
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ab initio mechanical and thermal properties of FeMnP1−xGax compounds as refrigerant for room-temperature magnetic refrigeration
Shuang Maa,
B. Wurentuyaab,
Xiaoxia Wuc,
Yongjing Jianga,
O. Tegusa,
Pengfei Guand and
B. Narsu *a
*a
aCollege of Physics and Electronic Information, Inner Mongolia Key Lab for Physics and Chemistry of Functional Materials, Inner Mongolia Normal University, Hohhot 010022, China. E-mail: nars@imnu.edu.cn
bShenyang National Laboratory of Materials Science, Institute of Metal Research, Chinese Academy of Sciences, Shenyang 110016, China
cDepartment of Physics, Inner Mongolia University of Science and Technology, Baotou 014022, China
dBeijing Computational Science Research Center, Beijing 100193, China
First published on 23rd May 2017
Abstract
Density functional theory was used to identify possible Fe2P-type giant magnetocaloric FeMnP1−xGax compounds. The calculated formation energies, elastic constants and phonon spectra confirm the energetic, mechanical and dynamical stability of hexagonal FeMnP1−xGax compounds in both the ferromagnetic and paramagnetic states. The predicted magnetic moment, elastic properties, and Curie temperature of FeMnP0.67Ga0.33 are close to those obtained for FeMnP0.67Ge0.33 compounds using the same calculation scheme. The entropy changes and latent heat of FeMnP0.67Ga0.33 are similar with those of FeMnP0.67Ge0.33. The electronic density of states and charge density analysis indicate that the FeMnP1−xGax compounds have similar electronic structures to those of FeMnP1−xGex. These results predict that FeMnP1−xGax is a possible candidate refrigerant for room-temperature magnetic refrigeration.
Introduction
Hexagonal Fe2P-type transition metal FeMn-based magnetocaloric materials (FeMnP1−xTx, T = As, Si, Ge) are driving tremendous research interest because of their promising application in solid-state refrigeration as energy efficient, environment friendly alternative to the traditional gas-compressor based cooling scheme.1–9 This family of materials exhibit a first-order magnetoelastic transition close to room temperature.10–12 Significant exchange striction, in which the hexagonal lattice parameter c decreases and a increases with a very small volume change, is observed during the paramagnetic (PM) to ferromagnetic (FM) transition.10,13 This leads to large local strain in the polycrystalline samples, and therefore good mechanical stability across the phase transition becomes very important for FeMn-based giant magnetocaloric (GMC) materials because they work under repeated magnetic and thermal cycles.5,6,10,14To obtain materials with good performance in magnetic refrigeration, extensive experimental investigation of FeMnP1−xSix compounds has been performed.8,9,11–17 The thermal hysteresis decreases to less than 1 K and the isothermal entropy change |Siso| increases to ∼9.0 J kg−1 K−1 around the Curie temperature,12 which can be continuously tailored.4,5,16,18 However, the weak mechanical stability across the phase transition restricts the applicability of FeMnP1−xSix compounds in practical devices. One way to optimize the mechanical stability of FeMnP1−xSix compounds is to dope them with interstitial B atoms.10,12,14,18–20 Another viable way to improve the mechanical properties of these compounds is to replace Si atoms with other elements to adjust the bonding between atoms.21,22 Si and Al have been suggested to be the most ideal substitutes for the toxic As element in MnFeP1−xAsx compounds,23,24 although experimentalists have failed to alloy Al into hexagonal Fe2P-type magnetocaloric materials. From the viewpoint of alloying, Ga is a possible candidate because it has a similar outermost electronic structure and atomic size to Ge. However, no theoretical investigations have been performed to determine the structural, magnetic, and electronic properties of FeMnP1−xGax and FeMnP1−xGex compounds.
Here, we report the possibility of developing GMC compounds by alloying Ga into hexagonal Fe2P-type compounds by quantum mechanical calculations. The static and dynamic stabilities of FeMnP1−xGax compounds in different states are verified by total energy and elastic constant calculations. In addition, the mechanical properties of FeMnP1−xTx (T = Ga, Ge) in different magnetic states are evaluated. The thermodynamic properties of FeMnP1−xGax compounds were calculated and they are compared with those of FeMnP1−xGex compounds to determine the possibility of using them in magnetic refrigeration.
Computational details
The Fe2P-type hexagonal structure has the symmetry of space group P6![[( with combining macron]](https://www.rsc.org/images/entities/char_0028_0304.gif) )2m, in which three atoms occupy four different crystal sites. Usually, the Fe atom prefers the 3f (x1, 0, 0) site and the Mn atom occupies the 3g (x2, 0, 0.5) site.25 There are two different atomic sites for the metalloid atoms, namely, 2c (0.333, 0.667, 0) and 1b (0, 0, 0.5). Local occupation disorder of P and Si atoms at 2c and 1b sites in FeMnP1−xSix compounds was observed in the experiment.26 However, site occupation order between 2c and 1b sites is expected for FeMnP1−xGex and FeMnP1−xGax compounds because of the larger atomic radii of these dopant atoms than P and Si atoms. Therefore, we chose the occupation of Ga (or Ge) atom at the 1b site for x = 0.33 and at the 2c for x = 0.67. The FM state was calculated self consistently with the primitive cell (half of the cell shown in Fig. 1), while the PM state was modeled by an antiferromagnetically ordered 1 × 1 × 2 supercell structure (Fig. 1). Antiferromagnetic model is usually used to simulate the paramagnetic state of transition metals and their alloys, and it can practically capture the physics observed in experiment.27–29 The magnetic moments of the transition metal atoms were set to be parallel in the primitive cells and an antiparallel alignment of magnetic moments between primitive cells was established to form the antiferromagnetic (AFM) order. In this case, the long-range FM order along the c axis is broken, but short-range magnetic order still exits,30 so this supercell structure models the magnetic state just above the Curie temperature.
)2m, in which three atoms occupy four different crystal sites. Usually, the Fe atom prefers the 3f (x1, 0, 0) site and the Mn atom occupies the 3g (x2, 0, 0.5) site.25 There are two different atomic sites for the metalloid atoms, namely, 2c (0.333, 0.667, 0) and 1b (0, 0, 0.5). Local occupation disorder of P and Si atoms at 2c and 1b sites in FeMnP1−xSix compounds was observed in the experiment.26 However, site occupation order between 2c and 1b sites is expected for FeMnP1−xGex and FeMnP1−xGax compounds because of the larger atomic radii of these dopant atoms than P and Si atoms. Therefore, we chose the occupation of Ga (or Ge) atom at the 1b site for x = 0.33 and at the 2c for x = 0.67. The FM state was calculated self consistently with the primitive cell (half of the cell shown in Fig. 1), while the PM state was modeled by an antiferromagnetically ordered 1 × 1 × 2 supercell structure (Fig. 1). Antiferromagnetic model is usually used to simulate the paramagnetic state of transition metals and their alloys, and it can practically capture the physics observed in experiment.27–29 The magnetic moments of the transition metal atoms were set to be parallel in the primitive cells and an antiparallel alignment of magnetic moments between primitive cells was established to form the antiferromagnetic (AFM) order. In this case, the long-range FM order along the c axis is broken, but short-range magnetic order still exits,30 so this supercell structure models the magnetic state just above the Curie temperature.
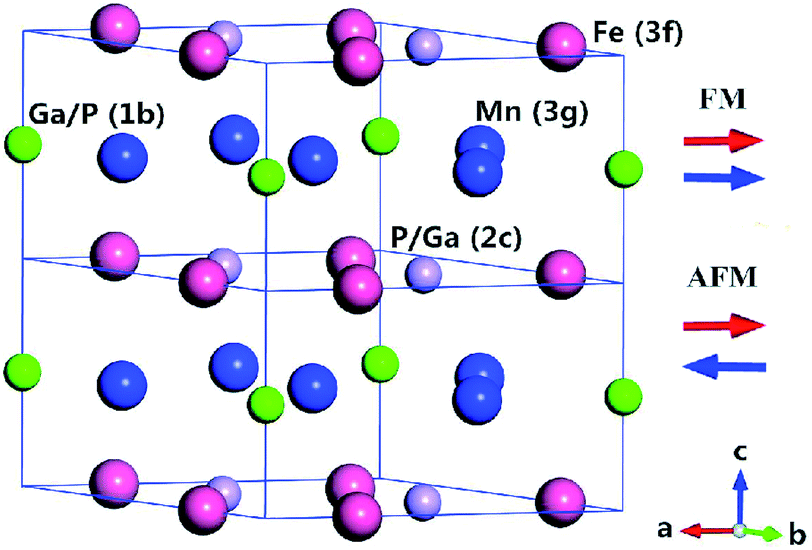 | ||
| Fig. 1 Cell structure of hexagonal phase FeMnP1−xGax compounds. For the case of x = 0.33, Ga occupies the 1b site, and for x = 0.67, Ga occupies the 2c site. For the FM state, the magnetic moments of the unit cells are aligned parallel, and for the AFM state the magnetic moments in the upper and lower unit cells are aligned antiparallel. | ||
The total electronic energy was calculated by the projector augmented wave method31 in the framework of density functional theory,32,33 as implemented in the Vienna ab initio simulation package (VASP).34,35 The exchange correlation effect was evaluated by the generalized gradient approximation parameterized by Perdew, Burke, and Ernzerhof.36 The accuracy of PBE functional for FeMn based Fe2P type alloys has previously been confirmed.17,51 The scalar relativistic scheme was used and the spin–orbit coupling effect was omitted. Mn (3d64s1), Fe (3d74s1), P (3s23p3), Ga (3d104s24p1), and Ge (3d104s24p2) electrons were treated as valance states.37 The cutoff energy of the plane wave basis was set to 530 eV, and the convergence criterion of self-consistent electronic relaxation was set to 10−5 eV per atom. For ionic relaxation, the force convergence criterion of 0.01 eV Å−1 was used. The Monkhorst–Pack scheme38 was used for Brillouin zone sampling, with an 11 × 11 × 17 k-point mesh for the FM state and an 11 × 11 × 9 k-point mesh for the AFM state. With these computational settings, the total energies of compounds with different magnetic states were calculated for different volume V and c/a values. The equation of state was fitted with the Murnaghan equation,39 and then the bulk modulus and the equilibrium volume were obtained. The five independent elastic constants were calculated by the energy-strain method used in ref. 40, with isochoric strain δ of 0.00 to ±0.05. The phonon spectra were calculated by phonopy code.41 For FM and AFM states, 72 atom and 144 atom 2 × 2 × 2 supercells were used, respectively. An accurate prediction of the thermodynamic properties should be based on the quasiharmonic approximation (QHA)42 with a direct ab initio phonon spectrum calculation.41 However, this is a very difficult task for multicomponent compounds with large unit cells. An alternative method is based on Debye theory.43 This method has been used to calculate the thermodynamic properties of the FeMnP1−xSix system44 and gives good agreement with the properties obtained from the QHA.26
Therefore, the thermodynamic properties of the FeMnP1−xGax compounds were calculated by the quasiharmonic Debye–Slater model implemented in the Gibbs2 code.45
Results and discussion
Structure and stability
FeMnP1−xTx (T = Si, Ge, As) compounds crystallize in a hexagonal structure with space group P6)2m. They exhibit a first-order FM to PM transition at the Curie temperature. Lattice constant a decreases and c increases with almost zero change in the volume during the phase transition. For FeMnP0.8Ge0.2 compounds, the experimentally observed lattice constants a and c are 6.18 and 3.31 Å for the FM state and 6.06 and 3.46 Å for the PM state, respectively. The calculated a and c values for 33 atom% Ge compounds are very close to the experimental results, as listed in Table 1. A small increase in the cell volume is observed in FeMnP0.8Ge0.2 compounds46 across the FM–PM transition, although the compounds with the AFM configuration have smaller cell volumes than those with FM order. This deviation can be understood by the anharmonic effect. Taking into account thermal expansion, the cell volume of FeMnP0.67Ge0.33 calculated with AFM order (i.e., the PM state) will increase to 36.93 Å3, which is larger than the value of 36.51 Å3 obtained for FM order at 0 K. The total and local magnetic moments of FM FeMnP0.67Ge0.33 obtained from the calculation are very close to those from experiments (Table 1). Unfortunately, no experimental local magnetic moments of PM FeMnP1−xGex have been reported, and thus quenching of the local moment at 3f (Fe) cannot be verified. DFT level calculations of PM state in Fe2P21 and FeMnP1−xSix (ref. 8) indicate that the magnetic moment at 3f sites will disappear in the PM state. Hence, our results are in agreement with those calculations. A recent experimental study of FeMnP0.5Si0.5 compounds suggested that DFT calculations underestimate the magnetic moment of PM compounds at the 3f site.19 However, we attribute this discrepancy between theory and experiment to local disorder of P and Si atoms in FeMnP0.5Si0.5 compounds. Because Fe moment quenching at 3f site is not observed in the AFM state for FeMnP0.33Ge0.67 compounds (Table 1), there is a strong local environment dependence of the magnetic moment at the 3f site in this class of compounds. The significantly lower, but not zero, Fe moment at 3f site in PM FeMnP0.5Si0.5 compounds19 can be easily understood. The predicted relative positions of 3f and 3g sites (x1 and x2) for FeMnP0.67Ge0.33 are significantly different from the experimental values shown in Table 1, which can be ascribed to Fe/Mn disorder and the lower concentration of Ge in Mn1.1Fe0.9P0.8Ge0.2 compounds.
| Mag. Ord. | a (Å) | c (Å) | V (Å3 per f.u.) | E0 (eV per f.u.) | μtot (μB per f.u.) | μFe (μB) | μMn (μB) | x1 | x2 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Experimental results for Mn1.1Fe0.9P0.8Ge0.2 compounds. FM are the values at 10 K and PM are those 295 K from ref. 46 and 47. | |||||||||||
| FeMnP1−xTx | |||||||||||
| x = 0.33 | Ga | FM | 6.14 | 3.36 | 36.61 | −22.74 | 4.38 | 1.49 | 2.96 | 0.274 | 0.588 |
| AFM | 5.83 | 3.67 | 35.90 | −22.68 | 0.00 | 0.00 | 2.83 | 0.262 | 0.570 | ||
| Ge | FM | 6.16 | 3.33 | 36.51 | −23.46 | 4.39 | 1.46 | 2.99 | 0.271 | 0.591 | |
| AFM | 5.86 | 3.61 | 35.71 | −23.38 | 0.00 | 0.00 | 2.80 | 0.262 | 0.575 | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||||||
| FeMnP0.8Ge0.2a | |||||||||||
| FM (10 K) | 6.18 | 3.31 | 36.43 | — | — | 1.71 | 3.01 | 0.2558 | 0.5956 | ||
| PM (295 K) | 6.06 | 3.46 | 36.70 | — | — | — | — | 0.2527 | 0.5916 | ||
| x = 0.67 | Ga | FM | 6.56 | 3.05 | 37.83 | −21.69 | 4.63 | 1.68 | 3.03 | 0.252 | 0.623 |
| AFM | 6.66 | 2.91 | 37.27 | −21.52 | 0.00 | 1.29 | 2.99 | 0.256 | 0.622 | ||
| Ge | FM | 6.42 | 3.15 | 37.39 | −22.99 | 4.44 | 1.55 | 2.98 | 0.250 | 0.612 | |
| AFM | 6.47 | 3.10 | 37.51 | −22.70 | 0.00 | 1.49 | 2.95 | 0.251 | 0.614 | ||
For the FeMnP1−xGax compounds considered in this study, the structural and magnetic parameters are very close to those obtained for FeMnP1−xGex compounds. This is in accordance with our expectation because of the similar atomic sizes and electronic configurations of Ga and Ge, with the only difference between these atoms being that Ga has one less p electron. Interestingly, a decrease in the c parameter and expansion is observed across the FM–PM transition in both FeMnP0.33Ge0.67 and FeMnP0.33Ga0.67 compounds. This has not been reported in experiment and should be further discussed in detail.
The atomic distances between the four components of the compounds were measured from the fully relaxed cell structures with different magnetic order, and they are listed in Table 2. Experimental observation shows that the interplane metal-to-metal and metal-to-metalloid distances D(Fe–Mn), D(Fe–Ge), and D(Mn–P) increase while the intraplane atomic distances D(Fe–P) and D(Mn–Ge) decrease across the FM–PM phase transition. Ab initio calculation and AFM modeling of the FeMnP0.67Ge0.33 compounds show the same trend for variation of the atomic distances with the phase transition. These conclusions are still valid even though the volume expansion effect was taken into account. For instance, the predicted Curie temperature Tc is 590 K and the corresponding coefficient of thermal expansion (CTE) is approximately 3.6 × 10−5 K−1 (see Fig. 5). Considering thermal expansion, D(Fe–P) at 590 K is 2.240 Å, which is still smaller than D(Fe–P) = 2.271 Å of the FM state at 0 K. Similarly, the relaxed D(Fe–Mn) of the AFM FeMnP0.67Ge0.33 compounds at 590 K will be larger than that obtained for the FM state at 0 K, even though the calculated D(Fe–Mn) for AFM is smaller at 0 K. The predicted evolution of the atomic distances of the FeMnP1−xGax compounds shows almost the same features as those of FeMnP1−xGex. Interestingly, FeMnP0.33Ga0.67 and FeMnP0.33Ge0.67 compounds show the opposite atomic distance evolution trend during the phase transition. This result can be attributed to c decreasing with the phase transition.
| Mag. Ord. | D(Fe–Mn) | D(Fe–P) | D(Fe–Ga/Ge) | D(Mn–P) | D(Mn–Ga/Ge) | D(P–Ga/Ge) | ||
|---|---|---|---|---|---|---|---|---|
| a Experimental results for Mn1.1Fe0.9P0.8Ge0.2 compounds. FM are the values at 10 K and PM are those 295 K from ref. 46 and 47. | ||||||||
| FeMnP1−xTx | ||||||||
| x = 0.33 | Ga | FM | 2.561 | 2.254 | 2.376 | 2.502 | 2.530 | 3.925 |
| AFM | 2.565 | 2.180 | 2.384 | 2.520 | 2.505 | 3.830 | ||
| Ge | FM | 2.581 | 2.271 | 2.358 | 2.500 | 2.522 | 3.927 | |
| AFM | 2.575 | 2.192 | 2.367 | 2.511 | 2.486 | 3.832 | ||
|
||||||||
| FeMnP0.8Ge0.2a | ||||||||
| FM (10 K) | 2.672 | 2.358 | 2.2874 | 2.5026 | 2.499 | — | ||
| PM (295 K) | 2.686 | 2.311 | 2.3039 | 2.5225 | 2.476 | — | ||
| x = 0.67 | Ga | FM | 2.662 | 2.247 | 2.496 | 2.473 | 2.560 | 4.081 |
| AFM | 2.617 | 2.236 | 2.519 | 2.518 | 2.531 | 4.094 | ||
| Ge | FM | 2.694 | 2.246 | 2.451 | 2.491 | 2.534 | 4.024 | |
| AFM | 2.622 | 2.244 | 2.476 | 2.501 | 2.523 | 4.011 | ||
To evaluate the stability of the predicted FeMnP1−xGax compounds, the formation energies were calculated by
| ΔEFeMnP1−xGax = EFeMnP1−xGax − EFe − EMn − (1 − x)EP − xEGa |
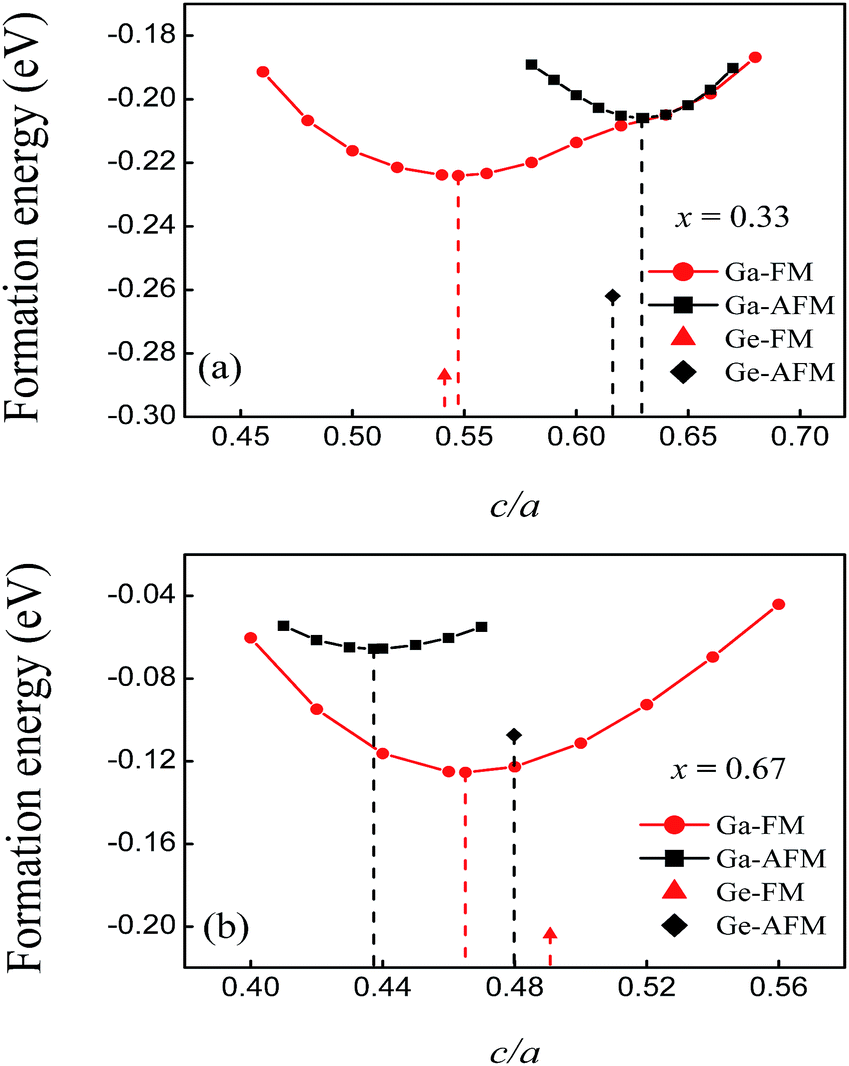 | ||
| Fig. 2 c/a dependence of the formation energies of FeMnP1−xGax compounds in different magnetic states. The formation energies of the FeMnP1−xGex compounds at equilibrium are also shown for comparison. (a) x = 0.33. (b) x = 0.67. | ||
The negative formation energies of all of the compounds in the different magnetic states indicate that alloying of these elements is an exothermic reaction. The formation energies of the FM and AFM states of FeMnP0.67Ga0.33 are −0.224 and −0.206 eV per atom, respectively. These values are close to those obtained for the FeMnP0.67Ge0.33 compounds, as shown in Fig. 2(a). The absolute values of the formation energies of FeMnP0.33Ga0.67 are smaller than those of FeMnP0.67Ga0.33. The formation energy curves of the FM and AFM states of FeMnP0.67Ga0.33 overlap around c/a of the equilibrium AFM state, which suggests that the FM to AFM transition could occur by simple axial elongation. The curves of the FeMnP0.33Ga0.67 compounds show different characteristics. According to the analysis presented above, the predicted FeMnP1−xGax compounds are energetically stable.
Mechanical properties
Magnetocaloric materials work in a repeated thermal and magnetization cycle, so good mechanical stability around their phase transition temperature is required. In addition, for crystals with hexagonal symmetry, a dynamical stability condition of C44 > 0, C11 > C12, C11C33 > (C13)2, and C33 (C11 + C12) > 2(C13)2 must be satisfied.41 The calculated elastic constants of the FeMnP1−xGax and FeMnP1−xGex compounds are listed in Table 3. According to the elastic constants, the predicted FeMnP1−xGax compounds are mechanically stable. In combination with the formation energy analysis, we can conclude that the hexagonal phases of the FeMnP1−xGax compounds are stable in both the FM and AFM states. Namely, this class of compounds could exhibit a FM to PM transition similar to those of FeMnP1−xGex and FeMnP1−xSix compounds.| FeMnP1−xTx | Mag. Ord. | C11 | C12 | C13 | C33 | C44 | B | R | Cs | C66 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| x = 0.33 | Ga | FM | 219.3 | 68.96 | 114.5 | 150.2 | 107.7 | 130.7 | −0.18 | 130.6 | 75.2 |
| AFM | 240.6 | 94.74 | 88.86 | 227.1 | 95.1 | 139.1 | −0.04 | 434.2 | 72.9 | ||
| Ge | FM | 282.4 | 108.1 | 129.0 | 197.0 | 112.4 | 162.5 | −0.24 | 268.6 | 87.2 | |
| AFM | 273.5 | 103.2 | 102.8 | 260.1 | 104.5 | 158.2 | −0.03 | 484.7 | 85.3 | ||
| x = 0.67 | Ga | FM | 216.0 | 79.61 | 76.97 | 169.6 | 90.3 | 117.1 | −0.15 | 327.0 | 68.2 |
| AFM | 214.0 | 97.57 | 87.06 | 152.3 | 97.92 | 120.5 | −0.27 | 267.9 | 58.2 | ||
| Ge | FM | 265.4 | 95.28 | 105.5 | 198.2 | 108.6 | 146.9 | −0.17 | 335.3 | 85.1 | |
| AFM | 229.2 | 91.53 | 117.7 | 168.9 | 104.1 | 141.0 | −0.18 | 187.9 | 68.8 | ||
For both the FeMnP0.67Ga0.33 and FeMnP0.67Ge0.33 compounds, there are marginal decreases in the bulk modulus B and shear moduli C44 and C66 with the FM to AFM transition. However, there are large differences in the Cs and c axial compressibility values of the hexagonal crystals of both the FeMnP0.67Ga0.33 and FeMnP0.67Ge0.33 compounds in the FM and AFM states, suggesting that the magnetic order has a strong effect on the crystal structure (c/a) of this class of compounds. For FeMnP0.33Ga0.67 and FeMnP0.33Ge0.67, the elastic constants show the opposite trend for the FM and AFM states compared with those for the compounds with lower Ga and Ge concentration. This can be ascribed to the opposite trends of the lattice parameters during the phase transition.
Exchange strain (lattice parameter changes) is always accompanied by the FM–PM phase transition. Therefore, magnetocaloric materials should be sufficiently mechanically stable to bear a large internal strain. The polycrystalline bulk and shear moduli obtained by different averaging schemes,40,41 Young's modulus E, Poisson's ratio v, the Debye temperature θD, and the Grüneisen constant γ are listed in Table 4. According to the Pugh criteria,48 the FeMnP1−xGax and FeMnP1−xGex compounds with x = 0.33 are ductile (B/G > 1.75 and v > 0.26) in the FM state and brittle (B/G < 1.75 and v < 0.26) in the AFM state. The situation is reversed for x = 0.67, where the FM state is brittle and AFM state is ductile.
| FeMnP1−xTx | Mag. Ord. | BV | BR | GV | GR | AVR | GH | BH | BH/GH | E | v | θD | γ | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| x = 0.33 | Ga | FM | 131.6 | 130.7 | 77.48 | 54.68 | 0.17 | 66.08 | 131.2 | 1.985 | 169.7 | 0.284 | 441.7 | 1.82 |
| AFM | 139.3 | 139.1 | 81.69 | 80.27 | 0.01 | 80.98 | 139.2 | 1.718 | 203.5 | 0.256 | 486.5 | 1.83 | ||
| Ge | FM | 166.0 | 162.5 | 88.77 | 78.68 | 0.06 | 83.72 | 164.3 | 1.962 | 214.7 | 0.282 | 493.6 | 1.85 | |
| AFM | 158.3 | 158.2 | 92.08 | 90.96 | 0.01 | 91.52 | 158.3 | 1.729 | 230.2 | 0.258 | 515.3 | 1.85 | ||
| x = 0.67 | Ga | FM | 118.7 | 117.1 | 74.31 | 71.36 | 0.02 | 72.83 | 117.9 | 1.692 | 182.0 | 0.244 | 445.0 | 1.85 |
| AFM | 124.9 | 120.5 | 71.83 | 64.11 | 0.05 | 67.74 | 122.7 | 1.811 | 171.6 | 0.267 | 427.0 | 1.83 | ||
| Ge | FM | 149.0 | 146.9 | 88.63 | 83.21 | 0.03 | 85.92 | 148.0 | 1.722 | 216.0 | 0.257 | 480.3 | 2.22 | |
| AFM | 142.3 | 141.0 | 75.43 | 62.10 | 0.10 | 68.77 | 141.7 | 2.060 | 177.6 | 0.291 | 432.2 | 1.98 | ||
The cases investigated in this study are the two extreme cases where Ga (or Ge) only occupies the 1b or 2c site. According to the current result, we speculate that site occupation disorder of P and Ga/Ge elements may increase the ductility of compounds with x = 0.33 in the AFM state.
Phonon spectra
The phonon band structures and densities of states of FeMnP0.67Ga0.33 compounds in different magnetic states are shown in Fig. 3. No imaginary frequency was observed in phonon modes for both FM (Fig. 3(a)) and AFM (Fig. 3(b)) states, confirming the dynamical stability of the compounds in different magnetic state. Comparing the phonon DOS of compounds in different magnetic state, the acoustic modes in the low frequency (0–2 THz) part are similar. The optical modes are reasonably different for FM and AFM states, especially the optical mode between frequency range 9.5–14 THz. In the moderate temperature, the difference between low frequency part of optical phonon modes for FM and AFM states determines the evolution of thermal properties of the compounds across FM–PM (AFM) phase transition. From Fig. 3(c) one can find that the phonon of AFM state is a bit softer than that of FM state for frequency larger than 2 THz. A small difference in optical modes of FM and AFM states for frequency above 2 THz, thus a small difference in the vibrational entropy and the heat capacity Cv is expected in the isothermal process. The quantitative analysis on thermal properties of the compounds is given in next section.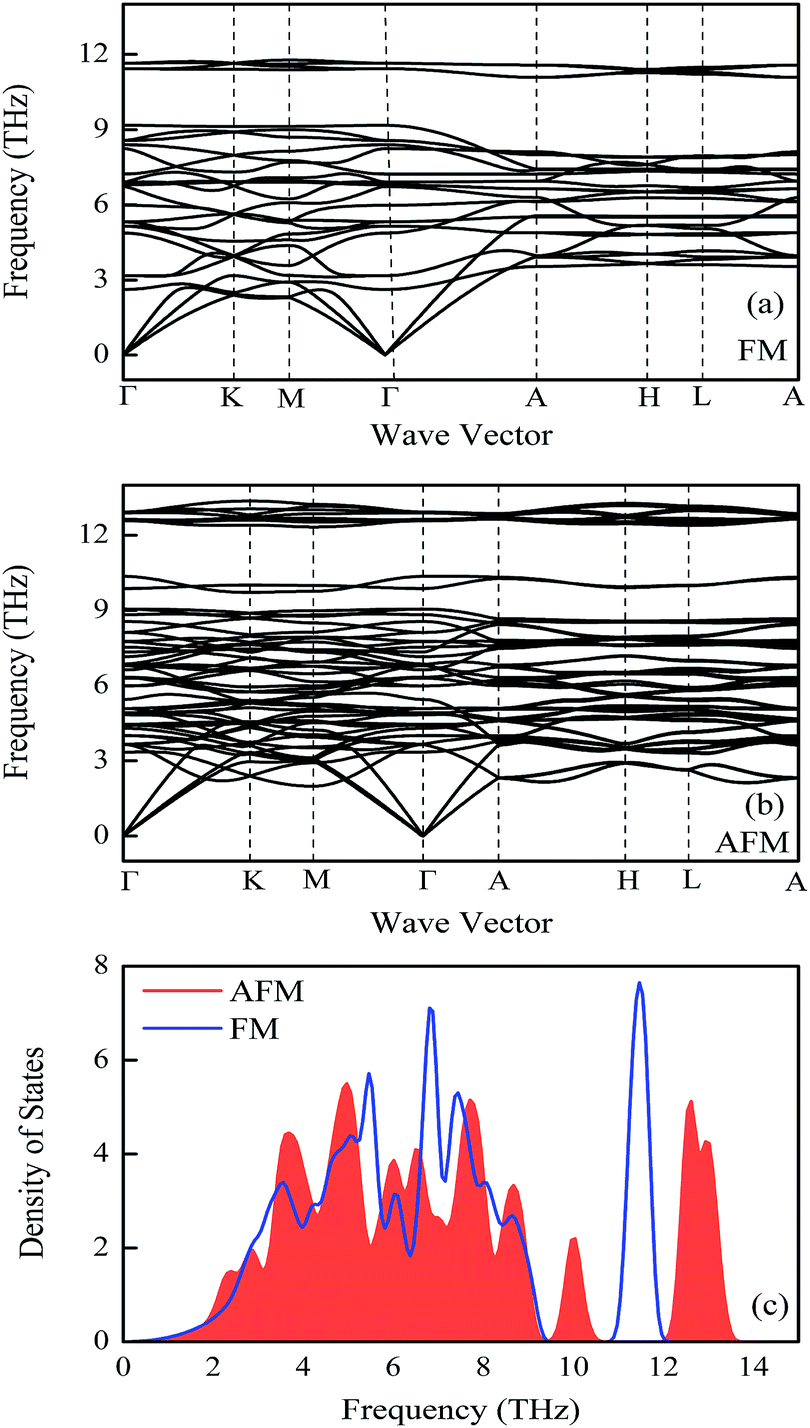 | ||
| Fig. 3 Phonon spectra of FeMnP0.67Ga0.33 compounds. (a) Ferromagnetic state, (b) antiferromagnetic state. (c) Phonon densities of states. | ||
Electronic structure
To understand the structural and magnetic properties calculated above, the electronic density of states (DOS) and charge density difference (CDD) were analyzed. In the total DOS (TDOS) of FeMnP1−xGax and FeMnP1−xGex compounds with different magnetic states shown in Fig. 4, the overall shapes of the TDOS are similar for FeMnP1−xGax and FeMnP1−xGex compounds, although the Fermi level of FeMnP1−xGax moves towards a lower energy state because Ga has one less electron than Ge. This leads to a larger DOS value at the Fermi level for FeMnP1−xGax compounds, and thus results in smaller shear moduli compared with those of FeMnP1−xGex (Table 3).49 According to the force theorem,50 alloy system having larger density of state at Fermi level is more sensitive to lattice strain and endows the system with smaller elastic modulus. Comparing TDOS of FeMnP0.67Ga0.33 in different magnetic states, one can find that AFM state always has larger DOS value at Fermi level than that of FM state, namely AFM state should have smaller shear moduli. This qualitative analysis is in line with the self consistent results of shear moduli C44 and C66 for FM and AFM states.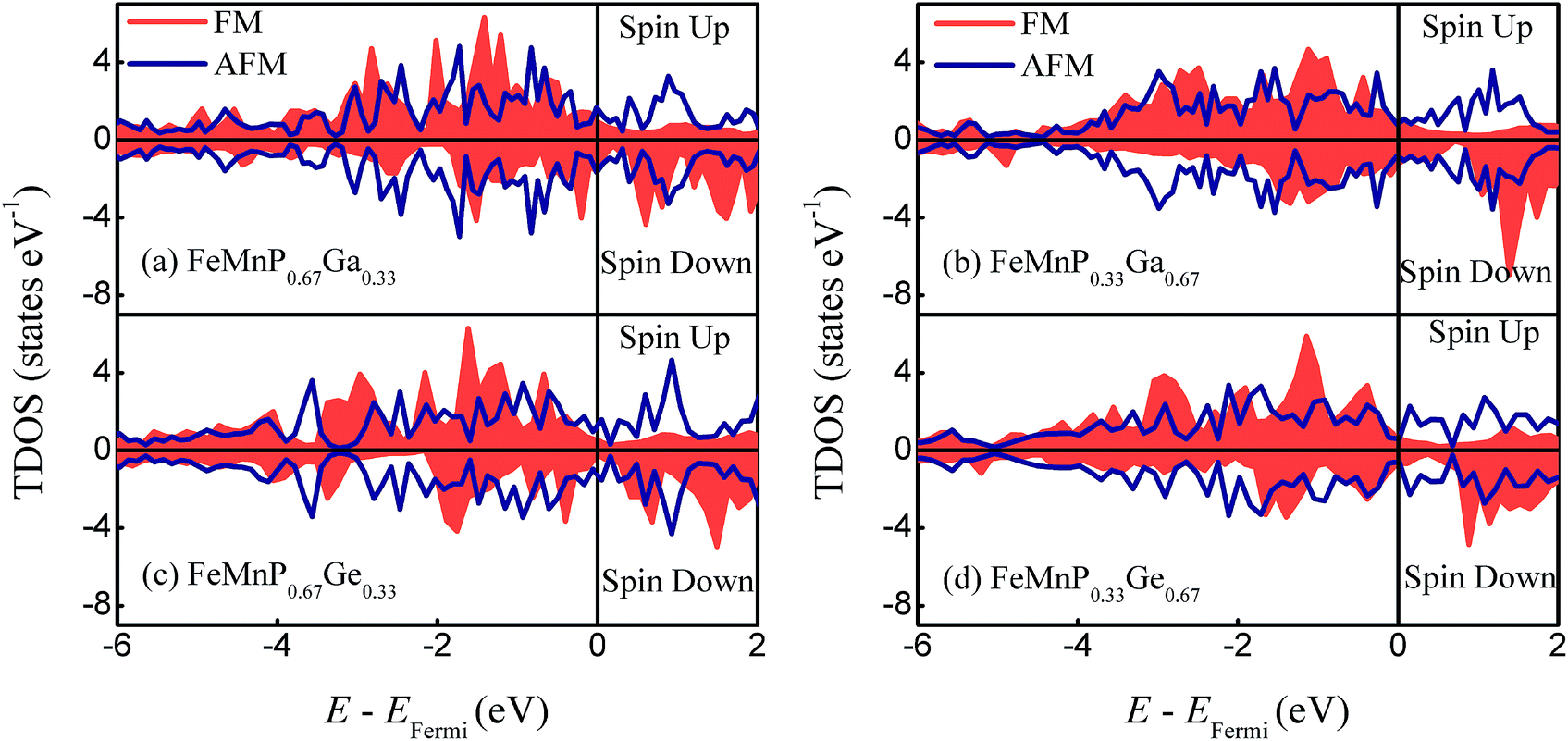 | ||
| Fig. 4 Calculated TDOS of the FeMnP1−xGax and FeMnP1−xGex compounds. (a) FeMnP0.67Ga0.33, (b) FeMnP0.33Ga0.67. (c) FeMnP0.67Ge0.33. (d) FeMnP0.33Ge0.67. | ||
The two spin channels are symmetric for the AFM state of all of the compounds, indicating disappearance of the overall magnetic moment of the AFM cell. Because of the breaking symmetry, several bands split and new states appear close to the Fermi level as the compounds undergo the FM to PM transition. Comparing the electronic DOS of the AFM and FM states, significant electronic redistribution occurs during the phase transition.
To estimate the evolution of the bonding characteristics and local magnetic moment with the phase transition, the site-projected density of states (pDOS) of FeMnP1−xGax are plotted in Fig. 5. In the FM phase, Mn and Fe share almost all of the pDOS peak for both x = 0.33 and 0.67, that means there is strong covalent-like bonding between Fe and Mn. As the FM order breaks, the pDOS behaves differently. For FeMnP0.67Ga0.33, the spin-up and spin-down channels of Fe become symmetric, indicating disappearance of the local magnetic moment at the 3f Fe site. For FeMnP0.33Ga0.67, the pDOS of Fe is still asymmetric and has a certain local moment. While the bonding characteristics between Mn and Fe significantly change during the phase transition, only the spin-down channels of Fe and Mn have obvious overlap in the pDOS peaks.
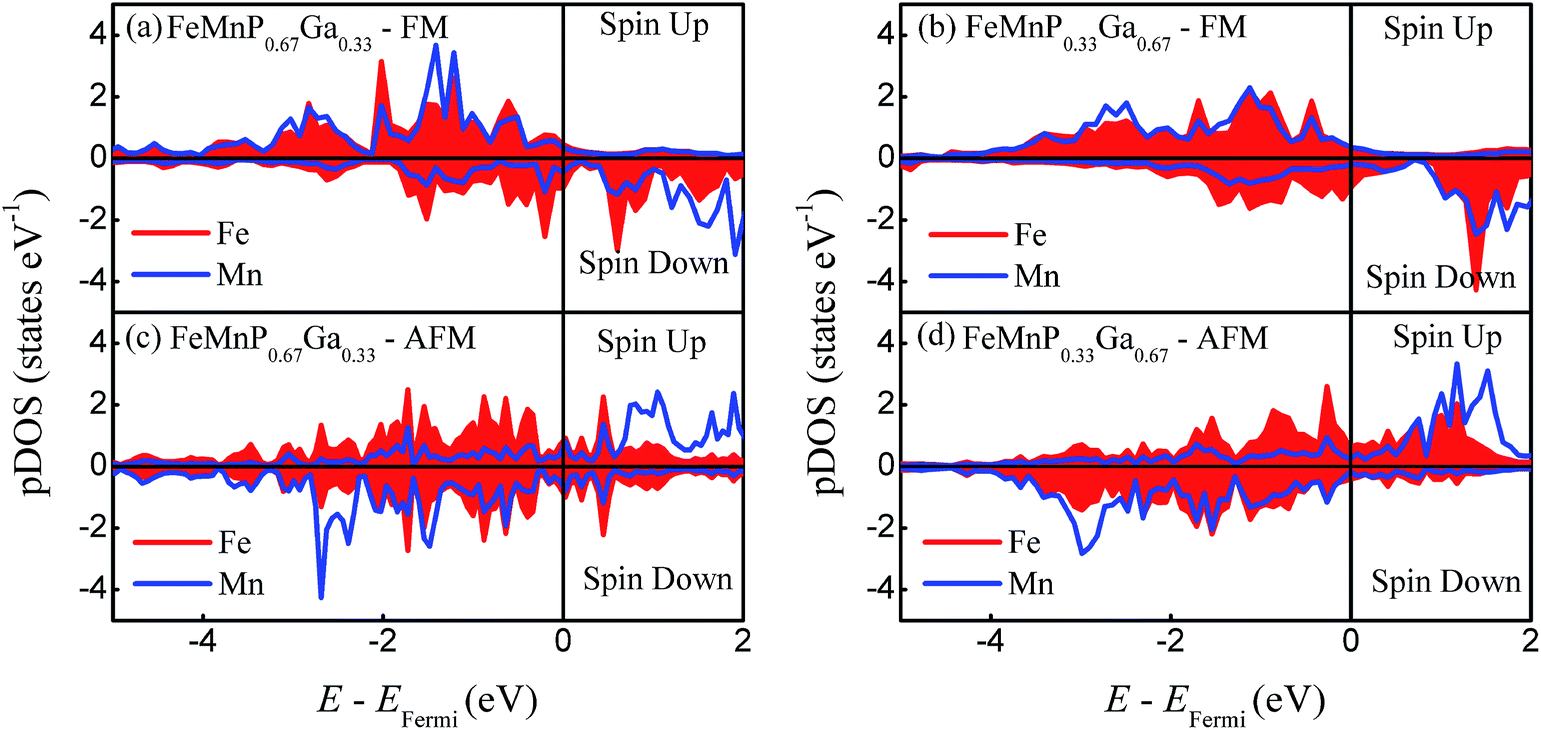 | ||
| Fig. 5 Calculated pDOS of the FeMnP1−xGax compounds. (a) Ferromagnetic state of FeMnP0.67Ga0.33, (b) ferromagnetic state of FeMnP0.33Ga0.67. (c) Antiferromagnetic state of FeMnP0.67Ga0.33. (d) Antiferromagnetic state of FeMnP0.33Ga0.67. | ||
Fig. 6 shows the charge redistribution characteristics with the FM–PM transition. Comparing the CDD in the ac plane of FeMnP0.67Ga0.33 in the FM (Fig. 6(a)) and AFM (Fig. 6(c)) states, there is significant redistribution of electrons around Fe. However, no significant electronic redistribution close to Fe is detected for FeMnP0.33Ga0.67. These results indicate that occupation of P atoms on different sites is crucially important for the magnetic properties of this family of compounds. Surprisingly, the electronic distribution between Fe(↓) and Mn atoms with different magnetic polarization is symmetric for AFM FeMnP0.67Ga0.33 (Fig. 6(c)). However, asymmetric electronic distribution between Fe(↓) atoms and Mn(↑) and Mn(↓) atoms is observed for FeMnP0.33Ga0.67, where the magnetic moment of 3f Fe survives during the phase transition. There is more electron density between Fe(↓) and Mn(↓) atoms than between Fe(↓) and Mn(↑) atoms. This picture indicates that the 3f-Fe site of FeMnP0.67Ga0.33 cannot show spin polarization in the AFM states.
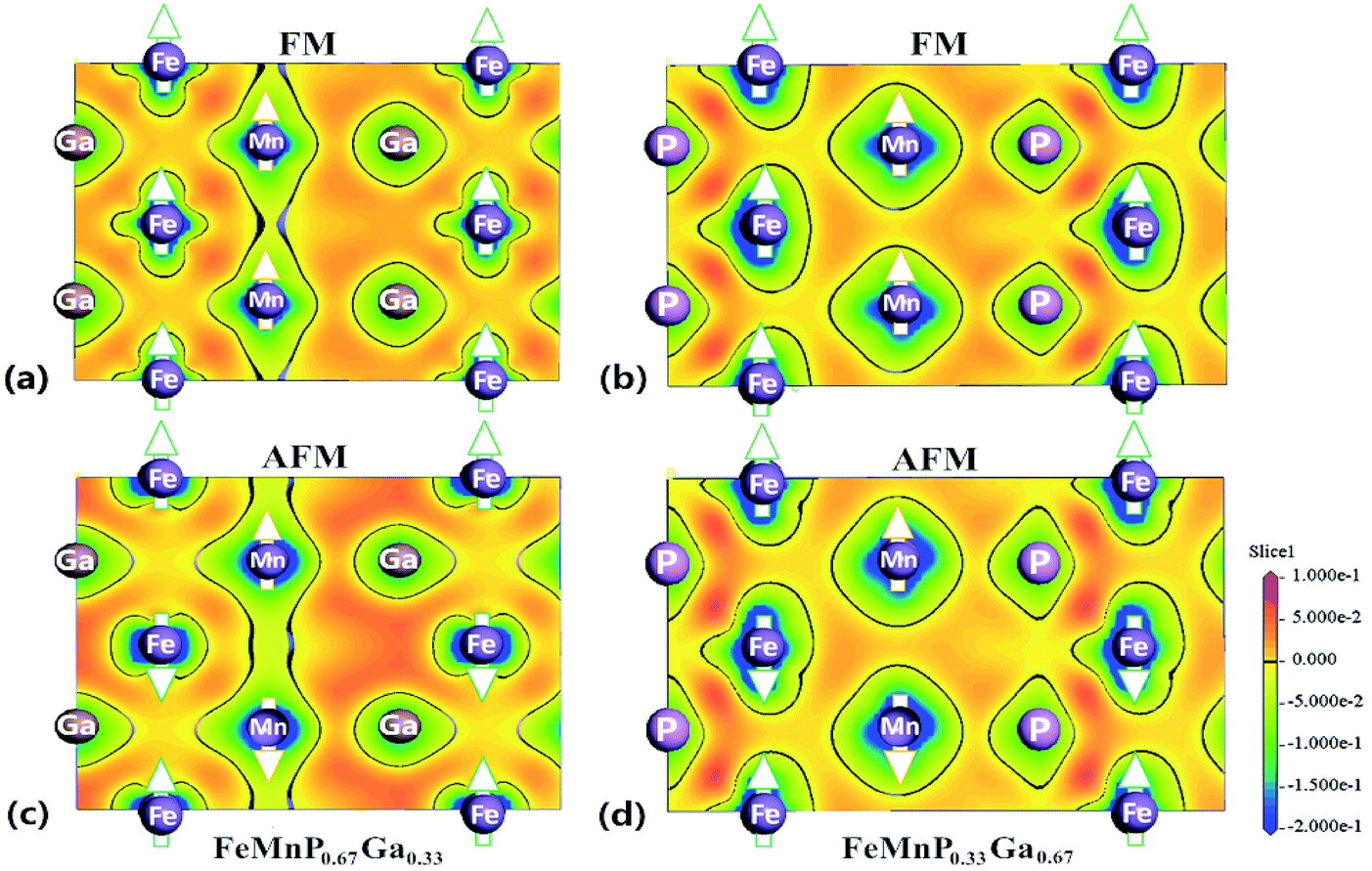 | ||
| Fig. 6 CDD of FeMnP1−xGax compounds in the FM and AFM states. (a) Ferromagnetic state of FeMnP0.67Ga0.33, (b) ferromagnetic state of FeMnP0.33Ga0.67. (c) Antiferromagnetic state of FeMnP0.67Ga0.33. (d) Antiferromagnetic state of FeMnP0.33Ga0.67. | ||
From the viewpoint of the energetics,17,51 FeMnP0.67Ga0.33 in the AFM state shows shrinkage in the basal plane, and thus a large overlap in the electron cloud between Fe and P occurs. Hence, the kinetic energy, which is lower for the spin-degenerate states and leads to a decrease of the magnetic moment, becomes the dominant term in the total energy. Asymmetric electronic distribution in the rectangle formed by two Mn atoms and two P atoms is also detected, indicating the strong influence of the magnetic order in the electronic distribution.
Comparing CDD in Fig. 6(a) and (c), denser charge is observed around Ga for AFM state, indicating that Ga–Fe and Ga–Mn bonding is strengthened. In other words, the redistribution of charge around Ga is responsible for the splitting and right shift of optical mode (∼10 THz) of Ga in AFM state. Due to the shrinkage of Fe–P distance in AFM state, the covalent like interaction between Fe and P become stronger (CDD between Fe and P become much denser for AFM state) and thus the vibration frequency of optical mode of P and Fe increased reasonably from ∼11.5 THz for FM state to ∼13 THz for AFM state.
Thermodynamic properties
Materials for magnetic refrigeration usually work under constant pressure and elevated temperature. Therefore, the Gibbs free energy of compounds with different magnetic order are calculated and compared for elevating temperature to determine Tc.26 The Gibbs free energy is expressed aswhere m represents the FM or AFM magnetic state. The vibrational free energy was estimated by the Debye model,45 and the free energy of thermal electronic excitation was calculated within the single electron excitation picture.41
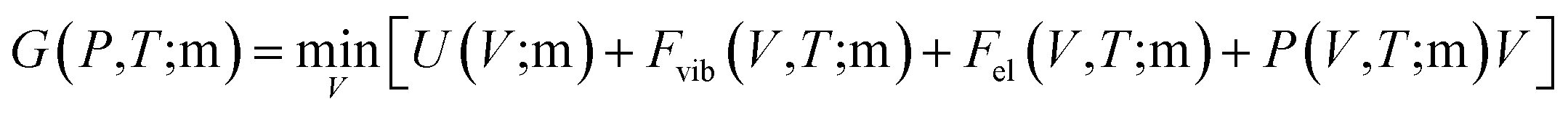
Another term that should be taken into account for the PM state is the magnetic entropy. The static energy of the PM state was simulated with an ordered AFM cell and did not include the entropy contribution from magnetic disorder. The magnetic entropy of the PM state can be estimated using the mean-field approximation:52 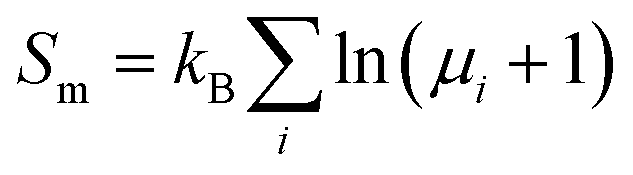 , where μi is the magnetic moment at the ith site. The total Gibbs free energy of the PM state then becomes Gtot(P,T) = G(P,T,AFM) − SmT.
, where μi is the magnetic moment at the ith site. The total Gibbs free energy of the PM state then becomes Gtot(P,T) = G(P,T,AFM) − SmT.
The Gibbs free energy difference ΔG(P,T) = G(P,T,AFM) − G(P,T,FM) − SmT was calculated for FeMnP1−xGax as a function of temperature. ΔG(P,T) for FeMnP0.67Ge0.33 is also shown in Fig. 7(a) for comparison. ΔG(P,T) gradually decreases and intersects with the abscissa axis at a certain temperature, which defines Tc, as shown in Fig. 7(a). The decrease of ΔG(P,T) at elevated temperature is mainly from magnetic entropy, but the non-constant slope of ΔG(P,T) suggests that the anharmonic effect is still playing a considerable role.
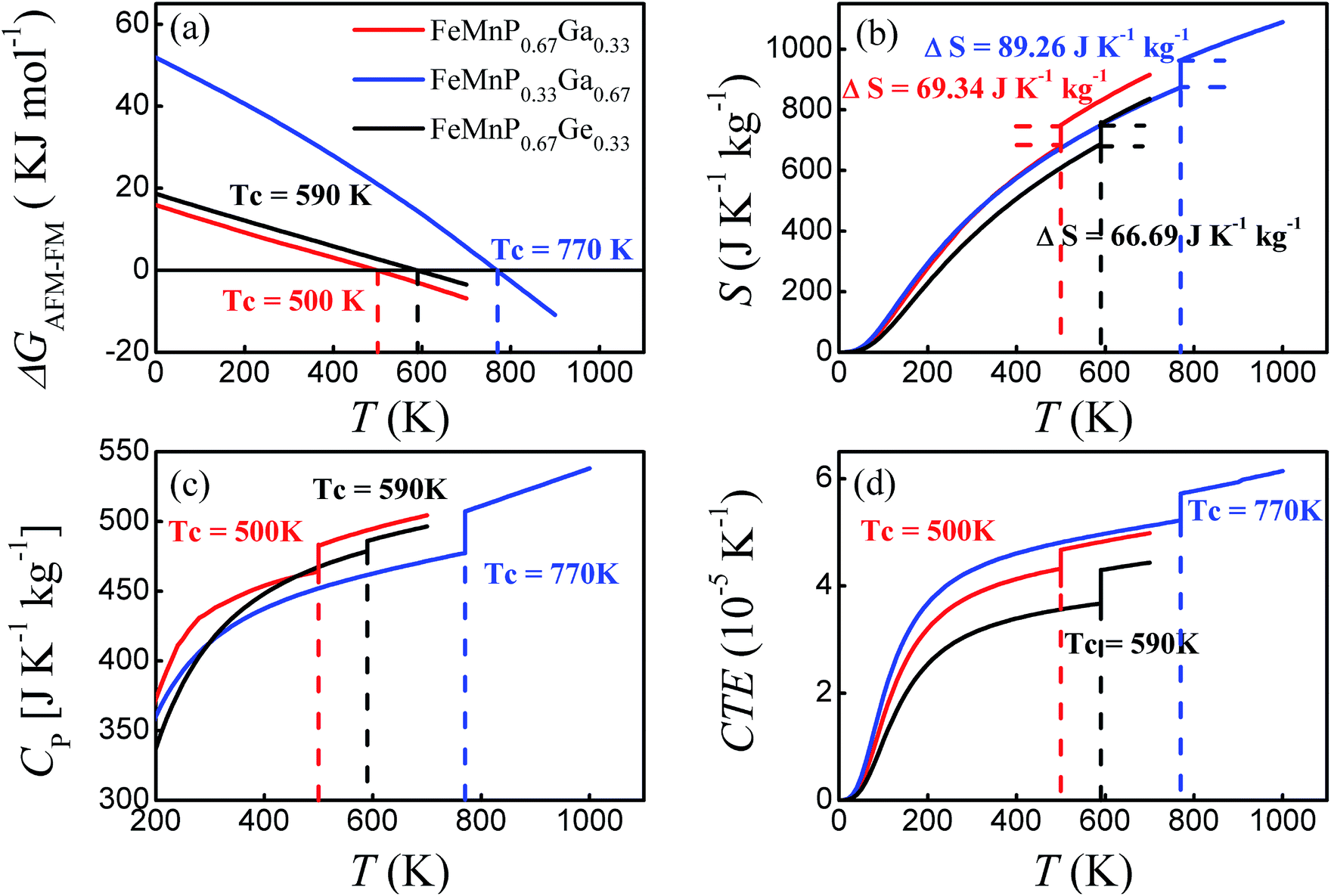 | ||
| Fig. 7 Calculated Gibbs free energy G (a), entropy S (b), specific heat at constant pressure CP (c), and linear CTE (d) as a function of temperature. | ||
The predicted Tc for FeMnP0.67Ge0.33 is 590 K, which is close to the measured Tc = 405 K for FeMnP0.67Ge0.33 compounds and Tc = 580 K for FeMnP0.5Ge0.5 compounds,44,53 verifying that the present scheme can give a reliable prediction of Tc. The difference between experiment and theory could be ascribed to the missing entropy contributions from spin wave excitation and phonon–phonon interactions. The Tc values of FeMnP1−xGax are 500 and 770 K for x = 0.33 and 0.67, respectively, indicating Tc increases as a function of the Ga concentration. Similar characteristics have also been found for Fe2P1−xSix,54 FeMnP1−xSix,16 and FeMnP1−xGex (ref. 46 and 47) compounds.
The temperature dependence of the total entropy of the compounds is plotted in Fig. 7(b). The entropy change of FeMnP0.67Ge0.33 around the phase transition is ΔS = 66.69 J K−1 kg−1, which is comparable with the experimental value of 49.74 J K−1 kg−1 for Fe1.1Mn0.9P0.8Ge0.2 at Tc = 278 K.46 FeMnP0.67Ga0.33 exhibits an entropy change of ΔS = 69.34 J K−1 kg−1 around Tc, which is similar in magnitude to ΔS of FeMnP0.67Ge0.33 and close to the experimental value for FeMnP0.67Si0.33 of ΔS = 58.6 J K−1 kg−1.55 FeMnP0.33Ga0.67 shows a much larger entropy change than FeMnP0.67Ga0.33 of ΔS = 89.26 J K−1 kg−1.
The entropy change can be divided into the vibrational and magnetic entropy changes: ΔS = ΔSvib + ΔSm. Here, the contribution of thermal electronic excitation is omitted because it is too small to have a measurable effect. For FeMnP0.67Ga0.33 and FeMnP0.67Ge0.33, the ΔSvib values are −2.78 and −4.56 J K−1 kg−1 for the FM–PM transition, respectively. And ΔSvib = −20.4 J K−1 kg−1 for FeMnP0.33Ga0.67, and thus adding the magnetic entropy term ΔSm the total entropy change becomes 89.26 J K−1 kg−1. Therefore, the magnetic entropy is playing major role on the entropy change of the compounds. And further, the decrease in the vibrational entropy is partially account for the relatively higher Tc for FeMnP1−xGax and FeMnP1−xGex compounds than that of FeMnP1−xSix.26,27
Fig. 7(c) and (d) shows the temperature dependence of the heat capacity under constant pressure and the linear CTE, respectively. They are discontinuous at Tc, which is a feature of first-order phase transitions. For first-order phase transition, the latent heat is defined as L = ΔS × Tc. The estimated latent heats for FeMnP1−xGax are 34.67 (x = 0.33) and 68.73 kJ kg−1 (x = 0.67), and the value for FeMnP0.67Ge0.33 is 39.35 kJ kg−1.
The jump in the CTE does not contradict with experimental observations of a very small volume change of ∼1% at Tc for FeMnP1−xSix and FeMnP1−xGex compounds.24 Assuming that FeMnP0.67Ge0.33 is in full FM order just before the phase transition and in the PM state just above Tc, the volume per unit formula at Tc = 590 K for FM order is 38.83 Å3 and that for AFM order is 38.57 Å3. The change in volume is at most ∼1%, which is still in line with experimental findings. Similar results were obtained for the FeMnP1−xGax compounds.
Conclusions
Density functional theory was used to investigate the mechanical and thermophysical properties of Fe2P-type FeMnP1−xTx (T = Ga, Ge; x = 0.33, 0.67) compounds, and thus identify the possibility of substituting phosphorus with gallium rather than silicon or germanium and obtaining GMC compounds with better mechanical stability with FM to PM phase transitions. The predicted formation energies, elastic constants and phonon spectra show that the different magnetic states of the hexagonal phase of FeMnP1−xGax are energetically and mechanically stable. The predicted magnetic moment, Curie temperature, and entropy change of FeMnP0.67Ga0.33 are very close to those of FeMnP0.67Ge0.33, a well-known GMC compounds. Therefore, it is expected that FeMnP1−xGax could be a candidate for room temperature magnetic refrigeration.The relatively large single crystalline elastic constants show the mechanical stability of this family of compounds in both the FM and AFM states. This ensures the long-term applicability of FeMnP1−xGax in magnetic refrigeration facilities under repeated thermal and magnetic cycles. Both FeMnP0.67Ga0.33 and FeMnP0.67Ge0.33 show good ductility in the FM state. However, their AFM states are brittle. Conversely, the FM states of FeMnP0.33Ga0.67 and FeMnP0.33Ge0.67 are brittle and their AFM states exhibit good ductility. However, it is expected that the occupation disorder of P and Ga/Ge could increase the ductility of the compounds in the AFM states. Our DFT calculations indicate that elements like Ga with three valence electrons could also be alloyed into the hexagonal phase of FeMn-based Fe2P type GMC compounds. Experimental synthesis of FeMnP1−xGax compounds, and thus verification of their magnetocaloric properties, is expected.
Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (grant numbers 11464037 and 50901039) and the Inner Mongolia Natural Science Foundation (grant number 2016MS0113), and the support from NJYT-12-B07.Notes and references
- K. A. Gschneidner Jr, V. K. Pecharsky and A. O. Tsokol, Rep. Prog. Phys., 2005, 68, 1479–1539 CrossRef.
- A. Smith, C. Bahl, R. Bjork, K. Engelbrecht, K. K. Nielsen and N. Pryds, Adv. Energy Mater., 2012, 2, 1288–1318 CrossRef CAS.
- O. Tegus, E. Brück, K. H. J. Buschow and F. R. de Boer, Nature, 2002, 415, 150–152 CrossRef CAS PubMed.
- E. Brück, O. Tegus, D. T. C. Thanh and K. H. J. Buschow, J. Magn. Magn. Mater., 2007, 310, 2793–2799 CrossRef.
- E. Brück, O. Tegus, L. Zhang, X. W. Li, F. R. de Boer and K. H. J. Buschow, J. Alloys Compd., 2004, 383, 32–36 CrossRef.
- L. Song, G. F. Wang, Z. Q. Ou, O. Haschaolu, O. Tegus, E. Brück and K. H. J. Buschow, J. Alloys Compd., 2009, 474, 388–390 CrossRef CAS.
- N. T. Trung, Z. Q. Ou, T. J. Gortenmulder, O. Tegus, K. H. J. Buschow and E. Brück, Appl. Phys. Lett., 2009, 94, 102513 CrossRef.
- N. H. Dung, Z. Q. Ou, L. Caron, L. Zhang, D. T. C. Thanh, G. A. de Wijs, R. A. de Groot, K. H. J. Buschow and E. Brück, Adv. Energy Mater., 2011, 1, 1215–1219 CrossRef CAS.
- V. Hoglin, M. S. Andersson, T. Sarkar, P. Nordblad and M. Sahlberg, J. Magn. Magn. Mater., 2015, 374, 455–458 CrossRef.
- Z. Gercsi, E. K. Delczeg-Czirjak, L. Vitos, A. S. Wills, A. Daoud-Aladine and K. G. Sandeman, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 024417 CrossRef.
- N. H. Dung, L. Zhang, Z. Q. Ou and E. Brück, Appl. Phys. Lett., 2011, 99, 092511 CrossRef.
- F. Guillou, G. Porcari, H. Yibole, N. van Dijk and E. Brück, Adv. Mater., 2014, 26, 2671–2675 CrossRef CAS PubMed.
- S. Berri, J. Supercond. Novel Magn., 2016, 29, 1309–1315 CrossRef CAS.
- P. Roy, E. Torun and R. A. de Groot, Phys. Rev. B, 2016, 93, 094110 CrossRef.
- L. Caron, M. Hudl, N. H. Hoglin, C. P. Gomez, M. Sahlberg, E. Brück, Y. Andersson and P. Nordblad, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 094440 CrossRef.
- V. Hoglin, J. Cedervall, M. S. Andersson, T. Sarkar, M. Hudl, P. Nordblad, Y. Andersson and M. Sahlberg, RSC Adv., 2015, 5, 8278–8284 RSC.
- M. Hudl, P. Nordblad, T. Bjorkman, O. Eriksson, L. Haggstrom, M. Sahlberg, Y. Andersson, E. K. Delczeg-Czirjak and L. Vitos, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 134420 CrossRef.
- X. F. Miao, L. Caron, P. Roy, N. H. Dung, L. Zhang, W. A. Kockelmann, R. A. de Groot, N. H. van Dijk and E. Brück, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 174429 CrossRef.
- H. Yibole, F. Guillou, L. Caron, E. Jimenez, F. M. F. de Groot, P. Roy, R. de Groot and E. Brück, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 014429 CrossRef.
- F. Guillou, K. Ollefs, F. Wilhelm and A. Rogalev, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 224427 CrossRef.
- H. Wang, Z. D. Zhang, R. Q. Wu and L. Z. Sun, Acta Mater., 2013, 61, 2919–2925 CrossRef CAS.
- S. Berri, J. Magn. Magn. Mater., 2016, 401, 667–672 CrossRef CAS.
- E. Brück, Handb. Magn. Mater., 2007, 17, 235–291 Search PubMed.
- E. Brück, J. Phys. D: Appl. Phys., 2005, 38, 381–391 CrossRef.
- M. J. Neish, M. P. Oxley, J. Guo, B. C. Sales, L. J. Allen and M. F. Chisholm, Phys. Rev. Lett., 2015, 114, 106101 CrossRef CAS PubMed.
- D. T. Cam Thanh, E. Brück, O. Tegus, J. Klaasse and K. Buschow, J. Magn. Magn. Mater., 2007, 310, e1012–e1014 CrossRef.
- X. Moya, S. Kar-Narayan and N. D. Mathur, Nat. Mater., 2014, 13, 439–450 CrossRef CAS PubMed.
- P. Roy, E. Torun and R. A. de Groot, Phys. Rev. B, 2016, 93, 165101 CrossRef.
- M. E. Gruner, W. Keune, B. Roldan Cuenya, C. Weis, J. Landers, S. I. Makarov, D. Klar, M. Y. Hu, E. E. Alp, J. Zhao, M. Krautz, O. Gutfleisch and H. Wende, Phys. Rev. Lett., 2015, 114, 057202 CrossRef CAS PubMed.
- X. F. Miao, L. Caron, J. Cedervall, P. C. M. Gubbens, P. D. de Réotier, A. Yaouanc, F. Qian, A. R. Wildes, H. Luetkens, A. Amato, N. K. van Dijk and E. Brück, Phys. Rev. B, 2016, 94, 014426 CrossRef.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Berri, Chin. Phys. B, 2017, 55, 195–202 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- F. D. Murnaghan, Proc. Natl. Acad. Sci. U. S. A., 1994, 244–247 Search PubMed.
- L. Vitos, Computational Quantum Mechanics for Materials Engineers: the EMTO Method and Applications, Springer-Verlag, 2007, pp. 107–113 Search PubMed.
- G. Grimvall, Thermophysical Properties of Materials, Elsevier Science B V, 1999, pp. 27–40 Search PubMed.
- A. Togo, L. Chaput, I. Tanaka and G. Hug, Phys. Rev. B, 2010, 81, 174301 CrossRef.
- J. Slater, Introduction to Chemical Physics, McGraw-Hill, 1939, vol. 152, pp. 488–489 Search PubMed.
- B. Wurentuya, S. Ma and O. Tegus, et al., J. Phys.: Condens. Matter Search PubMed , submitted.
- A. Otero-de-la-Roza, D. Abbasi-Perez and V. Luana, Comput. Phys. Commun., 2011, 182, 2232–2248 CrossRef CAS.
- M. Yue, H. G. Zhang, D. M. Liu and J. X. Zhang, Chin. Phys. B, 2015, 24, 017505 CrossRef.
- D. Liu, M. Yue, J. X. Zhang, T. M. McQueen, J. W. Lynn, X. L. Wang, Y. Chen, J. Y. Li, R. J. Cava, X. B. Liu, Z. Altounian and Q. Huang, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 014435 CrossRef.
- S. F. Pugh, Philosophical Magazine, 1954, 45, 823–843 CrossRef CAS.
- H. L. Zhang, M. P. J. Punkkinen, B. Johansson, S. Hertzman and L. Vitos, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 184105 CrossRef.
- A. R. Mackintosh and O. K. Anderson, Electrons at the Fermi Surface, Cambridge University Press, 1980, pp. 149–224 Search PubMed.
- G. J. Li, W. Li, S. Schönecker, X. Q. Li, E. K. Delczeg-Czirjak, Y. O. Kvashnin, O. Eriksson, B. Johansson and L. Vitos, Appl. Phys. Lett., 2014, 105, 262405 CrossRef.
- G. Grimvall, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 12300–12301 CrossRef CAS.
- X. W. Li, O. Tegus, L. Zhang, W. Dagula, E. Brück, K. H. J. Buschow and F. R. de Boer, IEEE Trans. Magn., 2003, 39, 3148–3150 CrossRef CAS.
- P. Jernberg, A. A. Yousif, L. Häggström and Y. Andersson, J. Solid State Chem., 1984, 53, 313–322 CrossRef CAS.
- Z. Ou, Magnetic structure and phase formation of magnetocaloric Mn-Fe-P-X compounds, Delft University of Technology, 2013 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |