
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bio-mimicking TiO2 architectures for enhanced photocatalytic activity under UV and visible light†
Iman Hashemizadeha,
Daniel C. W. Tsang b,
Yun Hau Ngcf,
Zhijie Wud,
Vladimir Golovko*e and
Alex C. K. Yip*af
b,
Yun Hau Ngcf,
Zhijie Wud,
Vladimir Golovko*e and
Alex C. K. Yip*af
aDepartment of Chemical and Process Engineering, University of Canterbury, Christchurch, New Zealand. E-mail: alex.yip@canterbury.ac.nz
bDepartment of Civil and Environmental Engineering, Hong Kong Polytechnic University, Hong Kong, China
cSchool of Chemical Engineering, University of New South Wales, Australia
dState Key Laboratory of Heavy Oil Processing, The Key Laboratory of Catalysis of CNPC, China University of Petroleum, Beijing, 102249, China
eDepartment of Chemistry, University of Canterbury, Christchurch, New Zealand. E-mail: vladimir.golovko@canterbury.ac.nz
fThe Joint Laboratory for Energy and Environmental Catalysis, City University of Hong Kong, Hong Kong, China
First published on 10th August 2017
Abstract
Green leaves are responsible for natural photosynthesis in plants and their unique structures offer the most efficient blueprint for artificial materials in terms of solar energy capture and utilisation. The full architecture of the leaf photosystem was successfully replicated at both the nano and micro levels using biotemplating with TiO2. This approach resulted in a highly porous structure that can be used as a photocatalyst with enhanced properties such as improved visible light-harvesting ability. Scanning and transmission electron microscopy images of the final products confirmed that the detailed microscale framework and nanostructures, such as the chloroplast and the thylakoids were well replicated. Biotemplated artificial TiO2 leaves with the architecture of Camellia tree leaves outperformed well-known P25 TiO2 in photocatalytic degradation of methylene blue dye under visible light: more than twofold in the case of blue (440 nm) and ca. one and a half times under green (515 nm) light. Also, the carbon dioxide yield of photocatalytic oxidation of ethanol catalysed by the biotemplated TiO2 material was approximately 1.3 times higher than the CO2 produced by P25 under green light. We attributed this enhanced visible light photocatalytic performance to the light-harvesting features and to the high surface area imparted by the interconnected nanosheets (replicating the thylakoids) resulting from our improved biotemplating method. The method reported in this work presents a facile route for the production of synthetic inorganic materials which possess morphologies similar to that present in the natural template materials.
1 Introduction
The efficiency of solar-assisted reactions strongly depends on the activity of the photocatalysts. Semiconductors possess a band structure in which the conduction band is separated from the valence band by a band gap with a suitable width for the absorption of photons. When the energy from incident light is greater than that of the band gap, electron hole pairs are formed.1–4 Currently, more than 130 materials and derivatives are known to function as photocatalysts, including simple metal oxides, niobates, tantalates, metal nitrides, metal sulphides and covalent network solids such as C3N4. Among all of these materials, titanium dioxide (TiO2)-based photocatalysts have been considered as one of the best materials due to their wide availability, low toxicity, good durability, high stability and, most importantly, favourable energies of photogenerated electrons and holes, which results in high photocatalytic efficiency.5–7A vast range of TiO2 structures, including nanosheets, nanowires, nanotubes, and hierarchical architectures, have been developed to improve both light harvesting efficiency and visible light photocatalytic activity via structuring of titania.8–13 Commonly studied nanoscale TiO2 can be synthesised via a number of different methods, including sol–gel, micellar and solvothermal syntheses, direct oxidation, chemical or physical vapour deposition, electrodeposition or anodization, and microwave synthesis.14 The sol–gel method is widely used to produce small, well-defined particles even at relatively low temperatures. This method also enables the introduction of secondary components (e.g., active metals) into the reaction mixture, which allows the synthesis of application-oriented TiO2-based materials.15 Although titanium tetrachloride (TiCl4) was used as a precursor in the initial studies of TiO2 synthesis, this approach resulted in the formation of chloride ions as impurities in the product. Recent synthetic methods have focused on the use of titanium alkoxides, Ti(OR)4, where R is an alkyl group, as precursors. The precursor is dissolved in water or in a mixture of solvents and is hydrolysed to give Ti(OH)xR4−x, which subsequently undergoes condensation through either substitution or addition reactions to produce TiO2. Both halide and alkoxide compounds of titanium hydrolyse rapidly in the presence of water, causing difficulty in controlling the end product. Thus, post-synthesis modifications are necessary for tuning the properties of the end product. The most common technique is to use a solution containing acids and alcohol, in which the former serve as a catalyst and peptising agent. The use of alcohol as a solvent, on the other hand, can substantially reduce the temperature required in the TiO2 synthesis process.16,17 However, using structural facilitating agents to control the shape, size and crystallinity of the end product still remains as a challenge. For example, the use of fluoride and other fluorine-containing ions to control the particle shape of TiO2 often results in surface-bound fluorine in the end product, which negatively influences the photocatalytic properties of TiO2.18,19
Recently, bio-inspired materials have emerged as a potential area of research for developing advanced functional systems with a higher standard of environmental compatibility, recyclability and energy efficiency.20 Natural materials offer a combination of advantageous properties, such as sophistication, miniaturization, hierarchical organization, hybridization, resistance and adaptability, which have evolved through billions of years of the natural selection processes. The morphologies of the biological structures ranging from the nanometre to the millimetre scale inspire the design of artificial materials that aim at advanced applications, such as energy capture, storage and conversion.21,22
Many bio-mimicking methods have recently been developed, including the following approaches:
(a) Replication, which involves use of biological templates to replicate the morphology via chemical transformations or physical processes, such as nanocasting and nanolithography;
(b) Self-assembly, which relies on using biological systems (usually at the nanoscale) as building blocks for a controllable assembly into complex structures through electrostatic, metal–ligand and inter-biomolecular interactions;
(c) Encapsulation, whereby biomolecules or living cells are isolated from their superstructures and encapsulated into manmade matrices with complex structures to form bioreactors.22,23
The templating method has recently emerged as an effective approach to overcome the difficulty in controlling the morphology of the end product.24–26 The main advantage of this technique is that the final structure can be easily tuned through selection of a template with an appropriate morphology. By contrast, the conventional sol–gel synthesis is strongly affected by the conditions of the synthesis, including the pH of the solution, drying, subsequent thermal treatments etc. Importantly, controlled self-assembly of the primary nanoparticulate building blocks into larger-scale structures is a major challenge in this case. In efforts to develop TiO2-based photocatalysts with improved light-harvesting functions, researches have recently proposed a few biotemplating techniques for reproduction of the natural hierarchical structures.16,27–29 However, further research on the details of the biotemplating process and the relationship between the morphology of obtained product and its photocatalytic performance is required.
The photosystem of the green leaves naturally evolved both at the molecular level and at the structural level (nanoscale and microscale frameworks), making it overall a superior light-harvesting structure.23,30 The lens-like epidermal cells focus incident light, and cylinder cells of palisade parenchyma serve as channels for the focused light, where the channels are arranged parallel to the incident light direction. The less-regular arrangement of spongy mesophyll leads to light scattering and long effective light path lengths in the intercellular air spaces of the leaf.31–34 Although such complex porous architectures trap sunlight within the leaves, the main antenna of plants' photosystem is the chloroplast. The thylakoid cylindrical stacks (granum) inside the chloroplast are 3-dimensional scaffolds made of interconnected nanolayers. These structures harvest the light efficiently by directing the sunlight to the chlorophyll molecules which are the reactive centres of the plant photochemical machine.35–37 The overall structure of the leaf photosystem is schematically illustrated in Fig. 1.
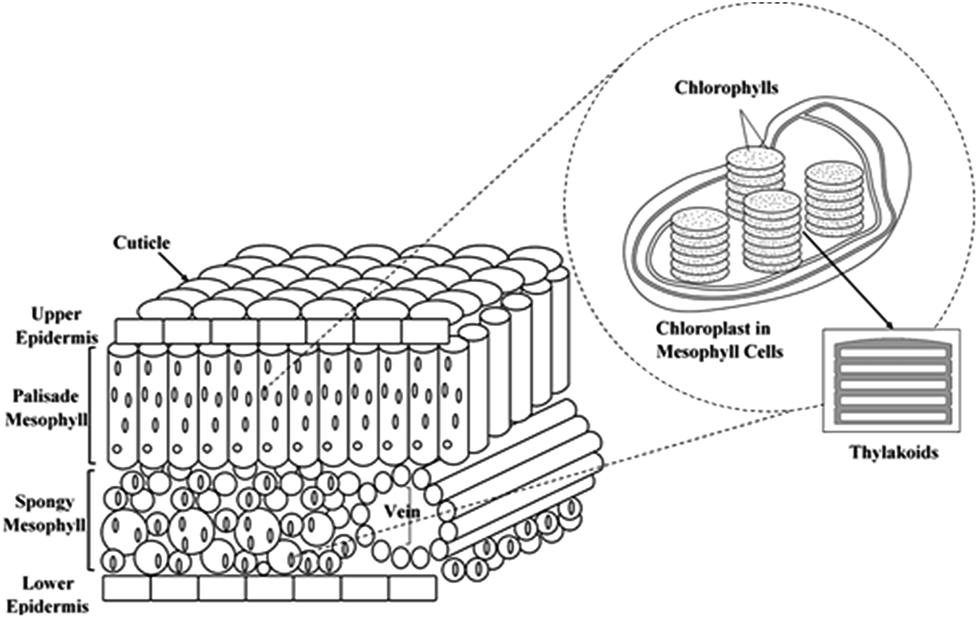 | ||
| Fig. 1 The microscale-to-nanoscale structures of leaf photosystem. | ||
In this research, the structure of Camellia sinensis tree leaves from the family Theaceae, belonging to C4 plants possessing the Kranz anatomy, has been replicated using TiO2 from the microscale to the nanoscale. The photocatalytic activity of the artificial TiO2 leaves at different level of biomimicked structures was studied through methylene blue degradation and the photocatalytic oxidation of ethanol under UV and visible light irradiation. We aim to demonstrate that presence of the structures analogous to the stack-layered nanostructures and mesoporous architectures present in natural leaves offer a unique opportunity for efficient use of novel TiO2 structures under visible light.
2 Experimental
2.1 Materials
All reagents were of analytical grade and were purchased from Sigma-Aldrich. Methylene blue was supplied by Sekolah Tinggi Teknologi Tekstil, Indonesia. All reagents were used as received. All glassware was oven dried and experiments were performed using Schlenk line technique to avoid contact with ambient air when required.2.2 Synthesis procedure of bio-mimicked TiO2 architectures
The replication of nano- to micro-architectures of leaf photosystem was done by ion-exchange method to mimic the natural chlorophyll breakdown process. Fresh leaves were first washed with MilliQ water, dried and cut into ca. 2 by 2 cm pieces. Then, 2.00 g of cut leaves was acid treated with a 5 v/v% HCl solution under inert atmosphere (Schlenk line technique) with vigorous stirring (250 rpm) using magnetic stirrer at room temperature for 3 h until the leaves changed the colour to yellow-brown. Ion-exchange was subsequently carried out in a 5 v/v% TiCl3 solution in water with stirring (250 rpm) under inert atmosphere at ambient temperature overnight. The Ti3+ exchanged leaves were then coated with titania using a sol–gel approach modified from the previously reported method.38 The treated leaves were collected on the Büchner funnel, washed with MilliQ water (3 × 50 mL) and dried in the vacuum desiccator over P2O5. Resulting dry leaves were suspended with stirring (250 rpm) in dry isopropanol (98 mL) and the mixture was stirred for 12 h to ensure soaking/exchange. After water exchange or soaking step, the leaves were filtered off and quickly cut into smaller pieces (ca. 1 × 1 mm) to allow more efficient diffusion of titanium isopropoxide within the leaf structure. Titanium isopropoxide (1.8 mL, 6.5 mmol) was then added using a syringe to the suspension of dry leaves (stirred at 250 rpm) in dry isopropanol (98 mL) as sol–gel precursor. The mixture was kept stirring at room temperature for another 12 h. Finally, the mixture was refluxed with stirring for 6 h, cooled and the product was filtered off, washed with isopropanol (10 mL), and dried in an aerated oven at 353 K for 12 h. The sample was then heat treated under static air at 773 K for 2 h (ramping rate of 1 K min−1 to avoid thermal shock damage) to remove all organic components and to crystallize the TiO2 leaves.Intact chloroplasts were isolated from Spinacia oleracea leaves using the method proposed by Robinson et al.39 as described in Scheme S1.† The resulted chloroplast cells were used as biotemplates for the replication of the nanostructure of leaf photosystem using the abovementioned Ti3+ exchange method with no cutting of leaves required. The product was filtered off, washed with isopropoxide, dried and heat treated at 773 K for 2 h (ramping rate of 1 K min−1) under static air.
2.3 Characterization
The morphology of the TiO2 architectures synthesized via the bio-templating method was imaged using a JEOL 7000F scanning electron microscope (SEM). The samples were sputtered with carbon, and the working distance was varied between 10 mm and 4 mm to give the best image quality; the accelerating voltage was maintained at 10 kV.Transmission electron microscopy (TEM) images were recorded using a Philips CM-200 transmission electron microscope operating at 200 kV. The samples were prepared by sonication in ethanol until fully dispersed followed by drop-casting onto a 150 square mesh copper TEM grid coated with carbon film.
The crystal phase of TiO2 was identified by X-ray diffraction (XRD) on a Bruker APEXII X-ray diffractometer equipped with a Mo Kα radiation source and a graphite monochromator.
The surface area was estimated by nitrogen adsorption measurements using the BET equation. The measurements were conducted at 77 K using a Micromeritics Gemini VI 2385-C Surface Area Analyser. All of the samples were degassed at 473 K under vacuum overnight before the measurements.
The surface compositions were measured by X-ray photoelectron spectroscopy (XPS) using a Thermo Scientific ESCALAB250Xi probe with monochromated Al Kα radiation (hν = 1486.68 eV). The binding energy was calibrated using the C 1s peak at 284.8 eV as reference. Measured spot was defined as a 500 μm square. The survey scans and the high-resolution (HR) XPS spectra were collected at the pass energy of 100 eV and 20 eV, respectively, and the photoelectron take off angle at 90° with respect to the surface plane. The XPS data was analysed using the Avantage software.
Dynamic light scattering (DLS) measurements were performed using a Microtrak Zetatrak DLS instrument. The samples were homogeneously dispersed in Milli-Q water (1 g L−1) via sonication for 15 min followed by 30 min of stirring (same conditions as used in the dye degradation reaction before UV/visible light irradiation) in order to evaluate of the particle size effect on the reaction.
2.4 Photocatalytic activity under UV and visible lights
Diffuse-reflectance UV-vis spectra were recorded using a Citra 4040 spectrophotometer equipped with an integrating sphere and operated in reflectance mode. The catalyst samples were diluted with BaSO4, and the resulting mixture was uniformly mixed, ground and loaded into the sample-holder. UV-vis spectra were recorded in reflectance mode, and the obtained reflectance values (R) were transformed to the Kubelka–Munk function F(R) according to the eqn (1):
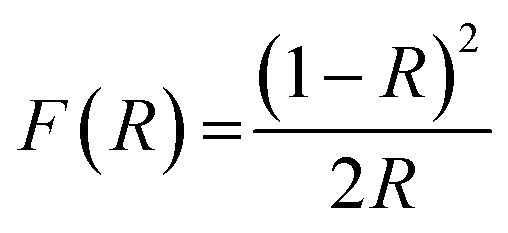 | (1) |
The band gap was estimated by creating a Tauc plot of (hνF(R))1/n against hν (where h is Planck's constant, ν is the frequency of vibration and n = 1/2 for direct transitions).40
The photocatalytic activity of the bio-mimicked TiO2 architectures under ultraviolet (UV) and visible light was studied in methylene blue degradation in a batch mode. An enclosed photocatalytic chamber equipped with two 50 W LEDs with maximum intensities at 370 nm (UV), 440 nm (blue) or 515 nm (green) was used in these experiments with remote sampling performed using syringe equipped with a long needle. The light flux was monitored to ensure that it was consistent across all experiments. The experimental conditions were set according to the literature.41–44 The solution pH was maintained at 7 to exclude the effects of acidity and alkalinity on the photocatalytic activity. The reaction solution was prepared by dispersing 15 mg of catalyst in 100 mL of dye solution (6.5 mg of methylene blue in 1 L deionised water) in a clean, dry quartz tube. The slurry was sonicated for 15 min to ensure that the catalyst was homogeneously dispersed before the reaction. The dye-catalyst slurry in the quartz tube was placed into the sealed photolysis chamber and stirred for 30 min in the dark to reach adsorption equilibrium. A 3 mL initial sample was collected before irradiation started. The reaction was timed immediately after the irradiation began and the temperature of the solution increased only marginally (up to 303 K maximum) during irradiation. Samples were collected at different reaction time and were centrifuged twice (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 3 min) and analysed by UV-vis spectroscopy within a wavelength range from 500 to 750 nm. The dye conversion was determined by eqn (2) on the basis of the maximum absorption.
000 rpm, 3 min) and analysed by UV-vis spectroscopy within a wavelength range from 500 to 750 nm. The dye conversion was determined by eqn (2) on the basis of the maximum absorption.
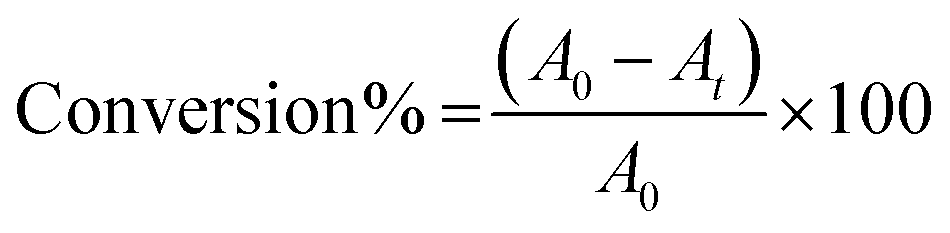 | (2) |
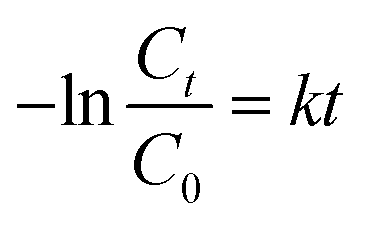 | (3) |
Control experiments without irradiation were performed to make sure that adsorption equilibrium was established after 30 min. The result of the dark experiments are provided in Fig. S1.† Blank reactions were also conducted to ensure that the decomposition of methylene blue was due to the photocatalytic degradation. No significant dye conversion (<3%) was observed under UV and visible lights in the absence of photocatalyst. The result of blank reactions are included in the dye conversion figures.
The photocatalytic oxidation of ethanol was carried out in a stirred batch annular reactor made from quartz with an internal volume of 150 mL and wall thickness of 0.5 cm. The catalyst powder (50 mg) was suspended in 50 mL of 20 v/v% ethanol solution for typical batches. The reaction slurry was stirred vigorously at 500 rpm for 60 min using magnetic stirrer to ensure homogeneity before the reaction. The reactor was purged with an inert gas using Schlenk line technique and was closed tightly. The reactor was placed inside an enclosed photolysis chamber and the mixture was stirred at 250 rpm to prevent sedimentation of the catalyst. Green LEDs with a maximum intensity at 515 nm were used to irradiate the reactor. The solution temperature and the gas phase pressure were continuously monitored. A gas sample was collected at the end of each reaction using a gas-tight syringe (10 mL). The samples were immediately analysed using a gas chromatograph (SRI Instruments, methanizer FID and TCD detectors, Haysep-D column). The photocatalytic performance in ethanol oxidation was assessed by the CO2 yield.
3 Results and discussion
3.1 Improved fabrication of the bio-templated TiO2 leaf
The templated materials with micro- and nano-architecture of the leaf photosystem are referred to as a “micro- and nano-structure templated artificial leaf” (MNAL). The natural enzymatic reactions of chlorophyll breakdown generally include breakdown of the chlorophyll chains by the chlorophyllase enzyme, followed by the Mg-dechelatase step and then oxidation of pheophorbide.46 In this study, the colour change of the acid treated leaves to the yellow-brown indicates the conversion of chlorophyll into pheophorbide.47,48 The ion-exchange procedure was performed under inert atmosphere to prevent Ti3+ ions from oxidizing. Energy-dispersive X-ray spectroscopy (EDS) results in Table S1† clearly demonstrate that there are no detectable metal contents of Mg2+ and other metal ions present in the leaf template. This finding indicates that the acid treatment was effective in extracting such metals during the artificial leaf synthesis process. Trace amounts of a vast range of metals including Mg, Zn, Fe, Pb etc. were observed in the non-treated samples. The extraction of metal ions is significant because the presence of metals may affect the crystal phase formation of the final TiO2 catalyst. In addition, the presence of metal ion impurities could make the final product composition difficult to control because the leaf contents change over the seasons.49In this research, an important improvement of the synthesis method was made by a solvent exchange step using dry isopropanol. This step was introduced to exchange or extract water present within leaves and replace it with isopropanol and also washing out any remaining salts from ion-exchange procedure. Minimizing water content inside the leaves allows titanium isopropoxide to diffuse deep inside the leaves, allowing better replication of the structure. Attempts to perform extraction using Soxhlet technique resulted in visible structural deterioration of the leaves, most likely due to higher temperature of the process. It is well known that water is critically important for the start of titanium isopropoxide hydrolysis. Therefore, the sol–gel was carried out open to ambient air to allow very gradual diffusion of water into the system, which resulted in noticeable hydrolysis and formation of titania. When this step was performed under inert dry atmosphere (Ar, Schlenk line) no visible signs of titania sol formation can be detected, thus confirming success of the earlier water extraction step. Noteworthy, the optimum size of leaf cuts accelerate infiltration of titanium ions through the leaf structure. Preliminary experiments using larger sizes of leaf cuts (ca. 5 mm by 5 mm) produced material which had titania structures formed only at the periphery of the leaf fragments.
To investigate the relationship between the morphology of the bio-templated product and its photocatalytic performance, Ti3+ exchange step was excluded to produce templated materials with only the microstructure of leaf photosystem. According to Li et al.,27 it is hypothesised that the Ti3+ ions get trapped within the thylakoids via ion-exchange with the hydrogen in the chlorophylls. During the heat treatment, the titania coat produced from titanium isopropoxide hydrolysis act as seeding sites for crystallization of TiO2 to form the nano-scale interconnected stacked-sheets structure. Hence, the sample synthesized via the sol–gel method alone does not possess the nano-architecture of leaf photosystem and is referred to as a “micro-structure mimicked artificial leaf” (MAL).
In our preliminary experiments, the sol–gel coating step of the Ti3+-exchanged leaves was omitted to replicate the nano-architecture of leaf photosystem alone. The produced templated materials showed no photocatalytic activity for degradation of methylene blue under UV light which could be attributed to the titania crystalline phase. The XRD pattern of non-coated sample (Fig. S2†) shows that the crystalline phase is mostly rutile. These results show that eliminating of sol–gel step favours the anatase-to-rutile phase transformation during crystallization of the TiO2 leaves. Therefore, intact chloroplast cells were isolated from Spinacia oleracea leaves using the method involving mechanical breaking of the cell wall and the plasma membrane, filtration of cell debris and unbroken leaf tissues, collection of chloroplasts by centrifugation, and separation of the intact chloroplasts from the broken ones. The resulted replicas of isolated chloroplast cells are referred to as “artificial chloroplast” (AChl).
Several samples from different synthesis batches were used to repeat the catalyst characterizations and photocatalytic activity experiments to ensure that the developed replication approach is reproducible. All characterizations were in an acceptable criteria and the photocatalytic activity results obtained from different batch of same catalysts under UV/visible light are within the error bars.
3.2 Catalyst characterization
Fig. 2a–c shows the scanning electron microscopy images of the final catalysts after calcination. These images demonstrate that the porous microstructures of the photosystem, including the palisade and spongy layers of the mesophylls, are replicated and the removal of the organic components of the leaves was successfully accomplished. The well-replicated layered nanostructures of the thylakoids in the chloroplast are shown in the higher resolution TEM images of the MNAL and AChl samples (Fig. 2d–f). The average length of the bio-templated TiO2 chloroplasts is 1.5–2 μm, which is in a good agreement with the size of natural chloroplast cells.50–52 The results show that the developed method provides a reliable approach to synthesize TiO2 hierarchical architectures similar to natural materials. This improved templating method could address the issues in controlling the morphology of final product associated with conventional procedures of synthesis of TiO2 structures.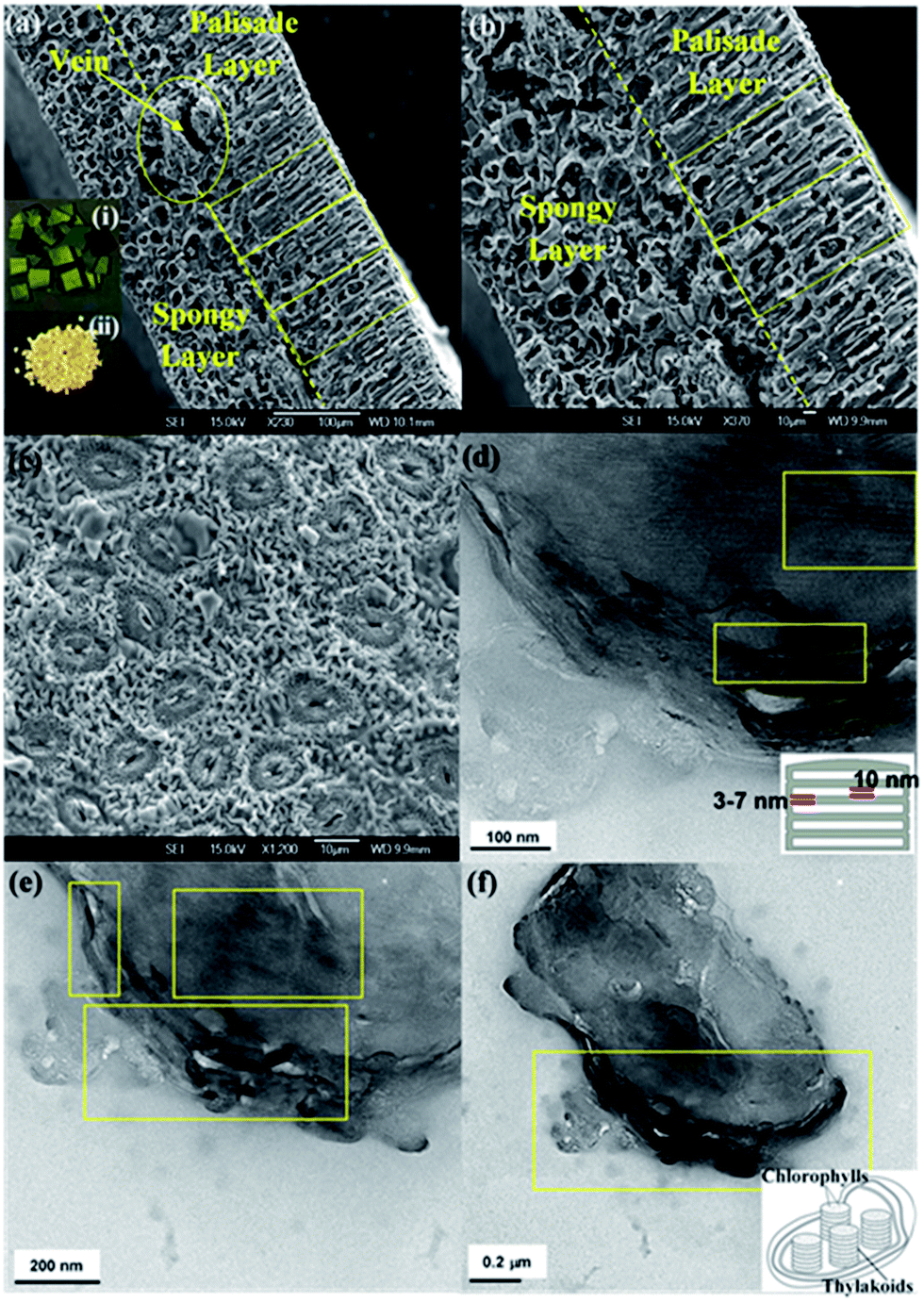 | ||
| Fig. 2 (a and b) Cross-section SEM images of final catalysts, showing the well-replicated microarchitecture of the leaf photosystem. Inset: (i) photos of natural camellia sinensis leaves and (ii) TiO2 artificial leaves. (c) Top-view SEM image of final catalysts, illustrating that the porous structure of the stomata is retained after the removal of the leaves. (d and e) TEM images of MNAL samples showing that the stack-layered nanostructures of the thylakoids in the chloroplast are retained after the removal of the leaves. (f) TEM images of AChl samples illustrating the well-replicated nano-architecture of the thylakoid membranes. | ||
The results of energy-dispersive X-ray spectroscopy (EDS) measurements summarized in Table S1† demonstrate that the templated materials are composed mostly from titanium and oxygen. Given a minimum of 2 wt% doping has been reported to be required to influence the efficiency of TiO2 photocatalysts,53 the trace amounts of other elements (N, P and C) retained within the synthesized catalysts from the biotemplates are considered insufficient to contribute to the photocatalytic performance of these materials. In addition, lack of detectable C within the templated samples based on the EDS analysis confirms the effective removal of the organic template which corroborates the observations made under SEM.
Although a mixture of anatase and rutile phases has been commonly reported for the room-temperature sol–gel synthesis of TiO2 followed by a heat treatment, the phase formation strongly depends on several factors, including the nature and volume of the solution, the reaction atmosphere and time, the presences of impurities, the morphology of the sample, and also the rate and thermal homogeneity of the post-synthesis heat treatment.54–56 The XRD patterns in Fig. 3 show that the MNAL and MAL consist predominantly of the anatase crystalline phase, whereas the AChl is a combination of anatase and rutile TiO2 phases. The results show that our biotemplating method generally favours the formation of a pure anatase phase (the MNAL and MAL) after the samples underwent a heat treatment at 500 °C for 2 h under static air. This is consistent with the synthesis of TiO2 films in which the initial crystalline TiO2 phase formed is usually anatase.57,58 On the other hand, mixed phases of anatase and rutile in the AChl could be caused by a high ratio between the organic components from natural cells and TiO2 during heat treatment. Many researches have attempted to study the effects of impurities on the anatase-to-rutile phase transformation. A high content of organic impurities can change the level of oxygen vacancies in the TiO2 lattice, thus favouring the anatase to rutile phase transformation.59 In addition, the faster phase transformation rate observed in the AChl also suggests that there are more potential nucleation sites available for the anatase-to-rutile transformation at low temperatures compared with the MNAL and MAL. As a result, the AChl is finely crystalline with small crystallite sizes.
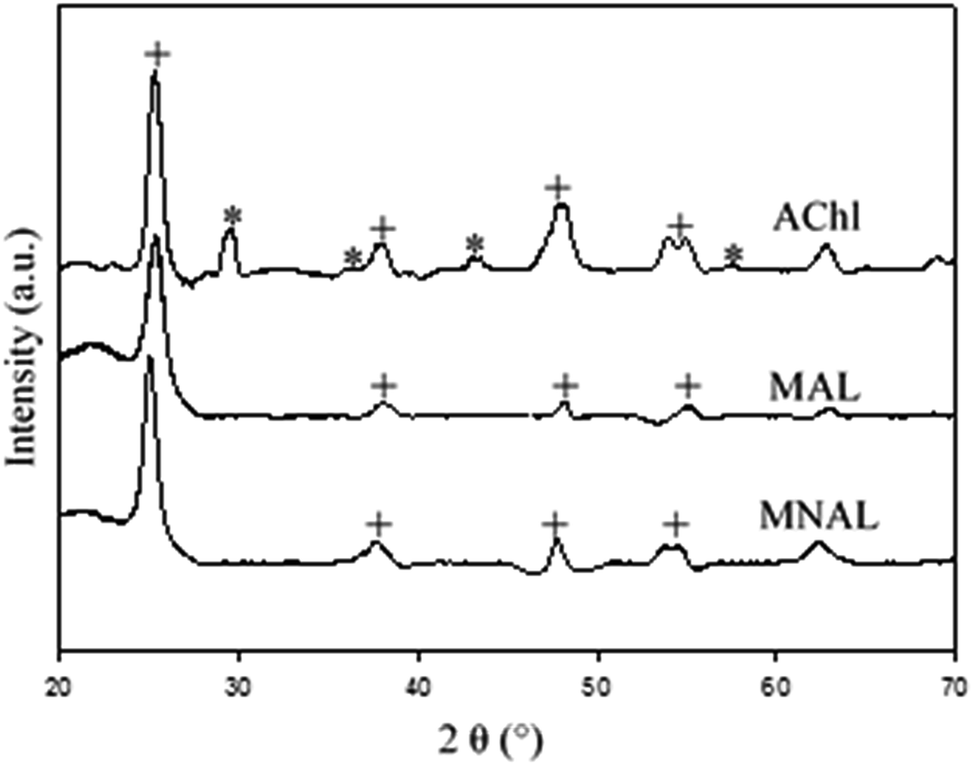 | ||
| Fig. 3 XRD patterns of the final catalysts. Corresponding peaks of the anatase phase and the rutile phase TiO2 are marked by “+” and “*”, respectively. | ||
Nitrogen (N2) physisorption experiment shows that the specific surface area of the MAL and the MNAL are 29.9 m2 g−1 and 66.1 m2 g−1, respectively (Fig. S3†). While both samples exhibited mesoporosity in their structures according to the hysteresis observed in the adsorption isotherms, MNAL specifically gave a H3 hysteresis loop indicating possible random pore network and irregular pore shape such as the “ink-bottle” geometry.60 The stepwise desorption which occurred at the relative pressure of 0.55 suggests the characteristic desorption mechanism via cavitation and, thereby, confirms the presence of pore necks that are smaller than the critical size of N2 (ca. 5–6 nm) at 77 K. In addition, the amount of N2 adsorbed (mmol g−1) on the MNAL is significantly higher than that adsorbed on the MAL (approximately 2.5 and 2.6 times higher on the MNAL at P/Po of 0.3 and 0.9, respectively, as shown in Fig. S3†), demonstrating that the biotemplated nano-architecture derived from the thylakoid in chloroplasts present within the MNAL gives rise to a larger surface area.
The general XPS spectrum of the MNAL sample is shown in Fig. 4a. The P and Ca peaks centred at the binding energy (BE) of 133.7 and 347.7 eV could be assigned to the inorganic phosphate61 and the calcium carbonate or phosphate species,62 respectively. The small peak corresponding to N at BE of 400.1 eV could be attributed to the Ti nitride species.63,64 The trace amounts of phosphorus, calcium and nitrogen detected in the MNAL sample are presumably residues remaining from the original biotemplate. The symmetric peak at the BE of 458.9 eV in the Ti 2p high resolution (HR) spectrum (Fig. 4b) is identified as TiO2.65,66 The BE for the Ti 2p peak is expected to decrease if chemical doping occurs, necessitating curve-fitting using peaks corresponding to several complex species.63,67 However, in the MNAL sample only one symmetrical peak is observed, confirming that doping with other elements (N, C or P) is negligible. The HR spectrum of O 1s was deconvoluted using three peaks (Fig. 4c). From the reported TiO2 models,68,69 the first peak at the BE of 530.2 eV can be attributed to oxygen of the TiO2 crystal lattice, the second at 531.6 eV can be attributed to the OH groups and the third one (BE = 532.8 eV) corresponds to Ti–O–N species.
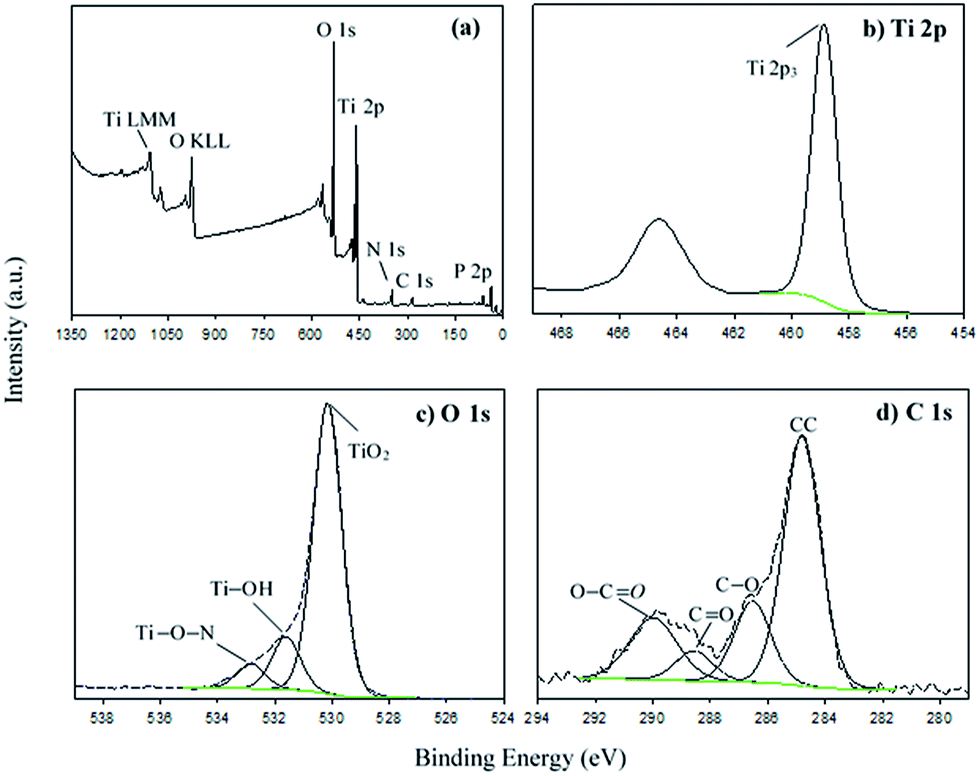 | ||
| Fig. 4 (a) General XPS spectrum of the MNAL and high resolution deconvoluted XPS spectra of (b) Ti 2p, (c) O 1s and (d) C 1s. | ||
Four carbon species with BE of 284.8 eV (A), 286.5 eV (B), 288.6 eV (C) and 290 eV (D) were observed in the C 1s spectrum (Fig. 4d). The species “A” and “B” could be assigned as carbon backbone (aromatic and aliphatic) and C–O bonds70,71 within the adventitious carbon contaminants which are commonly reported in samples that have been exposed to the atmosphere.65,71 The peaks “C” and “D” could be characteristic for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO groups due to the residual oxidized organic species of biotemplate, respectively.71–73 Similar peaks were also observed in the C 1s spectra reported by Peng et al.74 for the activated carbon derived from pomelo peel. Thus, carbon species “C” and “D” are probably related to the carbon species remaining after removal the leaf via calcination.75,76
O and O–CO groups due to the residual oxidized organic species of biotemplate, respectively.71–73 Similar peaks were also observed in the C 1s spectra reported by Peng et al.74 for the activated carbon derived from pomelo peel. Thus, carbon species “C” and “D” are probably related to the carbon species remaining after removal the leaf via calcination.75,76
No significant C content was detected in the bulk composition analysis by EDS (Table S1†), proving that the observed carbon species originated predominantly from contamination with adventitious carbon. The surface composition of the MNAL was specifically characterized because it exhibited the best photocatalytic performance among the three bio-templated samples both under UV (Fig. 5) and blue (Fig. 7) light. The elemental surface composition (atom%) of the MNAL is summarized in Table S2.†
 | ||
| Fig. 5 Conversion of methylene blue under UV light with a maximum intensity at 370 nm. The experimental conditions are 6.5 ppm dye solution, a catalyst loading of 150 mgcat L−1, a pH of 7, 30 °C. The accuracy of all experiments was verified by series of repeated measurements and the relative error of less than 10% was determined. | ||
3.3 Photocatalytic activity under UV and visible light
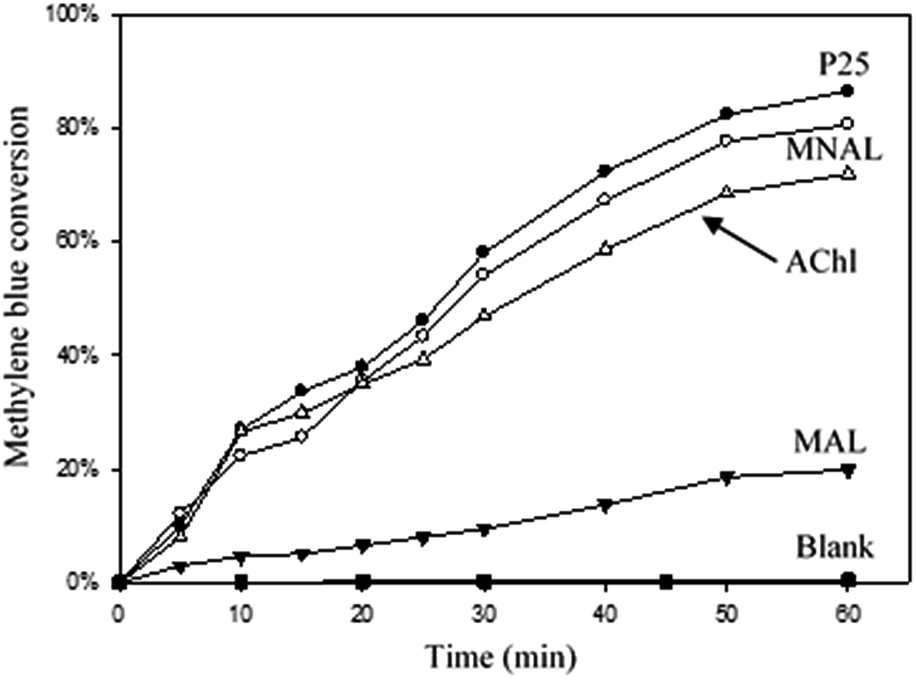
The efficiency of a photocatalytic reaction is an overall outcome of various factors: light absorption, transport of photogenerated charge carriers, electron/hole recombination rate and mass transfer of the reactant(s) or reactive radical species to/from the catalyst surface. The morphology of the photocatalyst, and particularly the particle size and shape, strongly influences the aforementioned factors.77–79 Fig. 5 shows that the MNAL exhibited photocatalytic activity (80 ± 8%) comparable to that of the commercial P25 catalyst (86 ± 8.5%) after 60 min of irradiation under UV light (370 nm). The slightly higher conversion given by the commercial P25 (composed by approximately 80% anatase and 20% rutile) could be mainly due to the enhanced charge carrier transfer between anatase and rutile (preventing undesirable recombination) caused by the energy difference between the conduction band edges of the two phases.80 On the other hand, nonporous TiO2 catalysts are not capable of exhibiting favoured excitation behaviour in parallel with a high specific surface area, implying that photogenerated electron/hole pairs within the bulk of the semiconductor predominantly recombine before reaching the surface, causing a low quantum yield.81–83In contrast to the nanoparticulate reference P25, prolonged residence time of the methylene blue within the internal interconnected membrane-like structures and unique hierarchical pore networks of MNAL could explain observed comparatively high activity. Thus, improved reactant-catalyst contact offsets the theoretically lower photocatalytic activity in the MNAL made of pure anatase phase, which is expected to have higher electron/hole recombination rates compared to P-25. In addition, Howe and Grätzel suggested that photo-induced charge carriers in TiO2 nanomaterials can be efficiently trapped at different defect sites in the bulk and on the surface.84,85 The electron/hole recombination rate could be reduced by providing charge traps at the defect sites located at the surface of hierarchical structures MNAL TiO2.86–88 Here, we demonstrate that the structure that is responsible for the photocatalytic activity of the MNAL is the stacked titania nano-sheets (thylakoid membranes replica) as the micro-architecture (palisade and spongy layers of the mesophylls) of the leaf photosystem tends to disintegrate under stirring. The DLS measurements showed that the synthesized MNAL and MAL catalysts break down to relatively small particles with an average diameter of 2.3 ± 0.2 μm during the reaction. The total organic carbon (TOC) measurement (Fig. S4†) shows that the commercial P25 and the MNAL catalysts decreased the TOC from 3.5 ± 0.2 to 2.5 ± 0.25 ppm (28.5 ± 3% decrease) and from 3.4 ± 0.2 to 2.7 ± 0.3 mg L−1 (20.5 ± 2% decrease), respectively. This is consistent with the results reported by Houas et al.89 that the rate of mineralization of organic carbon by P25 TiO2 is slow over the first hour of photocatalytic reaction despite fast decolourization of the dye. The TOC removal is expected to proceed further during a more prolonged reaction.
The AChl structures gave a slightly lower photocatalytic activity (72 ± 7% methylene blue conversion after 60 min) than the MNAL possibly due to insufficient mesoporous channels for fast adsorption and desorption of the reactants and oxidized products, respectively. This finding indicates that the porous structure, remaining from the broken mesophyll cells replica, could improve the photocatalytic activity. However, the light harvesting by the micro-architectures of the artificial leaf photosystem could not resemble that of the natural leaf due to structural breakdown in the slurry reaction system. Poor photocatalytic performance was observed in the case of using the MAL (only 20 ± 2% conversion) under UV light irradiation. This low activity in methylene blue degradation probably results from the low surface area of the MAL made of predominantly anatase crystalline phase; furthermore, nano-membrane structures (thylakoids replica) are absent in this catalyst. Moreover, the dye degradation measured for MAL (20%) after 120 min is 4 times lower than that measured for MNAL (80%). The difference in dye degradation efficiency is far greater than the 2.5-times factor due to the difference surface areas of these materials. The normalized results of photocatalytic degradation of methylene blue are presented in Table S3.† Both the MNAL and AChl catalysts exhibited photocatalytic dye degradation performance (34.7 and 31.2 mg gcat−1) similar to that of the nanoparticulate P25 (37.3 mg gcat−1) under UV light in contrast to the poor photocatalytic performance of the MAL (8.7 mg gcat−1). Thus, the layered nanostructures replicated from the thylakoid nano-membranes could be considered as the key contributing factor to the photocatalytic performance of the biotemplated materials under UV light.
Reported here easy multi-step chemical replication method for producing micro- and nanostructure-templated artificial leaves (MNAL) is better suited for large-scale synthesis compared with the AChl route relying on the isolation of intact chloroplast cells which yields only small quantity of material (average yield ∼ 40 mg chloroplasts per g spinach leaf39,90). Thus, the AChl was not included in the study of visible light activity. The diffuse-reflectance UV-vis spectra shown in Fig. 6 demonstrate two important features. Firstly, the MNAL exhibits greater absorption in the UV region over a broad UV-vis wavelength (ca. 350–600 nm) compared to the reference P25 material. We hypothesise that this is due to the improved light trapping and light scattering properties through the layered nano-architectures. Interestingly, this is consistent with the excellent light trapping properties of the parent biotemplating materials (i.e. natural chloroplasts) reported in literature,35–37 which are capable of maximizing light scattering through the layered structures. Secondly, the absorption efficiency has been improved in the visible light region (particularly between 400–600 nm). Such improvement of the visible light absorbance can be attributed to the presence of localized states within the TiO2 compact layers in the MNAL. There are two types of trap centres in TiO2 nanostructures. First, the lattice defects, such as oxygen vacancies, that can form localised Ti3+ centres, which may lead to additional electronic states within the TiO2 band gap, allowing sub-band gap transitions and thus light absorbance in the visible region. However, these were not observed in noticeable quantity in the XP spectra in this study. Second, higher surface area of such materials (66.1 m2 g−1 in MNAL) may offer larger proportion Ti atoms at the surface which could act as trap centres, explaining improved light absorbance.87,91 The advanced optical properties of titania nano-architectures have been observed by many research groups over the past few years.14 For instance, Bavykin et al.92 revealed the improved optical properties in TiO2 nanotubes; Sato and Sakai et al. showed that the effective wavelengths for light harvesting red shifted when titania is fabricated in the form of nanosheets.93,94
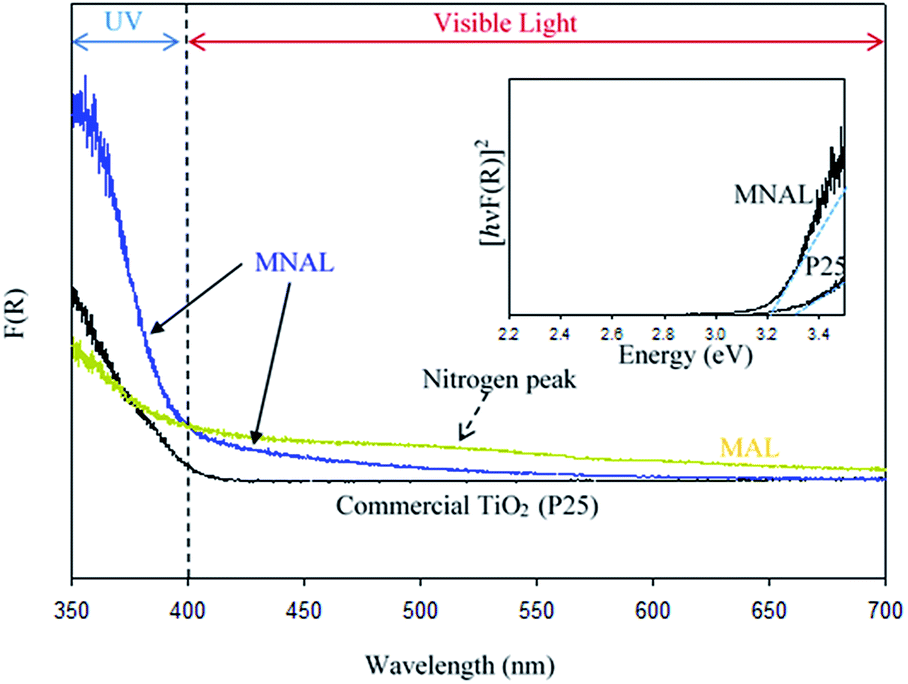 | ||
| Fig. 6 Diffuse-reflectance UV-vis spectra of the MNAL, MAL and the reference P25 (inset: Tauc plot showing fits used for the estimation of band gap values; the MNAL spectrum was used to estimate the bandgap in order to exclude the effects from the nitrogen peak). | ||
The observed peak centred at ca. 500 nm in the spectrum of the MAL sample could be due to the absorption of light by N content.95,96 However, the amount of N is insufficient to contribute to the photocatalytic efficiency of biotemplated catalyst as discussed earlier. There is no detectable absorption peak associated with N content in the MNAL spectrum, suggesting that the amount of nitrogen in the catalyst retained from the leaf is negligible (corroborating results in Table S1†). This shows that the extrinsic excitation behaviour of TiO2 photocatalyst can potentially be augmented by modifying its morphology without changing its composition. The Tauc plot in Fig. 6 (inset) allows to roughly estimate an energy band gap of 3.2 eV for the MNAL, which is lower than that of the reference P25 TiO2 (Eg = 3.3 eV). Although only a minor reduction of bandgap is evidenced by the Tauc plot, the result is in good agreement with reported band gap narrowing by only 0.2 eV via structural modification for TiO2 nanomaterials.97
The results of the methylene blue photo-degradation in Fig. 7 and 8a show that the MNAL gave a significantly better photocatalytic activity than P25 under both visible blue and green lights. MNAL and P25 achieved 13 ± 1.5% and 7 ± 0.9% of methylene blue conversion, respectively, under blue light at 440 nm which is near the edge of the visible region (Fig. 7). Under green light irradiation at 515 nm (Fig. 8a), the methylene blue conversion achieved by MNAL (9 ± 1.2%) is also higher than that achieved by P25 (6.3 ± 0.8%). In addition, the MAL exhibited no apparent photocatalytic activity in methylene blue degradation under blue light (not shown here). The improved visible-light activity of the MNAL could be attributed to several key factors, large surface area and high efficiency of light absorption provided by the morphology of the chloroplast-like structures (see earlier discussion of UV-vis spectra). We hypothesise that the 3-D TiO2 nanocrystalline structure of the MNAL allows rapid injection of charge carriers from bulk into both internal and external surfaces. One of the carriers is then captured on a localised state at the surface while the other one is free for reaction. The net result is that the electron/hole recombination rate is reduced significantly by both the 3-D layered structure and the charge trapping phenomena resulted from defect points located within the volume of the MNAL. The 3-D construction of the interconnected nanolayers (replicated from thylakoids) in the MNAL also allows efficient trapping of the incident light via multiple light scattering within structure of MNAL. Thus, a longer optical path35–37 of light travelling through MNAL could result in the greater chance of the eventual photoexcitation events during the photocatalytic reaction. This is consistent with other reported results on the 3-D crystalline materials.25,98,99 For example, Cheng et al.24 showed a high photocurrent density of 3-D TiO2 inverse opals under simulated solar-light illumination. Attempts to measure the TOC removal under blue and green lights were not successful owing to low conversion of methylene blue under the tested conditions. The change in TOC concentration was very small after 2 h of reaction under visible lights and was within the uncertainty of the TOC measurement.
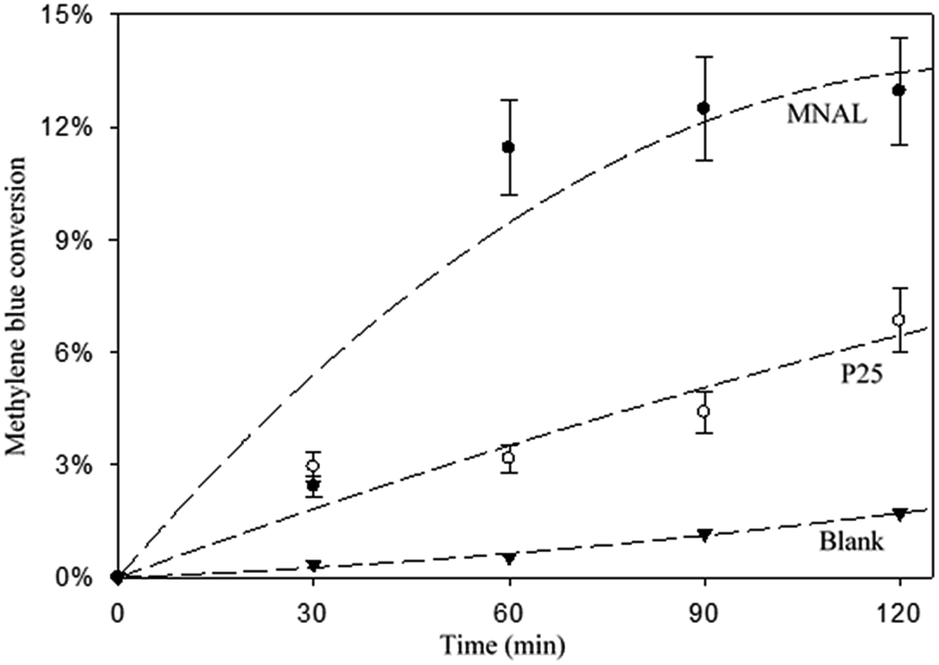 | ||
| Fig. 7 Conversion of methylene blue under blue LEDs with a maximum intensity at 440 nm (the edge of the visible light region). The experimental conditions are 6.5 ppm dye solution, a catalyst loading of 150 mgcat L−1, a pH of 7, 30 °C. Notes: (i) the MAL exhibited no photocatalytic activity under blue light (result not shown). (ii) A much lower conversion of methylene blue was recorded here due to the use of a lower energy light source; (iii) lines are guide to eyes. | ||
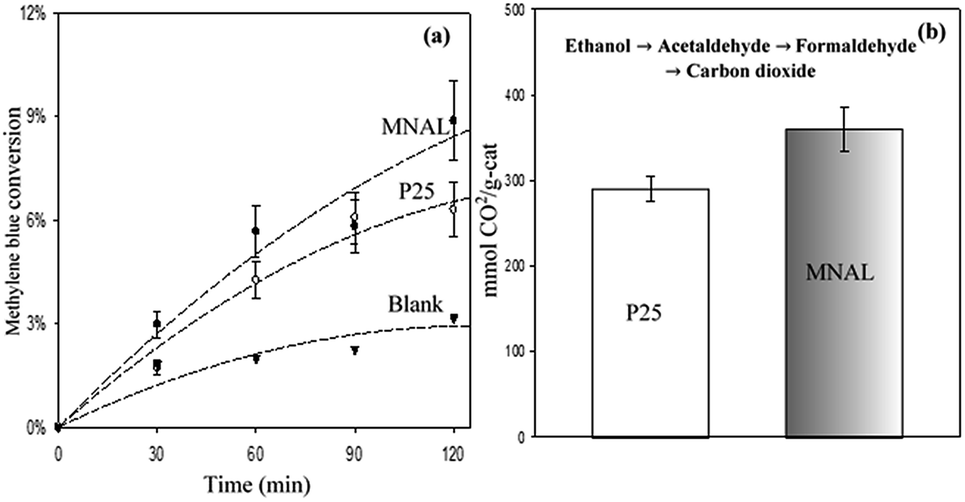 | ||
| Fig. 8 (a) Conversion of methylene blue under green LEDs with a maximum intensity at 515 nm. The experimental conditions are 6.5 ppm dye solution, a catalyst loading of 150 mgcat L−1, a pH of 7, 30 °C (note: lines are guide to eyes). (b) Carbon dioxide yield of photocatalytic oxidation of ethanol under green LEDs with a maximum intensity at 515 nm. The experimental conditions are 20 v/v% ethanoic solution, a catalyst loading of 1 gcat L−1, 30 °C. | ||
The apparent rate constants are estimated using data reported in Fig. S5.† The MNAL catalyst gave a rate constant approximately 2.4 and 1.3 times higher than those observed in the case of P25 under blue and green light irradiation, respectively (Fig. S5b and c†). Given that nitrogen species were evidenced by EDS and XPS analysis (Tables S1 and S2†), the estimated rate constants are also compared with the rate constant given by N-doped TiO2 in the work of Burda et al.100 Burda et al. reported that the rate constants of methylene blue photo-degradation by N-doped TiO2 (8 atomic%) are approximately 3 and 1.5 times higher than those given by the benchmark P25 titania under near blue (390 nm) and green (540 nm) light irradiation, respectively. The superior visible-light photocatalytic activity in heavily N-doped TiO2 compared with the MNAL could be attributed to the band gap narrowing of titania resulting from substitution of O for N in the TiO2 lattice. As stated earlier in the discussion of the XPS spectra, the amount of N dopant observed in this work is insufficient to form localized centres by mixing N 1s states with O 1s states in the MNAL. Hence, the trace amount of N dopant is less likely to have influence on the photocatalytic activity of the MNAL.
Photocatalytic oxidation of ethanol (Fig. 8b) was also conducted as an alternative probe reaction to verify the superior photocatalytic activity of MNAL under green light (515 nm). Ethanol oxidation on TiO2 catalysts are initiated by oxidizing the adsorbed ethanol molecules to acetaldehyde and proceed through several intermediate reactions.101 The results show that the yield of CO2 generated by MNAL (360 ± 25 μmol gcat−1) is almost 1.3 times higher than the yield of CO2 given by P25 (290 ± 15 μmol gcat−1) under green light. The MNAL was specifically tested against the reference TiO2 under green light because it gave the best photocatalytic performance among the three bio-templated samples under UV and blue wavelengths (as shown in Fig. 5 and 7). The electron transfer from rutile crystals to the trapping sites of anatase lattice extends the light absorption efficiency of P25 into visible range.80 On the other hand, the pure anatase (single crystalline phase) with a crystalline domain diameter below the critical radius of approximately 10 nm may exhibit an increase in band gap, causing low photocatalytic efficiency under the visible light.102,103 We demonstrate that sub-band gap transitions caused by the localized states within the compact, layered nanostructure in the MNAL could lead to a stable charge separation over a broad range of visible wavelengths, despite its pure anatase phase. The detailed mechanisms of the photocatalytic oxidation of ethanol over the biotemplated MANL titania will be examined further in our follow-up study.
4 Conclusions
In this study, we developed a multi-step chemical replication method using micro- to nano-architectures of the green leaves as templates to prepare advanced visible-light active TiO2-based photocatalyst. Novel micro- and nano-structured porous pure anatase-based architecture enabled by biotemplating (MNAL) exhibited photocatalytic dye degradation performance similar to that of commercial nanoparticulate mixed-phase P25 catalyst under UV light. A substantially improved photocatalytic activity was observed with the MNAL under visible (blue and green) light. The enhanced photocatalytic performance of the MNAL was interpreted as arising due to the complex 3-D morphology of the templated macro- and nano-structured light-harvesting system. This catalyst contains extensive pore network and has high surface area resulting from the interconnected titania nanosheets derived from the thylakoid membranes of the natural leaves. This work demonstrated that the extrinsic excitation behaviour of TiO2 photocatalyst can be potentially augmented by modifying its morphology alone. The synthesis method reported in this work provides a reliable and promising procedure to synthesize TiO2 hierarchical structures which could overcome the issues in controlling the structure and crystallinity of end product associated with conventional procedures. The unique advantages of the layered TiO2 structure made using biotemplating method are confirmed by the performance of reported here materials in two model photocatalytic reactions under visible light. Given that visible light with wavelengths longer than 400 nm makes up more than 95% of the solar spectrum, the novel TiO2 structure obtained using biotemplating method could be extremely attractive for solar-assisted chemical reactions.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the Royal Society of New Zealand for funding this project under the Catalyst: Seeding General grant (16-UOC-002-CSG).References
- D. Abbott, Proc. IEEE, 2010, 98, 42–66 CrossRef CAS.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed.
- E. B. Stechel and J. E. Miller, J. CO2 Util., 2013, 1, 28–36 CrossRef CAS.
- A. Corma and H. Garcia, J. Catal., 2013, 308, 168–175 CrossRef CAS.
- K. Mori, H. Yamashita and M. Anpo, RSC Adv., 2012, 2, 3165–3172 RSC.
- G. Liu, L. Wang, H. G. Yang, H.-M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- M. Kitano, M. Matsuoka, M. Ueshima and M. Anpo, Appl. Catal., A, 2007, 325, 1–14 CrossRef CAS.
- E. Serrano, G. Rus and J. García-Martínez, Renewable Sustainable Energy Rev., 2009, 13, 2373–2384 CrossRef CAS.
- M. Woodhouse and B. A. Parkinson, Chem. Soc. Rev., 2009, 38, 197–210 RSC.
- M. Gao, L. Zhu, W. L. Ong, J. Wang and G. W. Ho, Catal. Sci. Technol., 2015, 5, 4703–4726 CAS.
- B. Liu, Y. Fang, Z. Li and S. Xu, J. Nanosci. Nanotechnol., 2015, 15, 889–920 CrossRef CAS PubMed.
- S. Khanchandani, S. Kumar and A. K. Ganguli, ACS Sustainable Chem. Eng., 2016, 4, 1487–1499 CrossRef CAS.
- B. Sarkar, N. Singhal, R. Goyal, A. Bordoloi, L. N. Sivakumar Konathala, U. Kumar and R. Bal, Catal. Commun., 2016, 74, 43–48 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- D. P. MacWan, P. N. Dave and S. Chaturvedi, J. Mater. Sci., 2011, 46, 3669–3686 CrossRef CAS.
- A. Chen, J. Qian, Y. Chen, X. Lu, F. Wang and Z. Tang, Powder Technol., 2013, 249, 71–76 CrossRef CAS.
- P. H. Mutin and A. Vioux, Chem. Mater., 2009, 21, 582–596 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- J.-Y. Ruzicka, F. A. Bakar, L. Thomsen, B. C. Cowie, C. McNicoll, T. Kemmitt, H. E. A. Brand, B. Ingham, G. G. Andersson and V. B. Golovko, RSC Adv., 2014, 4, 20649–20658 RSC.
- C. Sanchez, H. Arribart and M. M. Giraud Guille, Nat. Mater., 2005, 4, 277–288 CrossRef CAS PubMed.
- F. Xia and L. Jiang, Adv. Mater., 2008, 20, 2842–2858 CrossRef CAS.
- A. H. Lu and F. Schüth, Adv. Mater., 2006, 18, 1793–1805 CrossRef CAS.
- H. Zhou, T. Fan and D. Zhang, ChemSusChem, 2011, 4, 1344–1387 CrossRef CAS PubMed.
- C. Cheng, S. K. Karuturi, L. Liu, J. Liu, H. Li, L. T. Su, A. I. Y. Tok and H. J. Fan, Small, 2012, 8, 37–42 CrossRef CAS PubMed.
- G. I. N. Waterhouse and M. R. Waterland, Polyhedron, 2007, 26, 356–368 CrossRef CAS.
- X. H. Li, W. M. Liu and H. L. Li, Appl. Phys. A, 2005, 80, 317–320 CrossRef CAS.
- X. Li, T. Fan, H. Zhou, S. K. Chow, W. Zhang, D. Zhang, Q. Guo and H. Ogawa, Adv. Funct. Mater., 2009, 19, 45–56 CrossRef CAS.
- X. Ye, Q. Yang, Y. Zheng, W. Mo, J. Hu and W. Huang, Mater. Res. Bull., 2014, 51, 366–371 CrossRef CAS.
- A. Chen, J. Qian, Y. Chen, X. Lu, F. Wang and Z. Tang, Powder Technol., 2013, 249, 71–76 CrossRef CAS.
- Ü. Niinemets and L. Sack, in Progress in Botany, ed. K. Esser, U. Lüttge, W. Beyschlag and J. Murata, Springer, Berlin, Heidelberg, 2006, pp. 385–419 Search PubMed.
- R. A. Bone, D. W. Lee and J. M. Norman, Appl. Opt., 1985, 24, 1408–1412 CrossRef CAS PubMed.
- M. E. Poulson and T. C. Vogelmann, Plant, Cell Environ., 1990, 13, 803–811 CrossRef.
- T. C. Vogelmann and G. Martin, Plant, Cell Environ., 1993, 16, 65–72 CrossRef.
- T. C. Vogelmann, J. F. Bornman and D. J. Yates, Physiol. Plant., 1996, 98, 43–56 CrossRef CAS.
- L. Mustárdy, Oxygenic Photosynthesis: The Light Reactions, Kluwer, Dordrecht, Netherlands, 1996 Search PubMed.
- A. V. Ruban, M. P. Johnson and C. D. P. Duffy, Energy Environ. Sci., 2011, 4, 1643–1650 CAS.
- I. Rumak, K. Gieczewska, B. Kierdaszuk, W. I. Gruszecki, A. Mostowska, R. Mazur and M. Garstka, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 1736–1748 CrossRef CAS PubMed.
- Z. He, W. Que and Y. He, Mater. Lett., 2013, 94, 136–139 CrossRef CAS.
- S. P. Robinson, Photosynth. Res., 1983, 4, 281–287 CrossRef CAS PubMed.
- S. Adachi, Optical Properties of Crystalline and Amorphous Semiconductors, Kluwer, Norwell MA, 1999 Search PubMed.
- R. J. Tayade, T. S. Natarajan and H. C. Bajaj, Ind. Eng. Chem. Res., 2009, 48, 10262–10267 CrossRef CAS.
- F. Abu Bakar, J.-Y. Ruzicka, I. Nuramdhani, B. E. Williamson, M. Holzenkaempfer and V. B. Golovko, Aust. J. Chem., 2015, 68, 471–480 CrossRef CAS.
- S. Bae, S. Kim, S. Lee and W. Choi, Catal. Today, 2014, 224, 21–28 CrossRef CAS.
- J. Yao and C. Wang, Int. J. Photoenergy, 2010, 643182 Search PubMed.
- X. Yan, C. Zou, X. Gao and W. Gao, J. Mater. Chem., 2012, 22, 5629–5640 RSC.
- S. Hörtensteiner and B. Kräutler, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 977–988 CrossRef PubMed.
- A. Drzewiecka-Matuszek, A. Skalna, A. Karocki, G. Stochel and L. Fiedor, J. Biol. Inorg Chem., 2005, 10, 453–462 CrossRef CAS PubMed.
- H. Küpper, F. Küpper and M. Spiller, Photosynth. Res., 1998, 58, 123–133 CrossRef.
- J. Yuan, D. Yuan, X. Tan, F. Zou and S. Xiao, Open Biotechnol. J., 2015, 9, 170–177 CrossRef CAS.
- J. R. Ellis and R. M. Leech, Planta, 1985, 165, 120–125 CrossRef CAS PubMed.
- Ł. Rudowska, K. Gieczewska, R. Mazur, M. Garstka and A. Mostowska, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1380–1387 CrossRef PubMed.
- E. Shimoni, O. Rav-Hon, I. Ohad, V. Brumfeld and Z. Reich, Plant Cell, 2005, 17, 2580–2586 CrossRef CAS PubMed.
- S. Rehman, R. Ullah, A. M. Butt and N. D. Gohar, J. Hazard. Mater., 2009, 170, 560–569 CrossRef CAS PubMed.
- S. D. Sharma, D. Singh, K. K. Saini, C. Kant, V. Sharma, S. C. Jain and C. P. Sharma, Appl. Catal., A, 2006, 314, 40–46 CrossRef.
- Y. Takahashi and Y. Matsuoka, J. Mater. Sci., 1988, 23, 2259–2266 CrossRef CAS.
- N. Smirnova, A. Eremenko, V. Gayvoronskij, I. Petrik, Y. Gnatyuk, G. Krylova, A. Korchev and A. Chuiko, J. Sol-Gel Sci. Technol., 2004, 32, 357–362 CrossRef CAS.
- K. Okada, N. Yamamoto, Y. Kameshima, A. Yasumori and K. J. D. MacKenzie, J. Am. Ceram. Soc., 2001, 84, 1591–1596 CrossRef CAS.
- H. Shin, H. S. Jung, K. S. Hong and J.-K. Lee, J. Solid State Chem., 2005, 178, 15–21 CrossRef CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- K. Sing and R. Williams, Adsorpt. Sci. Technol., 2004, 22, 773–782 CrossRef CAS.
- S. Caporali, U. Bardi and A. Lavacchi, J. Electron Spectrosc. Relat. Phenom., 2006, 151, 4–8 CrossRef CAS.
- Z. Zhao, W. Shan, Y. Zhang, X. Li, J. Ma and Y. Yan, J. Appl. Polym. Sci., 2012, 125, 2502–2509 CrossRef CAS.
- Y. Cong, J. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2007, 111, 6976–6982 CAS.
- G. A. Battiston, R. Gerbasi, A. Gregori, M. Porchia, S. Cattarin and G. A. Rizzi, Thin Solid Films, 2000, 371, 126–131 CrossRef CAS.
- P. Górska, A. Zaleska, E. Kowalska, T. Klimczuk, J. W. Sobczak, E. Skwarek, W. Janusz and J. Hupka, Appl. Catal., B, 2008, 84, 440–447 CrossRef.
- N. Drnovšek, N. Daneu, A. Rečnik, M. Mazaj, J. Kovač and S. Novak, Surf. Coat. Technol., 2009, 203, 1462–1468 CrossRef.
- M. Paszkiewicz, J. Łuczak, W. Lisowski, P. Patyk and A. Zaleska-Medynska, Appl. Catal., B, 2016, 184, 223–237 CrossRef CAS.
- E. György, A. Pérez del Pino, P. Serra and J. L. Morenza, Surf. Coat. Technol., 2003, 173, 265–270 CrossRef.
- J. Yu, X. Zhao and Q. Zhao, Thin Solid Films, 2000, 379, 7–14 CrossRef CAS.
- G. Xue, Q. Dai and S. Jiang, J. Am. Chem. Soc., 1988, 110, 2393–2395 CrossRef CAS.
- J. J. Lagowski, J. Chem. Educ., 1993, 70, A25 Search PubMed.
- L. Calliari, S. Fanchenko and M. Filippi, Carbon, 2007, 45, 1410–1418 CrossRef CAS.
- G. Milczarek, A. Ciszewski and I. Stepniak, J. Power Sources, 2011, 196, 7882–7885 CrossRef CAS.
- C. Peng, J. Lang, S. Xu and X. Wang, RSC Adv., 2014, 4, 54662–54667 RSC.
- J. Chen, G. Zhang, B. Luo, D. Sun, X. Yan and Q. Xue, Carbon, 2011, 49, 3141–3147 CrossRef CAS.
- J.-w. Lang, X.-b. Yan, W.-w. Liu, R.-t. Wang and Q.-j. Xue, J. Power Sources, 2012, 204, 220–229 CrossRef CAS.
- H. Cheng, J. Wang, Y. Zhao and X. Han, RSC Adv., 2014, 4, 47031–47038 RSC.
- A. Testino, I. R. Bellobono, V. Buscaglia, C. Canevali, M. D'Arienzo, S. Polizzi, R. Scotti and F. Morazzoni, J. Am. Chem. Soc., 2007, 129, 3564–3575 CrossRef CAS PubMed.
- M. Schiavello, Heterogeneous Photocatalysis, Wiley, 1997, pp. 117–125 Search PubMed.
- D. C. Hurum, A. G. Agrios, K. A. Gray, T. Rajh and M. C. Thurnauer, J. Phys. Chem. B, 2003, 107, 4545–4549 CrossRef CAS.
- H. Lin, C. P. Huang, W. Li, C. Ni, S. I. Shah and Y.-H. Tseng, Appl. Catal., B, 2006, 68, 1–11 CrossRef CAS.
- W. C. Hao, S. K. Zheng, C. Wang and T. M. Wang, J. Mater. Sci. Lett., 2002, 21, 1627–1629 CrossRef CAS.
- J. Jiang, G. Oberdörster, A. Elder, R. Gelein, P. Mercer and P. Biswas, Nanotoxicology, 2008, 2, 33–42 CrossRef CAS.
- R. F. Howe and M. Grätzel, J. Phys. Chem., 1985, 89, 4495–4499 CrossRef CAS.
- R. F. Howe and M. Grätzel, J. Phys. Chem., 1987, 91, 3906–3909 CrossRef CAS.
- M. K. Nowotny, L. R. Sheppard, T. Bak and J. Nowotny, J. Phys. Chem. C, 2008, 112, 5275–5300 CAS.
- R. Krishnan, in Encyclopedia of Electrochemistry, Wiley, 2007, vol. 6, pp. 34–39 Search PubMed.
- I. R. Macdonald, S. Rhydderch, E. Holt, N. Grant, J. M. D. Storey and R. F. Howe, Catal. Today, 2012, 182, 39–45 CrossRef CAS.
- A. Houas, H. Lachheb, M. Ksibi, E. Elaloui, C. Guillard and J.-M. Herrmann, Appl. Catal., B, 2001, 31, 145–157 CrossRef CAS.
- M. Nishimura, D. Graham and T. Akazawa, Plant Physiol., 1976, 58, 309–314 CrossRef CAS PubMed.
- I. Abayev, A. Zaban, V. G. Kytin, A. A. Danilin, G. Garcia-Belmonte and J. Bisquert, J. Solid State Electrochem., 2007, 11, 647–653 CrossRef CAS.
- D. V. Bavykin, S. N. Gordeev, A. V. Moskalenko, A. A. Lapkin and F. C. Walsh, J. Phys. Chem. B, 2005, 109, 8565–8569 CrossRef CAS PubMed.
- N. Sakai, Y. Ebina, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2004, 126, 5851–5858 CrossRef CAS PubMed.
- H. Sato, K. Ono, T. Sasaki and A. Yamagishi, J. Phys. Chem. B, 2003, 107, 9824–9828 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- K. Yang, Y. Dai and B. Huang, J. Phys. Chem. C, 2007, 111, 12086–12090 CAS.
- V. Etacheri, C. Di Valentin, J. Schneider, D. Bahnemann and S. C. Pillai, J. Photochem. Photobiol., C, 2015, 25, 1–29 CrossRef CAS.
- G. I. N. Waterhouse, J. B. Metson, H. Idriss and D. Sun-Waterhouse, Chem. Mater., 2008, 20, 1183–1190 CrossRef CAS.
- I. Paramasivam, H. Jha, N. Liu and P. Schmuki, Small, 2012, 8, 3073–3103 CrossRef CAS PubMed.
- C. Burda, Y. Lou, X. Chen, A. C. S. Samia, J. Stout and J. L. Gole, Nano Lett., 2003, 3, 1049–1051 CrossRef CAS.
- D. S. Muggli, J. T. McCue and J. L. Falconer, J. Catal., 1998, 173, 470–483 CrossRef CAS.
- K. Kočí, L. Obalová, L. Matějová, D. Plachá, Z. Lacný, J. Jirkovský and O. Šolcová, Appl. Catal., B, 2009, 89, 494–502 CrossRef.
- S. M. Gupta and M. Tripathi, Chin. Sci. Bull., 2011, 56, 1639–1657 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chloroplast isolation procedure, results of the dye degradation experiments without irradiation, XRD pattern of the non-coated Ti3+ exchanged samples, energy-dispersive X-ray spectroscopy (EDS) measurements, nitrogen adsorption–desorption isotherm, elemental surface composition of the MNAL, normalized results of methylene blue dye degradation, results of the total organic carbon (TOC) measurements and estimation of apparent kinetic constants of methylene blue dye degradation. See DOI: 10.1039/c7ra04185c |
| This journal is © The Royal Society of Chemistry 2017 |