
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ambient temperature induced Diels–Alder crosslinked networks based on controlled methacrylate copolymers for enhanced thermoreversibility and self-healability†
Sungmin Junga,
So Young Kimb,
Jin Chul Kimb,
Seung Man Nohb and
Jung Kwon Oh *a
*a
aDepartment of Chemistry and Biochemistry, Concordia University, Montreal, Quebec, Canada H4B 1R6. E-mail: john.oh@concordia.ca
bResearch Center for Green Fine Chemicals, Korea Research Institute of Chemical Technology, Ulsan 44412, Republic of Korea
First published on 17th May 2017
Abstract
The development of covalently-crosslinked dynamic (reversible) networks has been extensively explored due to their built-in ability to self-repair damages, which prevents catastrophic failure of high-performance materials. Here, we report effective thermoreversible crosslinked networks fabricated at room or mild temperature from reactive blends consisting of a controlled methacrylate copolymer having maleimide pendants (CoPMA) and a trifunctional furan (TFu). Well-defined CoPMA is synthesized by controlled radical polymerization, ensuring predetermined chain lengths with narrow molecular weight distribution, and following post-modification through deprotection of pendant furan-protected maleimide groups, ensuring conversion to the corresponding maleimide pendants. The well-defined CoPMA is reactive to different amounts of TFu as a model polyfuran to form dynamic networks at ambient temperature through a click type Diels–Alder (DA) reaction. Comprehensive analyses indicate that the mole ratio of furan/maleimide group is an important parameter that significantly influences network flexibility. The developed dynamic networks crosslinked with thermo-labile DA cycloadduct linkages enable the dissociation and recombination through retro DA (rDA)/DA reactions at elevated temperatures. Such thermoreversibility, combined with balanced flexibility, allows for the development of dynamic network exhibiting effective self-healability at as low as 110 °C with great mechanical property restoration as well as healing elasticity through reversible restoration of viscoelastic properties.
Introduction
Thermosets are crosslinked polymers that exhibit enhanced mechanical performance, thermal stability, dimensional stability, and solvent resistance. Due to these unique properties, polymeric thermosets have been a choice of effective coating and composite materials useful in a variety of industrial and biomedical fields that require high performance, such as transportation, aerospace, electronics, and tissue engineering.1–3 Having been designed with irreversibility, however, conventional thermosets could not be melted or dissolved in organic solvents for further processing even at elevated temperatures. This drawback has limited the recyclability of irreversibly-crosslinked thermosets. Further to the recyclability, the self-healability is highly desired because the property allows for the cracks or cuts of thermosets to be autonomously self-repaired. A promising solution that have been extensively explored is to develop dynamic (or reversible) crosslinked networks that retains self-healing upon external damages, thus improving their lifetime, energy efficiency, and environmental impact.4–6 Taking the benefits from an initial approach based on microcapsule or vascular network technologies,7–9 the promising approach involves the incorporation of reversible covalent or supramolecular crosslinkages into the design of dynamic thermosets. In addition to non-covalent interactions (or reversible physical crosslinking),10–30 the dynamic covalent bonds (or reversible chemical crosslinking)31–42 and recently their combination43–45 have shown the repeatable and multiple occurrence of self-healing in intrinsic self-healing systems.Typical dynamic chemical crosslinkages based on disulfide, hindered urea, and alkoxyamine chemistries are composed of flexible bonds. In contrast, the cycloadducts formed by DA reaction are based on newly-formed carbon–carbon bonds between an electron-rich diene and an electron-poor dienophile. Consequently, the thermally-induced DA-crosslinked networks provide enhanced mechanical properties, compared with other networks.46,47 Further, the formed DA linkages in the networks can be dissociated to the corresponding diene and dienophile through rDA reaction within the networks at elevated temperatures. Utilizing rDA and following DA reactions, the DA-crosslinked networks can be displaced by their dissociation and reformed by reconnection through void filling-thermoreversible process upon controlled heating. Because of the maleimide/furan pair being highly reactive to cycloaddition as well as being shifted of its DA/rDA equilibrium at low temperatures, thermally-reversible DA/rDA chemistry utilizing maleimide as a dienophile and furan as a diene has been extensively explored to develop thermally-induced self-healable networks. Most of DA-crosslinked networks were fabricated through step-growth polyaddition or coupling reaction of polymaleimides and polyfurans.48–57
Of our interests are thermo-labile networks based on reactive methacrylate copolymers. The nature of the polymethacrylate backbones of carbon–carbon single bonds, the high molecular weight of polymeric chains, and the facile copolymerization with soft monomers allow for tuning the balance of rigidity and flexibility of methacrylate networks. Early reports describe the synthesis of methacrylate copolymers having furan pendants by chain-growth polymerization58–60 or post-modification;61 these copolymers then reacted with a bismaleimide crosslinker to form DA-crosslinked networks. Recently, the synthesis of methacrylate copolymers having both furan and furan-protected maleimide pendants62–64 as well as a methacrylate homopolymer having pendant furan-protected maleimide groups65 has been reported. To exhibit enhanced mechanical properties and thermoreversible self-healability, however, these systems required elevated temperatures (>120 °C) for the formation of DA-crosslinked networks. This is mainly because of the deprotection of furan-protected maleimide groups to generate the corresponding maleimide groups in the copolymers, which in situ react with polyfurans. We have envisioned that the synthesis of methacrylate copolymers with bare maleimide pendants could allow for the fabrication of DA-crosslinked networks with polyfurans at ambient or even room temperatures. This approach could be beneficial in constructing high-performance materials for industrial applications; more specifically our polymers could be considered as self-healable adhesives in surface coating formulations that can protect automobiles and heavy-duty metals.
In this work, we have explored an approach utilizing controlled radical polymerization and post-modification to synthesize a series of methacrylate copolymers having different densities of reactive maleimide and inert ethylhexyl pendants (CoPMAs). As a model polyfuran, a new tri-functional furan (TFu) was also synthesized by a click-type thermally-induced thiol–ene reaction. The occurrence of DA-crosslinking reactions of the maleimide and the furan groups in reactive blends composed of controlled CoPMA with TFu at ambient temperatures was characterized with not only gel content measurement and sol–gel transition but also spectroscopic and thermal analysis. The resultant DA-crosslinked networks formed at different mole ratios of furan/maleimide groups were further analyzed for thermoreversibility and self-healability as well as self-healing viscoelastic and mechanical properties through DA/rDA reactions (Scheme 1).
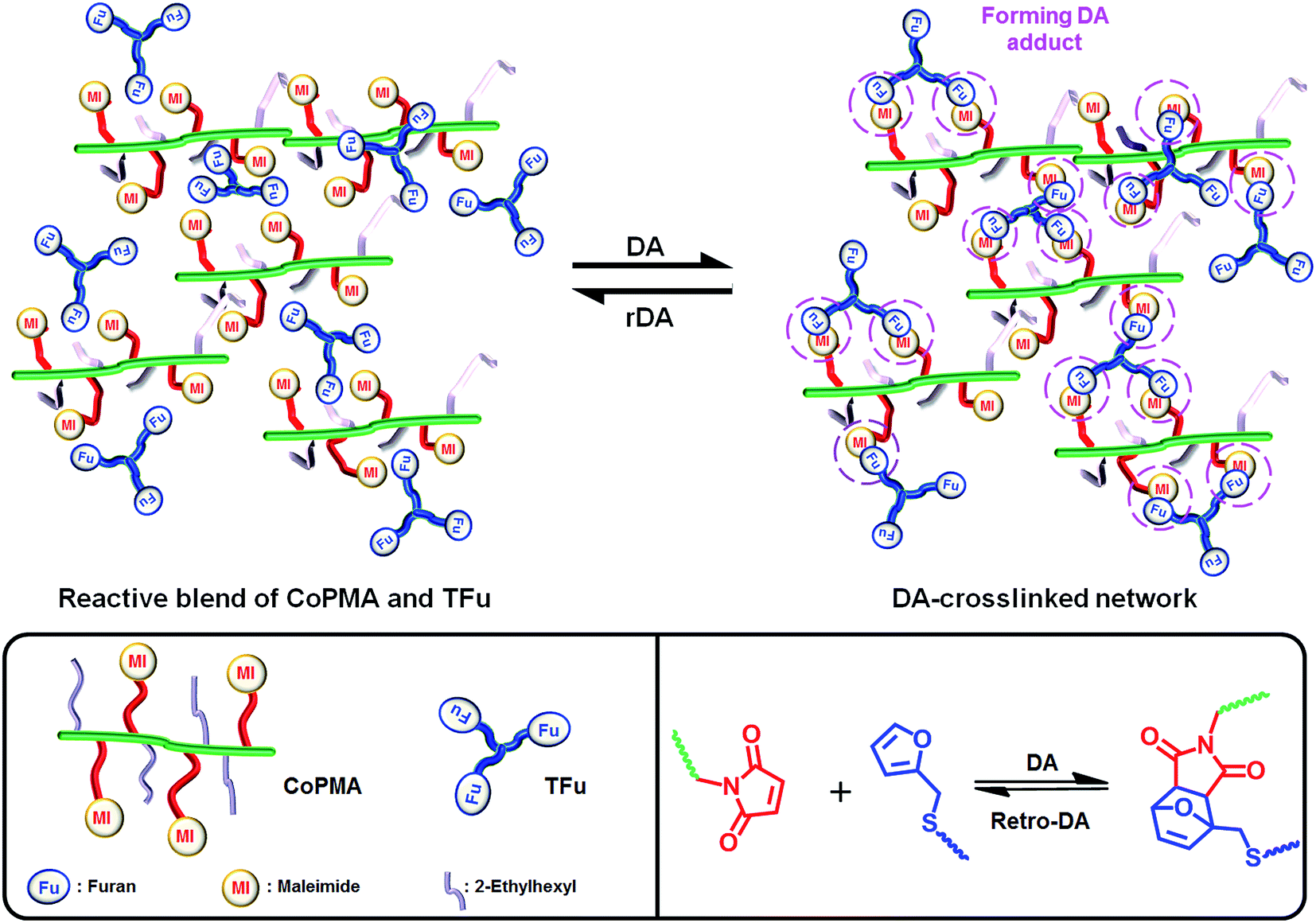 | ||
| Scheme 1 Thermoreversible DA/rDA reactions for self-healing of dynamic networks prepared from a reactive mixture containing CoPMA having reactive maleimide pendants and TFu. | ||
Experimental
Instrumentation and analyses
1H-NMR spectra were recorded using a 500 MHz Varian spectrometer. The CDCl3 singlet at 7.26 ppm and DMSO-d6 quintet at 2.5 ppm were selected as the reference standards. Molecular weight and molecular weight distribution were determined by gel permeation chromatography (GPC) with a Viscotek VE1122 pump and a refractive index (RI) detector. Two PolyAnalytik columns (PAS-103L and 105L, designed to determine molecular weight up to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 g mol−1) were used with THF as eluent at 30 °C at a flow rate of 1 mL min−1. Linear poly(methyl methacrylate) standards were used for calibration. Aliquots of polymer samples were dissolved in THF. The clear solutions were filtered using a 0.25 μm PTFE filter to remove any solvent-insoluble species. A drop of anisole was added as a flow rate marker.
000000 g mol−1) were used with THF as eluent at 30 °C at a flow rate of 1 mL min−1. Linear poly(methyl methacrylate) standards were used for calibration. Aliquots of polymer samples were dissolved in THF. The clear solutions were filtered using a 0.25 μm PTFE filter to remove any solvent-insoluble species. A drop of anisole was added as a flow rate marker.
Differential scanning calorimetry (DSC)
Thermal properties including glass transition temperature (Tg) of polymers and crosslinked films were measured with a TA Instruments DSC Q20 differential scanning calorimeter. Samples were dried under vacuum for 24 h at room temperature to remove residual solvents. Temperature range was from −70 to 200 °C with heating and cooling cycles conducted at a rate of 10 °C min−1 (cycles: cool to −70 °C, heat up to 200 °C (1st run), cool to −70 °C, heat up to 200 °C (2nd run), and cool to 25 °C). The Tg values were determined from the 2nd heating run.Thermogravimetric analysis (TGA)
TGA measurements were carried out using a TA instruments Q50 analyzer. Typically, dried samples (5–10 mg) were placed into a platinum pan and heated from 25 to 800 °C at a heating rate of 20 °C min−1 under nitrogen flow.Materials
3-Phenyl-1-propanol (PP-OH, 98%), α-bromoisobutyryl bromide (Br-iBuBr, 98%), tin(II) 2-ethylhexanoate (Sn(II)EH2, >92.5%), triethylamine (Et3N, 99.5%), furfuryl mercaptan (Fu-SH, 99%), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, ≥98.0%), 4-(dimethylamino)pyridine (DMAP, ≥99%), 2,2′-azobis(2-methylbutyronitrile) (AMBN, >98%), trimethylolpropane trimethacrylate (TMA, technical grade), and furan (≥99%) from Sigma Aldrich as well as maleic anhydride (>98%) from Fluka were used as received. 2-Ethylhexyl methacrylate (EHMA, 98%) purchased from Aldrich was passed through basic alumina column to remove inhibitors before polymerization. Tris(2-pyridylmethyl)amine (TPMA) was synthesized as described elsewhere.66 A methacrylate having pendant furan-protected maleimide (MIMA)64 was synthesized with a slight modification as described in ESI.†Synthesis of 3-phenylpropyl 2-bromo-2-methylpropanoate (PP-Br)
Br-iBuBr (9.3 g, 40.4 mmol) was added to a clear solution consisting of PP-OH (5.0 g, 35.7 mmol), DMAP (0.45 g, 3.67 mmol), and Et3N (5.6 g, 55.0 mmol) in methylene chloride (128 mL) at room temperature. The resulting mixture was stirred at room temperature for 14 h, and then diluted with diethyl ether. The yellow precipitates as byproducts were removed by vacuum filtration and organic solvents were removed using a rotary evaporation. The residues were purified by silica column chromatography with a mixture of ethyl acetate/hexane (1/5 v/v). The product was collected as the first of a total three bands from a silica gel column: yield = 9.3 g (89%) and Rf = 0.3 on silica (1/5 ethyl acetate/hexane).Synthesis of CoPMIMA by ATRP
For the synthesis of CoPMIMA-2 as a typical example, PP-Br (0.12 g, 0.43 mmol), MIMA (1.5 g, 5.41 mmol), EHMA (1.07 g, 5.41 mmol), [Cu(II)TPMABr]Br (11.1 mg, 22 μmol), TPMA (12.6 mg, 43 μmol), and anisole (7.76 mL) were mixed in a 25 mL Schlenk flask. The resulting mixture was deoxygenated by purging under nitrogen for 40 min. A nitrogen-prepurged Sn(II)EH2 solution in anisole (0.5 mL, 0.234 g mL−1) was injected into reaction mixture and placed in an oil bath preheated at given temperature (35 °C) to initiate polymerization. Aliquots were taken from polymerization mixture at regular intervals to determine conversion using 1H-NMR. Polymerization was stopped by cooling the reaction mixture in an ice bath and exposing to air.For purification, the as-synthesized polymer solution was precipitated from MeOH three times to remove unreacted monomers. The precipitate was dissolved in minimal amount of chloroform and then passed through a column filled with basic alumina to remove residual copper species. Solvents were removed by rotary evaporation and the residues were dried in a vacuum oven at room temperature for 24 h.
Similar procedure was applied with the use of PP-Br (123.4 mg, 433 μmol), MIMA (1.5 g, 2.7 mmol), EHMA (1.61 g, 8.1 mmol), [Cu(II)TPMABr]Br (11.1 mg, 22 μmol), TPMA (12.6 mg, 43.3 μmol), and anisole (9.3 mL) for the synthesis of CoPMIMA-1, and PP-Br (123.4 mg, 433 μmol), MIMA (1.5 g, 8.1 mmol), EHMA (0.54 g, 2.7 mmol), [Cu(II)TPMABr]Br (11.1 mg, 22 μmol), TPMA (12.6 mg, 43.3 μmol), and anisole (6.1 mL) for synthesis of CoPMIMA-3.
Synthesis of CoPMA
The purified, dried CoPMIMA (2 g) dissolved in DMSO (5 mL) was heated at 120 °C under stirring. After 3 h, the polymer solution was cooled down to room temperature and then precipitated from a mixture of MeOH/water (1.5/1 v/v). The precipitates were dried in vacuum oven at room temperature for 16 h.Synthesis of TFu
TMA (3.0 g, 16 mmol), AMBN (0.52 g, 3.2 mmol), Fu-SH (20.9 mL, 207 mmol), and anhydrous DMF (31 mL) were placed in a Schlenk flask. The resulting mixture was then stirred at 75 °C for 24 h, and then cooled down to room temperature. Solvents and excess Fu-SH were removed under reduced pressure. Residues were precipitated from a mixture of deionized water/methanol (1/1.5 v/v) twice. The precipitates were purified by silica column chromatography using a mixture of ethyl acetate/hexane (1/5 v/v). The product was collected as the third of a total five bands from a silica gel column; yield = 2.6 g (40%) and Rf = 0.3 on silica (1/5 ethyl acetate/hexane).Preparation of crosslinked films through DA reactions
A series of reactive blends of CoPMA-2 and TFu at the mole equivalent ratio of pendant furan/maleimide group = 1/1, 2/1, and 3/1 were prepared by mixing the fixed amount of CoPMA-2 (100 mg, corresponding to 0.24 mmol of maleimide group) with different amounts of TFu (54, 81, and 162 mg, corresponding to 0.08 mmol, 0.12 mmol, and 0.24 mmol of furan group) in anisole (0.24, 0.27, and 0.4 mL). Their aliquots were dropped on glass slides placed at room temperature for 24 h and in an oven preset at 60 °C for 12 h. The resultant crosslinked films were scraped using a knife for further characterization.Gel content measurements
Pieces of dried films prepared above (approximately 10 mg) were mixed with THF (10 mL) for 48 h. THF was carefully decanted and precipitates were dried in vacuum oven for 2 days. Gel content was calculated as the weight ratio of dried precipitates to initial films.Sol–gel–sol transitions
Mixtures of CoPMA-2 (90 mg, 0.21 mmol of maleimide group) with different amounts of TFu in anisole at 43 wt% were sealed and placed at room temperature for 12 h to form free-standing gels in vials. For the gel–sol transition experiment, a piece of the dried gel (110 mg) was mixed with DMF (1 mL) at 11 wt% and heated up at 150 °C for 5 h, and then cooled down to room temperature.Optical microscopy
Fresh cuts were made using a sharp blade on surfaces of the crosslinked films and annealed in an oven at given temperatures (85, 110, and 150 °C) for 5 h to 72 h depending on samples. The occurrence of self-healing in cross-linked networks was followed by an optical microscope (Olympus BX51) coupled with a digital camera.Rheology measurements
Viscoelastic properties (G′ and G′′ moduli) of DA-crosslinked films were measured on a DHT-2 rheometer (TA Instruments, USA) in amplitude oscillatory shear mode with parallel plate geometry (8 mm diameter). The elasticity of the crosslinked materials were tested by applying cyclic changes of amplitude oscillatory from 5% strain for 1500 s to 100% strain for 500 s. The gap was set to obtain an axial force of 1.5 N at room temperature and this cycle is repeated three times.Tensile measurements
Mechanical properties of polymer samples before and after healing were measured using a tensile testing machine (MCT-2150, AND, Japan) at a crosshead speed of 50 mm min−1 at room temperature. To prepare dumbbell-shaped specimens (10 mm × 30 mm × 3 mm) by a conventional solution casting method, a reactive mixture of CoPMA-2 and TFu (furan/maleimide mole ratio = 3/1) in anisole at 40 wt% was poured into a mold and placed in a convection oven preset at 60 °C for 24 h. After being recovered from the mold, the cast specimens were tested for tensile measurements. For quantitative analysis of self-healing, a single cut of 20 μm wide and 3 mm long on the center of the specimens was made using a sharp cutting blade (Dorco, Korea). The resultant specimens were placed at 110 °C for 2 h and then 60 °C for 24 h (a self-healing condition) and then subjected to tensile tests.Results and discussion
Synthesis and thermal properties of CoPMA having maleimide pendants
Fig. 1a illustrates our approach consisting of two steps to synthesize new multivalent methacrylate copolymers bearing pendant maleimide groups (CoPMA).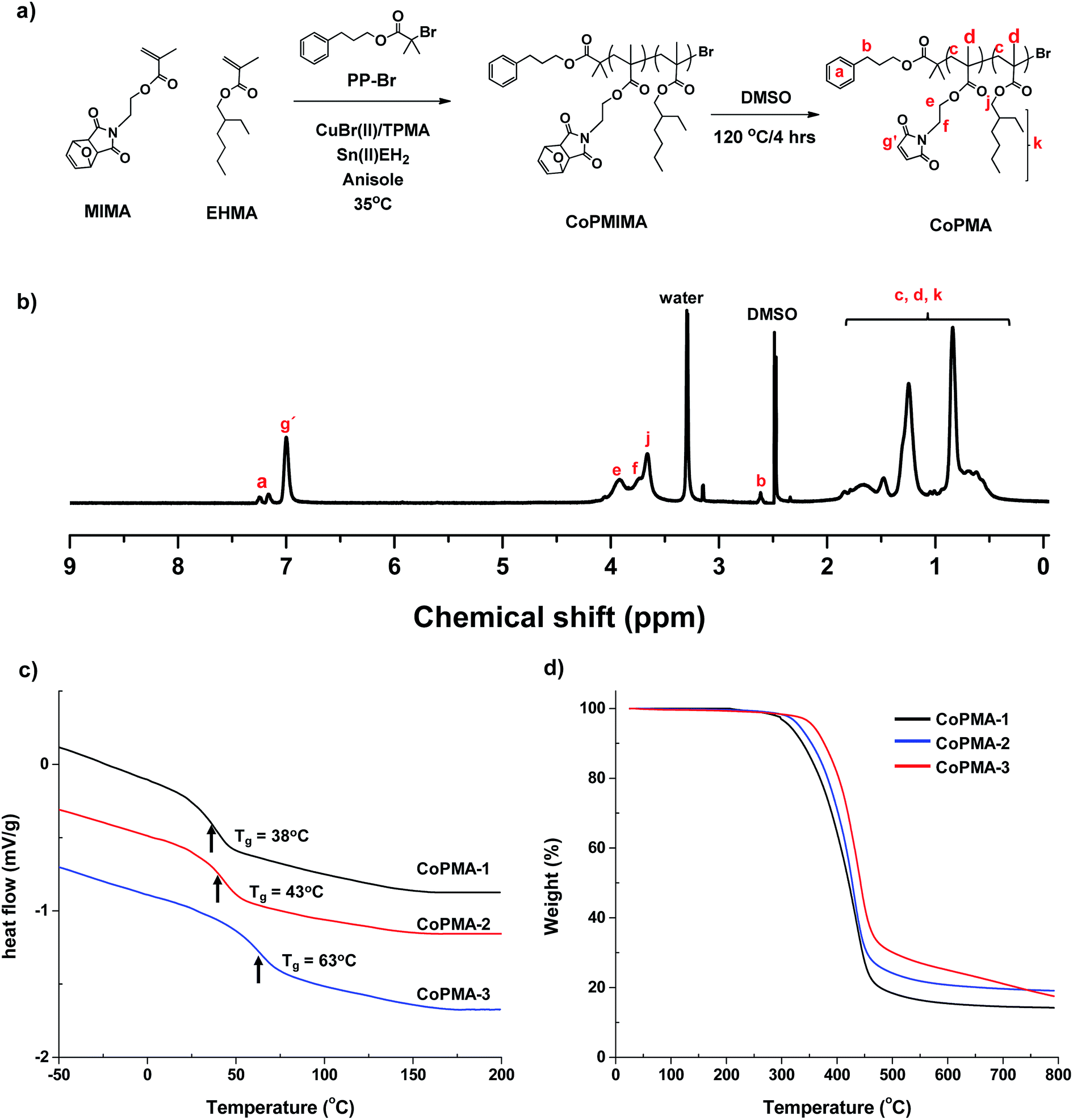 | ||
| Fig. 1 Synthetic route to CoPMA bearing pendent maleimide groups (a), 1H-NMR spectrum of CoPMA-2 as an example in DMSO-d6 (b), DSC (c), and TGA (d) histograms of CoPMA. | ||
The first step is the synthesis of random copolymers having furan-protected maleimide groups. First, PP-Br as an initiator was synthesized by a facile coupling reaction of PP-OH with Br-iBuBr in a high yield. Its chemical structure was analyzed with 1H and 13C-NMR spectra (Fig. S1†). Then, Activators ReGenerated by Electron Transfer (ARGET) process for Atom Transfer Radical Polymerization (ATRP) was examined for a monomer mixture of MIMA and EHMA at different mole ratios in the presence of PP-Br and Cu(II)/TPMA active complexes. The degree of polymerization (DP) as the initial mole ratio of [MIMA + EHMA]0/[PP-Br]0 was designed to be 50/1. The polymerization conditions include [PP-Br]0/[CuBr(II)]0/[TPMA]0/[Sn(II)EH2]0 = 1/0.05/0.15/0.4 in anisole at 35 °C. Polymerization was stopped after 2–3 h when conversion of MIMA reached 50–60%, based on 1H-NMR analysis. After the purification by extensive precipitation from MeOH and following filtration with a basic alumina column, the resulting CoPMIMA copolymers were characterized for chemical structures by 1H-NMR spectroscopy and molecular weight by GPC. As a typical example with CoPMIMA-2, 1H-NMR spectrum in Fig. S2† shows the presence of furan-protected maleimide groups at 6.5, 5.1, and 2.9 ppm, ethylhexyl ester groups at 3.6 ppm, and phenyl groups in initiating moieties at 7.1–7.3 ppm. The DP was determined by their integral ratio to be 16 for MIMA and 16 for EHMA units. GPC analysis indicates molecular weight as the number average molecular weight (Mn) = 5.9 kg mol−1, with molecular weight distribution as Mw/Mn = 1.5. In similar ways, CoPMIMA-1 and CoPMIMA-3 were analyzed (Fig. S3 and S4† for 1H-NMR spectra and Fig. S5† for GPC traces). Table 1 summarizes the characteristics and results of CoPMIMA copolymers.
| Entry no. | MIMA (mol%) | DPb | Mn (kg mol−1) | Mw/Mnc | CoPMA Tgd (°C) | |||
|---|---|---|---|---|---|---|---|---|
| Feed | Copolymerb | MIMA | EHMA | 1H-NMR | GPCc | |||
| a Condition for ARGET ATRP: [MIMA + EHMA]0/[PP-Br]0/[CuBr(II)]0/[TPMA]0/[Sn(II)EH2]0 = 50/1/0.05/0.15/0.4, [monomer] = 1.76 M; 35 °C.b Determined by 1H-NMR.c Determined by THF GPC with PMMA standards.d Determined by DSC. | ||||||||
| CoPMIMA-1 | 25 | 40 | 13 | 20 | 7.7 | 5.6 | 1.5 | 38 |
| CoPMIMA-2 | 50 | 50 | 16 | 16 | 7.3 | 5.9 | 1.5 | 43 |
| CoPMIMA-3 | 75 | 75 | 20 | 7 | 7.1 | 7.4 | 1.7 | 63 |
The second step is the deprotection of furan groups from furan-protected maleimide pendants to synthesize CoPMA. This step was simply achieved by heating the solution of the purified CoPMIMA dissolved in DMSO at 120 °C for 3 h. 1H-NMR spectrum of CoPMA-2 in Fig. 1b shows the disappearance of characteristic peaks corresponding to furan aromatic protons and the appearance of new peak (g′) corresponding to maleimide double bond protons, suggesting >99% efficiency. Similar results were observed for CoPMA-1 and CoPMA-3 (Fig. S3 and S4†). These results confirm the successful synthesis of CoPMA with different densities of pendant maleimide groups.
The synthesized CoPMIMA and CoPMA were examined for their thermal properties using DSC and TGA. For all series of CoPMIMA, their glass transitions could not be clearly detected. However, their DSC diagrams exhibit a strong thermal transition at 100–180 °C, which is due to the loss of furan groups from furan-protected maleimide pendants. Such loss was also clearly observed in their TGA traces at 150–180 °C (Fig. S6†). In contrast, DSC traces of CoPMA copolymer in Fig. 1c exhibit single transitions at 38, 43, and 63 °C; these Tg values increased with an increasing density of maleimide pendants in copolymers. TGA traces in Fig. 1d show a progressive decomposition of the copolymers to 500 °C. Similarly, the on-set temperature of starting thermal decomposition increased from 300 to 350 °C, suggesting that the enhanced thermal stability with an increasing amount of maleimide pendants in the copolymers.
Synthesis of a trifunctional furan (TFu)
As illustrated in Fig. 2a, a thermally-induced radical addition of Fu-SH to TMA in the presence of AMBN, an azo-type radical initiator, was examined to synthesize TFu. Excess Fu-SH should be used to minimize undesired side reaction through free radical homopolymerization of methacrylate groups in TFu. 1H-NMR spectrum in Fig. 2b shows the appearance of new peaks corresponding to furan groups (a, b, c) and the complete disappearance of peaks corresponding to methacrylate groups at 5–6 ppm. In addition to 1H-NMR, 13C-NMR spectrum in Fig. S7† also confirms the successful synthesis of TFu.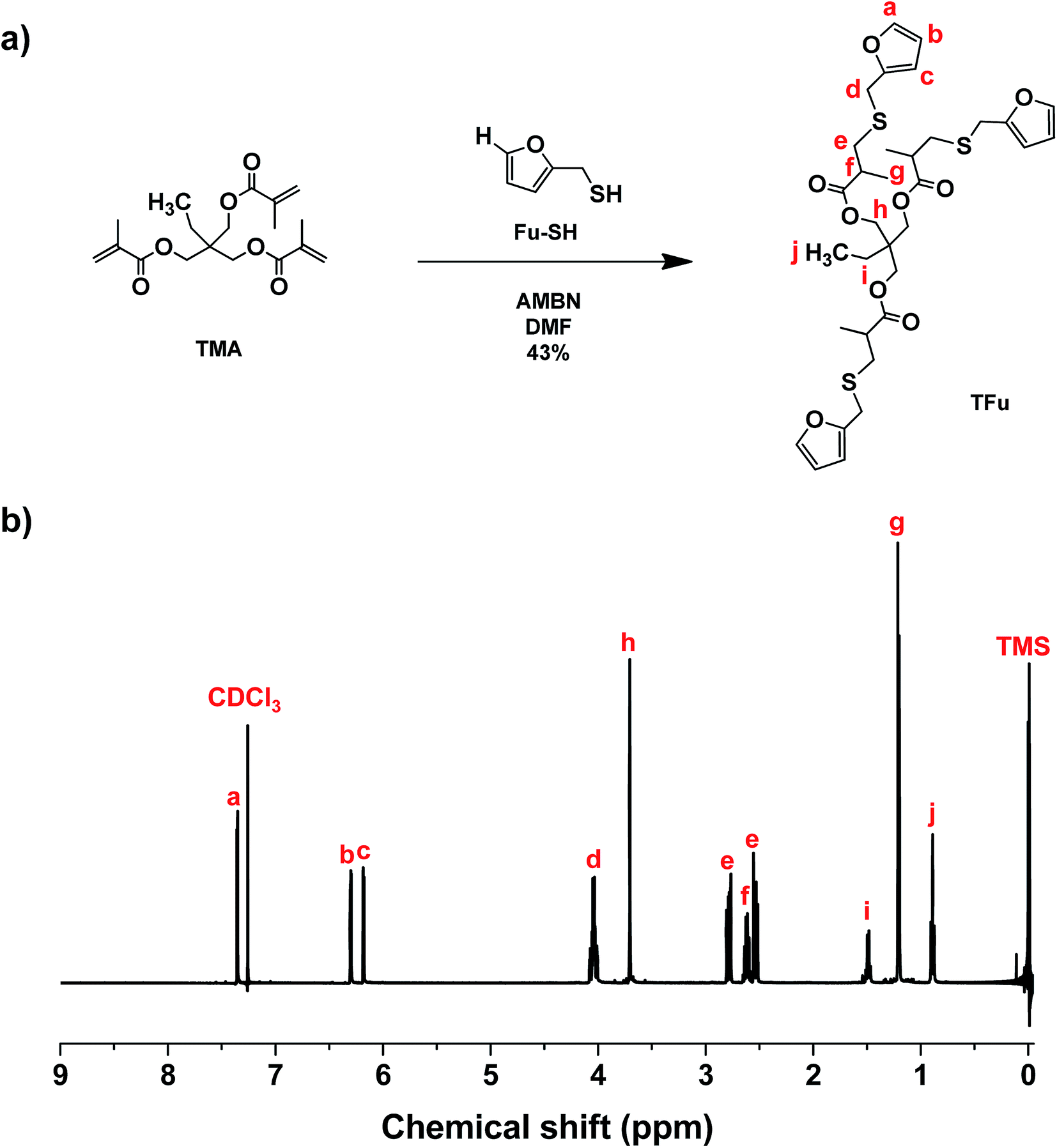 | ||
| Fig. 2 For TFu, synthetic scheme (a) and 1H-NMR spectrum in CDCl3 (b). | ||
Preparation and characterization of DA-crosslinked networks
As illustrated in Scheme 1, thermally-induced DA reactions between pendant maleimide groups in CoPMA and furan groups in TFu yields DA-crosslinked networks. With the selected CoPMA-2 (50 mol% maleimide group), the different mole equivalent ratios of furan to maleimide group in reactive blends of CoPMA and TFu were varied to adjust the mobility of polymeric chains and flexibility of crosslinked networks, thus effective self-healing and enhanced mechanical properties.A series of reactive blends consisting of CoPMA-2 and TFu at the different mole equivalent ratios of furan/maleimide group = 1/1, 2/1, and 3/1 were prepared in anisole at 43 wt%. Aliquots of the mixtures were cast on glass plates at room temperature and 60 °C. Note that the formed films denote as F1M1, F2M1, and F3M1 for 1/1, 2/1, and 3/1 furan/maleimide ratio. They were then characterized with gel content measurements, spectroscopic and thermal analysis. For gel content determination based on solubility in an organic solvent, aliquots of the cast films were soaked in THF for 2 days and insoluble species were isolated and dried. Note that gels here can be defined as insoluble species in THF. As seen in Fig. 3d, the gel content was 97% for the cast film with 1/1 furan/maleimide at 60 °C, which could be attributed to the occurrence of DA-crosslinking reactions between pendant maleimide groups of CoPMA and furfuryl groups of TFu in the reactive mixtures. It decreased to 92% and further to 78% as the furan/maleimide ratio increased to 2/1 and further to 3/1 (excess furan). Promisingly, the films cast at room temperature had the gel content to be 93, 87, and 67% corresponding to 1/1, 2/1, and 3/1 ratio. These quantities appeared to be smaller, compared to those for the films cast at 60 °C, presumably due to greater efficiency of DA reactions at 60 °C than room temperature. Nevertheless, our system consisting of CoPMA and TFu enabled to the formation of thermo-labile networks induced even at room temperature. Further, the feasibility to the formation of DA-crosslinked networks at room temperature was confirmed by sol–gel transition experiments. As seen in Fig. 3a–c, the reactive solutions consisting of CoPMA-2 and TFu turned to standing gels at room temperature in 12 h or earlier.
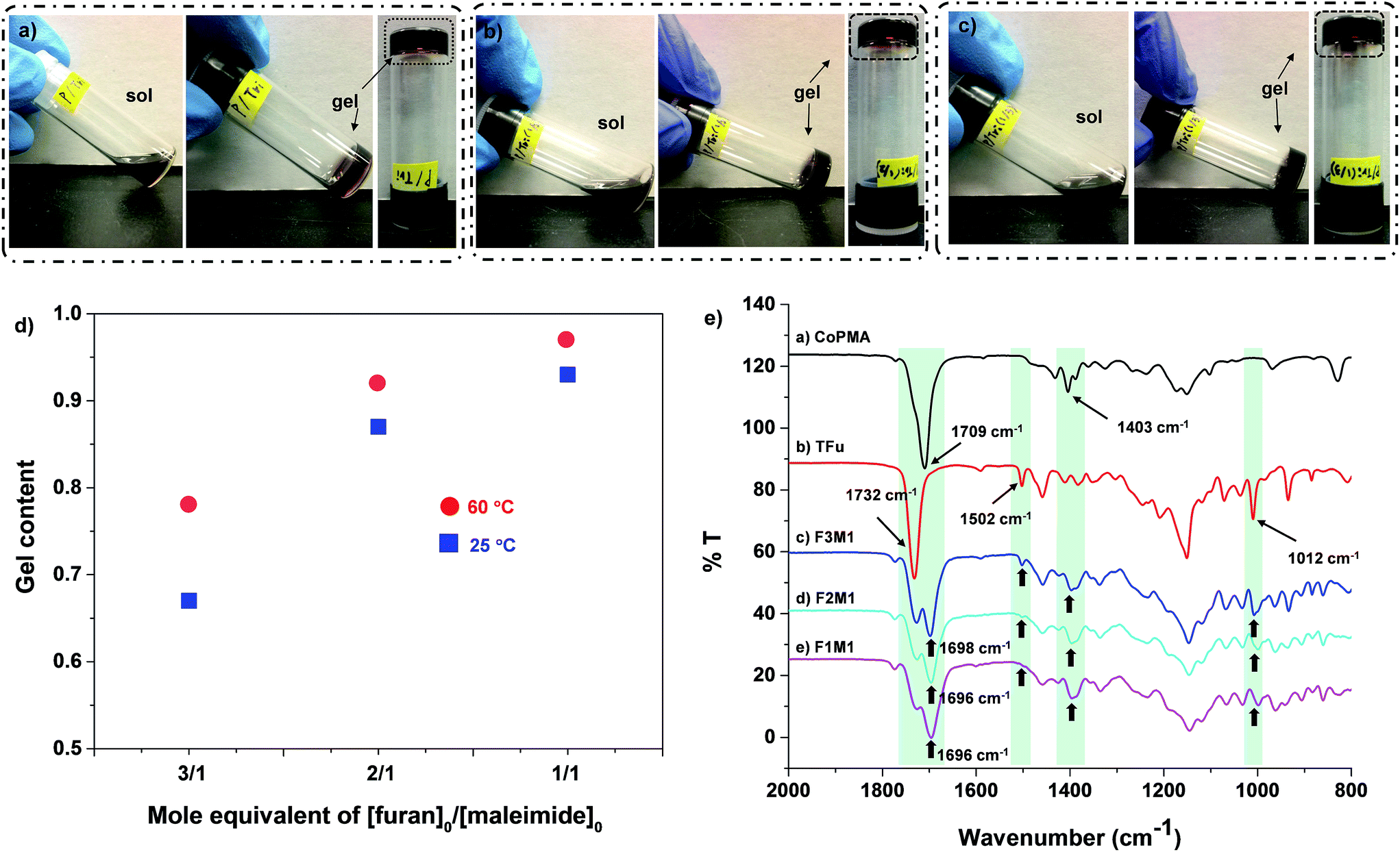 | ||
| Fig. 3 Digital images illustrating gelation of a series of reactive mixtures of CoPMA-2 and TFu at different mole equivalent ratios of furan/maleimide = 1/1 (a), 2/1 (b), and 3/1 (c) in anisole at 43 wt% at room temperature for 12 h as well as characterization of DA-crosslinked films from reactive blends of CoPMA-2 and TFu at different mole ratios of furan/maleimide = 3/1 (F3M1), 2/1 (F2M1), and 1/1 (F1M1); gel contents cast at room (25 °C) and moderate temperature (60 °C) (d) and FT-IR spectra of films cast at room temperature, compared with CoPMA and TFu (e). | ||
Spectroscopic and thermal analysis of DA-crosslinked networks
FT-IR was used to characterize the DA-crosslinked films denoted as F1M1, F2M1, and F3M1 cast at room temperature. Fig. 3e shows their FT-IR spectra, compared with those of individual CoPMA and TFu. First, the peak at 1502 cm−1 and 1012 cm−1 corresponding to furan rings decreased to disappeared as the furan/maleimide ratio decreased from 3/1 (excess furan) to 1/1. Second, the characteristic peak at 1403 cm−1 for maleimide groups in CoPMA was not detected; rather, broad peaks appeared in DA-crosslinked films. Third, the peaks corresponding to ester carbonyl groups appeared to be broad with one relatively small peak at 1732 cm−1 corresponding to residual furan groups and the other new peak at 1696–1698 cm−1. Such changes are attributed to the formation of DA cycloadducts in the cast films.Further, DSC and TGA were used to examine the thermal property of the DA-crosslinked films cast at 25 and 60 °C (Fig. S8†). DSC diagrams of the films cast at 60 °C show a large endotherm peak between 90 and 200 °C for all films, suggesting the occurrence of rDA reaction which can be originated from the cleavage of DA-linkage. Similar DSC results were obtained for the films cast at room temperature. In addition, TGA traces suggest that all the DA-crosslinked films are thermally stable up to 300 °C, regardless of the composition of furan and maleimide groups and casting temperature.
Thermoreversibility and self-healing studies
First, the occurrence of thermally-induced rDA reaction in DA-crosslinked networks was examined with the standing gels formed from F1M1 in anisole at 43% (see Fig. 4a). As seen in Fig. 4b–d, a piece of the gel was mixed with DMF in a vial. The resulting mixture was heated at 150 °C for 5 h to form sol, suggesting the occurrence of rDA reaction. After being cool down to room temperature and placed for 48 h, the sol solution turned to free-standing gel again through DA reaction, proving thermoreversibility of our system.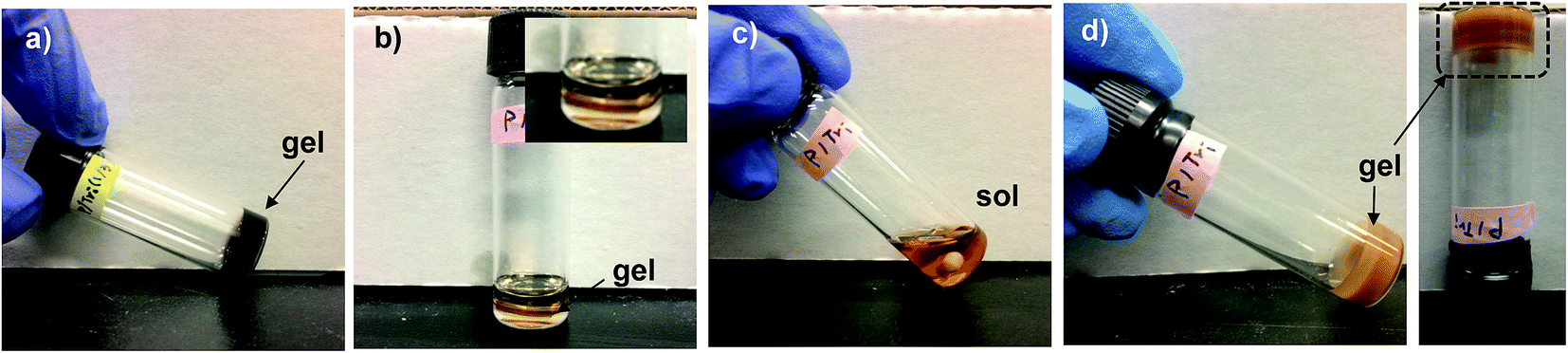 | ||
| Fig. 4 To illustrate the occurrence of rDA/DA reaction in F1M1 gel, digital images of standing gel formed at 25 °C/12 h (a), a mixture of a piece of gel with DMF (b), sol formed upon heating at 150 °C for 5 h (c), and gel formed upon cooling to room temperature (d). | ||
Given promising results, three films cast at 60 °C were studied for self-healing at 150 °C. First, micron-sized cuts were made using a blade. They were placed in an oven preset at 150 °C for 5 h, promoting the occurrence of rDA reactions, and then 80 °C for 2 h, allowing for the occurrence of DA reactions. Fig. 5 shows optical microscope images of the cuts on the films before and after self-healing. Note that cuts were presented as black-colored straight parts in all the images before healing. For the F1M1 and F2M1 films, the cuts with 20–30 μm wide did not appeared to be completely healed; however, note that F2M1 film shows better healing, compared with F1M1 film (Fig. 5a and b). For the F3M1 film, promisingly, the cuts with 50–100 μm wide (larger than those in F1M1 and F2M1 films) completely healed through void-filling and following rDA/DA crosslinking (Fig. 5c). As seen in Fig. 5c (inset), the F3M1 film is tough, but elastic to be vented and recovered in shape. This property could be attributed to a combination of dangling chains and crosslinking density through the formulation of excess furan groups in the networks. These results suggest the better approach in the design of self-healing networks with excess functional groups in soft components. Furthermore, the efficiency (or kinetics) of self-healing rely on the width and length of cuts.
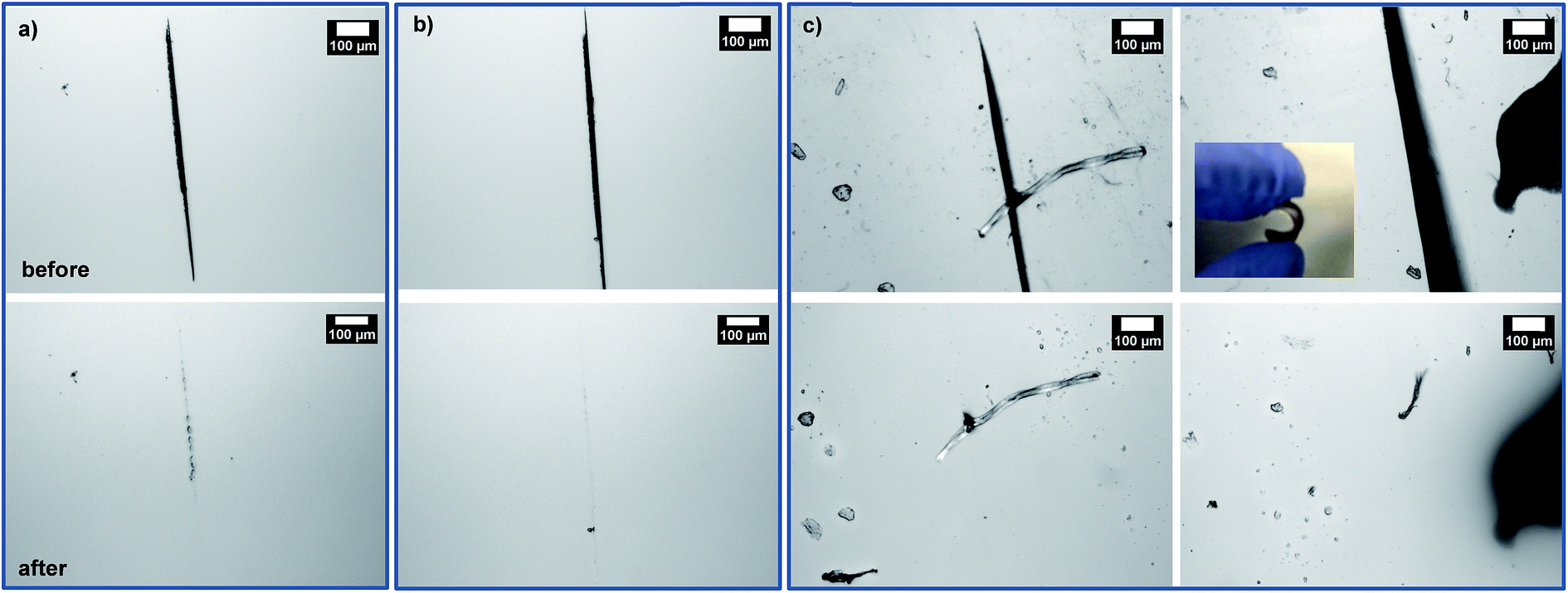 | ||
| Fig. 5 Optical microscope images of cuts on DA-crosslinked F1M1 (a), F2M1 (b), and F3M1 (c) films before and after being annealed at 150 °C for 5 h and following 80 °C for 2 h. Inset of (c) digital image of F3M1 film. | ||
In another set, the feasibility to self-healing through the occurrence of dynamic rDA and DA reactions at low temperatures was examined, with the selected F3M1 film. As seen in Fig. 6a and b, when the films were annealed at 110 °C for 5 h, the occurrence of healing was observed. Further, the extent of thermal healing was relied on the width of cuts. At temperature as low as 85 °C, the important healing was not observed over 3 days (Fig. S9†), suggesting that enough thermal energy could be required for the occurrence of rDA reactions in our system consisting of F3M1.
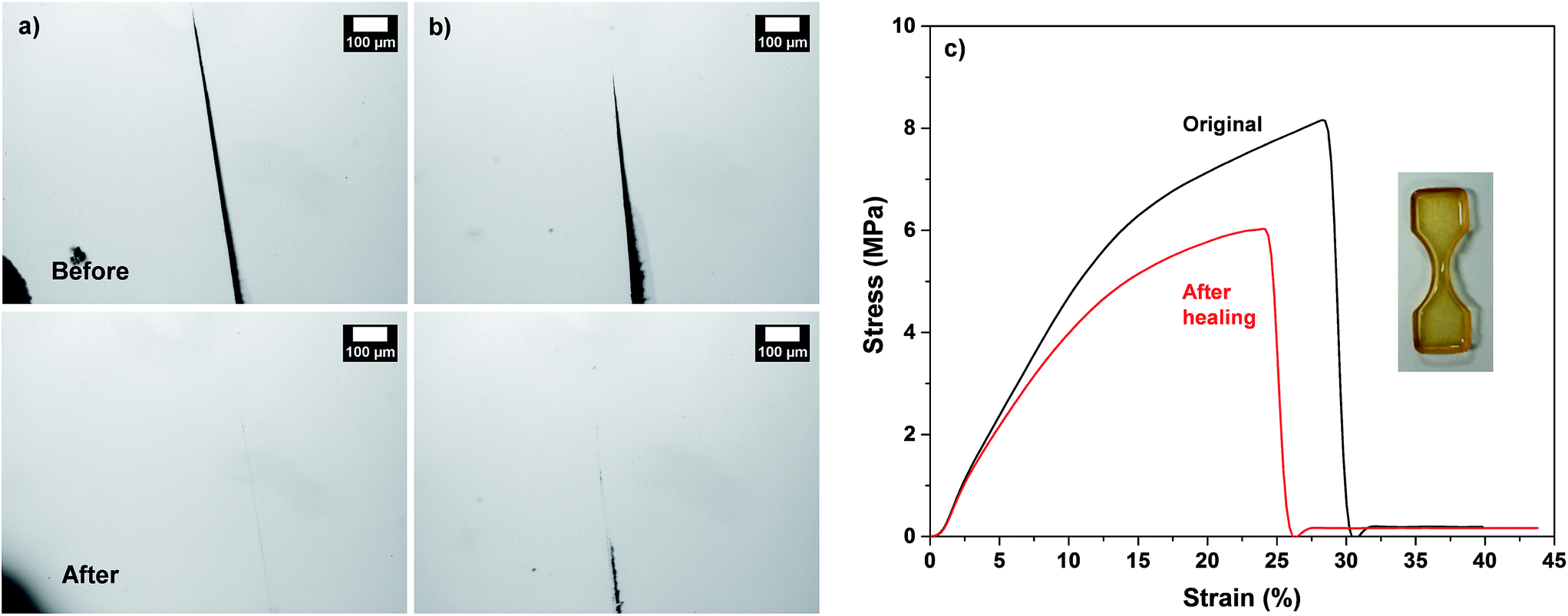 | ||
| Fig. 6 For DA-crosslinked F3M1 films cast at 60 °C, optical microscope images with different sizes of cuts (a and b) and stress–strain curves (c) before and after being annealed at 110 °C and following 80 °C. | ||
Self-healing viscoelastic and mechanical properties
Quantitative analysis of self-healing behavior was followed with tensile measurements. Dumbbell-shaped specimens were prepared from F3M1 reactive mixtures (inset in Fig. 6c). A small micron-sized crack was made using a blade on the center of the specimens. The scratched specimens were subjected to thermal annealing process at 110 °C for 2 h and then 60 °C for 24 h to promote self-healing through rDA/DA reactions. Fig. 6c compares stress and strain curves of DA-crosslinked films before and after self-healing. The tensile strength at break decreased from 8 to 6 MPa as well as its elongation also decreased from 30 to 26% by 4%, indicating >85% healing efficiency upon one healing event. The decrease in tensile strength and elongation at break is presumably attributed to the existence of invisible defects in cuts that could be caused by rearrangement of polymeric chains (void-filling) and incomplete rDA/DA reactions during self-healing process at 110 °C.Further to get an insight into elasticity for self-healing of the crosslinked polymeric networks, their viscoelastic properties were measured using a rheometer with a cyclic change of oscillation force: 5% strain for 1500 s to 100% strain for 500 s. As seen in Fig. 7, the elastic modulus (G′) of the crosslinked films was 4.5 × 105 Pa when a 5% strain was applied. Upon the change of oscillation force to 100% strain, the G′ modulus dropped to 1.2 × 104 Pa. Then, the G′ modulus was restored when the oscillation force was recovered back to 5% strain. Such a reversible restoration of G′ modulus was able to be repeated several times, exhibiting self-healing elasticity of the crosslinked polymeric networks.
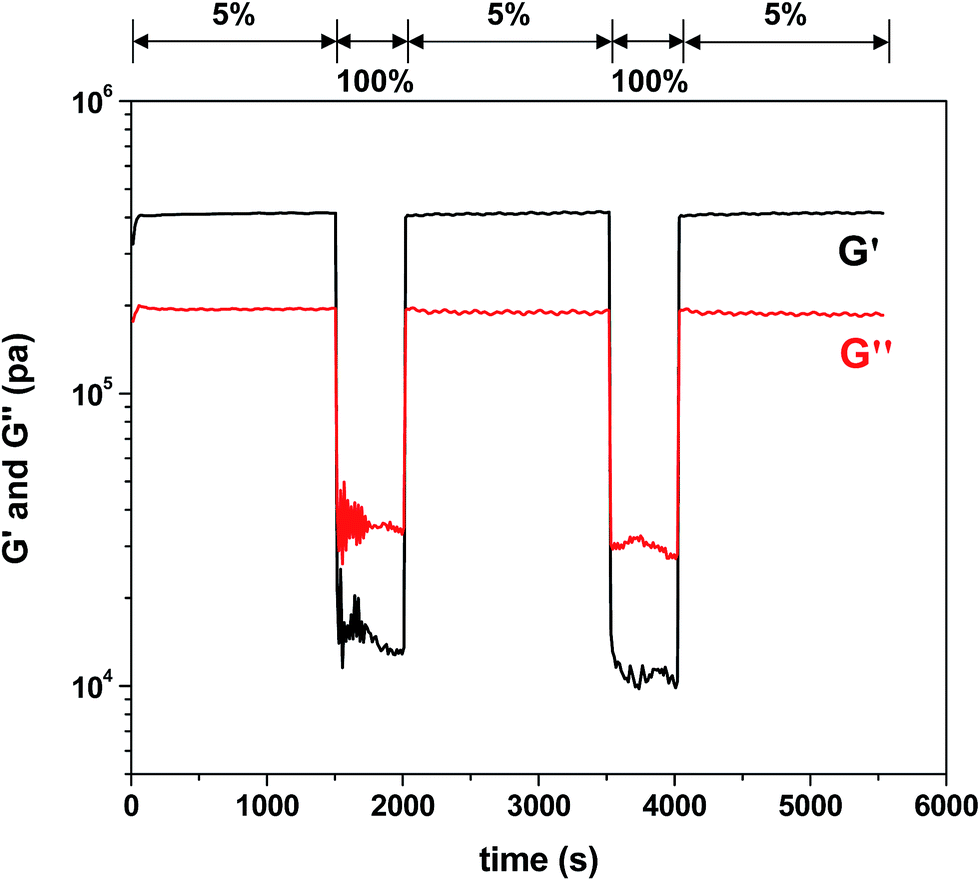 | ||
| Fig. 7 Viscoelastic properties upon a cyclic change of oscillation force: 5% strain for 1500 s to 100% strain for 500 s for crosslinked F3M1 films cast at 60 °C. | ||
Conclusion
New random CoPMA with different densities of pendant maleimide and ethylhexyl groups were synthesized by two steps, including ARGET ATRP to CoPMIMA precursors and following deprotection of pendant furan-protected maleimide groups to the corresponding maleimide groups. Their structural and thermal analysis indicate that the synthesized CoPMA copolymers had the molecular weights mostly ranging at 7–8 kg mol−1 by 1H-NMR, while their thermal transitions varied from 38 to 63 °C with an increasing amount of maleimide groups from 40 to 75 mol% in the copolymers. Reactive blends of the resultant CoPMA with TFu at different mole ratios of furan/maleimide groups allowed for the fabrication of thermo-labile networks with adjustable network flexibility at room and moderate temperatures (60 °C), confirmed by gel content, sol–gel transition, spectroscopic and thermal analysis. Thermoreversible self-healing studies through optical microscopy, tensile and viscoelastic measurements suggest that the use of excess of TFu enhanced the mobility and flexibility of the networks; ultimately, the thermoreversible networks had excellent self-healability with great mechanical strength recovery as well as self-healing elasticity with reversible restoration of viscoelastic properties.Acknowledgements
Authors greatly thank for financial supports from Canada Research Chair (CRC) Award and Korea Research Institute of Chemical Technology (KRICT). Additionally, a partial support from The Ministry of Trade, Industry & Energy Korea (Industrial Technology Innovation Program No. 10067082) is greatly acknowledged. JKO is a Tier II CRC (renewed) in Nanobioscience. Authors thank So Young An for her helpful discussions.References
- M. Cabodi, N. W. Choi, J. P. Gleghorn, C. S. D. Lee, L. J. Bonassar and A. D. Stroock, J. Am. Chem. Soc., 2005, 127, 13788 CrossRef CAS PubMed.
- N. W. Choi, M. Cabodi, B. Held, J. P. Gleghorn, L. J. Bonassar and A. D. Stroock, Nat. Mater., 2007, 6, 908 CrossRef CAS PubMed.
- M. Ooka and H. Ozawa, Prog. Org. Coat., 1994, 23, 325 CrossRef CAS.
- B. J. Blaiszik, S. L. B. Kramer, S. C. Olugebefola, J. S. Moore, N. R. Sottos and S. R. White, Annu. Rev. Mater. Res., 2010, 40, 179 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973 RSC.
- S. Y. An, D. Arunbabu, S. M. Noh, Y. K. Song and J. K. Oh, Chem. Commun., 2015, 51, 13058 RSC.
- D. G. Shchukin, Polym. Chem., 2013, 4, 4871 RSC.
- Y.-K. Song and C.-M. Chung, Polym. Chem., 2013, 4, 4940 RSC.
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223 CrossRef CAS.
- R. Vaiyapuri, B. W. Greenland, H. M. Colquhoun, J. M. Elliott and W. Hayes, Polym. Chem., 2013, 4, 4902 RSC.
- L. R. Hart, J. L. Harries, B. W. Greenland, H. M. Colquhoun and W. Hayes, Polym. Chem., 2013, 4, 4860 RSC.
- Z. Wei, J. He, T. Liang, H. Oh, J. Athas, Z. Tong, C. Wang and Z. Nie, Polym. Chem., 2013, 4, 4601 RSC.
- J. Zheng, P. Xiao, W. Liu, J. Zhang, Y. Huang and T. Chen, Macromol. Rapid Commun., 2016, 37, 265 CrossRef CAS PubMed.
- X. Yan, D. Xu, J. Chen, M. Zhang, B. Hu, Y. Yu and F. Huang, Polym. Chem., 2013, 4, 3312 RSC.
- M. Krogsgaard, M. A. Behrens, J. S. Pedersen and H. Birkedal, Biomacromolecules, 2013, 14, 297 CrossRef CAS PubMed.
- Z. Wang and M. W. Urban, Polym. Chem., 2013, 4, 4897 RSC.
- S. Basak, J. Nanda and A. Banerjee, Chem. Commun., 2014, 50, 2356 RSC.
- F. Potier, A. Guinault, S. Delalande, C. Sanchez, F. Ribot and L. Rozes, Polym. Chem., 2014, 5, 4474 RSC.
- Z. Wang, W. Fan, R. Tong, X. Lu and H. Xia, RSC Adv., 2014, 4, 25486 RSC.
- C.-H. Li, C. Wang, C. Keplinger, J.-L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X.-Z. You and Z. Bao, Nat. Chem., 2016, 8, 618 CrossRef CAS PubMed.
- Y. Chen and Z. Guan, Polym. Chem., 2013, 4, 4885 RSC.
- Y. Chen and Z. Guan, Chem. Commun., 2014, 50, 10868 RSC.
- Y. Lin and G. Li, J. Mater. Chem. B, 2014, 2, 6878 RSC.
- J. Hentschel, A. M. Kushner, J. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2012, 51, 10561 CrossRef CAS PubMed.
- S. Chen, N. Mahmood, M. Beiner and W. H. Binder, Angew. Chem., Int. Ed., 2015, 54, 10188 CrossRef CAS PubMed.
- X. Yang, H. Yu, L. Wang, R. Tong, M. Akram, Y. Chen and X. Zhai, Soft Matter, 2015, 11, 1242 RSC.
- P. Michael, D. Doehler and W. H. Binder, Polymer, 2015, 69, 216 CrossRef CAS.
- D. Zhu, Q. Ye, X. Lu and Q. Lu, Polym. Chem., 2015, 6, 5086 RSC.
- C. Xu, L. Cao, B. Lin, X. Liang and Y. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 17728 CAS.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 4218/1 CAS.
- H. Ying and J. Cheng, J. Am. Chem. Soc., 2014, 136, 16974 CrossRef CAS PubMed.
- S. Y. An, S. M. Noh, J. H. Nam and J. K. Oh, Macromol. Rapid Commun., 2015, 36, 1255 CrossRef CAS PubMed.
- Z. P. Zhang, M. Z. Rong, M. Q. Zhang and C.-e. Yuan, Polym. Chem., 2013, 4, 4648 RSC.
- C. e. Yuan, M. Z. Rong, M. Q. Zhang, Z. P. Zhang and Y. C. Yuan, Chem. Mater., 2011, 23, 5076 CrossRef CAS.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138 CrossRef CAS PubMed.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098 CrossRef CAS.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492 CrossRef CAS PubMed.
- H. Meng, P. Xiao, J. Gu, X. Wen, J. Xu, C. Zhao, J. Zhang and T. Chen, Chem. Commun., 2014, 50, 12277 RSC.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536 CrossRef CAS.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955 RSC.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142 CrossRef CAS.
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jiang and Y. Chen, ACS Macro Lett., 2012, 1, 275 CrossRef CAS.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabanero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237 RSC.
- D. Doehler, H. Peterlik and W. H. Binder, Polymer, 2015, 69, 264 CrossRef CAS.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133 RSC.
- M. Glassner, G. Delaittre, M. Kaupp, J. P. Blinco and C. Barner-Kowollik, J. Am. Chem. Soc., 2012, 134, 7274 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698 CrossRef CAS PubMed.
- E. B. Murphy, E. Bolanos, C. Schaffner-Hamann, F. Wudl, S. R. Nutt and M. L. Auad, Macromolecules, 2008, 41, 5203 CrossRef CAS.
- R. J. Sheridan and C. N. Bowman, Polym. Chem., 2013, 4, 4974 RSC.
- J. Zhao, R. Xu, G. Luo, J. Wu and H. Xia, J. Mater. Chem. B, 2016, 4, 982 RSC.
- Y. Heo and H. A. Sodano, Adv. Funct. Mater., 2014, 24, 5260 CrossRef CAS.
- G. B. Lyon, A. Baranek and C. N. Bowman, Adv. Funct. Mater., 2016, 26, 1477 CrossRef CAS.
- S. Wu, J. Li, G. Zhang, Y. Yao, G. Li, R. Sun and C. Wong, ACS Appl. Mater. Interfaces, 2017, 9, 3040 CAS.
- Y. Heo, M. H. Malakooti and H. A. Sodano, J. Mater. Chem. A, 2016, 4, 17403 CAS.
- L.-T. T. Nguyen, T. T. Truong, H. T. Nguyen, L. Le, V. Q. Nguyen, T. Van Le and A. T. Luu, Polym. Chem., 2015, 6, 3143 RSC.
- J. Zhao, R. Xu, G. Luo, J. Wu and H. Xia, Polym. Chem., 2016, 7, 7278 RSC.
- A. A. Kavitha and N. K. Singha, Macromolecules, 2010, 43, 3193 CrossRef CAS.
- A. A. Kavitha and N. K. Singha, ACS Appl. Mater. Interfaces, 2009, 1, 1427 CAS.
- N. B. Pramanik, G. B. Nando and N. K. Singha, Polymer, 2015, 69, 349 CrossRef CAS.
- S. Jung, J. T. Liu, S. H. Hong, D. Arunbabu, S. M. Noh and J. K. Oh, Polymer, 2017, 109, 58 CrossRef CAS.
- R. K. Bose, J. Koetteritzsch, S. J. Garcia, M. D. Hager, U. S. Schubert and S. van der Zwaag, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1669 CrossRef CAS.
- J. Koetteritzsch, M. D. Hager and U. S. Schubert, Polymer, 2015, 69, 321 CrossRef CAS.
- J. Koetteritzsch, S. Stumpf, S. Hoeppener, J. Vitz, M. D. Hager and U. S. Schubert, Macromol. Chem. Phys., 2013, 214, 1636 CrossRef CAS.
- D. Arunbabu, S. M. Noh, J. H. Nam and J. K. Oh, Macromol. Chem. Phys., 2016, 217, 2191 CrossRef CAS.
- G. J. P. Britovsek, J. England and A. J. P. White, Inorg. Chem., 2005, 44, 8125 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04222a |
| This journal is © The Royal Society of Chemistry 2017 |