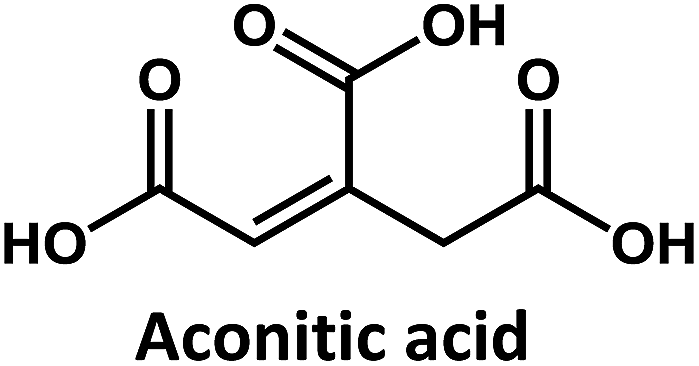
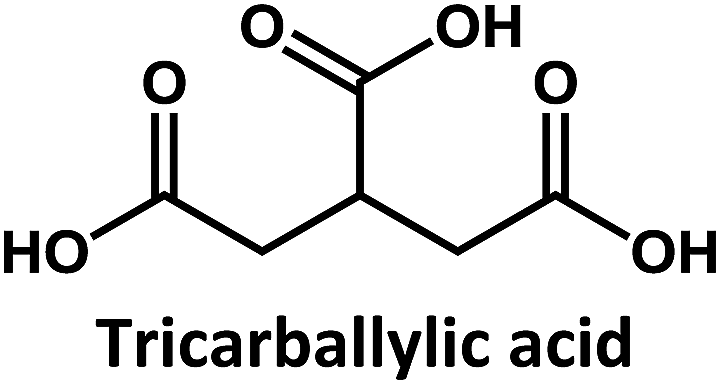
Considering that citric acid and diamines are widely used as carboxyl- and amino-containing CD precursors, carboxylic acids such as citric, aconitic, and tricarballylic acid were used to synthesize CDs, as shown in Table 1.
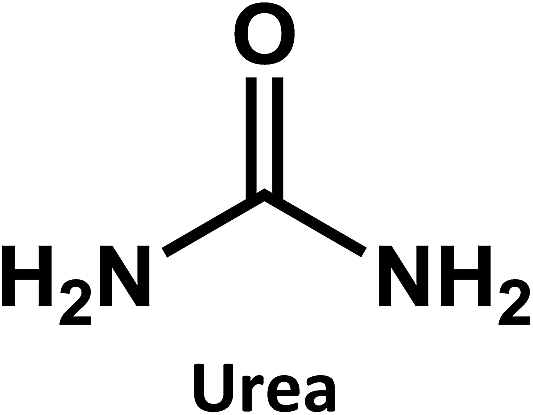
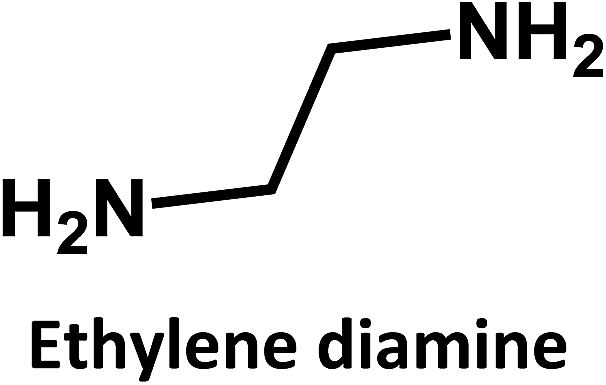
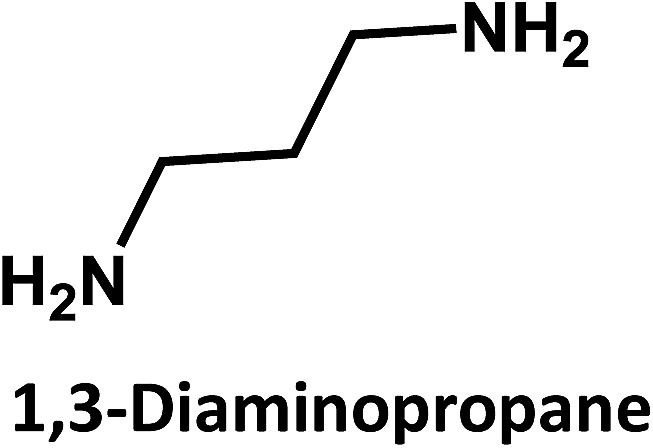

In addition, ethylene diamine, 1,3-diaminopropane, and 1,8-diaminooctane were studied as amino-containing precursors. However, none of these precursors exhibits any appreciable fluorescence in solution. After hydrothermal treatment under identical reaction temperatures and times, the products exhibit different fluorescent features. On the one hand, low fluorescence intensities are observed when longer amino precursor chains are used, most likely due to fast radiationless deactivation of the excited state facilitated by the long and flexible alkyl chains.
On the other hand, the results obtained from reactions of the diamines with different carboxylic acids allow conclusions to be drawn about the formation mechanism. The reaction of the carboxylic group with one of the two amino groups in the diamines to form an amino-terminated amide group is the initial step in the proposed CD formation mechanism as illustrated in Fig. 3.3–6 All three different carboxylic acid precursors may undergo this condensation reaction. As the next step the formation of dimers and trimers of the citric acid amides by hydrogen bonds is discussed.3–5 Fluorescent graphene-like CDs containing pyrrolic, pyridinic and graphitic nitrogen are formed from the citric acid amide aggregates by condensation reaction.3–5 In our work the molar ratio between urea and citric acid is lower by the factor of 2 in comparison to.3–5 Thus, instead of an intermolecular reaction of the citric acid amides a ring-closing reaction to form a pyridone based structure is likely to take place. Both citric and aconitic acid possess the ability to undergo ring closure to form a fluorescent precursor when reacted with diamino compounds.
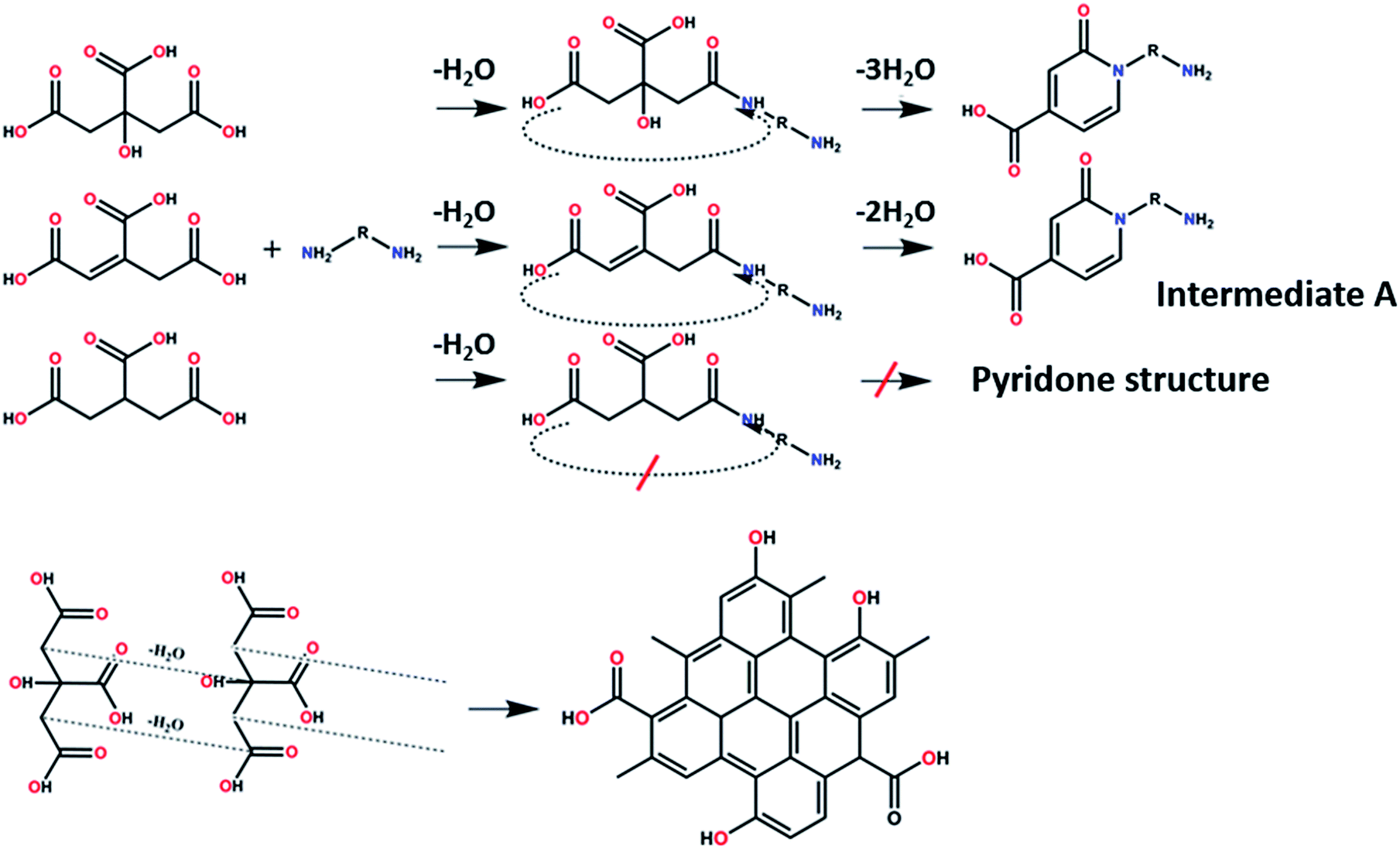 | ||
| Fig. 3 Proposed formation routes of a peripheral effective fluorophore structure and π-conjugated domain structure en-route towards CDs. | ||
Tricarballylic acid, however, lacks the alcohol or the additional double-bond required for the formation of an aromatic ring with the amino-terminated amide. As a matter of fact, in the case of tricarballylic acid no such aromatic ring structure is formed.
Interestingly, CDs formed from both citric acid and its dehydration product, namely aconitic acid, display char-acteristic blue CD fluorescence. When citric acid is replaced by tricarballylic acid, the resulting product is not fluorescent. These results support the hypothesis that the formation of a fluorescent structure is connected to the ability of the central carbon atom in the carboxylic acid molecule to undergo ring closure. Citric and aconitic acid, which contain a suitable functional group, but not tricarballylic acid, may form a pyridone ring structure, such as 1-carbamoyl-2-oxo-1,2-dihydropyridine-4-carboxylic acid (intermediate A in Fig. 3).
Density functional theory (DFT) simulations of intermediate A as shown in Fig. 4 were used to shed light on the possible relationship between UV/Vis-absorption, Fourier transform infrared (FT-IR) spectra, and CD structure. The model structure used for the DFT calculations was geometrically optimized by the semiempirical PM3 method, see Fig. 4. The energy gap between the HOMO and LUMO at the B3LYP/3-21G level calculated by DFT is about 3.8 eV, which could be the source of fluorescence after energy decay to the emissive state. HOMO and LUMO levels of −6.6 and −2.8 eV, respectively, are in good accordance with the experimental values of −6.2 and −2.9 eV determined by cyclic voltammetry and absorption edge measurements (Fig. S6 in ESI†). The experimental CD absorption spectrum in suspension is in reasonable agreement with that simulated for intermediate A (Fig. 5a). The simulated FT-IR spectrum also matches the experimental results (Fig. 5b). The main peaks in the range of 700–1800 cm−1 are ascribed to C–O, C–N, and carboxyl groups. This is an important indication that model intermediate A reliably reflects the CD effective fluorescent structure.
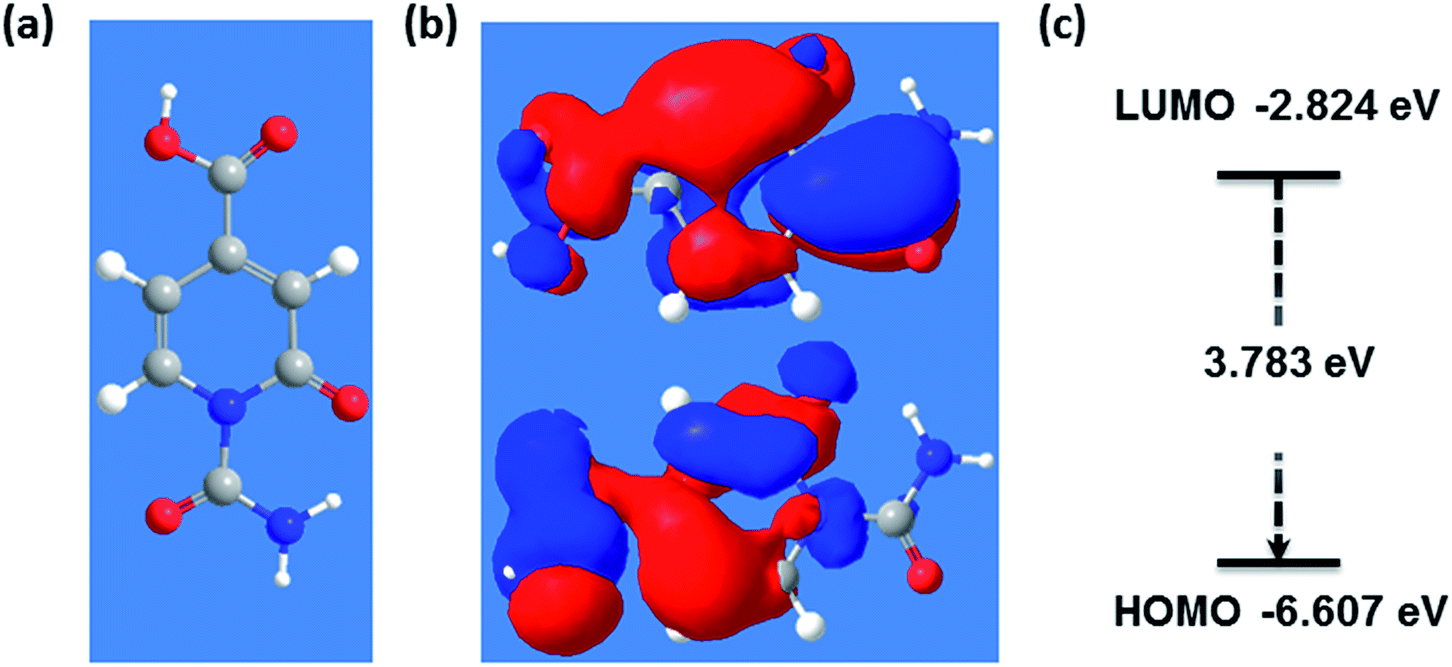 | ||
| Fig. 4 (a) PM3 optimized geometry of effective fluorescent structure, (b) exemplary molecular orbitals corresponding to LUMO and HOMO and (c) its energy gap. | ||
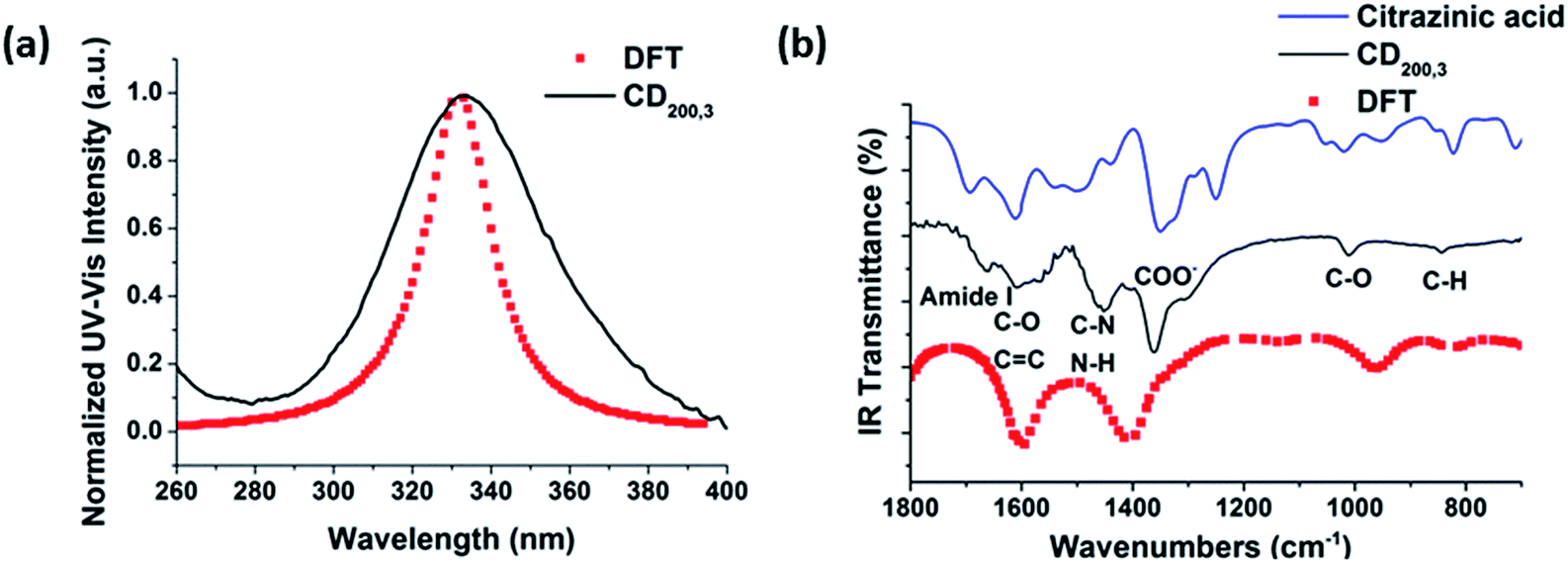 | ||
| Fig. 5 The comparisons in (a) UV-Vis absorption and (b) FT-IR spectra of experimental results and DFT/B3LYP calculated results. | ||
Pyridone-like structures are involved in other condensation reactions between urea and citric acid. Initial research dates back to 1893 and was carried out by Sell and Easterfield.29 They proposed that 2-pyridone, 4-carboxyl is produced upon heating dry urea and citric acid. In 2015, Kasprzyk and co-workers utilized heteronuclear single quantum coherence spectroscopy (HSQC) and electrospray ionization mass spectrometry (ESI-MS) to prove that the 2-pyridone based structure is the efficient fluorophore in products synthesized from citric acid and α,β- or α,γ-bifunctional amines.30 Additionally, theoretical calculations on pyridone based structures have predicted a HOMO/LUMO gap of ∼3.5 eV.30 The latter could conceivably increase to 4 eV, when, for example, hydrogen bonding is strengthened by the existence of OH- or NH-containing functional groups in CDs. 3.5–4 eV energy gaps correspond to transitions in the 310 to 355 nm absorption wavelength, which is consistent with the 340 nm absorption peak observed for CDs. As far as the fluorescence is concerned, the 450 nm feature occurs after vibrational relaxation and, in turn, red shifted relative to the calculated HOMO–LUMO gap.
In addition to the formation of a pyridone structure, the model, which is hypothesized in Fig. 3, implies a pyrolytic reaction between citric acid molecules.31 Accordingly, an aromatic carbon skeleton, namely a CD π-conjugated domain, is formed. Its size is governed by both pressure and ion concentration in the reaction medium.32 The CD π-conjugated domain is a subunit of graphene or graphene oxide composed of inhomogeneous sp2 domains with an energy gap in the range from 1.8 to 7 eV.17,33–36
The CDs in this report are 1–2 nm in diameter as previously determined by analytical ultracentrifugation.37 Thus, the π-conjugated domains contain only a few aromatic rings and a theoretically as well as experimentally determined energy gap of around 4 eV.16,26 In accordance with literature reports, none of the π-conjugated domains have high fluorescence QYs. Their contribution to the overall fluorescence is insignificant relative to that of the low-molecular fluorophores bound to the CD surface.33
The fluorescence pattern of the π-conjugated domains depends on the excitation wavelength as a result of edge effects.15 In strong contrast, the CDs studied in our current work have QYs as high as 50% - Fig. 2 – and their spectral response is excitation wavelength independent. Moreover, fluorescent material can be removed from the CD suspension by dialysis. All these findings serve as further support for the idea that CD fluorescence and its subsequent photobleaching originates from low-molecular fluorophores bound to the CD surfaces rather than from π-conjugated domains. It is worth noting that pyridone based fluorophores (intermediate A in Fig. 3) may easily bind via condensation reactions to form fluorescent CDs, because of the presence of carboxyl and hydroxyl groups on the surface of their π-conjugated domains. In other words, the properties of such a carbon system are a consequence of both pyridone structures and sp2 clusters.
Excited states of a different nature are proposed for the CDs studied in this work.38,39 In contrast to the previous assignment, the core and trap states might relate to π-conjugated domains and pyridone structures, respectively. Hence, femtosecond transient absorption spectroscopy with an excitation wavelength of 320 nm was used to obtain further insights into the excited state dynamics of both freshly synthesized and photobleached CDs. In the case of pristine CDs (Fig. S7(a) and (b) in the ESI†), the initially populated excited state exhibits an absorption feature at around 600 nm, resembling that observed upon 387 nm excitation.38 This absorption feature, which is assigned to the excited state of the π-conjugated domains, decays during the first few picoseconds with the concomitant formation of a 450 nm absorption. For this process, which is assigned to an energy transfer (ET) from the π-conjugated domain to the peripheral effective fluorophores, a lifetime of around 4 ps was derived. Following its formation, the 450 nm absorption decays biphasic. The short-lived component with a 163.6 ± 13.8 ps lifetime is tentatively assigned to a fluorescence process from pyridone-like peripheral effective fluorophores, and the long-lived component with a 2671.6 ± 664.8 ps lifetime is ascribed to fluorescence from other trap states. In parallel, we conducted 387 nm excitation measurements, shown in Fig. S8 in the ESI.† Initially, the π-conjugated domains are populated, but their absorption features around 550–600 nm are weaker than in the case of 320 nm excitation. This is related to the lower extinction coefficient of the sample at 387 nm vs. 320 nm. Later, an absorption feature around 470 nm increases biexponentially and two lifetimes could be derived separately. The first one (1.3 ± 0.2 ps) corresponds to the population of the shallow trap states and the second one (12.6 ± 4.8 ps) to the population of the deep trap states. After this, ground state recovery proceeded with a lifetime of 3.2 ± 0.2 ns, which is assigned to the fluorescence from trap states and correlated with the lifetime of 2671.6 ± 664.8 ps we obtained before. This result is independently confirmed in time-correlated single photon counting (TCSPC) measurements using 361 nm excitation wavelength and is shown in Fig. S9 in the ESI.† It is worth to note that population of peripheral effective fluorophores states from the core states might occur upon 387 nm excitation, but are probably hidden due to the very low absorptivity of these states.
The presence of ultra-short-lived components masked by the instrument response function should not be ruled out.38 In the case of photobleached CDs (Fig. S7(c) and (d) in the ESI†), the early differential absorption spectra are similar to those of freshly synthesized CDs, suggesting the population of the same initial excited state, that is, an excited state of the π-conjugated domains. A 6 ps lifetime was derived for an energy transfer process to the peripheral effective fluorophores upon 320 nm excitation, and a lifetime of 1.5 ± 0.2 ps could be derived for the energy transfer process to the trap states upon 387 nm excitation. Importantly, the intensity of the excited state absorption feature of the peripheral effective fluorophores is in photobleached CDs much lower than in freshly synthesized CDs. This suggests that photodegradation strongly affects these surface structures. Nevertheless, lifetimes of 98.3 ± 79.7 ps and 830.0 ± 653.9 ps were determined for the fluorescence processes originating from the peripheral effective fluorophores and the trap states, respectively, although the low signal to noise ratios impact the fitting accuracy. The corresponding lifetimes are similar or lower to those obtained for the freshly synthesized CDs due to additional or more effective deactivation pathways within the peripheral effective fluorophores and the π-conjugated domains upon degradation. The photophysical and photobleaching processes in CDs and photobleached CDs are schematically shown in Fig. 6.
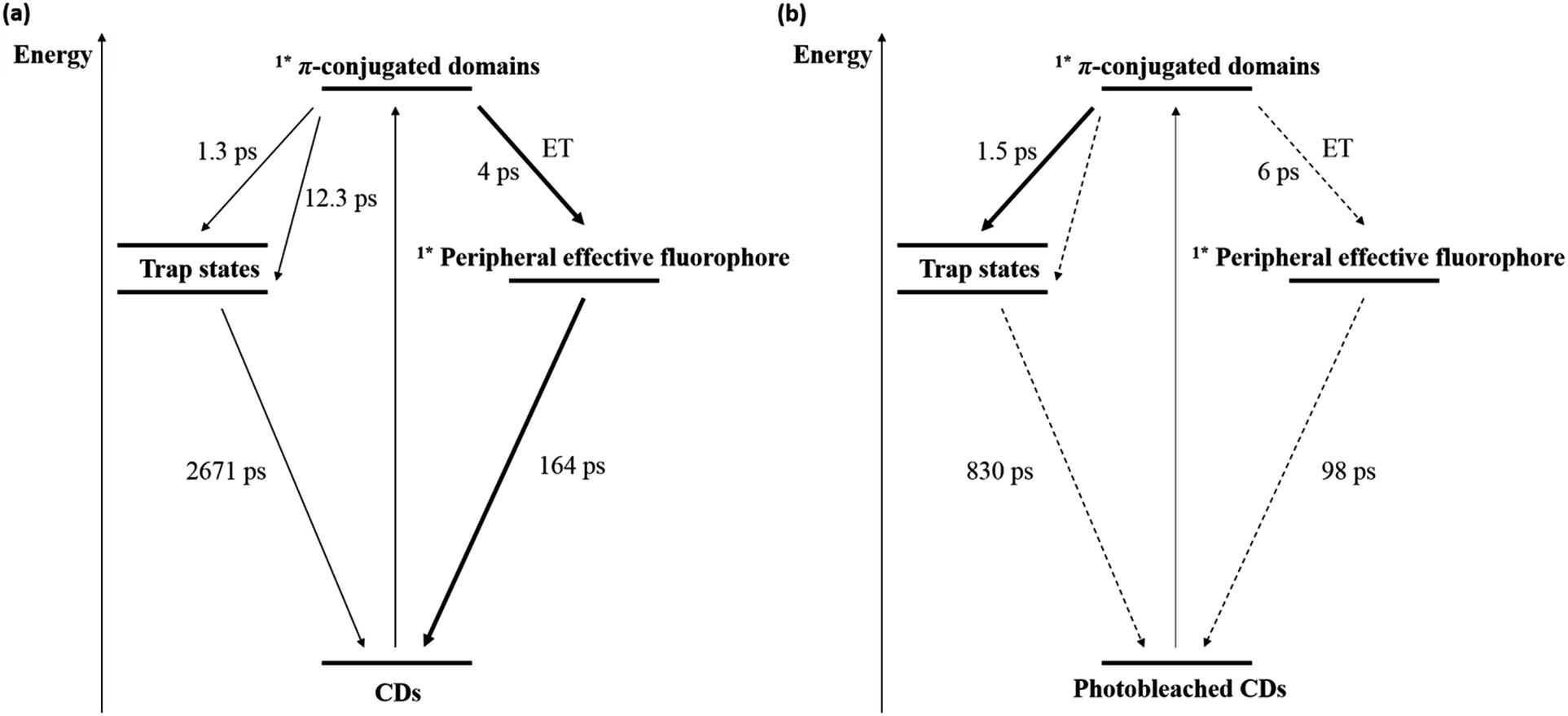 | ||
| Fig. 6 Energy level diagram and excited state deactivation pathways for (a) CDs and (b) photobleached CDs derived from time resolved spectroscopy. | ||
We propose that the peripheral effective fluorophores are pyridone based structures because of our findings from UV/Vis and FTIR and data from literature.6 To validate our hypothesis, 2,6-dihydroxypyridine-4-carboxylic acid (citrazinic acid) was used as a pyridone model. Citrazinic acid was chosen because its tautomer, 6-hydroxy-2-oxo-1,2-dihydropyridine-4-carboxylic acid, contains several 4-carboxylic-2-pyridone structures, similar to intermediate A (see Fig. 3). Citrazinic acid exhibits blue fluorescence, which is excitation wavelength-independent – Fig. S10(c) in the ESI.† Its absorption and fluorescence pattern is similar to that of the CDs synthesized in this work, namely a 365 nm absorption peak and a 440 nm fluorescence peak.38 Moreover, citrazinic acid is sensitive to UV exposure and undergoes photobleaching. As a matter of fact, the intensity of the 340 nm absorption decreased after exposure to UV, while the fluorescence intensity reduced by 80%. This is similar to what has been observed for our CDs. Transient absorption measurements with citrazinic acid (Fig. S11 in the ESI†) render results similar to those observed for CDs. For example, the Franck–Condon singlet excited state of citrazinic acid absorbs at around 600 nm and decays quickly via internal conversion with a lifetime of 1.2 ± 0.09 ps. Meanwhile, the absorption features of the vibrationally cooled singlet excited state of citrazinic acid grow-in around 450 nm and decay with a lifetime of 270.5 ± 5.9 ps. A summary of the energy level diagram and the excited state deactivation pathways for citrazinic acid is shown in Fig. S11(a) in the ESI.† For photobleached citrazinic acid, similar excited state deactivation pathways are concluded with a lifetime of 0.8 ± 0.1 ps for internal conversion and 272.3 ± 11.9 ps for fluorescence and ground state recovery. Notably, the intensities of the excited state absorption signals of photobleached citrazinic acid are weak. This behaviour is in sound agreement with that of CDs.
Our results suggest that the photobleaching of citric acid-derived CDs is caused by damage of their pyridone structures during UV exposure. This hypothesis is supported by XPS (Fig. S12 in the ESI†): the N 1s spectrum reveals a peak at 400 eV which can be assigned to C–N-bonds from pyridine carboxylic acids (Fig. S12a in the ESI†).40 Reduction of the nitrogen content of the CDs from about 9.5% to 8.5% due to photobleaching was detected. Moreover, the C 1s spectrum reveals reduction of CO bonds (peak at 289 eV) and formation of C–C and CC bonds (growing peak at ∼285 eV) due to photobleaching (Fig. S12b in the ESI†). These findings hint to damage of the fluorophore by carbonization. Similar molecular structures have been studied by Murdock and co-workers, who focused on the photo-isomerization dynamics of aqueous N-methyl-2-pyridone.41 They observed that, during the excited state decay, a considerable fraction of CDs undergo photoinitiated rearrangement and carbonization. Furthermore, they demonstrated that the photochemically formed radicals contribute to destroying the pyridone and, in turn, the effective fluorescent structure. Such a phenomenon was studied: the photobleaching rate of CDs increases when they are exposed to hydrogen peroxide during UV exposure (Fig. S13 in the ESI†). This trend of accelerated photobleaching in the presence of hydrogen peroxide is also observed in citrazinic acid solutions. The radicals in solution inherently damage the peripheral effective fluorophore structure of the CDs. This is consistent with the supressed photobleaching observed upon encapsulation of CDs into solid phase films and upon addition of radical scavengers.37 In summary, the steady-state and transient optical properties of citrazinic acid are very similar to those of freshly synthesized CDs, supporting the notion that it is the peripheral effective CD fluorophore.
Conclusions
In situ UV/Vis absorption and PL spectroscopy were used to investigate the photobleaching of CDs in aqueous solutions as a function of synthesis conditions, namely reaction temperature and time. CDs synthesized at lower reaction temperatures and/or shorter reaction times exhibit high initial QYs, but suffer from comparatively fast photobleaching. CDs synthesized at high reaction temperatures and longer reaction times show the opposite effect. It can be concluded that untreated “bottom-up” CD suspensions are characterized by a so far unavoidable trade-off between high QY and low photostability. By analysing the fluorescence properties of CDs resulting from different precursors, we were able to identify the peripheral effective fluorophore of CDs as a pyridone structure. The presence of a pyridone in the primary structure is expected to be responsible for a variety of fluorescence properties such as photobleaching, high QY, and excitation wavelength independent fluorescence. DFT simulation was employed to theoretically support the pyridone structure. Finally, the energy levels and excited state deactivation pathways of CDs were studied using ultrafast spectroscopy. Results from both transient and steady state absorption measurements supported the hypothesis that photobleaching could be the result of the photoinitiated carbonization of a peripheral effective fluorophore structure. The identification of an effective fluorophore structure in the CDs will facilitate product optimization, advance model simulation, and contribute to a better understanding of the synthesis pathway and fluorescence mechanism.Acknowledgements
This work was supported financially by the German Science Foundation (DFG), Collaborative Research Centre SFB953: “Synthetic Carbon Allotropes”. Moreover, the authors would like to thank Prof. H.-P. Steinrueck, Dr. C. Papp, and F. Spaeth from the Institute of Physical Chemistry II of the Friedrich-Alexander-University Erlangen-Nürnberg for doing XPS measurements. Many thanks go to Thomas J. Nacken for the fruitful discussions.References
- O. Kozak, M. Sudolská, G. Pramanik, P. Cigler, M. Otyepka and R. Zboril, Photoluminescent carbon nanostructures, Chem. Mater., 2016, 28, 4085–4128 CrossRef CAS.
- V. Strauss, A. Roth, M. Sekita and D. M. Guldi, Efficient energy-conversion materials for the future: understanding and tailoring charge-transfer processes in carbon nanostructures, Chem, 2016, 1, 531–556 CAS.
- T. Ogi, H. Iwasaki, K. Aishima, F. Iskandar, W.-N. Wang, K. Takimiya and K. Okuyama, Transient nature of graphene quantum dot formation via a hydrothermal reaction, RSC Adv., 2014, 4, 55709–55715 RSC.
- T. Ogi, K. Aishima, F. A. Permatasari, F. Iskandar, E. Tanabe and K. Okuyama, Kinetics of nitrogen-doped carbon dot formation via hydrothermal synthesis, New J. Chem., 2016, 40, 5555–5561 RSC.
- F. A. Permatasari, A. H. Aimon, F. Iskandar, T. Ogi and K. Okuyama, Role of C–N configuration in the photoluminescence of graphene quantum dots synthesized by a hydrothermal route, Sci. Rep., 2016, 6, 21042 CrossRef CAS PubMed.
- L. Shi, J. H. Yang, H. B. Zeng, Y. M. Cheng, S. C. Yang, C. Wu, H. Zeng, O. Yoshihito and Q. Zhang, Carbon dots with high fluorescence quantum yield: the fluorescence originates from organic fluorophores, Nanoscale, 2016, 8, 14374–14378 RSC.
- J. Tang, B. Kong, H. Wu, M. Xu, Y. Wang, Y. Wang, D. Zhao and G. Zheng, Carbon nanodots featuring efficient FRET for real-time monitoring of drug delivery and two-photon imaging, Adv. Mater., 2013, 25, 6569–6574 CrossRef CAS PubMed.
- C. Han, R. Wang, K. Wang, H. Xu, M. Sui, J. Li and K. Xu, Highly fluorescent carbon dots as selective and sensitive “on–off–on” probes for iron(III) ion and apoferritin detection and imaging in living cells, Biosens. Bioelectron., 2016, 83, 229–236 CrossRef CAS PubMed.
- S. Hu, Q. Chang, K. Lin and J. Yang, Tailoring surface charge distribution of carbon dots through heteroatoms for enhanced visible-light photocatalytic activity, Carbon, 2016, 105, 484–489 CrossRef CAS.
- Y. Liu, Y. Tian, Y. Tian, Y. Wang and W. Yang, Carbon-dot-based nanosensors for the detection of intracellular redox state, Adv. Mater., 2015, 27, 7156–7160 CrossRef CAS PubMed.
- V.-D. Dao, P. Kim, S. Baek, L. L. Larina, K. Yong, R. Ryoo, S. H. Ko and H.-S. Choi, Facile synthesis of carbon dot-Au nanoraspberries and their application as high-performance counter electrodes in quantum dot-sensitized solar cells, Carbon, 2016, 96, 139–144 CrossRef CAS.
- Z. Liu, X. Chen, X. Zhang, J. J. Gooding and Y. Zhou, Carbon-quantum-dots-loaded mesoporous silica nanocarriers with pH-switchable zwitterionic surface and enzyme-responsive pore-cap for targeted imaging and drug delivery to tumor, Adv. Healthcare Mater., 2016, 5, 1401–1407 CrossRef CAS PubMed.
- H. Peng, Y. Li, C. Jiang, C. Luo, R. Qi, R. Huang, C.-G. Duan and J. Travas-Sejdic, Tuning the properties of luminescent nitrogen-doped carbon dots by reaction precursors, Carbon, 2016, 100, 386–394 CrossRef CAS.
- Z. Song, F. Quan, Y. Xu, M. Liu, L. Cui and J. Liu, Multifunctional N, S co-doped carbon quantum dots with pH- and thermo-dependent switchable fluorescent properties and highly selective detection of glutathione, Carbon, 2016, 104, 169–178 CrossRef CAS.
- S. K. Pal, Versatile photoluminescence from graphene and its derivatives, Carbon, 2015, 88, 86–112 CrossRef CAS.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Highly photoluminescent carbon dots for multicolor patterning, sensors, and bioimaging, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- D. Qu, M. Zheng, L. Zhang, H. Zhao, Z. Xie, X. Jing, R. E. Haddad, H. Fan and Z. Sun, Formation mechanism and optimization of highly luminescent N-doped graphene quantum dots, Sci. Rep., 2014, 4, 5294 CrossRef CAS PubMed.
- S. Khan, N. C. Verma, A. Gupta and C. K. Nandi, Reversible photoswitching of carbon dots, Sci. Rep., 2015, 5, 11423 CrossRef CAS PubMed.
- A. M. Chizhik, S. Stein, M. O. Dekaliuk, C. Battle, W. Li, A. Huss, M. Platen, I. A. Schaap, I. Gregor and A. P. Demchenko, Super-resolution optical fluctuation bio-imaging with dual-color carbon nanodots, Nano Lett., 2015, 16, 237–242 CrossRef PubMed.
- A. Sharma, T. Gadly, A. Gupta, A. Ballal, S. K. Ghosh and M. Kumbhakar, Origin of excitation dependent fluorescence in carbon nanodots, J. Phys. Chem. Lett., 2016, 7, 3695–3702 CrossRef CAS PubMed.
- D. Li, M. B. Mueller, S. Gilje, R. B. Kaner and G. G. Wallace, Processable aqueous dispersions of graphene nanosheets, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- S. Zhu, Y. Song, X. Zhao, J. Shao, J. Zhang and B. Yang, The photoluminescence mechanism in carbon dots (graphene quantum dots, carbon nanodots, and polymer dots): current state and future perspective, Nano Res., 2015, 8, 355–381 CrossRef CAS.
- S. Khan, A. Gupta, N. C. Verma and C. K. Nandi, Time-resolved emission reveals ensemble of emissive states as the origin of multicolor fluorescence in carbon dots, Nano Lett., 2015, 15, 8300–8305 CrossRef CAS PubMed.
- X. T. Zheng, A. Ananthanarayanan, K. Q. Luo and P. Chen, Glowing graphene quantum dots and carbon dots: properties, syntheses, and biological applications, Small, 2015, 11, 1620–1636 CrossRef CAS PubMed.
- L. Wang, S.-J. Zhu, H.-Y. Wang, S.-N. Qu, Y.-L. Zhang, J.-H. Zhang, Q.-D. Chen, H.-L. Xu, W. Han and B. Yang, Common origin of green luminescence in carbon nanodots and graphene quantum dots, ACS Nano, 2014, 8, 2541–2547 CrossRef CAS PubMed.
- K. Lingam, R. Podila, H. Qian, S. Serkiz and A. M. Rao, Evidence for edge-state photoluminescence in graphene quantum dots, Adv. Funct. Mater., 2013, 23, 5062–5065 CrossRef CAS.
- M. Fu, F. Ehrat, Y. Wang, K. Z. Milowska, C. Reckmeier, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Carbon dots: a unique fluorescent cocktail of polycyclic aromatic hydrocarbons, Nano Lett., 2015, 15, 6030–6035 CrossRef CAS PubMed.
- H. Ding and H.-M. Xiong, Exploring the blue luminescence origin of nitrogen-doped carbon dots by controlling the water amount in synthesis, RSC Adv., 2015, 5, 66528–66533 RSC.
- W. Sell and T. Easterfield, LXXIII—Studies on citrazinic acid. Part I, J. Chem. Soc., Trans., 1893, 63, 1035–1051 RSC.
- W. Kasprzyk, S. Bednarz, P. Żmudzki, M. Galica and D. Bogdał, Novel efficient fluorophores synthesized from citric acid, RSC Adv., 2015, 5, 34795–34799 RSC.
- Y. Dong, J. Shao, C. Chen, H. Li, R. Wang, Y. Chi, X. Lin and G. Chen, Blue luminescent graphene quantum dots and graphene oxide prepared by tuning the carbonization degree of citric acid, Carbon, 2012, 50, 4738–4743 CrossRef CAS.
- L. Li, G. Wu, G. Yang, J. Peng, J. Zhao and J.-J. Zhu, Focusing on luminescent graphene quantum dots: current status and future perspectives, Nanoscale, 2013, 5, 4015–4039 RSC.
- G. Eda, Y. Y. Lin, C. Mattevi, H. Yamaguchi, H. A. Chen, I. Chen, C. W. Chen and M. Chhowalla, Blue photoluminescence from chemically derived graphene oxide, Adv. Mater., 2010, 22, 505–509 CrossRef CAS PubMed.
- T.-F. Yeh, W.-L. Huang, C.-J. Chung, I.-T. Chiang, L.-C. Chen, H.-Y. Chang, W.-C. Su, C. Cheng, S.-J. Chen and H. Teng, Elucidating quantum confinement in graphene oxide dots based on excitation-wavelength-independent photoluminescence, J. Phys. Chem. Lett., 2016, 7, 2087–2092 CrossRef CAS PubMed.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S. T. Lee, Water-soluble fluorescent carbon quantum dots and photocatalyst design, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed.
- R. Zhang, E. Bertran and S.-T. Lee, Size dependence of energy gaps in small carbon clusters: the origin of broadband luminescence, Diamond Relat. Mater., 1998, 7, 1663–1668 CrossRef CAS.
- W. Wang, C. Damm, J. Walter, T. J. Nacken and W. Peukert, Photobleaching and stabilization of carbon nanodots produced by solvothermal synthesis, Phys. Chem. Chem. Phys., 2016, 18, 466–475 RSC.
- V. Strauss, J. T. Margraf, C. Dolle, B. Butz, T. J. Nacken, J. Walter, W. Bauer, W. Peukert, E. Spiecker and T. Clark, Carbon nanodots: toward a comprehensive understanding of their photoluminescence, J. Am. Chem. Soc., 2014, 136, 17308–17316 CrossRef CAS PubMed.
- V. Strauss, A. Kahnt, E. M. Zolnhofer, K. Meyer, H. Maid, C. Placht, W. Bauer, T. J. Nacken, W. Peukert and S. H. Etschel, Assigning electronic states in carbon nanodots, Adv. Funct. Mater., 2016, 26, 7975–7985 CrossRef CAS.
- T. Yoshida, An X-ray photoelectron spectroscopic study of several ligands in coordination compounds, Bull. Chem. Soc. Jpn., 1980, 53, 1327 CrossRef CAS.
- D. Murdock, S. J. Harris, I. P. Clark, G. M. Greetham, M. Towrie, A. J. Orr-Ewing and M. N. Ashfold, UV-induced isomerization dynamics of N-methyl-2-pyridone in solution, J. Phys. Chem. A, 2014, 119, 88–94 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Description of CD synthesis and characterization methods; fluorescence and UV/Vis spectra as well as time-resolved UV/Vis spectra of CDs synthesized under different conditions and of citrazinic acid; cyclic voltammogram for CDs; N 1s and C 1s XPS spectra of fresh and photobleached CDs; influence of hydrogen peroxide on photobleaching of CDs and citrazinic acid. See DOI: 10.1039/c7ra04421f |
| This journal is © The Royal Society of Chemistry 2017 |