
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpatial charge manipulated set-selective apatite deposition on micropatterned piezoceramic
Jinxia Zhai†
af,
Qiyou Wang†b,
Jinquan Zengc,
Junqi Chenaf,
Xin Yidf,
Zhifeng Shiaf,
Guoxin Tane,
Peng Yu *af and
Chengyun Ning*af
*af and
Chengyun Ning*af
aCollege of Materials Science and Engineering, South China University of Technology, Guangzhou, 510641, China. E-mail: imcyning@scut.edu.cn; imyup@scut.edu.cn
bDepartment of Spine Surgery, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, 510630, China
cGuangzhou Tieyi Middle School, Guangzhou, 510660, China
dSchool of Medicine, South China University of Technology, Guangzhou, 510641, China
eSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou, 51006, China
fKey Laboratory of Biomedical Sciences and Engineering, South China University of Technology, Guangzhou 510006, China
First published on 29th June 2017
Abstract
Mineralization plays a crucial role in the formation and remodeling of bone and charge has a positive influence on the mineralization by manipulating apatite deposition. In this paper, micropatterned piezoelectric K0.5Na0.5NbO3 (MPK) was constructed via laser-irradiation induced micro-zonal phase transition to manipulate set-selective apatite deposition. Two types of piezoelectric micro-zones on polarized MPK showed different charge densities, namely, higher charge density zone (H-zone) and lower charge density zone (L-zone). The micro-zonal piezoelectric difference on negatively polarized MPK (N-MPK) was ascertained by scanning Kelvin probe force microscopy (SKPM). With the manipulation of spatial charge, apatite selectively deposited on H-zone rather than L-zone, forming stripped zonal distribution. The furry platelet morphology of apatite was observed by scanning electron microscopy (SEM). Energy-dispersive spectroscopy (EDS) mapping observation indicated selective distribution of Ca and P elements in apatite. Phase composition of deposited apatite also can be regulated effectively via X-ray diffraction (XRD), which confirmed the existence of hydroxyapatite phase. Meanwhile, the apatite was further identified as carbonated hydroxyapatite by X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FTIR). The feasible and convenient way of manipulating apatite deposition selectively with spatial charge realized by micro-zonal piezoelectric materials will inspire future development in biomimetic materials designation for biomedical applications.
1. Introduction
During the reconstruction of bone, bone mineral is induced to spatially deposit on the previous bone matrix, resulting in a hierarchical anisotropic structure of natural bone.1–3 To mimic the bone-remoulding process, numerous researchers have fabricated micropatterned materials which could regulate apatite deposition on their surfaces. The key to the development of these kinds of micropatterned material relies on the modulation of physical and chemical properties on their surfaces, which could regulate apatite deposition selectively.4,5 Considerable efforts have been devoted to achieve extremely a difference of micro-zonal chemical properties on a sample surface.6,7 For example, a surface with hydrophobic/hydrophilic difference was employed to modify surface chemicals properties. Apatite was clearly and selectively deposited on the hydrophilic zone in simulated body fluid (SBF).8,9 Other treatments were applied to obtain certain different topological structures impacting on the deposition of micro-zonal apatite. In our laboratory, two zones of nanoneedle zones and buffer zones were fabricated on the substrate realizing the micro-zonal difference of physical properties.10 More apatite deposited on nanoneedle zones than buffer zones in SBF. Meanwhile, further studies have been launched on the charge in SBF.11 Kim et al. introduced that negative charges were acquired by the surface modified with hydroxyl groups for accelerating bone-like apatite formation.12–14 Negative charges attracted calcium ions preferentially and thereby accelerated apatite formation in SBF.15 However, it has not been reported so far that the surface of micro-zonal charge difference was fabricated to regulate apatite deposition.Bone mineralization in vivo is the process of apatite deposition on piezoelectric collagen fibers.16–18 The piezoelectricity could control the size and three-dimensional distribution of bone mineral.19,20 Kakimoto demonstrated that a piezoelectric substrate had a huge potentiality in promoting apatite deposition.11,21 Meanwhile, apatite growth on negatively polarized surfaces of piezoelectric ceramics preferentially has also been reported.22,23 The development of piezoelectric materials for orthopedic implant applications has evinced more interest and attention owing to the piezoelectricity similar to piezoelectricity of natural bone.24 Though barium titanate (BT) ceramic possesses piezoelectricity and promotes bone-like crystal after poling treatment, BT piezoceramic has a poor thermal stability and cytotoxic barium ions. Meanwhile, PVDF films can be used to culture cells and promote a high proliferation of osteoblasts cells. However, PVDF has inferior physical properties, including the dielectric, ferroelectric, and piezoelectric, compared with the piezoceramics. The well-known environment-friendly lead-free piezoelectric ceramic potassium sodium niobate (K0.5Na0.5NbO3, KNN) as an electroactive bioceramic possesses good temperature stability, reasonably high piezoelectric constant and good biocompatibility. Currently, KNN piezoceramic has been used as a bone implant. Chen et al. have proved that the surface charge on polarized KNN has the influence on protein adsorption and cell proliferation.25 Navarrete et al. also reported the superior cellular response on niobium coatings.26 Therefore, in this context, KNN has been suggested as a potential alternative for biomimetic bone due to excellent biocompatibility and stable inherent piezoelectric properties.27–30
In our paper, micropatterned piezoelectric K0.5Na0.5NbO3 (MPK) was constructed via laser-irradiation induced micro-zonal phase transition. By adopting the laser irradiation, two types of piezoelectric zones could be processed on polarized MPK, which presented different charge densities, namely, higher charge density zone (H-zone) and lower charge density zone (L-zone) after the polarization of MPK sample in Fig. 1. Apatite selectively deposited on H-zone of negatively polarized MPK (N-MPK) in the left image of Fig. 1. Calcium ions were prior to accumulating at H-zone rather than L-zone selectively. Due to the different quantity of different zones absorbed calcium ions/phosphate ions/carbonate ions, crystal growth mentioned above was observed on H-zone in the right image of Fig. 1. We hypothesized that the polarized MPK could selectively manipulate apatite deposition by spatial charge. To test this hypothesis, we studied the mineralized process of polarized MPK samples in SBF.
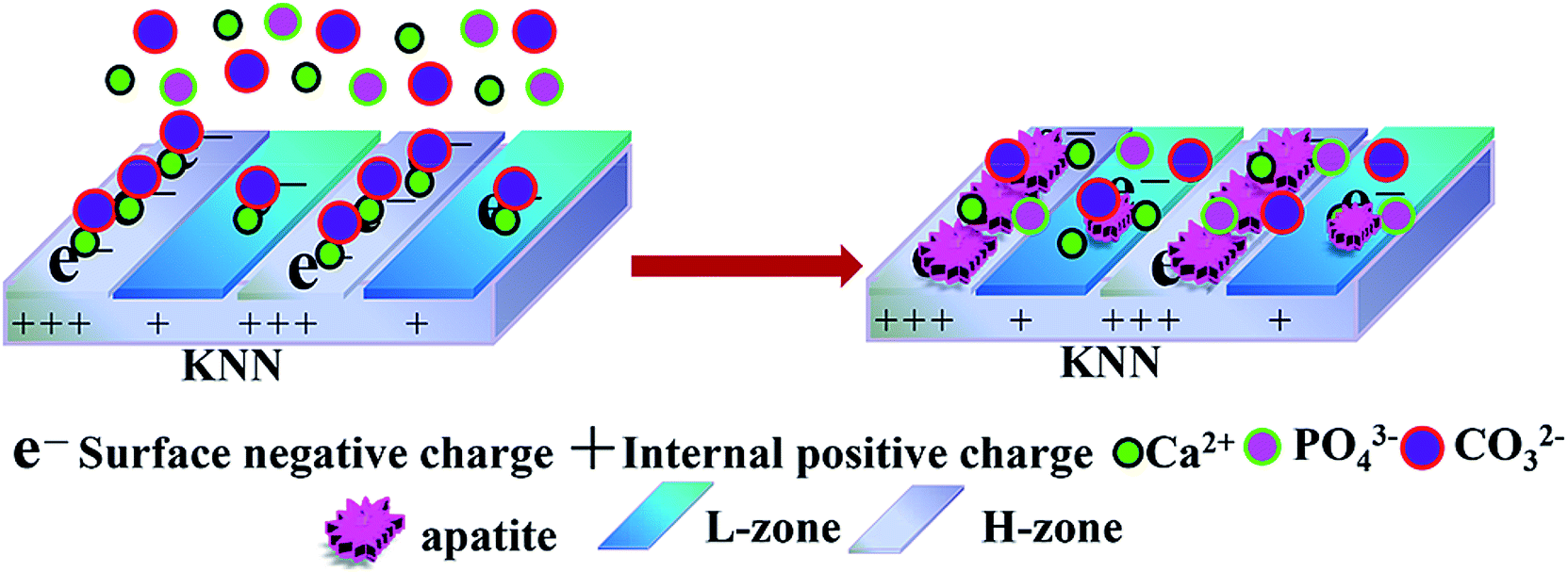 | ||
| Fig. 1 Schematic illustration of set-selective apatite nucleation and growth on negatively polarized MPK in SBF. MPK fabricated by laser-irradiation induced micro-zonal phase transition was composed by two types of negatively polarized piezoelectric zones: H-zone and L-zone. The nucleation of apatite occurred the surface of H-zone whereas L-zone (left image). Along with the growth of crystal nucleus, apatite was deposited densely on H-zone forming set-selective distribution (right image). | ||
2. Experimental
2.1 Materials
K0.5Na0.5NbO3 (KNN) ceramics were consolidated via a conventional solid state reaction method described in our laboratory.25 The raw materials were K2CO3 (AR, 99.0%, Aladdin), Na2CO3 (AR, 99.8%, Aladdin) and Nb2O5 (AR, 99.9%, Aladdin). These raw materials were weighed according to the stoichiometric ration, uniformly mixed with milling media of agate ball and ethanol, and well ball milled for 8 h. Then the slurry was dried in vacuum oven at 70 °C for 10 h and calcined at 750 °C for 2 h into a muffle furnace. The calcined KNN powder mixed with proportional polyvinyl alcohol (PVA) as a binder was grinded uniformly, and pressed into chips (10 mm diameter and 2.0 mm thickness) by tableting, isostatic pressing and an embedding sintering process at sintering temperature 1100 °C for 2 h.2.2 Fabrication of MPKs
MPKs were fabricated using laser-irradiation induced micro-zonal phase transition (HM20, Handslaser, Shenzhen, China) after gradient grinding at a power of 4 W and a scanning speed 120 mm s−1. The micro-zonal types and intervals were easily and uniquely modulated by AutoCAD software (HL software, Handslaser). Subsequently, MPK samples were polarized at an electric field of 1 kV mm−1 for 5 min. The polarized MPK samples were cleaned and dried.2.3 Charged manipulated apatite deposition on MPKs
It is generally acknowledged that SBF with ion concentration, pH and temperature closely resembles human blood plasma. The solution with Ca/P ratio deviated from the stoichiometry can accelerate the formation of calcium phosphates. The apatite was conducted via directly immersing samples in SBF, which serves as a source of Ca2+ and PO43−. 1.5× SBF was prepared in accordance to Kokubo and Takadama's classic formulation:31 chemicals of NaCl, NaHCO3, KCl, K2HPO3·3H2O, HCl, MgCl2·6H2O, CaCl2, Na2SO4 were dissolved in the de-ionized water and buffered at pH 7.4 with tris (hydroxymethyl)-aminomethane ((CH2OH)3CNH2) and hydrochloric acid (HCl) at 37 °C. The pH value of SBF solutions was monitored using a pH meter (PB-10, Sartorious). The whole samples were immersed in SBF for 7 days.Meanwhile, to keep the ion concentration stable, SBF solution was refreshed every two days. According to the predetermined time period of immersion, mineralized samples were removed from the solution, then rinsed gently with de-ionized water, and then dried in a vacuum desiccator for subsequent analysis.
2.4 Characterization of apatite deposited on MPKs
The micro-topographies and chemical constituents were examined by scanning electron microscopy (SEM, ZEISS EVO18, Carl Zeiss Company, Germany) equipped with energy dispersive spectroscopy (EDS, OxFord instruments X-Max Extreme). Scanning Kelvin probe force microscopy (SKPM, multimode8, Bruker, USA) was used to detect the surface potential on negatively polarized MPK in two adjacent areas of 5 × 5 μm2. In the process of testing, we can observe the location through the computer screen. Then, the probe can be translated from one zone to the adjacent zone. The crystalline phases of deposited apatite were characterized via X-ray diffraction (XRD, D8 Advance Bruker Company, Germany). The chemical bonding states and atomic concentration of apatite were checked via X-ray photoelectron spectroscopy (XPS, Axis Ultra DLD Kratos Company). Fourier transform infrared (FTIR, VERTEX 70, Bruker Company, Germany) was used to ascertain what types of the apatite crystals formed on N-MPK after immersion in SBF. The FTIR spectra were measured in transmittance mode using the KBr technique in the range from 4000 to 490 cm−1.3. Results and discussion
3.1 Deposited apatite on KNNs in SBF
The apatite precipitation process on KNN under different polarized modes were evaluated by immersion these samples in SBF. After 7 days of immersion, no apatite was found on unpolarized KNN (U-KNN) in Fig. 2(a). However, unlike the case with U-KNN sample, apatite deposited was observed on negatively KNN (N-KNN) and polarized KNN (P-KNN) in Fig. 2(b) and (c). Especially N-KNN sample, it has more deposited apatite. The different polarized mode of KNN sample exhibited different apatite deposition levels. The layers are composed of furry platelets forming spherical aggregates. Obviously, the crystal growth was dependent upon the charge. Especially, negative charge can significantly enhance apatite deposition as a result of the electrostatic interaction. And charge contributed to mineralization by stabilizing initiating nucleation.32 It was consistent with which Yoshinori Kuboki reported that acidic proteins with negative charges could bind calcium ions, induce the nucleation of apatite, and control their speed of growth.33 However, the different apatite deposition rate between the U-KNN and N-KNN inspired us to manipulate the distribution of apatite under the action of charge. It provided the basis to build a micro-zonal piezoelectricity on KNN samples. | ||
| Fig. 2 Apatite deposition on KNNs of different polarized modes. SEM micrographs of apatite deposited on (a) unpolarized KNN (U-KNN), (b) negatively polarized KNN (N-KNN) and (c) positively polarized KNN (P-KNN) in SBF for 7 days. The result indicated that N-KNN has a higher deposited rate compared to U-KNN and P-KNN. | ||
3.2 The construction and characterization of MPKs
In Fig. 3, SEM images taken at 500× magnification confirmed that MPK was fabricated by solid reaction process and laser irradiation induced micro-zonal phase transition, which formed two types of micro-zones about 50 μm in width, namely, higher charge density zone (H-zone) and lower charge density zone (L-zone). Besides, the technology showed the feasibility that a variety of surface patterns such as dots, stripes, circles, as well as squares were designed discretionarily and easily. After polarization of MPK samples, the zone of high piezoelectricity displayed the higher charge densities. Therefore, two types of piezoelectric zones were presented on polarized MPK. In order to evaluate the successful construction of different piezoelectric zones, the potential of the two zones on MPK was characterized by SKPM in Fig. 3(c) and (d). Obviously, the average potential of H-zone was ∼140 mV higher than that of L-zone.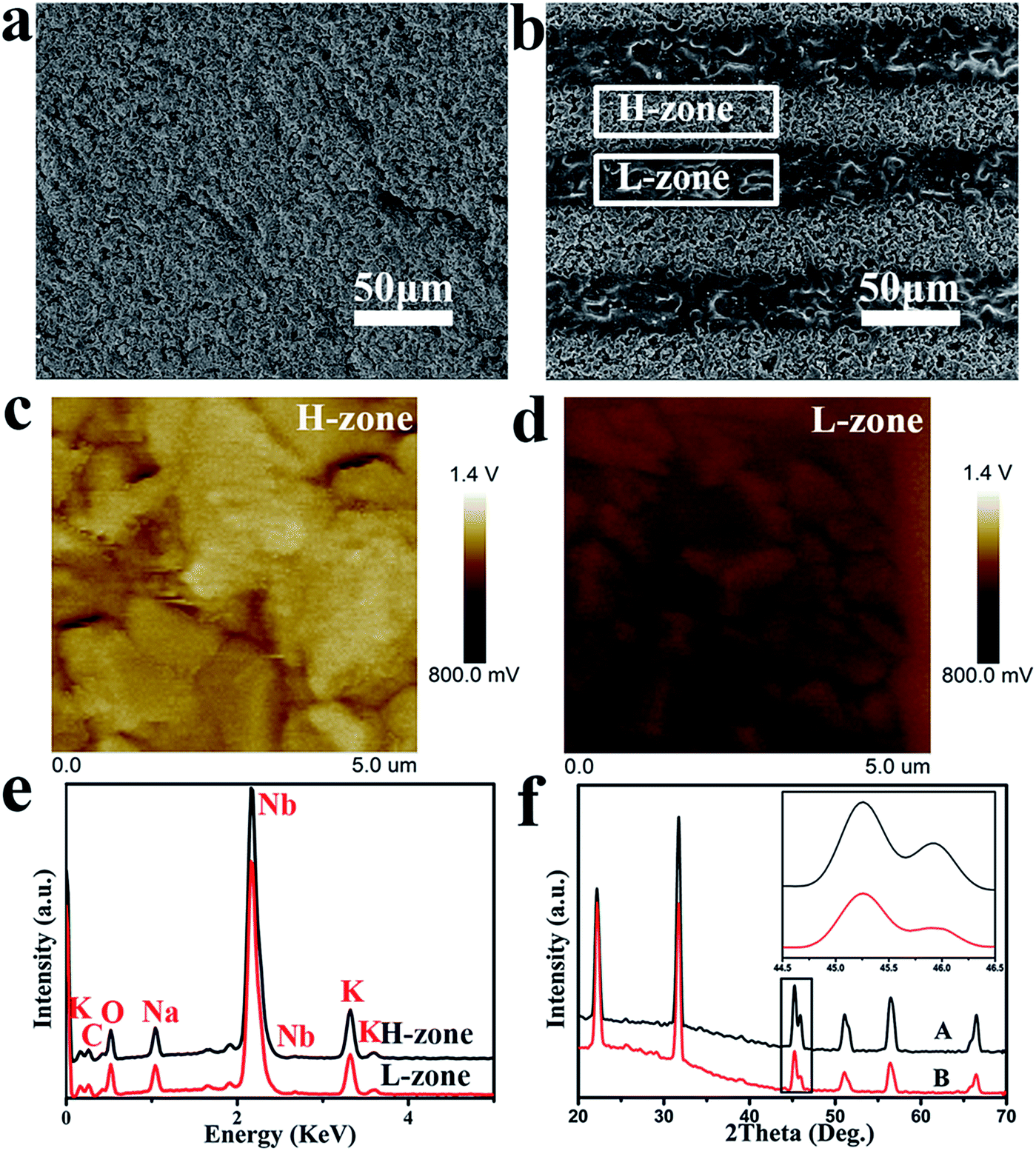 | ||
| Fig. 3 MPK fabricated by laser-irradiation induced micro-zonal phase transition. SEM micrographs of (a) KNN and (b) MPK. Apparent view showed the distribution of H-zone and L-zone on polarized MPK; SKPM characterization of the surface potential of the two adjacent zones, (c) H-zone and (d) L-zone of polarized MPK, respectively. The average potential of H-zone is ∼140 mV higher than that of L-zone. (e) EDS spectra of H-zone and L-zone on polarized MPK. (f) XRD patterns of A (KNN) and B (laser-irradiated KNN). An enlarged view of two peaks around 45° was shown in the right corner. In the illustration, the laser-irradiation caused a difference of the (002) and (200) peaks around 45°. The results indicated that the different piezoelectric zones on polarized MPK were fabricated by laser-induced micro-zonal phase transition successfully. | ||
Furthermore, EDS and XRD could be used to account for the reason of different piezoelectricity in Fig. 3(e) and (f). EDS spectra indicated that both zones were mainly composed of carbon, oxygen, potassium, sodium and niobium. However, potassium content and sodium content on H-zone was higher than on L-zone respectively, as shown in Table 1. Because laser-irradiation was realized via focusing vast energy, high temperature gave rise to the volatilization of potassium and sodium elements. Meanwhile, studies have been reported that for KNN ceramics, the difference of (002) and (200) peaks around 45° in XRD pattern is usually used as an evidence for the phase transition in literature.34 Relative intensities of two peaks can be used to distinguish orthorhombic from tetragonal phase. In general, the coexistence of orthorhombic phase and tetragonal phase can enhance the piezoelectricity of KNN compared to single phase.35,36 In our experiment, the relative intensity of (002) peak was nearly equal to that of (200) peak around 45° in the original KNN (A), indicating the coexistence of two phases. However, in the laser-irradiated KNN (B), the relative density of (200) peak was much lower than that of (002), representing mainly the single phase of the orthorhombic perovskite structure in the inset of Fig. 3(e). So, different phases of two zones on polarized MPK caused the different piezoelectricity. The composition and phase structure of both zones on polarized MPK elucidated the success of build two piezoelectric micro-zones. Then spatial distribution of charges was presented on the basis of micro-zonal piezoelectricity of polarized MPK, which became the main factor to regulate selectively apatite deposition in the next experiment. Some studies manifested that a negative charge was acquired on material surfaces to accelerate the formation of apatite.11,37 Therefore, spatial obtained with the micro-zonal piezoelectricity played significant role to regulate apatite deposition.
| Element | Atomic percentage (%, H-zone) | Atomic percentage (%, L-zone) |
|---|---|---|
| C | 28.43 | 32.71 |
| O | 40.10 | 39.99 |
| Na | 5.18 | 4.74 |
| K | 6.42 | 5.33 |
| Nb | 19.87 | 17.22 |
3.3 The apatite deposition on MPKs in SBF
Through the above experiments, we successfully realized the distribution of spatial charge. In order to evaluate the effect of spatial charge on apatite deposition, MPK samples were immersed in SBF for 7 days at 37 °C. These images in Fig. 4(a), (b) and (c) presented surface morphologies of deposited apatite on unpolarized MPK (U-MPK), negatively polarized MPK (N-MPK) and positively polarized MPK (P-MPK), respectively. A striped pattern and furry platelets morphology was observed. P-MPK was covered by small spherical cells of apatite owing to the repulsive interaction with Ca2+ ions and U-MPK has almost no apatite deposition. However, N-MPK lead to quicker deposition of apatite in Fig. 4(b). These platelets grew longitudinally and laterally in width through the stripes exhibiting set-selective apatite deposition due to the periodic distribution of H-zone and L-zone. Based on the set-selective apatite deposition (Fig. 4(b)), the map scanning of calcium and phosphorous elements on N-MPK also presented a state of spatial distribution in Fig. 4(d) and (e). In general, the process of apatite precipitation includes mainly initial apatite nucleation and crystal growth. Apatite formation in SBF is a multistep mechanism. In this study, the mechanism for the preferential nucleation of apatite on N-MPK was revealed. Firstly, Ca2+ ions were thought to play a key role in the initial nucleation of apatite on N-MPK, because the preferential accumulation of Ca2+ ions caused by the attractive electrostatic interaction between ions and N-MPK, and sequentially attracted phosphate ions and carbonate ions to form apatite nuclei. Once the apatite nuclei are formed, they grow spontaneously by accumulating the calcium and phosphate ion ions in SBF to form apatite. However, under the different piezoelectricity, H-zone and the L-zone presented the distribution of set-selectively deposited apatite on N-MPK in SBF. Due to the different quantity of calcium ions/phosphate ions/carbonate ions absorbed on two zones, increasing the charge density, apatite was more quickly grown on H-zone than L-zone. Therefore, the enhancement of apatite precipitation regulated by spatially negative charge on N-MPK has great significant.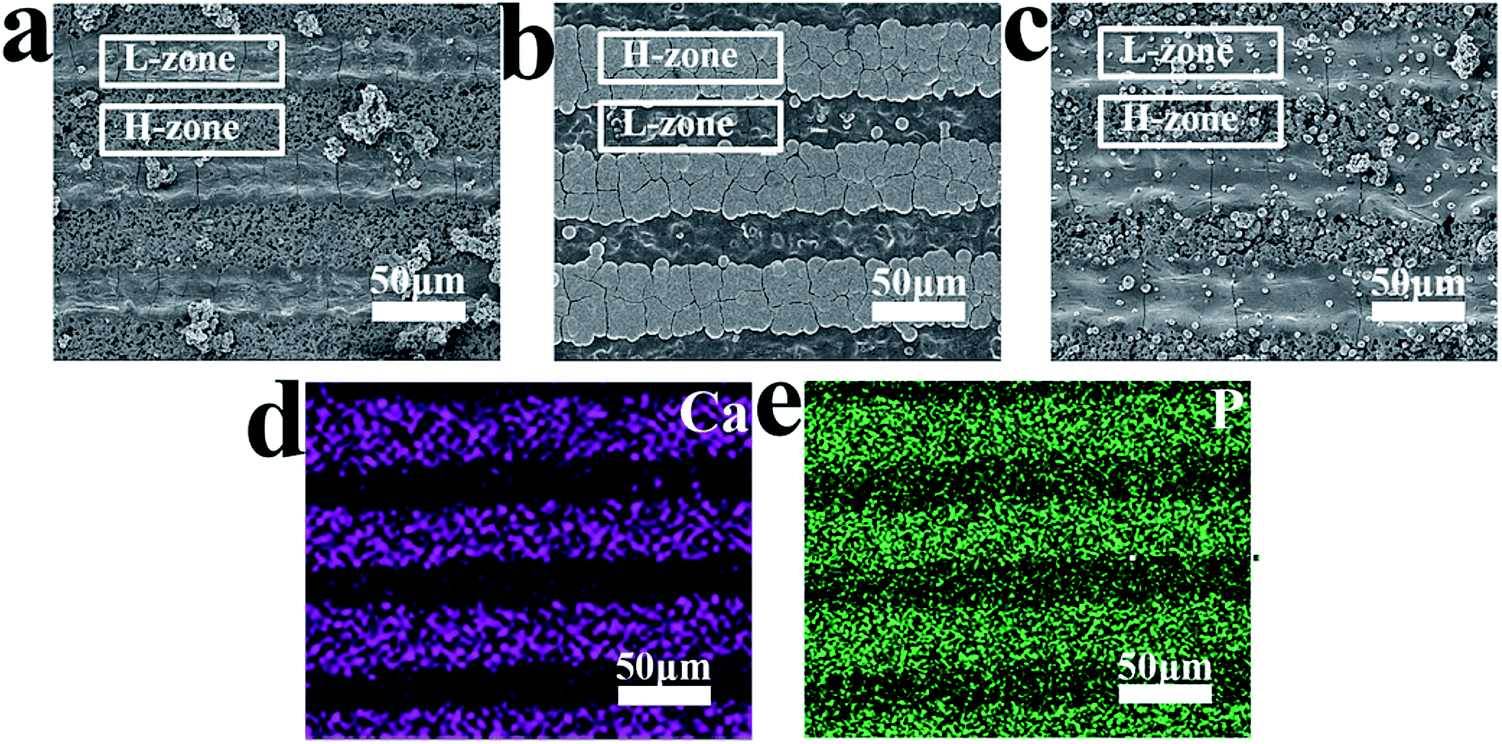 | ||
| Fig. 4 Set-selective apatite deposition on MPKs of different polarized modes. SEM micrographs of apatite deposition on (a) unpolarized MPK (U-MPK), (b) negatively polarized MPK (N-MPK) and (c) positively polarized MPK (P-MPK); element maps of (d) Ca and (e) P on N-MPK after apatite deposition. N-MPK presented more apatite deposition, whereas P-MPK and U-MPK have almost no apatite deposition. Obviously, spatial negative charge can preferably regulate set-selective apatite deposition. | ||
Hence, N-MPK achieved more set-selective apatite; P-MPK had small spherical cells of apatite and no selectivity; U-MPK had hardly any apatite, as illustrated in Fig. 4. It is visible that the existence of spatial charge provided a key way to form a set-selective apatite mimicking the apatite in natural bone. More apatite agglomeration can guide the formation of tissue that surrounds hard implant, and be beneficial to the construction and formation of osseous tissue.
In addition, Fig. 5 shows the morphologies of apatite deposition in SBF for (a) 4 days and (b) 7 days respectively. Biomineralization involves the controlled growth of apatite. At first, it did appear that the deposition of apatite was made up of tiny particles which were homogeneously distributed on H-zone. And over time, tiny particles grow up into the dense and thick apatite layer throughout the H-zone. However, there is almost no nucleation and growth of apatite deposition on L-zone. These results suggested that surface charges dominated the set-selective apatite nucleation and growth on N-MPK.
 | ||
| Fig. 5 The process of set-selective apatite deposition on N-MPK. SEM micrographs of (a) the sample immersed in SBF for 4 days and (b) the sample immersed in SBF for 7 days. The result indicated that the process of apatite deposition on H-zone has the nucleation and growth of crystals. | ||
For the set-selective deposited apatite, various analysis methods were used to the deposited apatite. The element compositions of calcium and phosphorous elements on N-MPK were presented by EDS in Fig. 6(a). The EDS detector was focused upon the apatite on H-zone. Besides, a small fraction of magnesium and sodium were also found by EDS analysis. Because natural bones are non-stoichiometric with trace components as Mg2+, Na+, CO32−, or Cl−. Mg2+ and Na+ ions which might substitute the Ca2+ ions in apatite were in agreement with the composition of biological apatite.38–40 The average Ca/P molar ratio of the apatite precipitated on H-zone of N-MPK was measured as 1.58, which indicated that the deposited apatite had a good state compared to typical Ca/P ratio (1.67). Fig. 6(b) shows typical XRD pattern obtained from deposited apatite on N-MPK after immersion in SBF for 7 days. Meanwhile, the standard pattern of natural hydroxyapatite (JCPDS card no. 09-0432) was used as a reference to analyze various peaks in our experiment. Herein a strong diffraction peak centred on 2θ values of 31.6° was detected corresponding to the characteristic peak of hydroxyapatite.41 A broad peak associated with 2θ values of 25.8° suggest that the deposited apatite formed on H-zone was extremely fine and has low crystalline. And a number of relatively weak peaks at 27.8°, 34° and 39° corresponded to the peaks of hydroxyl-apatite. Above all, the XRD result confirmed that the inorganic hydroxyapatite phase was obtained in the mineralization.
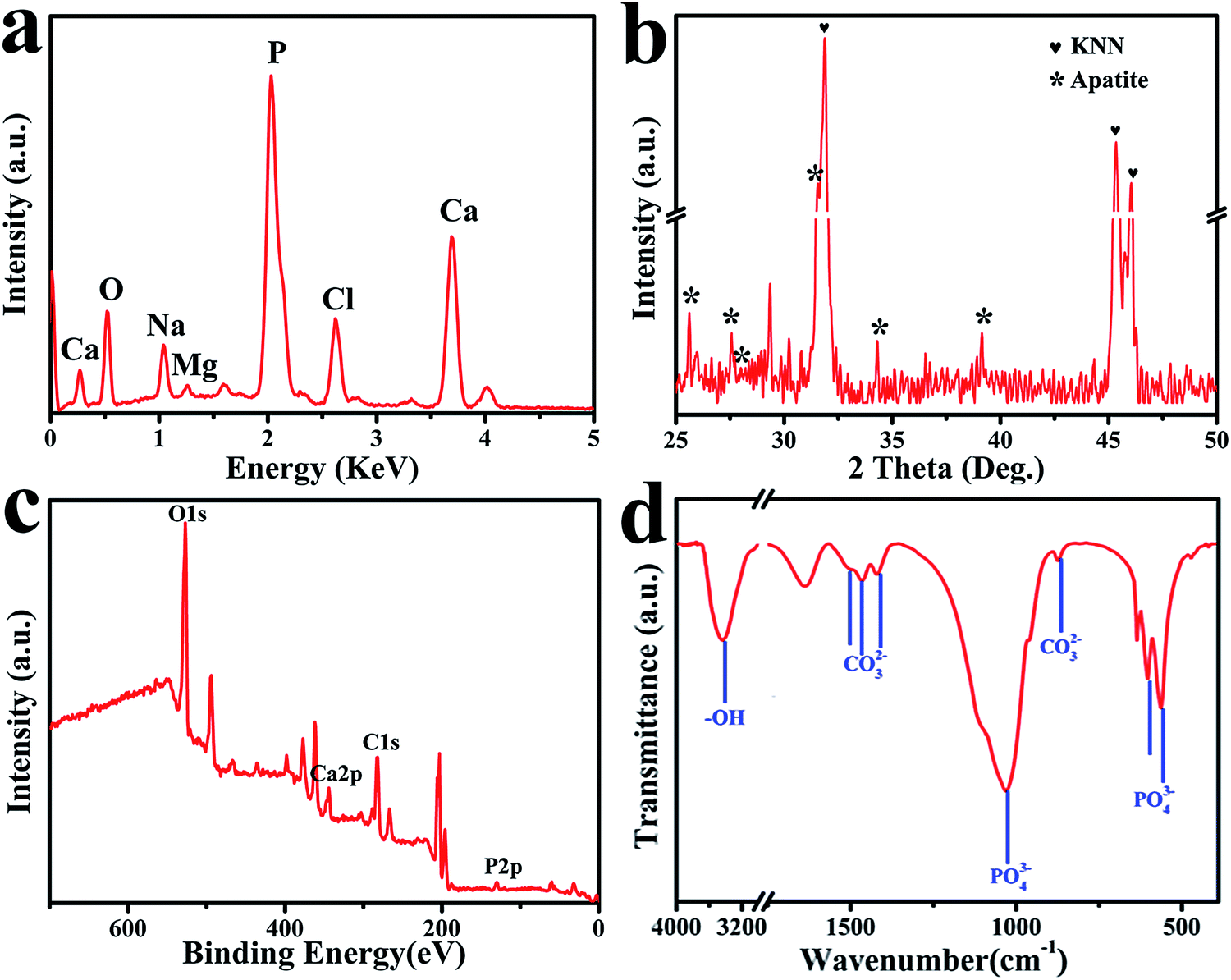 | ||
| Fig. 6 Characterization of set-selectively deposited apatite regulated with spatially negative charge on N-MPK after immersion in SBF for 7 days including (a) EDS analysis; (b) XRD pattern; (c) XPS spectrum; (d) FTIR spectrum. | ||
Fig. 6(c) presents the XPS spectrum obtained from the deposited apatite formed on N-MPK in SBF for 7 days. It indicated that Ca, P, O and C appear on N-MPK. In addition, the Ca/P molar ratio of deposited apatite from quantitative element analysis was in accordance with that calculated from the EDS. FTIR measurements confirmed the groups of deposited apatite formed on N-MPK. Fig. 6(d) exhibited sharp P–O bands at 1090 cm−1, 1030 cm−1, and 963 cm−1 for the P–O stretching vibration mode (ν3). The two P–O bands at 561 cm−1 and 603 cm−1 assigned to P–O bending vibration (ν4).42 In addition, FTIR spectra also displayed that the weak bands at 1497 (ν3) cm−1, 1466 (ν3) cm−1 assigned to C–O stretching vibration of CO32− group were attributed to A-type carbonate-containing apatite, where CO32− substituted for –OH sites.43,44 And the two bands at 1419 (ν3) cm−1 and 863 (ν2) cm−1 of CO32− group corresponded to B-type carbonate-containing apatite, where partial PO43− sites were replaced by CO32− in the apatite lattice.45 Meanwhile, there was a broad absorption band in the 3000–3700 cm−1 range, originating from the replacement of PO43−, CO32− and the presence of lattice water in the solid, which is a common characteristic of apatite precipitates in aqueous solutions.46 Due to the hydrogen bonding between water and hydroxyl, especially the surface bounding water, the absence of the hydroxyl groups which were represented by stretching (3570 cm−1) and vibrational (635 cm−1) bands provided further evidence for the existence of carbonate apatite. Combined with XRD pattern (Fig. 6(b)), it indicated that the deposited mineral was composed of partially carbonated calcium hydroxyapatite. Therefore, these observations confirmed that the existence of carbonated hydroxyapatite as a mixed AB-type carbonate substitution similar to natural bone apatite formed on MPK after immersion in SBF.47,48
These analytical results indicated the formation of partially carbonated hydroxyapatite. Deposited apatite was in accorded with the apatite in natural bone. So, by modulating apatite precipitation here, MPK materials will help realize the great potential as bone implants. What's more, the formation of set-selective apatite is not only an imitation of bone composition, but also a measure of in vivo bioactivity. Spatial charge could afford an effective means to regulate the deposition of apatite selectively.
4. Conclusions
In summary, this study demonstrated the construction of spatial charge with different piezoelectricity generated on polarized MPK in regulating the set-selective apatite precipitation in SBF. The MPK was built via a simple laser-induced micro-zonal phase transition, forming two types of micro-zones on polarized MPK, H-zone and L-zone. Two zones displayed different piezoelectricity. H-Zone of N-MPK displayed the best growth of set-selective carbonated hydroxyapatite, which was in accorded with the apatite in natural bone. Overall, the concept of manipulating the set-selective apatite deposition via micropatterned piezoelectric materials will inspire future development in biomimetic materials designation. Therefore, spatial charge manipulating the set-selective apatite deposition has provided a method to design biomimetic materials for bone repair. The feasible concept will also inspire future development of biomimetic materials.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 51372087, 51232002, 51541201), the National High Technology Research and Development Program of China (863 Program) (Grant No. 2015AA033502), Science and Technology Planning Project of Guangdong Province, China (Grant No. 2014A010105048), the Natural Science Foundation of Guangdong Province (Grant No. 2015A030313493, 2016A030308014) and State Key Laboratory for Mechanical Behavior of Materials, China (Grant No. 20141607), Technological Projects of Guangzhou, China (No. 201604020110).Notes and references
- Y. Li, T. T. Thula, S. Jee, S. L. Perkins, C. Aparicio, E. P. Douglas and L. B. Gower, Biomacromolecules, 2012, 13, 49–59 CrossRef CAS PubMed
.
- W. X. He, A. K. Rajasekharan, A. R. Tehrani-Bagha and M. Andersson, Adv. Mater., 2015, 27, 2260–2264 CrossRef CAS PubMed
- J. P. Orgel, T. C. Irving, A. Miller and T. J. Wess, PNAS, 2006, 103, 9001–9005 CrossRef CAS PubMed
- C. J. Bettinger, R. Langer and J. T. Borenstein, Angew. Chem., Int. Ed., 2009, 48, 5406–5415 CrossRef CAS PubMed
- J. Kim, W.-G. Bae, H.-W. Choung, K. T. Lim, H. Seonwoo, H. E. Jeong, K.-Y. Suh, N. L. Jeon, P.-H. Choung and J. H. Chung, Biomaterials, 2014, 35, 9058–9067 CrossRef CAS PubMed
- A. Matsugaki, G. Aramoto, T. Ninomiya, H. Sawada, S. Hata and T. Nakano, Biomaterials, 2015, 37, 134–143 CrossRef CAS PubMed
- A. Kurella and N. B. Dahotre, J. Biomater. Appl., 2005, 20, 5–50 CrossRef PubMed
- L. Li, C. Y. Mao, J. M. Wang, X. R. Xu, H. H. Pan, Y. Deng, X. H. Gu and R. K. Tang, Adv. Mater., 2011, 23, 4695–4701 CrossRef CAS PubMed
- G. M. Luz, L. Boesel, A. del Campo and J. F. Mano, Langmuir, 2012, 28, 6970–6977 CrossRef CAS PubMed
- P. Yu, X. J. Zhu, X. L. Wang, S. Y. Wang, W. P. Li, G. X. Tan, Y. Zhang and C. Y. Ning, Adv. Healthcare Mater., 2016, 5, 364–372 CrossRef CAS PubMed
- A. K. Dubey, H. Yamada and K. Kakimoto, J. Appl. Phys., 2013, 114, 124701 CrossRef
- P. X. Zhu, Y. Masuda, T. Yonezawa and K. Koumoto, J. Am. Ceram. Soc., 2003, 86, 782–790 CrossRef CAS
- W. Cui, X. Li, C. Xie, H. Zhuang, S. Zhou and J. Weng, Biomaterials, 2010, 31, 4620–4629 CrossRef CAS PubMed
- T. Kokubo, F. Miyaji, H.-M. Kim and T. Nakamura, J. Am. Ceram. Soc., 1996, 79, 1127–1129 CrossRef CAS
- A. K. Dubey, B. Basu and A. Bandyopadhyay, J. Am. Ceram. Soc., 2014, 97, 481–489 CrossRef CAS
- A. H. Heuer, D. J. Fink, V. J. Laraia, J. L. Arias, P. D. Calvert, K. Kendall, G. L. Messing, J. Blackwell, P. C. Rieke and D. H. Thompson, et al., Science, 1992, 255, 1098–1105 CAS
- F. Nudelman, K. Pieterse, A. George, P. H. H. Bomans, H. Friedrich, L. J. Brylka, P. A. J. Hilbers, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS PubMed
- Y. Wang, T. Azais, M. Robin, A. Vallee, C. Catania, P. Legriel, G. Pehau-Arnaudet, F. Babonneau, M. M. Giraud-Guille and N. Nassif, Nat. Mater., 2012, 11, 724–733 CrossRef CAS PubMed
- B. Ye, X. Luo, Z. Li, C. Zhuang, L. Li, L. Lu, S. Ding, J. Tian and C. Zhou, Mater. Sci. Eng., C, 2016, 68, 43–51 CrossRef CAS PubMed
- X. Niu, R. Fan, F. Tian, X. Guo, P. Li, Q. Feng and Y. Fan, Mater. Sci. Eng., C, 2017, 73, 137–143 CrossRef CAS PubMed
- Y. Zhang, A. A. Gandhi, J. Zeglinski, M. Gregor and S. A. M. Tofail, IEEE Trans. Dielectr. Electr. Insul., 2012, 19, 1151–1157 CrossRef CAS
- S. Itoh, S. Nakamura, M. Nakamura, K. Shinomiya and K. Yamashita, Biomaterials, 2006, 27, 5572–5579 CrossRef CAS PubMed
- S. Nakamura, T. Kobayashi and K. Yamashita, J. Biomed. Mater. Res., Part A, 2004, 68, 90–94 CrossRef PubMed
- Y. F. Tang, C. Wu, Z. X. Wu, L. Hu, W. Zhang and K. Zhao, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed
- W. Chen, Z. Yu, J. Pang, P. Yu, G. Tan and C. Ning, Materials, 2017, 10, 1–12 Search PubMed
- R. Olivares-Navarrete, J. J. Olaya, C. Ramírez and S. E. Rodil, Coatings, 2011, 1, 72–87 CrossRef CAS
- L. Jiang, J. Xing, Z. Tan, J. Wu, Q. Chen, D. Xiao and J. Zhu, J. Mater. Sci., 2016, 51, 4963–4972 CrossRef CAS
- K. Xu, J. Li, X. Lv, J. Wu, X. Zhang, D. Xiao and J. Zhu, Adv. Mater., 2016, 28, 8519–8523 CrossRef CAS PubMed
- M. H. Lee, D. J. Kim, J. S. Park, S. W. Kim, T. K. Song, M. H. Kim, W. J. Kim, D. Do and I. K. Jeong, Adv. Mater., 2015, 27, 6976–6982 CrossRef CAS PubMed
- Y. He, X. Wang, L. Chen and J. D. Ding, J. Mater. Chem. B, 2014, 2, 2220–2227 RSC
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed
- M. Manso, C. Jiménez, C. Morant, P. Herrero and J. M. Martínez-Duart, Biomaterials, 2000, 21, 1755–1761 CrossRef CAS PubMed
- Y. Kuboki, R. Fujisawa, K. Aoyama and S. Sasaki, J. Dent. Res., 1979, 58, 1926–1932 CrossRef CAS PubMed
- K. Wang, F. Z. Yao, W. Jo, D. Gobeljic, V. V. Shvartsman, D. C. Lupascu, J. F. Li and J. Rodel, Adv. Funct. Mater., 2013, 23, 4079–4086 CrossRef CAS
- J. F. Li, K. Wang, B. P. Zhang and L. M. Zhang, J. Am. Ceram. Soc., 2006, 89, 706–709 CrossRef CAS
- F. Bortolani, A. del Campo, J. F. Fernandez, F. Clemens and F. Rubio-Marcos, Chem. Mater., 2014, 26, 3838–3848 CrossRef CAS
- A. K. Dubey, H. Yamada and K.-i. Kakimoto, J. Cryst. Growth, 2013, 382, 7–14 CrossRef CAS
- S. R. Radin and P. Ducheyne, J. Biomed. Mater. Res., 1993, 27, 35–45 CrossRef CAS PubMed
- B. Wopenka and J. D. Pasteris, Mater. Sci. Eng., C, 2005, 25, 131–143 CrossRef
- L. Müller and F. A. Müller, Acta Biomater., 2006, 2, 181–189 CrossRef PubMed
- H. Takadama, H. M. Kim, T. Kokubo and T. Nakamura, J. Biomed. Mater. Res., 2001, 55, 185–193 CrossRef CAS PubMed
- C. C. Ribeiro, I. Gibson and M. A. Barbosa, Biomaterials, 2006, 27, 1749–1761 CrossRef CAS PubMed
- J. Xu, L. L. Liu, P. Munroe and Z. H. Xie, J. Mater. Chem. B, 2015, 3, 4082–4094 RSC
- L. Müller, E. Conforto, D. Caillard and F. A. Müller, Biomol. Eng., 2007, 24, 462–466 CrossRef PubMed
- C. Rey, B. Collins, T. Goehl, I. R. Dickson and M. J. Glimcher, Calcif. Tissue Int., 1989, 45, 157–164 CrossRef CAS PubMed
- A. Roguska, M. Pisarek, M. Andrzejczuk, M. Dolata, M. Lewandowska and M. Janik-Czachor, Mater. Sci. Eng., C, 2011, 31, 906–914 CrossRef CAS
- E. Ajami and K.-F. Aguey-Zinsou, J. Funct. Biomater., 2012, 3, 327–348 CrossRef CAS PubMed
- R. N. Panda, M. F. Hsieh, R. J. Chung and T. S. Chin, J. Phys. Chem. Solids, 2003, 64, 193–199 CrossRef CAS
Footnote |
| † Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |