
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ferromagnetic-like behaviour in bismuth ferrite films prepared by electrodeposition and subsequent heat treatment†
Doga Bilicana,
Enric Menéndez*a,
Jin Zhanga,
Pau Solsonaa,
Jordina Fornella,
Eva Pellicer *a and
Jordi Sortab
*a and
Jordi Sortab
aDepartament de Física, Facultat de Ciències, Universitat Autònoma de Barcelona, E-08193 Bellaterra, Spain. E-mail: enric.menendez@uab.cat; eva.pellicer@uab.cat
bInstitució Catalana de Recerca i Estudis Avançats (ICREA), Pg. Lluís Companys 23, 08010 Barcelona, Spain
First published on 23rd June 2017
Abstract
Bismuth ferrite (BiFeO3) films (10 μm in thickness) are synthesized by electrodeposition from a dimethylformamide bath containing Bi and Fe nitrate salts as precursors followed by heat treatment in air. Thermal treatments are needed to induce crystallisation of the as-deposited amorphous-like films. A detailed morphological, structural and magnetic characterisation evidences that BiFeO3 forms in a rather narrow temperature window (around 600 °C). At lower and higher temperatures, secondary binary and ternary oxides are also formed. Soft ferromagnetic-like behaviour is observed in all films, which presumably arises from the obtained nanoscale structure, which favours the occurrence of spin canting in the BiFeO3 antiferromagnetic phase. The eventual contribution from secondary phases to the observed ferromagnetic-like response is also discussed. The obtained results are appealing for the integration of BiFeO3 into devices that may require relatively thick films with an enhanced surface area-to-volume ratio. The growth of this type of material by techniques other than electrodeposition is not straightforward.
Introduction
The primary physical mechanisms of ferroelectricity and magnetism are different and tend to exclude each other.1 However, both phenomena may coexist in the so-called multiferroic materials.1,2 These materials show coupled ferroelectric and magnetic properties. That is, magnetism can be controlled through electric fields and vice versa. From a technological viewpoint, this provides increased degrees of freedom in the design and energy-efficient manipulation of spintronic and memory devices.1,2 Since these devices are ultimately controlled by electric currents, their operation involves considerable energy loss by the Joule heating effect. Thus, the partial substitution of electric currents by electric fields (i.e., voltage-driven magnetic actuation) would significantly decrease the overall power consumption.3,4Among the few materials which exhibit intrinsic multiferroicity, BiFeO3 (BFO) is attracting huge interest since it is both ferroelectric and antiferromagnetic at room temperature (its ferroelectric Curie temperature is 770 °C, while its Néel temperature is around 370 °C (ref. 1, 2 and 5)). BFO crystallises in a rhombohedral (R3c) structure and belongs to the group of perovskites with the ABO3 formula.2 From a thermodynamic point of view, BiFeO3 is a metastable compound that exists between the stable Bi25FeO39 (sillenite) and Bi2Fe4O9 (mullite) compounds according to the equilibrium phase diagram of Bi2O3 and Fe2O3.2,6 It actually forms between 447 °C and 767 °C. Nevertheless, BFO has a high tendency to grow simultaneously with these other two stable phases. The quality of the end product depends on several factors, such as purity of the precursors, preparation method, chemical additives or heat treatment conditions.7–10 The properties of BFO are indeed very sensitive to oxygen stoichiometry and tiny deviations might lead to undesired properties, such as strong current leakages.11 Furthermore, although BFO is antiferromagnetic, it often shows soft ferromagnetism which originates from spin canting.2,12 Interestingly, larger saturation magnetisation is achieved due to size-effects (in samples with reduced lateral dimensions) since spin canting reinforces due to the partial suppression of the spiral spin structure.13 In this context, the development of nanoscale architectures becomes crucial to engineer novel magnetoelectric materials.14,15 Remarkably, besides its potential applications in spintronics and data storage, BFO is a suitable candidate to be used also in photovoltaic devices and related applications.16–18
A wide variety of growth techniques and post-synthesis treatments have been used to prepare BFO. Solid state reaction,9 sol–gel,19,20 pulsed laser deposition,21,22 sputtering,23 solution combustion,24 aqueous gel precursors,25 microemulsion,26 the Pechini method27 or microwave hydrothermal synthesis28 are amongst the most frequently employed. A fast-growing research has been devoted to the manufacture and characterisation of BFO nanoscopic shapes, such as nanofibers,29 nanowires,30 nanotubes,29,31 or nanoparticles.32 These nanostructures offer distinctive size effects which, as previously explained, can affect some of the physical properties of BFO.
Some of the aforementioned techniques (solid state reaction, sol–gel or solution combustion) typically render powdery BFO, thus hindering its eventual integration into devices since it would need further immobilization on a substrate. Usually, smooth coatings with compositional homogeneity are successfully obtained by pulsed laser deposition. However, this method is a costly method since it requires high vacuum conditions. Moreover, it does not allow the growth of micrometre-thick films, neither the deposition onto curved substrates nor the control of film's porosity.
Interestingly, electrodeposition enables the low-cost growth of relatively thick films directly on a substrate with a simple setup. Besides its obvious use for the growth of pure metals and metallic alloys, electrodeposition is a powerful methodology that can be utilized for the preparation of semiconducting metal oxides, hydroxides and chalcogenides, either in the form of films or nanostructures.33–36 So far, investigation on BFO electrodeposition has been very scarce and the results have not been fully satisfactory in the sense that they lack a clear and unambiguous structural characterisation.37–39 Moreover, in these previous works, emphasis was laid on the electrochemical performance of the materials (i.e., BFO nanoflake and thin films electrodes were applied in supercapacitor applications). The magnetic properties were overlooked.
To the best of our knowledge, here we report for the first time the formation of rhombohedral BFO phase by cathodic electrodeposition from a novel N,N-dimethylformamide (DMF) bath and subsequent thermal treatment. Fe(III) and Bi(III) nitrate salts have been solubilized in the organic electrolyte. Interestingly, the applied electrodeposition conditions render rough, not fully dense (porous-like), BFO films. Heat treatments at various annealing temperatures are performed in order to investigate the crystallisation behaviour of the as-prepared BFO. These materials are further studied from a magnetic point of view. Ferromagnetic-like properties stemming from finite size effects (spin canting) are observed.
Experimental
10 μm-thick, amorphous-like films with Bi![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe ≈ 1 were deposited on Si/Ti/Cu substrates by potentiostatic electrodeposition from a bath containing 6 g L−1 Bi(NO3)3·5H2O (Sigma-Aldrich, ≥98.0%) and 10 g L−1 Fe(NO3)3·9H2O (Sigma-Aldrich, ≥99.95%) dissolved in DMF (Sigma-Aldrich, ≥99.8%). A three-electrode cell with a Pt wire as counter electrode and a Ag|AgCl (3 M KCl) reference electrode (E = + 0.210 V NHE) was used. The configuration (i.e., arrangement and separation) of the electrodes was kept the same in all experiments (cyclic voltammetry (CV) and electrodeposition). Specifically, depositions were carried out under direct current conditions at 28 °C for 1800 s, while stirring the bath at 200 rpm. Unless otherwise stated, the cathodic potential applied was −1.3 V. The working area was kept between 0.250 and 0.275 cm2. After electrodeposition, the samples were rinsed in Milli-Q water and hot dried. Coating thickness was measured with a 3D Optical Surface Metrology System Leica DCM 3D. Subsequently, with the aim to investigate the crystallisation behaviour and ultimately to obtain crystalline BiFeO3, the as-deposited samples were subjected to different thermal treatments in a Carbolite MTF 9/15/130 tubular furnace in open atmosphere at 500, 600, 700 and 800 °C for 2 hours. The samples were heated at a rate of 1 °C min−1 and cooled down in air. A Merlin ZEISS scanning electron microscope was used to investigate the morphology of the samples using secondary electrons. The structural characterisation was done by means of θ/2θ X-ray diffraction (XRD) using Cu Kα radiation. Furthermore, Rietveld refinements of the XRD patterns using the MAUD software40,41 were carried out to determine the crystallite size (i.e., average coherently diffracting domain size) of the main phases (further details on the Rietveld refinements can be found in the ESI†). Transmission electron microscopy (TEM) analyses were done on a Jeol JEM-2011 microscope operated at 200 kV. Finally, the samples were magnetically characterised by recording the magnetisation (M) vs. applied magnetic field (Happlied) curves (i.e., hysteresis loops) by means of vibrating sample magnetometry (Microsense from LOT-QuantumDesign) at room temperature.
:Fe ≈ 1 were deposited on Si/Ti/Cu substrates by potentiostatic electrodeposition from a bath containing 6 g L−1 Bi(NO3)3·5H2O (Sigma-Aldrich, ≥98.0%) and 10 g L−1 Fe(NO3)3·9H2O (Sigma-Aldrich, ≥99.95%) dissolved in DMF (Sigma-Aldrich, ≥99.8%). A three-electrode cell with a Pt wire as counter electrode and a Ag|AgCl (3 M KCl) reference electrode (E = + 0.210 V NHE) was used. The configuration (i.e., arrangement and separation) of the electrodes was kept the same in all experiments (cyclic voltammetry (CV) and electrodeposition). Specifically, depositions were carried out under direct current conditions at 28 °C for 1800 s, while stirring the bath at 200 rpm. Unless otherwise stated, the cathodic potential applied was −1.3 V. The working area was kept between 0.250 and 0.275 cm2. After electrodeposition, the samples were rinsed in Milli-Q water and hot dried. Coating thickness was measured with a 3D Optical Surface Metrology System Leica DCM 3D. Subsequently, with the aim to investigate the crystallisation behaviour and ultimately to obtain crystalline BiFeO3, the as-deposited samples were subjected to different thermal treatments in a Carbolite MTF 9/15/130 tubular furnace in open atmosphere at 500, 600, 700 and 800 °C for 2 hours. The samples were heated at a rate of 1 °C min−1 and cooled down in air. A Merlin ZEISS scanning electron microscope was used to investigate the morphology of the samples using secondary electrons. The structural characterisation was done by means of θ/2θ X-ray diffraction (XRD) using Cu Kα radiation. Furthermore, Rietveld refinements of the XRD patterns using the MAUD software40,41 were carried out to determine the crystallite size (i.e., average coherently diffracting domain size) of the main phases (further details on the Rietveld refinements can be found in the ESI†). Transmission electron microscopy (TEM) analyses were done on a Jeol JEM-2011 microscope operated at 200 kV. Finally, the samples were magnetically characterised by recording the magnetisation (M) vs. applied magnetic field (Happlied) curves (i.e., hysteresis loops) by means of vibrating sample magnetometry (Microsense from LOT-QuantumDesign) at room temperature.
Results and discussion
Electrochemical deposition
Metals and metal matrix/nanoparticle composites have been deposited from DMF in the past.42,43 The examples available, although scarce compared to water-based electrolytes, mainly focus on cobalt, nickel and their alloys. Little is known about the electrodeposition of metal oxides from DMF baths. In our laboratory, attempts to produce Bi- and Fe-containing films with Bi:Fe ≈ 1 from aqueous solution (0.045 M Fe(NO3)3·9H2O, 0.01 M Bi(NO3)3·5H2O and 0.1 M triethanolamine) failed. In particular, only Bi-rich deposits were obtained at pH 12.5 and T = 65 °C for deposition potentials ranging from −0.8 V to −1.5 V. On the contrary, codeposition of both Bi and Fe elements in comparable amounts was possible from the DMF bath. This is because the deposition potentials in water and DMF media are different.
Fig. 1a shows the CV curve recorded from the DMF bath under stagnant conditions. Negative currents were detected from +0.25 V during the cathodic going sweep. Prior to reversing the scan toward anodic potentials, a diffusion-controlled peak was recorded at −1.1 V. Fig. 1b shows the j–t curves recorded between −1.25 V and −1.35 V under stagnant conditions. Whilst less negative currents were observed for an applied potential of −1.25 V during the first 200 s of deposition, fluctuations caused the currents to almost merge afterwards. A similar trend was observed under stirred conditions (see inset of Fig. 1b). In this case the j–t transients showed even more pronounced fluctuations. Since the currents were similar, −1.30 V was selected for the subsequent growth of BFO films. Fig. 1c shows a scanning electron microscopy (SEM) image of an as-deposited film. A rough morphology consisting of not fully dense nodules was noticed. At a low magnification, the coating displayed a cracked appearance, likely caused by layer dehydration in the SEM vacuum chamber. The presence of small amount of water is unavoidable when using DMF as solvent. These cracks are frequently observed in metal oxides produced by electrodeposition in aqueous solutions.34
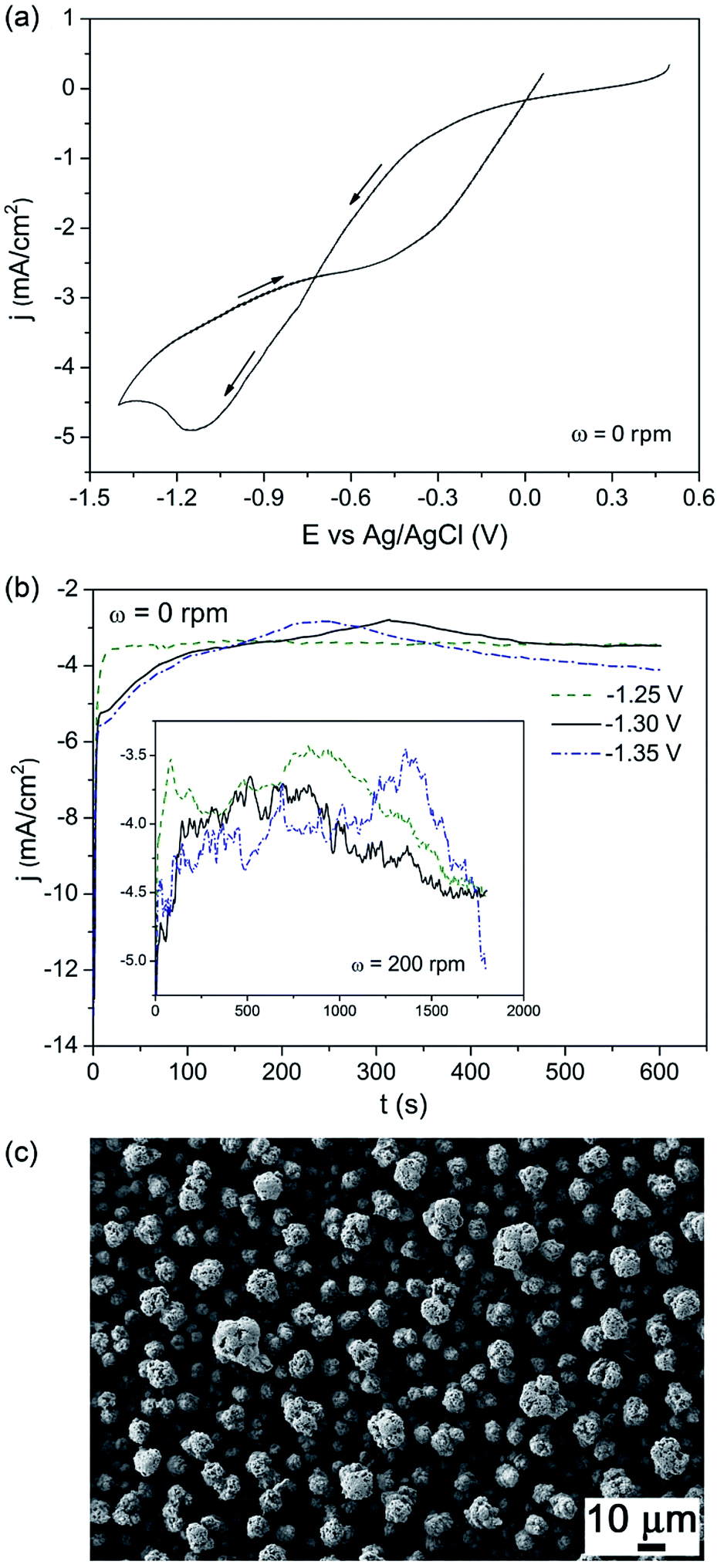 | ||
| Fig. 1 (a) CV curve recorded under quiescent conditions, initially scanning from +0.5 V toward negative potentials, at 50 mV s−1. (b) j–t curves recorded under stagnant conditions at −1.25 V, −1.30 V and −1.35 V for 600 s. The inset shows the j–t curves recorded at the same potentials but under stirred conditions (ω = 200 rpm) for longer deposition time (1800 s). (c) SEM image, taken with secondary electrons, of an as-electrodeposited film at −1.30 V for 1800 s. | ||
Structural and morphological characterisation
The XRD pattern of the as-deposited sample (not shown) is consistent with an amorphous-like phase since, besides peaks arising from the substrate, only wide halos are observable. As can be seen in Fig. 2, crystallisation takes place upon heat treatment. At 500 °C, rhombohedral BFO with a crystallite of around 42 nm is formed together with Bi2O3, Fe2O3, Fe3O4 and ternary oxides, such as Bi24Fe2O39 or Bi2Fe4O9. According to the phase diagram,2 the presence of these phases indicates that 500 °C is still a mild condition for the BFO formation.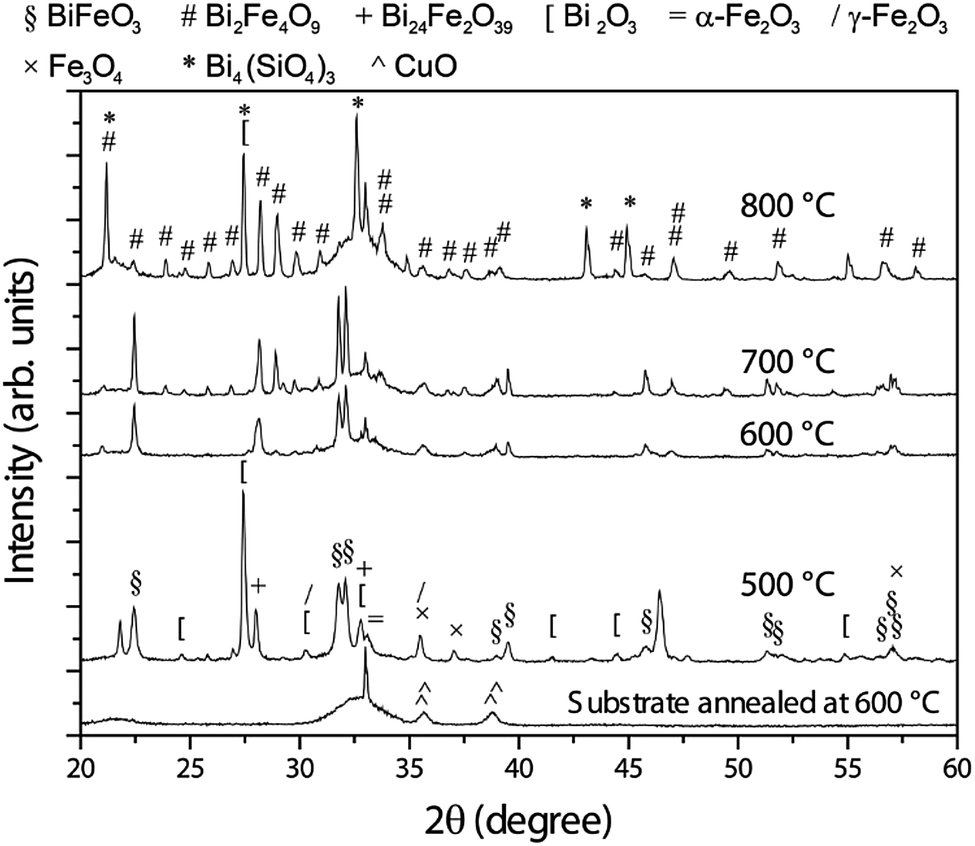 | ||
| Fig. 2 θ/2θ X-ray diffraction patterns of the samples annealed at 500, 600, 700 and 800 °C. To serve as reference, the pattern of the substrate annealed at 600 °C is also included. The peaks have been labelled according to the existing crystallographic phases (between parentheses, the used symbol to represent the phase together with its ICDD File Card no.): BiFeO3 (§, 01-072-2321), Bi2Fe4O9 (#, 01-072-1832), Bi24Fe2O39 (+, 00-042-0201), Bi2O3 ([, 00-045-1344), α-Fe2O3 (=, 01-085-0987), γ-Fe2O3 (/, 00-039-1346), Fe3O4 (×, 00-001-1111), Bi4(SiO4)3 (*, 01-088-0243) and CuO (^, 00-041-0254). | ||
From a thermodynamic point of view, BFO is formed from equal parts of Bi2O3 and Fe2O3. Upon annealing at 600 °C, the amount of BFO further increases in detriment mainly of Bi2O3 and Fe2O3, becoming nearly the only phase. The crystallite size grows up to around 80 nm, suggesting that ternary phases, which tend to grow at the grain boundaries of BFO, have strongly vanished, as suggested by the XRD characterisation. The thermal treatment at 700 °C leads to a significant formation of Bi2Fe4O9 at expenses of BiFeO3, which still shows an increased crystallite size (about 146 nm). In turn, the crystallite size of the Bi2Fe4O9 phase is around 100 nm. The relatively large crystallite size of BFO suggests that Bi2Fe4O9 grows in cluster-like form rather than precipitate-like at the grain boundaries of BFO, which would result in a decreased BFO crystallite size. Upon annealing at 800 °C, Bi2Fe4O9 becomes the main phase. In comparison with BFO, this ternary phase is an Fe-rich phase, thus, to keep the stoichiometry, Fe-free phases must be formed. As expected, the XRD pattern of the sample thermally treated at 800 °C shows that Bi2Fe4O9 forms along with Bi2O3 and Bi4(SiO4)3. At this temperature, the crystallite size of the Bi2Fe4O9 significantly grows, reaching a value above 200 nm which cannot be estimated by line profile analysis.44 At the same time, the amount of BFO strongly diminishes and the residual BFO is highly nanostructured as evidenced by its weak and broad XRD peaks, respectively (note that the BFO peaks located at 31.8 and 32.1 degrees nearly vanish upon annealing at 800 °C).
The film annealed at 700 °C was investigated by TEM (Fig. 3). The occurrence of both Bi2Fe4O9 and BiFeO3 phases was seen in the Fast Fourier Transform (FFT) patterns taken on different areas of the sample, in agreement with XRD results.
 | ||
| Fig. 3 (a) TEM image of a representative zone of the sample annealed at 700 °C. (b) HRTEM image of the red square depicted in panel (a). The inset corresponds to its FFT. The interplanar distances are characteristic of Bi2Fe4O9. (c) HRTEM image of the green square illustrated in panel (a). The inset corresponds to its FFT and the interplanar distances belong to BiFeO3 phase. | ||
As aforementioned, the as-prepared sample is not fully dense. Moreover, the film is very rough (Fig. 1c). Even though the thermal treatment at 500 °C leads to a somewhat denser film, the ‘open’ structure remains (Fig. 4a). It is at 600 °C where the morphology starts to evolve towards more particulate-like, although some voids are still preserved. The particles adopt prism morphologies, indicating the formation of rhombohedral BFO (Fig. 4b). This is in concordance with the XRD analysis, which reveals that 500 °C is still a mild condition for the formation of BFO. With further increase of the annealing temperature (i.e., 700 °C), the size of the particles slightly increases and the prism morphology becomes more well-defined (Fig. 4c). At 800 °C, even though the prism morphology remains, a significant particle coarsening occurs, resulting in a significant increase of the size of the particles. This is in agreement with the structural analysis which indicates that the crystallite size of the Bi2Fe4O9 phase increases upon annealing at 800 °C up to values that cannot even be estimated by Rietveld refinement.
 | ||
| Fig. 4 SEM images, taken with secondary electrons, of the samples annealed at (a) 500, (b) 600, (c) 700 and (d) 800 °C. | ||
Magnetic characterisation
Magnetisation (M) vs. applied magnetic field (Happlied) curves (i.e., hysteresis loops) are shown in Fig. 5. The as-prepared sample already exhibits traces of ferromagnetism which might be ascribed to Fe-rich areas in the amorphous oxide films.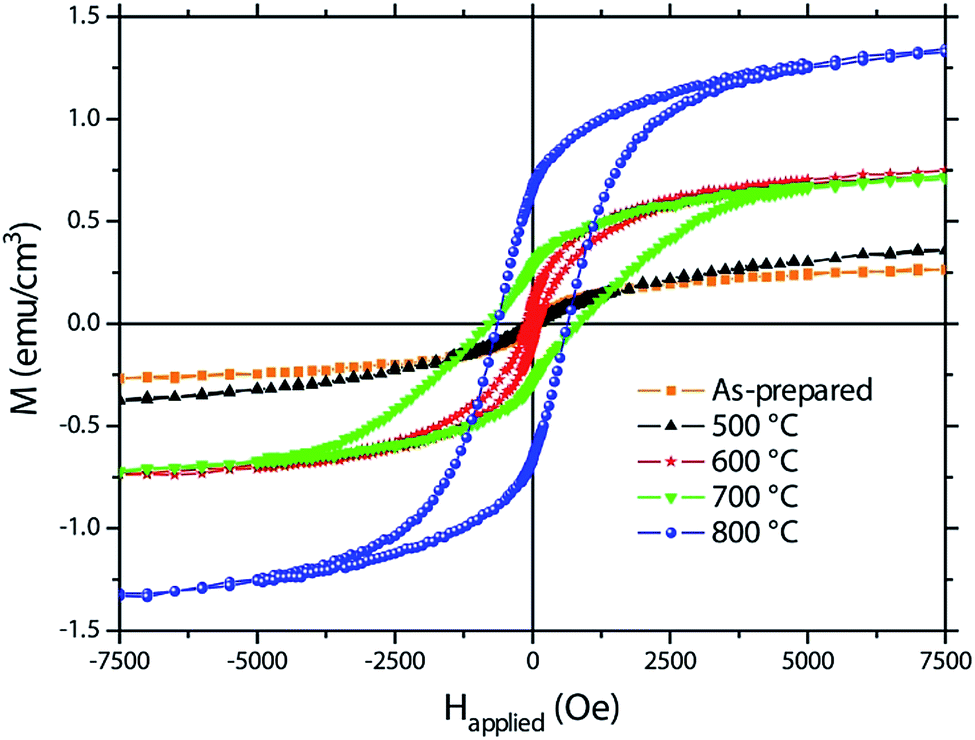 | ||
| Fig. 5 Hysteresis loops recorded by vibrating sample magnetometry of the as-prepared sample and the samples annealed at 500, 600, 700 and 800 °C. | ||
Crystallisation takes place upon heat treatment. Annealing at 500 °C results in a slightly increased saturation magnetization, which might be ascribed to the residual formation of iron oxides (see Fig. 5 and Table 1). At room temperature, α-Fe2O3 shows weak ferromagnetic-like behaviour due to its nanoscale nature45,46 and Fe3O4 and γ-Fe2O3 are ferromagnetic.47 On top of this, BFO may also contribute to ferromagnetism due to size effects which result in antiferromagnetic spin canting due to the suppression of the spiral spin structure.12–14,48 From a magnetic point of view, the ternary phases play a minor role since they are usually paramagnetic at room temperature.49,50 For instance, Bi2Fe4O9 becomes antiferromagnetic but only below 250 K.50 At 600 °C, the saturation magnetisation significantly increases. From the structural and morphological analyses, this is accompanied with the formation of a nearly pure BFO in detriment of both Fe-based oxides and ternary phases (Fig. 2). Therefore, the observed ferromagnetic response could be primarily ascribed to nanostructuring effects which reinforce antiferromagnetic spin canting, giving rise to a larger net magnetisation. While the sample annealed at 700 °C shows a similar MS than that of the sample annealed at 600 °C (Table 1), the saturation magnetisation of the sample annealed at 800 °C is considerably larger. The pronounced decomposition of BFO upon annealing at 800 °C yields a highly nanostructured phase with strong size effects which might be responsible for the increase in MS. Taking into account the described structural changes with temperature, the increase in HC upon annealing at 700 °C and 800 °C might also be related to the formation of Bi2Fe4O9 and the other paramagnetic phases, which probably tends to isolate the BFO grains, thus decreasing intergranular dipolar and exchange interactions, both of which are known to be detrimental for coercivity.51
| Sample | MS (emu cm−3) | HC (Oe) | MR/MS (%) |
|---|---|---|---|
| As-prepared | 0.275 | <40 | <2.5 |
| 500 °C | 0.390 | <40 | <2.5 |
| 600 °C | 0.770 | 85 | 11.0 |
| 700 °C | 0.745 | 822 | 38.3 |
| 800 °C | 1.360 | 640 | 48.9 |
Conclusions
The synthesis of bismuth ferrite (BiFeO3, BFO) has been successfully carried out by potentiostatic electrodeposition from a nitrate-based DMF bath followed by heat treatment in air. Since the electrodeposition results in amorphous-like films, thermal treatments are needed to induce crystallisation. Annealing procedures in air at 500, 600, 700 and 800 °C for 2 hours have been carried out. The heat treatment at 600 °C results in nearly pure nanostructured BFO with soft ferromagnetic-like properties. A detailed structural, morphological and magnetic characterisation as a function of annealing temperature evidences that BFO forms in a narrow temperature window. Moreover, the observed ferromagnetism is mainly ascribed to nanostructuration and is also sensitive to the presence of secondary phases, such as Bi–Fe–O ternary phases or iron oxides. Future investigations will focus on further optimization of the bath and annealing conditions in order to synthesize BFO with higher purity. It is envisaged that the electrodeposition of BFO in the form of films with increased surface area will emerge as a subject of interest that will inspire both new fundamental physics and device applications.Acknowledgements
Financial support by the European Research Council (SPIN-PORICS 2014-Consolidator Grant, Agreement No. 648454), the Spanish Government (Project MAT2014-57960-C3-1-R and associated FEDER), and the Generalitat de Catalunya (2014-SGR-1015) and the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreements No. 642642 and 665919 is acknowledged. E. P. is grateful to MINECO for the “Ramón y Cajal” contract (RYC-2012-10839). J. F. acknowledges the “Juan de la Cierva” fellowship from MINECO (IJCI-2015-27030).References
- N. Izyumskaya, Y. Aliyov and H. Morkoç, Crit. Rev. Solid State Mater. Sci., 2009, 34, 89 CrossRef CAS
.
- G. Catalan and J. F. Scott, Adv. Mater., 2009, 21, 2463 CrossRef CAS
- B. Dieny, R. C. Sousa, J. Herault, C. Papusoi, G. Prenat, U. Ebels, D. Houssameddine, B. Rodmacq, S. Auffret, L. D. Buda-Prejbeanu, M. C. Cyrille, B. Delaet, O. Redon, C. Ducruet, J.-P. Nozieres and I. L. Prejbeanu, Internet J. Nanotechnol., 2010, 7, 591 CrossRef CAS
- J.-M. Hu, Z. Li, L.-Q. Chen and C.-W. Nan, Nat. Commun., 2011, 2, 553 CrossRef PubMed
- T. Zhao, A. Scholl, F. Zavaliche, K. Lee, M. Barry, A. Doran, M. P. Cruz, Y. H. Chu, C. Ederer, N. A. Spaldin, R. R. Das, D. M. Kim, S. H. Baek, C. B. Eom and R. Ramesh, Nat. Mater., 2006, 5, 823 CrossRef CAS PubMed
- R. Palai, R. S. Katiyar, H. Schmid, P. Tissot, S. J. Clark, J. Robertson, S. A. T. Redfern, G. Catalan and J. F. Scott, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 014110 CrossRef
- M. Valant, A. K. Axelsson and N. Alford, Chem. Mater., 2007, 19, 5431 CrossRef CAS
- S. M. Selbach, M. A. Einarsrud and T. Grande, Chem. Mater., 2009, 21, 169 CrossRef CAS
- G. D. Achenbach, W. J. James and R. Gerson, J. Am. Ceram. Soc., 1967, 50, 437 CrossRef CAS
- T. Liu, Y. Xu and J. Zhao, J. Am. Ceram. Soc., 2010, 93, 3637 CrossRef CAS
- H. Yang, Y. Q. Wang, H. Wang and Q. X. Jia, Appl. Phys. Lett., 2010, 96, 012909 CrossRef
- M. Ramazanoglu, M. Laver, W. Ratcliff II, S. M. Watson, W. C. Chen, A. Jackson, K. Kothapalli, S. Lee, S.-W. Cheong and V. Kiryukhin, Phys. Rev. Lett., 2011, 107, 207206 CrossRef CAS PubMed
- F. Z. Huang, Z. J. Wang, X. M. Lu, J. T. Zhang, K. L. Min, W. W. Lin, R. X. Ti, T. T. Xu, J. He, C. Yue and J. S. Zhu, Sci. Rep., 2013, 3, 2907 CrossRef PubMed
- H. Béa, M. Bibes, S. Petit, J. Kreisel and A. Barthélémy, Philos. Mag. Lett., 2007, 87, 165 CrossRef
- T. E. Quickel, L. T. Schelhas, R. A. Farrell, N. Petkov, V. H. Le and S. H. Tolbert, Nat. Commun., 2015, 6, 6562 CrossRef CAS PubMed
- S. Y. Yang, L. W. Martin, S. J. Byrnes, T. E. Conry, S. R. Basu, D. Paran, L. Reichertz, J. Ihlefeld, C. Adamo, A. Melville, Y. H. Chu, C. H. Yang, J. L. Musfeldt, D. G. Schlom, J. W. Ager and R. Ramesh, Appl. Phys. Lett., 2009, 95, 062909 CrossRef
- S. Y. Yang, J. Seidel, S. J. Byrnes, P. Shafer, C. H. Yang, M. D. Rossell, P. Yu, Y. H. Chu, J. F. Scott, J. W. Ager, L. W. Martin and R. Ramesh, Nat. Nanotechnol., 2010, 5, 143 CrossRef CAS PubMed
- M. Alexe and D. Hesse, Nat. Commun., 2011, 2, 256 CrossRef
- D. Huang, H. Deng, P. Yang and J. Chu, Mater. Lett., 2010, 64, 2233 CrossRef CAS
- X. Wang, Y. G. Zhang and Z. B. Wu, Mater. Lett., 2010, 64, 486 CrossRef CAS
- J. Wang, J. B. Neaton, H. Zheng, V. Nagarajan, S. B. Ogale, B. Liu, D. Viehland, V. Vaithyanathan, D. G. Schlom, U. V. Waghmare, N. A. Spaldin, K. M. Rabe, M. Wuttig and R. Ramesh, Science, 2003, 299, 1719 CrossRef CAS PubMed
- K. Jiang, J. J. Zhu, J. D. Wu, J. Sun, Z. G. Hu and J. H. Chu, ACS Appl. Mater. Interfaces, 2011, 3, 4844 CAS
- P. S. Plyaka and G. N. Tolmachev, Tech. Phys. Lett., 2010, 36, 192 CrossRef CAS
- K. Sarkar, S. Mukherjee and S. Mukherjee, Process. Appl. Ceram., 2015, 9, 53 CrossRef CAS
- A. Hardy, S. Gielis, H. Van den Rul, J. D'Haen, M. K. Van Bael and J. Mullens, J. Eur. Ceram. Soc., 2009, 29, 3007 CrossRef CAS
- N. Das, R. Majumdar, A. Sen and H. S. Maiti, Mater. Lett., 2007, 61, 2100 CrossRef CAS
- E. A. V. Ferri, I. A. Santos, E. Radovanovic, R. Bonzaninic and E. M. Girotto, J. Braz. Chem. Soc., 2008, 19, 1153 CrossRef CAS
- C. Ponzoni, M. Cannio, D. N. Boccaccini, C. R. H. Bahl, K. Agersted and C. Leonelli, Mater. Chem. Phys., 2015, 162, 69 CrossRef CAS
- S. Mohan and B. Subramanian, RSC Adv., 2013, 3, 23737 RSC
- F. Gao, Y. Yuan, K. F. Wang, X. Y. Chen, F. Chen, J.-M. Liu and Z. F. Ren, Appl. Phys. Lett., 2006, 89, 102506 CrossRef
- T. J. Park, Y. B. Mao and S. S. Wong, Chem. Commun., 2004, 23, 2708 RSC
- C. Chen, J. R. Cheng, S. W. Yu, L. J. Che and Z. Y. Meng, J. Cryst. Growth, 2006, 291, 135 CrossRef CAS
- S. D. Sartale, C. D. Lokhande, M. Giersig and V. Ganesan, J. Phys.: Condens. Matter, 2004, 16, 773 CrossRef CAS
- A. Quintana, A. Varea, M. Guerrero, S. Suriñach, M. D. Baró, J. Sort and E. Pellicer, Electrochim. Acta, 2015, 173, 705 CrossRef CAS
- T. Yousefi, A. N. Golikand, M. H. Mashhadizadeh and M. Aghazadeh, Curr. Appl. Phys., 2012, 12, 193 CrossRef
- P. N. Bartlett, S. L. Benjamin, C. H. de Groot, A. L. Hector, R. M. Huang, A. Jolleys, G. P. Kissling, W. Levason, S. J. Pearce, G. Reid and Y. D. Wang, Mater. Horiz., 2015, 2, 420 RSC
- T. P. Gujar, V. R. Shinde, S. S. Kulkarni, H. M. Pathan and C. D. Lokhande, Appl. Surf. Sci., 2006, 252, 3585 CrossRef CAS
- V. V. Jadhav, M. K. Zate, S. Liu, M. Naushad, R. S. Mane, K. N. Hui and S. H. Han, Appl. Nanosci., 2016, 6, 511 CrossRef CAS
- T. P. Gujar, V. R. Shinde, C. D. Lokhande, R. S. Mane and S. H. Han, Electrochem. Solid-State Lett., 2007, 10, D1 CrossRef CAS
- L. Lutterotti and P. Scardi, J. Appl. Crystallogr., 1990, 23, 246 CrossRef CAS
- MAUD (Materials Analysis Using Diffraction), http://maud.radiographema.com/.
- C. Q. Cui, S. P. Jiang and A. C. C. Tseung, J. Electrochem. Soc., 1991, 138, 94 CrossRef CAS
- M. K. Tripathi, D. K. Singh and V. B. Singh, J. Electrochem. Soc., 2015, 162, D87 CrossRef CAS
- B. D. Cullity, Elements of X-ray Diffraction, Addison-Wesley Publishing Company, Inc., USA, 1956 Search PubMed
- Z. Y. Sun, H. Q. Yuan, Z. M. Liu, B. X. Han and X. R. Zhang, Adv. Mater., 2005, 17, 2993 CrossRef CAS
- S. Sakurai, A. Namai, K. Hashimoto and S. Ohkoshi, J. Am. Chem. Soc., 2009, 131, 18299 CrossRef CAS PubMed
- R. Masrour, E. K. Hlil, M. Hamedoun, A. Benyoussef, O. Mounkachi and H. El Moussaou, J. Magn. Magn. Mater., 2015, 378, 37 CrossRef CAS
- H. Modarresi, V. Lazenka, E. Menéndez, M. Lorenz, M. Bisht, A. Volodin, C. Van Haesendonck, M. Grundmann, M. J. Van Bael, K. Temst and A. Vantomme, J. Phys. D: Appl. Phys., 2016, 49, 325302 CrossRef
- A. A. Zatsiupaa, L. A. Bashkirov, I. O. Troyanchuk, G. S. Petrov, A. I. Galyas, L. S. Lobanovsky and S. V. Truhanov, J. Solid State Chem., 2014, 212, 147 CrossRef
- Z. Irshad, Z. H. Shah, M. A. Rafiq and M. M. Hasan, J. Alloys Compd., 2015, 624, 131 CrossRef CAS
- R. Skomski and J. M. D. Coey, Permanent Magnetism, Institute of Physics Publishing, Bristol and Philadelphia, 1999 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Further details on the Rietveld refinements. See DOI: 10.1039/c7ra04375a |
| This journal is © The Royal Society of Chemistry 2017 |