
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidized cyclodextrin-functionalized injectable gelatin hydrogels as a new platform for tissue-adhesive hydrophobic drug delivery†
Thai Thanh Hoang Thi‡
a,
Yunki Lee‡ab,
Seung Bae Ryua,
Hak-Joon Sungb and
Ki Dong Park *a
*a
aDepartment of Molecular Science and Technology, Ajou University, Suwon 443-749, Republic of Korea. E-mail: kdp@ajou.ac.kr
bDepartment of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
First published on 6th July 2017
Abstract
A practical approach toward developing dual-functional hydrogels with high adhesiveness and hydrophobic drug delivery is described. An additional Schiff base reaction was introduced into a phenol–phenol crosslinked gelatin hydrogel to markedly increase adhesiveness. Oxidized β-cyclodextrin (oβ-CD) functionalized with aldehyde groups and possessing a hydrophobic cavity was exploited as a crosslinker in the Schiff base reaction to solubilize the hydrophobic drug. Simply blending gelatin–tyramine (GTA) and oβ-CD in the presence of horseradish peroxidase (HRP)/H2O2 rapidly and controllably formed GTA–oβ-CD hydrogels in situ. The optimal composition of GTA–oβ-CD hydrogels was found to be 5 wt% GTA (GTA5) with 1 wt% oβ-CD. Their adhesiveness was 2.3- and 6.2-fold higher than those of GTA-only hydrogels and fibrin glue, respectively. Their elastic modulus and degradation rate were 1.8- and 1.5-fold higher than those of GTA hydrogels owing to additional imine bonds. Hydrophobic drugs (dexamethasone and curcumin) could be dissolved homogeneously in GTA–oβ-CD matrices with greater loading efficiencies than in GTA matrices. An in vitro test of cell viability using human dermal fibroblasts demonstrated that GTA–oβ-CD hydrogels were cytocompatible. In summary, dual-functional injectable GTA–oβ-CD hydrogels can be used as a promising platform to improve tissue adhesion and hydrophobic drug delivery.
Introduction
Tissue-adhesive biomaterials are a revolutionary platform that can be used for a wide range of medical applications, especially bleeding control and wound healing.1 Therefore, several types of tissue-adhesive biomaterials have been developed and applied in biomedical engineering. However, they still have limitations preventing their use in diverse applications. For example, cyanoacrylate-based adhesive materials work best on dry surfaces, but slowly degrade with toxic product release, and have a time-consuming exothermic polymerization reaction. Concerns have been raised regarding albumin and glutaraldehyde glues because of potential allergic reactions to albumin2 and glutaraldehyde toxicity. Fibrin-based adhesives have poor mechanical strength, induce allergic reactions, require long preparation times, and are relatively expensive.1,2 To overcome these limitations, researchers have explored various approaches to develop new tissue adhesives with high mechanical strength, high adhesiveness, and biocompatibility.As a representative example of on-going efforts toward developing bioadhesives, in situ forming hydrogels are gaining increasing interest. They exhibit outstanding adhesive abilities and fill the damaged area to stop bleeding and block body fluid leakage when applied to anastomosis of traumatic injuries or surgical incision.1,3 In situ forming hydrogels can be designed to accumulate a serofibrinous exudate to form a fibrin bridge, in which fibroblasts can migrate and secrete collagen to heal the injured tissues.4,5 Among others, horseradish peroxidase (HRP)-mediated reactions enable fast and controllable gel formation under mild experimental conditions. Using this method, Lih et al. successfully fabricated chitosan–poly(ethylene glycol)–tyramine (CPT) hydrogels as efficient tissue adhesives.6 Lee et al. also reported the adhesive function of gelatin-conjugated 3-(4-hydroxylphenyl)propionic acid hydrogels.7 As another promising option, a Schiff base reaction has been exploited to create in situ forming adhesive hydrogels. This reaction drives imine bond formation between aldehyde groups and amine groups in the polymer backbone or endogenous amines in the extracellular matrix (ECM). Many systems have been developed based on this Schiff base reaction, such as polydextran aldehyde/branched polyethylenimine,8 oxidized methacrylated alginate/8-arm poly(ethylene glycol) amine,9 PEG/dextran aldehyde10 and oxidized dextran/epsilon-poly-L-lysine.11 These hydrogels all demonstrated superior adhesive properties compared with commercially available fibrin glue. However, the high swelling characteristics caused by hydrophilic polymers are considered a disadvantage.2 To further improve adhesiveness, a combination of photocrosslinking and Schiff base reaction,9 or the formation of a catechol–Fe3+ complex and genipin-primed covalent crosslinking, have been attempted.12 However, using photo-initiators and oxidizing agents, such as like iron(III), often causes toxicity.13
To overcome these drawbacks and move towards clinical translation, we combined HRP-mediated and Schiff base reactions to develop in situ forming bioadhesive hydrogels with fast gelation, tunable material properties, improved adhesiveness, excellent biocompatibility, and ease of use. These hydrogels are also designed as a hydrophobic drug carrier, because the hydrophilic nature of hydrogels normally restricts hydrophobic drug loading. β-Cyclodextrin (β-CD) is well known as a non-toxic adjuvant for pharmaceutical and/or mucoadhesive applications.14 However, intramolecular hydrogen bonding limits its solubility in aqueous solution, preventing its expansion to a usage dose.15 Partly oxidized β-cyclodextrin (oβ-CD) was used to exploit the aldehyde groups for the Schiff base reaction and improve β-CD solubility, thereby improving the loading efficiency.
In this study, along with oβ-CD, gelatin was chosen because it is derived from collagen, a major ECM component, which is naturally non-cytotoxic, biodegradable, non-immunogenic, and adhesive. Simply blending the GTA and oβ-CD under oxidation with HRP/H2O2 resulted in the fast formation of GTA–oβ-CD hydrogels within a few seconds to several minutes. We tested the hypotheses that (i) their adhesive and mechanical strength would be reinforced by dual-crosslinking, including phenol–phenol and via the Schiff base reaction, and (ii) their hydrophobic drug loading efficacy would be higher compared with pure GTA hydrogels.
Experimental
Materials
Gelatin (type A from porcine skin, >300 bloom), tyramine hydrochloride (≥98%), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC, ≥98%), N-hydroxysuccinimide (NHS, 98%), horseradish peroxidase (HRP, type VI, lyophilized powder, 250–330 units per mg solid), hydrogen peroxide (H2O2, 30%), sodium periodate (NaIO4, 99%), ninhydrin (ACS reagent), and collagenase type II were obtained from Sigma-Aldrich (St. Louis, MO, USA). β-CD (Mw = 1134.98 g mol−1, ≥97%) was obtained from Tokyo Chemical Industry Co., Ltd. (T.C.I., Tokyo, Japan). Fibrin gel with two components (Tissucol Duo 500) was purchased from Baxter AG (Volkitswil, Switzerland). Dexamethasone (Dex, ≥97%) and curcumin (≥65%) were purchased from Sigma-Aldrich. Human dermal fibroblasts (hDFBs) were purchased from Lonza Inc. (Walkersville, MD, USA). Dulbecco's modified eagle's medium (DMEM), penicillin–streptomycin (P/S), and trypsin/EDTA were purchased from Gibco BRL (Grand Island, NY, USA). Fetal bovine serum (FBS) and Dulbecco's phosphate buffered saline (DPBS) were purchased from Wisent (Saint-Bruno, Canada). An EZ-Cytox enhanced cell viability assay kit (WST-1 assay reagent) was purchased from ITSBIO (Seoul, South Korea). A Live/Dead Viability/Cytotoxicity Assay Kit was obtained from Invitrogen (Carlsbad, CA, USA). All other chemicals and solvents were used as received.Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio). The resultant solution was dialyzed against deionized water using a dialysis membrane with an MWCO of 500 Da (Spectrum Labs) for 3 days, changing the deionized water (DIW) three times per day. The actual degrees of oxidation were calculated from the 1H NMR spectra16 (ESI, Fig. S2C†). The aldehyde groups in oβ-CD were observed at a wavelength of 1721 cm−1 in the IR spectrum17 (ESI, Fig. S2B†). However, the intensity of the peak was not high, making the peak inconspicuous, because of hemiacetal formation between the aldehyde groups and hydroxyl groups of unoxidized residues.18
:1 molar ratio). The resultant solution was dialyzed against deionized water using a dialysis membrane with an MWCO of 500 Da (Spectrum Labs) for 3 days, changing the deionized water (DIW) three times per day. The actual degrees of oxidation were calculated from the 1H NMR spectra16 (ESI, Fig. S2C†). The aldehyde groups in oβ-CD were observed at a wavelength of 1721 cm−1 in the IR spectrum17 (ESI, Fig. S2B†). However, the intensity of the peak was not high, making the peak inconspicuous, because of hemiacetal formation between the aldehyde groups and hydroxyl groups of unoxidized residues.18Gelation time, elastic modulus, swelling ratio, and in vitro proteolytic degradation
GTA solutions with final concentrations of 10 wt% and oβ-CD solutions with final concentrations in the range 0.2–6 wt% were prepared individually in phosphate buffered saline (PBS, pH 7.2). The GTA and HRP solutions were added into microtube A with a volume ratio of 9:1. The oβ-CD and H2O2 solutions were added into microtube B with a volume ratio of 9:1. The mixtures in microtubes A and B were combined to form the GTA–oβ-CD hydrogels. The detailed compositions of each hydrogel are presented in Table 1.
| Hydrogel code | GTA (wt%) | oβ-CD (wt%) | Phenol content (μmol mL−1) | Aldehyde content (μmol mL−1) | Amine content (μmol mL−1) | CHO/NH2 |
|---|---|---|---|---|---|---|
| GTA5 | 5 | 0 | 9 | 0 | 14.3 | 0 |
| GTA5–oβ-CD0.1 | 5 | 0.1 | 9 | 4.8 | 14.3 | 0.3 |
| GTA5–oβ-CD0.2 | 5 | 0.2 | 9 | 9.5 | 14.3 | 0.7 |
| GTA5–oβ-CD0.3 | 5 | 0.3 | 9 | 14.3 | 14.3 | 1.0 |
| GTA5–oβ-CD0.5 | 5 | 0.5 | 9 | 23.8 | 14.3 | 1.7 |
| GTA5–oβ-CD1 | 5 | 1.0 | 9 | 47.6 | 14.3 | 3.3 |
| GTA5–oβ-CD2 | 5 | 2.0 | 9 | 95.2 | 14.3 | 6.7 |
| GTA5/oβ-CD3 | 5 | 3.0 | 9 | 142.8 | 14.3 | 10.0 |
A vial tilting method was used to examine the gelation time of the GTA–oβ-CD hydrogels. The gelation time is the interval between when the two homogeneous solutions containing polymer precursors and HRP or H2O2 were mixed and when the mixture reached a state where it was no longer a liquid and could not flow.19–21 Each sample analysis was carried out in triplicate.
The elastic moduli (G′) of the GTA–oβ-CD hydrogels were measured using a rheometer (Advanced Rheometer GEM-150-050, Bohlin Instruments, Westborough, MA, USA) with a parallel plate (25 mm diameter, 0°) configuration at 37 °C in oscillatory mode. An HRP concentration of 5.0 μg mL−1 and 0.3% H2O2 were used. After carefully mixing the two types of prepared mixtures, 300 μL of the mixed solution was immediately loaded on the ground plate, and bubbles were removed if they appeared. The upper plate was then lowered to a measuring gap size of 0.5 mm, and the measurement started. The storage modulus (G′) was recorded using a strain of 0.01% and a frequency of 0.1 Hz. The measurement was repeated three times under the same conditions with a standard deviation typically below 20%.
For swelling ratio tests, GTA5 and GTA–oβ-CD hydrogels (500 μL) were kept at room temperature for 30 min to allow crosslinks to be completely formed. They were then immersed in PBS (10 mL) at 37 °C. The samples were removed from PBS after 24 h of incubation and the water residues outside the hydrogels were removed using filter paper. The swelling degree (DS) was calculated using the following formula:
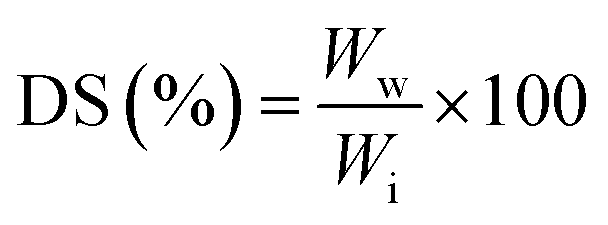
The degradation of GTA–oβ-CD hydrogels (200 μL) took place in the presence of collagenase (1.0 μg mL−1 in PBS), an enzyme that breaks the peptide bonds in gelatin and collagen. The control test was performed with the same hydrogel immersed in PBS without any enzymes. The remaining weight fraction was calculated using the following equation:
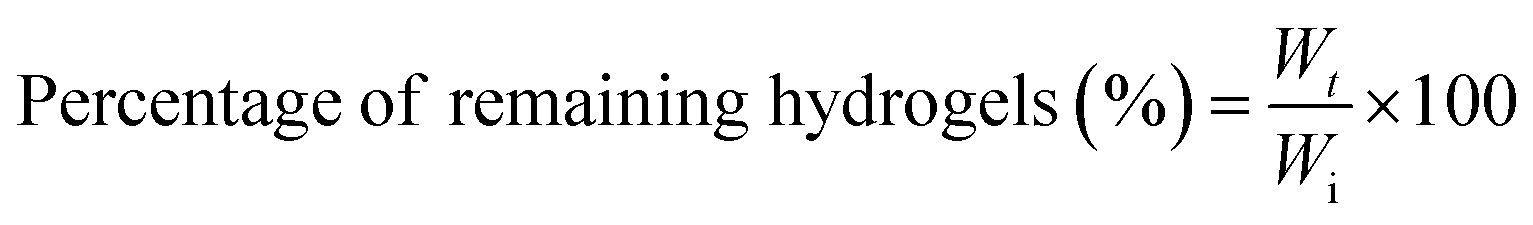
Tissue adhesive
To examine the adhesive strength of the GTA–oβ-CD hydrogels, a method modified from ASTM F2255-03 (“Test Method for Strength Properties of Tissue Adhesives in Lap-Shear by Tension Loading”) was used.6 Porcine skin tissue, directly obtained from a local slaughterhouse, was punched into round pieces with a diameter of 25 mm after removing subcutaneous layers using a surgical scalpel. As shown in Fig. 3A, the round skin pieces were stuck to an elastic substrate using cyanoacrylate glue. Before applying the hydrogels, fatty compositions on the skin surface were decreased by wiping with 70% isopropanol and drying under ambient conditions for 10 min. GTA–oβ-CD solution (100 μL) in the presence of 5.0 μg mL−1 HRP and 0.03% H2O2 was spread on the surface of a skin piece and quickly stuck to the other piece. They were cured in a humidified chamber at room temperature for 60 min under atmospheric conditions. Fibrin glue was used as a control and prepared according to manufacturer instructions using the same gel volume, curing time, and method of porcine skin preparation. The adhesive strength was measured using a UTM machine equipped with a 100 N load cell at room temperature using a constant strain rate of 5% s−1. The adhesive strength is defined as the maximum load observed in these measurements divided by the cross-sectional area of the porcine skin specimens. All samples were measured in triplicate.Cytotoxicity test
To evaluate the potential cytotoxicity of the GTA–oβ-CD hydrogel, 2D cultures of hDFBs were performed on the hydrogels containing various amounts of oβ-CD. Briefly, we fabricated GTA–oβ-CD hydrogels in the 24-well plate, and GTA5 hydrogels were used as a control. GTA5–oβ-CD1, GTA5–oβ-CD2, and GTA5–oβ-CD3 hydrogels were used to test the cytotoxicity as a function of oβ-CD concentration. An HRP concentration of 5.0 μg mL−1 and 0.03% H2O2 were used to form the hydrogels. All hydrogels were stabilized for 30 min before adding cell suspensions with a density of 104 cells per well and incubated at 37 °C in 5% CO2 atmosphere. After 3 days, cell viability was assessed using a Live/Dead Viability/Cytotoxicity Assay Kit (Invitrogen). Images of cells on the gels were recorded using a fluorescence microscope (TE2000, Nikon, Japan) after treating with 500 μL of a mixture containing 2 mM acetomethoxy derivate of calcein 35 (calcein AM) and 4 mM ethidium homodimer-1 (EthD-1) at 37 °C for 1 h. For the WST-1 assay, on the third day the medium was replaced with fresh medium in each hydrogel well and 100 μL of WST-1 solution was added, followed by incubation for 2 h at 37 °C under 5% CO2 atmosphere. After the reaction, 100 μL of the cell culture medium was transferred into a 96-well plate. Sample absorbance (OD) was measured at 450 nm using a microplate reader (VersaMax Tunable Microplate reader, Molecular Devices, Sunnyvale, CA, USA).In vitro drug release test
Solubilizing hydrophobic drug in oβ-CD solutions: Dex and curcumin were chosen as a hydrophobic drug model. Dex (5 mg) was weighed into a microtube containing 1 mL of 6 wt% oβ-CD solution, while another microtube containing PBS was used as a control. Similarly, curcumin (2 mg) was weighed into two microtubes containing 1 mL of 6 wt% oβ-CD solution and PBS, respectively. All drug suspensions were vortexed carefully to achieve the maximum solubility. All microtubes were centrifuged to obtain transparent supernatants, which were used to determine the solubilized drug concentration. The supernatant layers were measured using UV spectroscopy (Jasco V-670 Spectrophotometer) at wavelengths of 254 nm (for Dex) and 420 nm (for curcumin). The solubilized concentrations of Dex and curcumin were calculated based on a standard curve. The drug solubilizing capacity (DSC) was calculated according to the following formula:| DSC (%) = weight of solubilized drugs/(weight of polymer in hydrogels + weight of solubilized drugs) × 100% |
To test the releasing profile of GTA–oβ-CD, Dex was used as a hydrophobic drug model. Hydrogels encapsulating Dex were prepared as described for the gelation time measurement, but with oβ-CD-only solutions replaced by 6 wt% oβ-CD solution containing dissolved Dex (at 2 and 4 mg mL−1). After allowing 30 min for gel stabilization, DPBS (1 mL) was added to each sample. The samples were incubated at 37 °C. After 1.5 h, 5 h, 14 h, 1 d, 4 d, 6 d, 18 d, 24 d, and 29 d, aliquots of 1 mL were harvested from each solution to measure the release amount, and fresh PBS (1 mL) was added. Each analysis was replicated three times.
The released Dex concentration in PBS medium was quantified using high-performance liquid chromatography (HPLC) system (Agilent 1200 series, Waldbronn, Germany) with a UV detector. The mobile phase was acetonitrile/deionized water (ACN/DIW, 50:50 v/v). The automatic sampling injection was used to load 100 μL of sample into the instrument. A CAPCELL PACK C18 column (5 μm, 4.6 × 250 mm, Shiseido Co., Ltd., Tokyo, Japan) was used to separate pure Dex from the matrix at a flow rate of 1 mL min−1. Dex peaks were observed 2.1 min after injection using a UV detector at a wavelength of 254 nm. The standard curve was built by plotting the areas against known concentrations. Each analysis was repeated three times to obtain the mean and standard deviation of the measurements.
Statistical analysis
All data are presented as means ± standard errors. Statistical analyses were performed using Student's t test. A P-value of less than 0.05 was considered statistically significant.Results and discussion
Preparation and characterization of dual-functionalized GTA–oβ-CD hydrogels
The reaction of HRP and H2O2 is known to catalyze the in situ crosslinking of phenol-rich polymers for hydrogel formation. Therefore, tyramine containing a phenol moiety was conjugated to the carboxylic acid groups of the gelatin backbone using EDC/NHS chemistry, with the product named GTA. The phenol content of GTA quantitatively measured using a standard curve of the tyramine hydrochloride solutions was 180.2 ± 5.7 μmol g−1 GTA. For Schiff base formation, the residual amine groups on the gelatin backbones could interact with aldehyde groups. The vicinal diols of β-CD were partially oxidized using sodium periodate to create β-CD-containing aldehyde groups (named oβ-CD). Using 1H NMR (ESI, Fig. S2C†), the oxidation degree of oβ-CD was calculated to be 37.9%.GTA and oβ-CD were then mixed to form hydrogels via HRP/H2O2 and Schiff base reactions. HRP mediated the coupling of each phenol group in GTA, resulting in the formation of carbon–carbon and carbon–oxygen bonds in the ortho-positions, and generating phenoxy groups20 (Fig. 1A). Furthermore, aldehyde functional groups on the oβ-CD molecules reacted with amino groups on GTA to form imine bonds, which can reinforce hydrogel networks22,23 (Fig. 1A). The remaining aldehyde groups reacted with amines on tissue, which improved the hydrogel adhesiveness8,24 (Fig. 1B). Reacting GTA–oβ-CD hydrogels with a ninhydrin reagent (ESI†) enabled us to identify unreacted amine group-containing hydrogel samples because their color changed to purple25,26 (ESI, Fig. S3†). No purple color on the GTA5–oβ-CD0.5 hydrogels (CHO/NH2, n/n ratio of 1.7) indicated that all amine groups had reacted completely with aldehyde to form imine bonds. When the oβ-CD concentration was increased above 0.5 wt%, the GTA–oβ-CD hydrogels contained excess amounts of aldehyde groups, which would contribute to adhesive linkages.
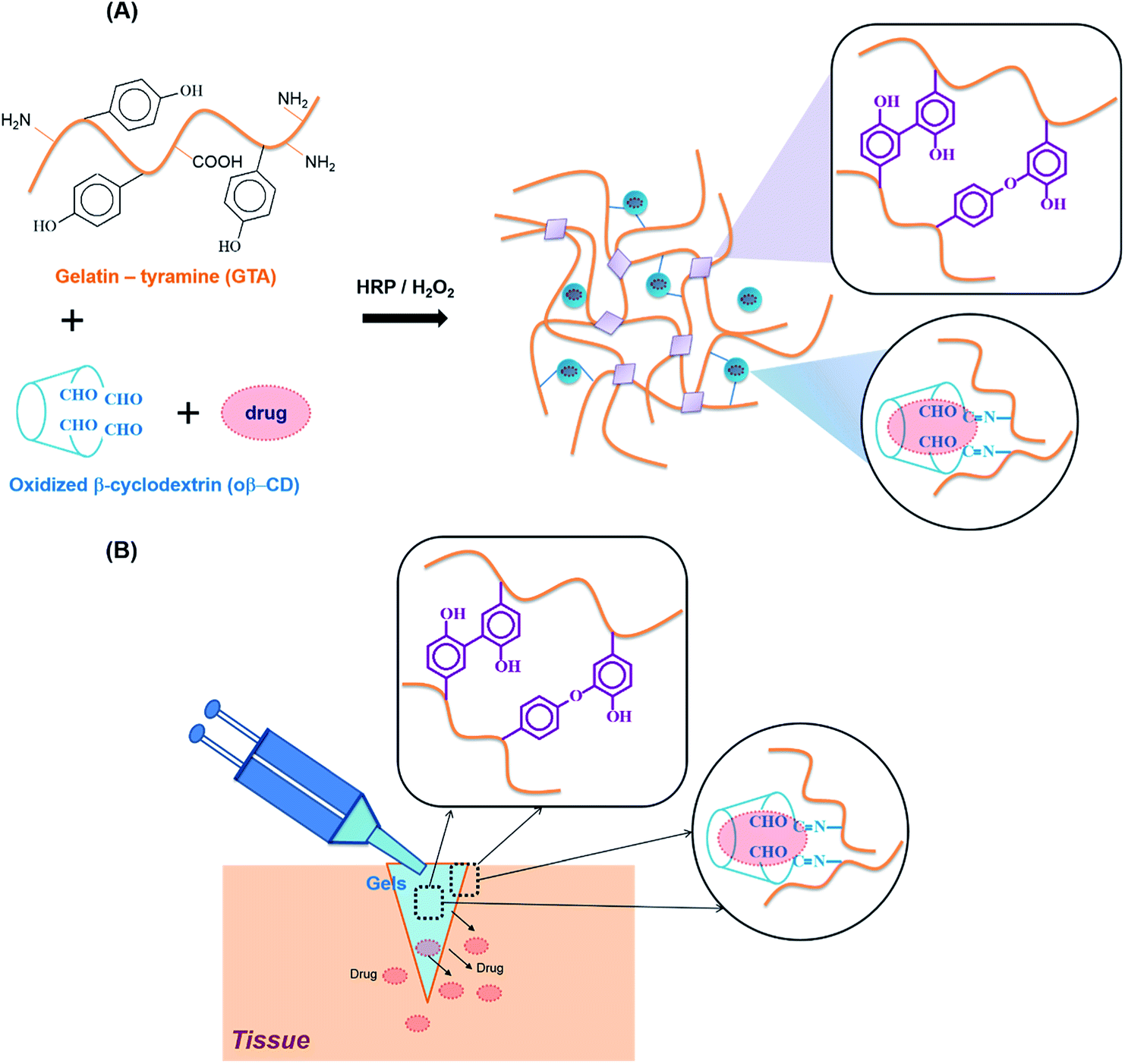 | ||
| Fig. 1 (A) Graphical illustration of the crosslinking methods used to obtain GTA–oβ-CD hydrogels, which can be used to load hydrophobic drugs. (B) Schematic representation of in situ forming adhesive GTA–oβ-CD hydrogels formed by combining HRP catalysis and the Schiff base reaction with therapeutic release. | ||
Gelation time is an important factor in tissue adhesive applications, which require not only a fast rate, to stop bleeding and close the wound effectively, but also tunable properties for applications in different cases. As shown in Fig. 2A, the gelation of GTA–oβ-CD hydrogels was fast and controllable (9–78 s) depending on the HRP concentration and CHO/NH2 n/n ratio. The effect of oβ-CD on the gelation time was negligible (P > 0.05) at low concentration (oβ-CD ≤ 0.5 wt% in GTA5; CHO/NH2 n/n ratio ≤ 1.7). However, the gelation time of GTA5–oβ-CD2 and GTA5–oβ-CD3 hydrogels (CHO/NH2 n/n ratios of 3.3 and 6.7) significantly increased by 1.9- and 2.7-fold (P < 0.05), respectively, when compared with those of GTA5 hydrogels formed using 2.5 μg mL−1 HRP and 0.03% H2O2. This result showed that oβ-CD influenced the velocity of the HRP-catalyzed crosslinking reaction due to the interaction between amine groups in HRP and the remaining aldehyde groups in oβ-CD via the Schiff base reaction, thereby inhibiting the HRP activity.27 In fact, Molin et al. have reported the kinetic reaction of glutaraldehyde with HRP.28 Glutaraldehyde reacted with thiol groups and amine groups in HRP macromolecules via Michael and Schiff base reactions. As a result, the polymerization of HRP increased the HRP molecular weight and size, generating steric hindrance and a consequent decrease in HRP activity. Another factor affecting the interaction between oβ-CD and HRP that can hinder HRP activity may include hydrogen bonding. The remaining hydroxyl groups of oβ-CD can form hydrogen bonds with polar amino acid groups in the HRP molecules. Although the addition of oβ-CD delayed gelation of the GTA hydrogels, the gelation time could still be controlled at around 10 s to a few minutes by increasing the HRP concentration, which was fast enough and tunable for tissue adhesive applications.
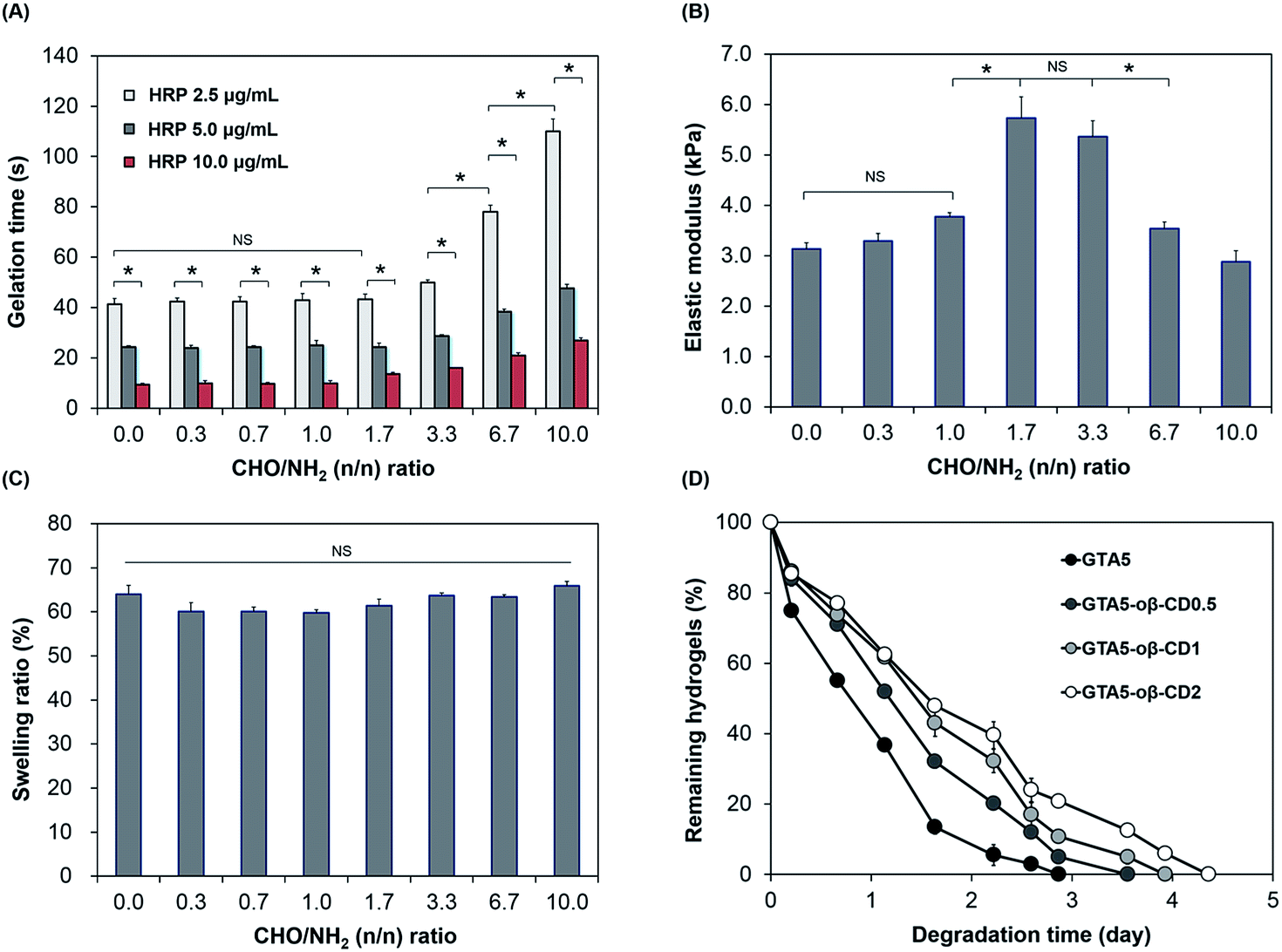 | ||
| Fig. 2 Characterization of GTA–oβ-CD hydrogels: (A) gelation time of GTA–oβ-CD hydrogels as a function of oβ-CD and HRP concentration, (B) elastic modulus, (C) swelling ratio of GTA–oβ-CD hydrogels as a function of aldehyde-to-amine ratio, and (D) in vitro proteolytic degradation of GTA–oβ-CD hydrogels with various oβ-CD content in 1.0 μg mL−1 collagenase. Hydrogel samples were formed by reacting with 5.0 μg mL−1 HRP and 0.03% H2O2 and incubated for 30 min to allow stabilization (*P < 0.05; NS P > 0.05). | ||
The mechanical properties of hydrogels are important design parameters for biomedical applications.29 Therefore, we investigated the mechanical strength of these hydrogels following exposure to factors able to impact them. As we previously reported,6,7,20,30 the stiffness of hydrogels formed using HRP-catalyzed crosslinking was significantly influenced by the concentration of H2O2 and the number of phenol groups available. An increase in H2O2 concentration led to an increase in the storage modulus of the formed hydrogels (ESI, Fig. S4†). In this study, the presence of oβ-CD in the GTA hydrogel system served as a crosslinker, which induced the Schiff base reaction and was, therefore, expected to reinforce the hydrogel network. As shown in Fig. 2B, when the CHO/NH2 ratio was increased from 0 to 1.0, their G′ was slightly increased from 3.1 to 3.8 kPa. This insignificant enhancement (P > 0.05) can be explained by the Schiff base crosslink not being fully formed. At a CHO/NH2 ratio of 1.7, the G′ was at a maximum value (5.7 kPa) due to the maximum formation of Schiff base crosslinks in these hydrogel systems of phenol–phenol crosslinking networks. Ninhydrin-dyed GTA–oβ-CD hydrogels showed no purple color for CHO/NH2 ratios of 1.7 or higher (ESI, Fig. S3†), demonstrating that most of the amine groups on the gelatin backbone were completely reacted with aldehydes in oβ-CD and that Schiff base cross-linking was saturated. However, the elastic modulus decreased significantly (P < 0.05) at CHO/NH2 ratios of 6.7 or higher due to an excess of aldehyde groups. The remaining aldehydes could influence the reaction with H2O2 and HRP, and the performance of the Schiff base reaction. Schöne et al. described that the reaction between aldehyde and H2O2 was possible, but slow.31 Molin et al. studied the kinetic reaction between glutaraldehyde and HRP, revealing that the formation of HRP polymers via Schiff base crosslinking between glutaraldehyde and amines in HRP led to steric hindrance, causing a reduction in the HRP activity.28 Another reason for the decrease in hydrogel stiffness with increasing aldehyde-to-amine ratio and large added amounts of solid content was a dilution effect imparted to excess oβ-CD. A similar trend was reported by Artzi et al.10 Dilution of the hydrogel system had a strong influence on hydrogel stiffness because the presence of excess CHO in GTA5–oβ-CD1 hydrogels (CHO/NH2 ratio of 3.3) did not significantly decrease their elastic moduli (P > 0.05). Based on the aforementioned results, the GTA5–oβ-CD0.5 and GTA5–oβ-CD1 hydrogels showed the highest mechanical strengths compared with pure GTA5 hydrogels due to additional Schiff base formation without the influence of solid dilution.
Swelling properties are among the most important characteristics for adhesive applications because a high swelling index causes compression of nerves or blood vessels, which can lead to life-threatening conditions.24 Fig. 2C shows the swelling values of GTA and GTA–oβ-CD hydrogels, which were immersed in PBS buffer (pH 7.2) at 37 °C. All swelling values of these hydrogels were 60–66%. The low degree of swelling indicated that they were suitable for use in vivo and can overcome the limitation13 of PEG or polysaccharide-based adhesives. Although the elastic moduli of GTA–oβ-CD hydrogels were significantly enhanced by increasing the CHO/NH2 ratio from 0 to 3.3, their swelling ratio was slightly decreased or comparable (P > 0.05). The hydrogel swelling ratio was dependent not only on G′, but also on the balance of hydrophilic and hydrophobic properties in the matrix.13 When an additional oβ-CD was attached to the gelatin backbone through Schiff base linkages, the hydrophilic/hydrophobic balance of the GTA–oβ-CD hydrogels shifted more toward the hydrophilic side than in pure GTA hydrogels due to the oβ-CD hydroxyl groups. Therefore, GTA–oβ-CD hydrogels allowed higher hydration within their matrix. Taken together, the compromise of hydration and elastic modulus within the GTA–oβ-CD hydrogel matrix led to a negligible change in its swelling ratio compared with GTA hydrogels.
Biodegradability is another advantage of gelatin-based hydrogels compared with their synthetic polymer-based counterparts. However, in some specific in vivo applications, gelatin-based biomaterials have shown rapid degradation, making them unsuitable for relevant applications. Therefore, developing strategies to increase their degradation time is needed to expand their various bio-applications. In terms of tissue adhesive applications, if the degradation time can be controlled over a wide range of times, the adhesive could be applied to various locations within the body. When the degradation rate of the GTA–oβ-CD hydrogels was tested, the GTA5–oβ-CD0.5, GTA5–oβ-CD1, and GTA5–oβ-CD2 hydrogels degraded after 3.5, 3.9, and 4.4 days, respectively, in 1 μg mL−1 collagenase solution (Fig. 2D). These results demonstrated that oβ-CD could prolong the degradation time of GTA hydrogels because of additional Schiff base crosslinks. Considering all the data, at a CHO/NH2 ratio of 6.7, the elastic modulus of the GTA–oβ-CD hydrogels decreased, but the degradation time slightly increased. To explain this degradation result, we considered the crosslinking density level between the samples as a major factor. If the crosslinking level was saturated when the oβ-CD amount increased, degradation would not change. Therefore, even with the same crosslinking density, if the total solid concentration increased, the elastic modulus should decrease.
Tissue adhesive strength
Tensile tests of the GTA–oβ-CD hydrogels were performed to determine their adhesive strength to porcine skin. Commercially available fibrin glue was used as a control. The results are presented in Fig. 3B. GTA5 exhibited 2.7-fold higher adhesive strength than fibrin glue. Gelatin-based hydrogels are known to be adhesive and have been studied extensively.32 Phenol moieties conjugated on the gelatin backbone were involved in the reaction between phenol in the hydrogel matrix and tyrosine on the tissue, resulting in the adhesive property of GTA. In previous reports, we demonstrated that GH (gelatin-conjugated 3-(4-hydroxyphenyl)propionic acid) and GHT (gelatin-conjugated 3-(4-hydroxylphenyl)propionic acid and gelatin-conjugated tyramine) presented a stronger adhesive strength (2–4-fold higher) than fibrin glue.7 In this study, GTA was chosen instead of GH or GHT to exploit the remaining amine groups in the gelatin backbone to create Schiff base bonding, which would increase cohesion, as discussed above. The aldehyde groups in the GTA–oβ-CD hydrogels were used to interact with the amino groups of lysine and hydroxylysine on tissues to form Schiff bases24 that were hypothesized to increase the adhesive strength of these hydrogels. In fact, the results in Fig. 3B show that GTA5–oβ-CD1 hydrogels (CHO/NH2 = 3.33) have a 6.2-fold higher adhesive strength than fibrin glue (P < 0.05) and a 2.3-fold higher adhesive strength than pure GTA5 hydrogels (P < 0.05). The adhesiveness of the GTA5–oβ-CD1 hydrogels was also significantly higher than that of pure GTA6, which had the same solid content (P > 0.05) with single phenol–phenol crosslinking. With a CHO/NH2 ratio less than 3.33, the adhesiveness of the GTA5–oβ-CD hydrogels did not significantly increase due to lacking aldehyde residues. Hydrogels with higher oβ-CD concentrations, corresponding to higher aldehyde amounts (CHO/NH2 = 6.66 and 9.98), showed slightly lower adhesive strengths than GTA–oβ-CD1 hydrogels. However, the difference was not significant (P > 0.05). From these data, we concluded that the adhesive strength of GTA hydrogels was improved by the additional Schiff base crosslinking and that saturated strength was achieved with the addition of 1 wt% oβ-CD.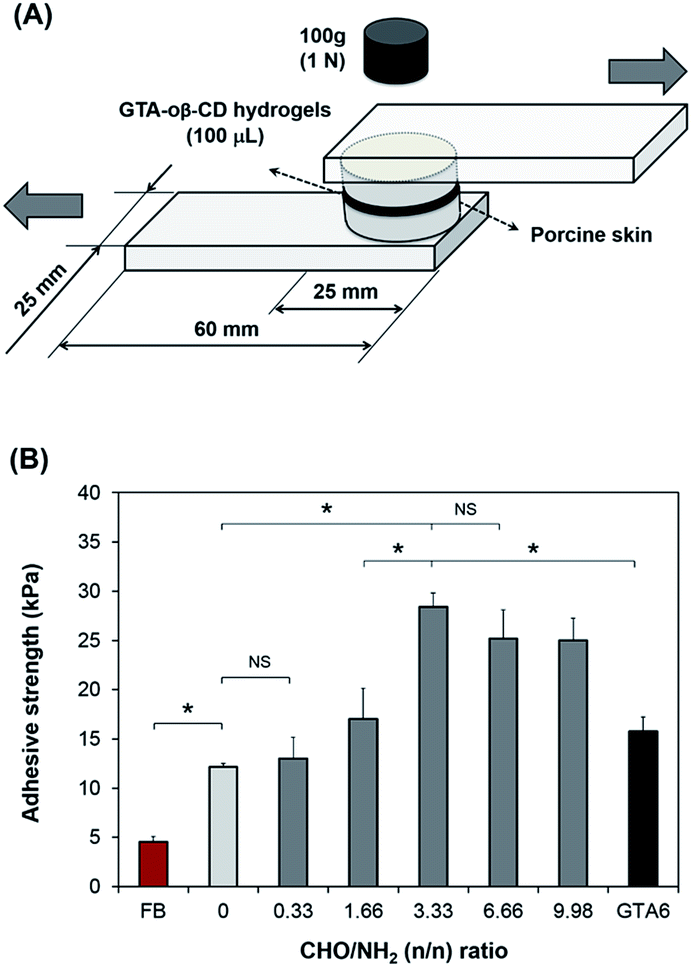 | ||
| Fig. 3 (A) Schematic illustration of tissue adhesive strength measurement. (B) Adhesive strength of GTA–oβ-CD composite hydrogels as a function of oβ-CD concentration using porcine skin. All GTA and GTA–oβ-CD hydrogels were formed by reacting with 5.0 μg mL−1 HRP and 0.03% H2O2. Fibrin glue (FB) was used as a control. *P < 0.05; NS P > 0.05. | ||
In vitro cell cytotoxicity
Gelatin-based hydrogels are cytocompatible and can be introduced into synthetic materials to support cell adhesion. CDs have long been used on the market due to their low toxicity and reduced immunogenicity.33 Depending on the route of administration, the toxic dose is different. For example, 0.79 g of β-CD per kg can cause 50% cell death via the intravenous route (LD50).33 The toxicological effects of β-CD are due to its complexation with cholesterol and membrane lipids through host–guest interactions. As oβ-CD has a similar structure, it is thought to have similar toxicity. The presence of aldehyde groups contributed to its cytotoxicity.2 Therefore, the aldehyde amount should be controlled to form hydrogel networks and enable integration with tissue, while forming imine bonds to attach to oβ-CD on the gelatin chains, limiting the toxicity of CD. In fact, hDFBs were cultured on the surface of GTA5, GTA5/β-CD1, and GTA5–oβ-CD hydrogels for 3 days, and the stained cell images from the Live/Dead assay (Fig. 4A) showed that most hDFBs were alive (green cells, >99%). These results demonstrated that 1–3 wt% oβ-CD did not impact the cytocompatibility of GTA hydrogels. The number of dead cells (in red) was not significant for all the tested gel surfaces. The cells spread and had similar spindle shapes on hydrogel surfaces with or without oβ-CD. To quantify the potential cytotoxic effects of the hydrogels, the WST-1 assay was performed. As shown in Fig. 4B, cell proliferation on all GTA–oβ-CD hydrogel surfaces was similar to that of the control GTA hydrogel. These results suggest that GTA–oβ-CD hydrogels do not affect cell health and that no cytotoxicity was observed when the oβ-CD concentration was below 3 wt%.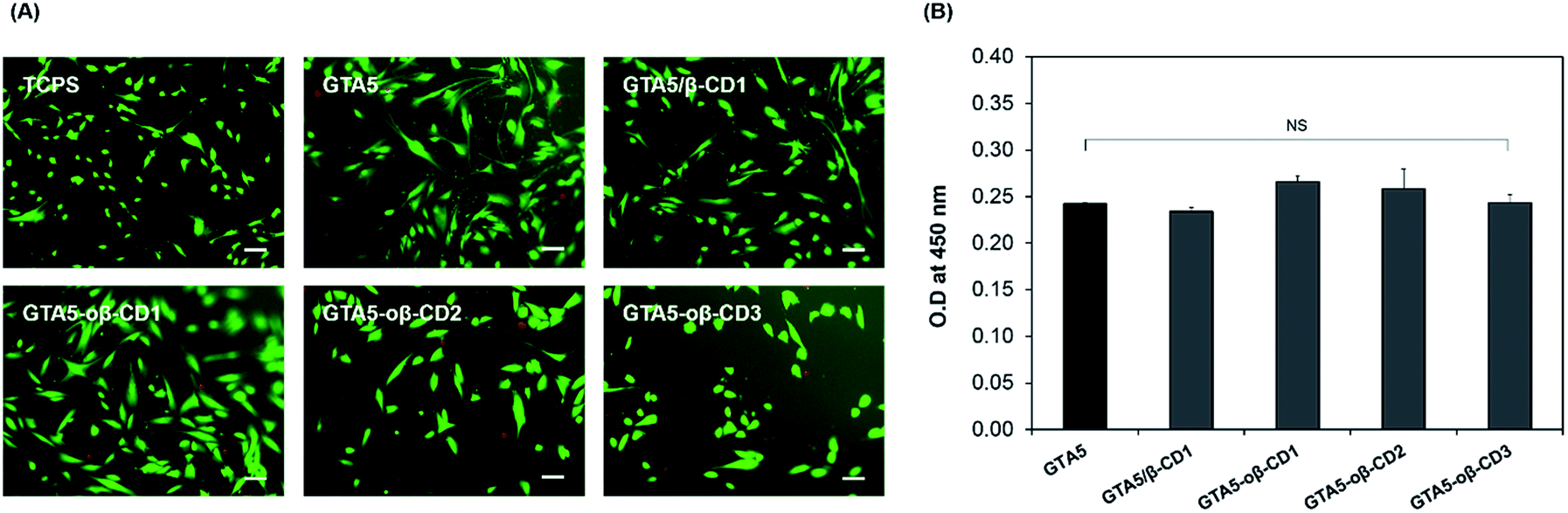 | ||
| Fig. 4 Cell viability and proliferation of hDFBs cultured on GTA and GTA–oβ-CD hydrogels. (A) Fluorescence images of hDFBs on the hydrogel surface after Live/Dead Viability/Cytotoxicity assaying (scale bars represent 100 μm). (B) WST-1 assay was performed to determine cell proliferation within 3 days. All hydrogels were fabricated using 5.0 μg mL−1 HRP and 0.03% H2O2, and incubated for 30 min before adding the cell suspension at a concentration of 104 cells per well (NS P > 0.05). | ||
In vitro drug delivery
Dex, an acidic anti-inflammatory agent, was incorporated within the bioadhesive because infection at an early stage of wound healing decreases the healing ability. Therefore, Dex within the bioadhesive was expected to prevent the entry of bacteria, and consequently accelerate the healing process. Curcumin, which has anti-inflammatory and antioxidant activities, is considered a good candidate for improving wound healing. Jia et al. demonstrated that curcumin can improve wound healing and decrease scar formation in a rabbit model at concentrations of 6–60 μg kg−1.34 Kant et al. used Pluronic F-127 gels loaded with 0.3% curcumin to treat diabetic rats. Their findings indicated that curcumin could induce many factors that expedited wound healing efficiently, including decreased pro-inflammatory cytokine production, reduced MMP-9 expression, increased antioxidant enzyme production, and early epithelialization.35 However, the solubility of curcumin in aqueous solution is extremely low, which limits its effective use as a treatment. Gelatin-based hydrogels, due to their hydrophilic properties in part, limit the topical delivery of Dex and curcumin, as well as other hydrophobic therapeutics. oβ-CD was expected to improve this limitation due to its hydrophobic cavity. Dex and curcumin were dissolved in 6 wt% oβ-CD to determine their maximum solubility. A solution of Dex in DPBS formed an opaque suspension, while the solution was transparent when Dex was mixed with 6 wt% oβ-CD solution. For curcumin, its color became more yellow in the presence of 6 wt% oβ-CD compared with DPBS (ESI, Fig. S5†). UV-Vis spectroscopy was used to determine the absorbance of each dissolved solution to compare the increased solubilities of Dex and curcumin in 6 wt% oβ-CD solution. The results showed that Dex and curcumin could be dissolved in 6 wt% oβ-CD at maximum concentrations of 3.46 mg mL−1 and 0.15 mg mL−1, respectively. Compared with their solubilities in DPBS (0.16 mg mL−1 and 0.02 mg mL−1, respectively), 6 wt% oβ-CD solution had enhanced Dex and curcumin solubility by 21.6- and 7.0-fold, respectively. Furthermore, these results confirmed that β-CD maintains the hydrophobic drug inclusion capability, even after partial oxidation. Therefore, GTA–oβ-CD hydrogels could improve the homogeneity/limited quantity of loaded hydrophobic drugs compared with GTA hydrogels. In fact, GTA5 hydrogels encapsulating 1 mg mL−1 of Dex had a high opacity (Fig. 5A(i)), while GTA5–oβ-CD3 hydrogels were transparent (Fig. 5A(ii)). When incorporating 0.15 mg mL−1 of curcumin, the GTA5–oβ-CD3 hydrogels became more yellow (Fig. 5A(iv)), due to soluble curcumin, than GTA hydrogels (Fig. 5A(iii)). Based on the maximum solubilized concentration of drugs and hydrogel contents, the DSC of GTA5 and GTA5–oβ-CD3 hydrogels was verified. The DSC of GTA5–oβ-CD3 hydrogels was 2.1% for Dex and 0.09% for curcumin, while the DSC of GTA5 was lower, showing 0.2% for Dex and 0.03% for curcumin. These results indicated that incorporating oβ-CD within GTA hydrogels significantly improved their hydrophobic drug solubilizing efficacy, leading to the enhanced availability of hydrophobic drugs in biological systems.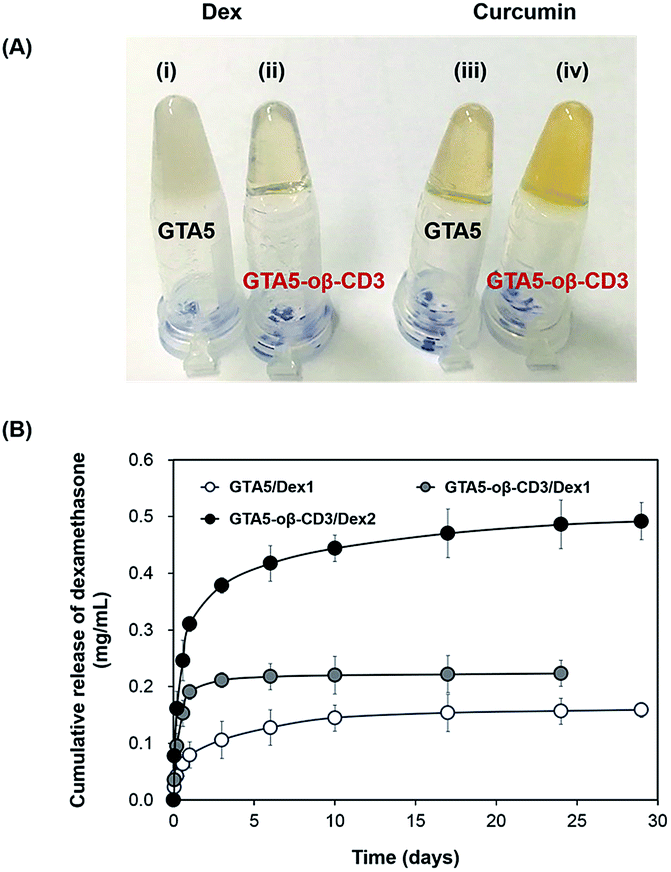 | ||
| Fig. 5 (A) Dex and curcumin were encapsulated in the GTA and GTA–oβ-CD3 hydrogels. (B) Release profiles of Dex from GTA5–oβ-CD3 hydrogels in DPBS (pH 7.4) at 37 °C as a function of Dex content. GTA5-only hydrogel was used as a control. | ||
To assess the release profile of GTA–oβ-CD hydrogels, we used two concentrations (1 and 2 mg mL−1) of Dex as a hydrophobic drug model. Dex was dissolved in 6 wt% oβ-CD at concentrations of 2 and 4 mg mL−1. Then, oβ-CD solutions were mixed with 10 wt% GTA solutions, and the hydrogels were formed by HRP/H2O2 reaction. These hydrogels were coded as GTA5–oβ-CD3/Dex1 and GTA5–oβ-CD3/Dex2, respectively. Pure GTA5 hydrogel was used to incorporate 1 mg mL−1 Dex (coded as GTA5/Dex1) as a control. Fig. 5B shows that Dex release was two-fold higher from GTA5–oβ-CD3/Dex1 hydrogels than from GTA5/Dex1 hydrogels. Due to the presence of oβ-CD, which enhanced the drug solubility (Fig. 5A), an increased amount of hydrophobic drugs could be encapsulated and released. This phenomenon implies that the presence of oβ-CD can improve the drug amount released from the GTA hydrogels because of the remaining hydrophobic cavities, allowing for the enhanced solubilization of hydrophobic drugs. GTA5–oβ-CD3/Dex2 hydrogels could release two-fold more Dex than GTA5–oβ-CD3/Dex1, indicating that increasing the loading dose of the drug can improve the releasing efficacy of the hydrogels. The release profile of GTA-only hydrogels or GTA–oβ-CD hydrogels containing various amounts of drug, showed that the hydrophobic drug could be sustainably released from the hydrogel matrices. After 1 day of incubation, the amount of Dex released from GTA–oβ-CD hydrogels was 2.7-fold higher than that from GTA hydrogels. Therefore, the presence of oβ-CD could support the use of GTA hydrogels for drug delivery in wound healing treatment with improved efficacy and sustainable release.
Conclusions
In this study, dual-functional GTA–oβ-CD hydrogels were successfully developed with controllable gelation times, high adhesiveness, improved stiffness, controlled drug release, and good cytocompatibility. The synergy of Schiff base formation with an enzyme-mediated reaction formed GTA–oβ-CD hydrogels with higher mechanical strengths and stabilities than GTA hydrogels. The adhesive strength of the GTA–oβ-CD hydrogels, measured using porcine skin, was significantly improved owing to additional covalent imine bonds between aldehyde residues of the hydrogel matrix and primary amines of the tissue in comparison with those of GTA hydrogels and commercially available fibrin glue. Due to the hydrophobic cavity of oβ-CD, hydrophobic drugs (Dex) could be released with 2.7-fold-higher efficacy than that of GTA-only hydrogels. With these advantages, the dual crosslinked GTA–oβ-CD hydrogels with twin functions could be promising bioadhesives for various biomedical applications, such as wound healing, topical drug delivery, and tissue engineering.Acknowledgements
This work was supported by the Materials and Components Technology Development Program (Strategic Core Material Technology Development Program) of MOTIE/KEIT [10062079, Development of tissue adhesive/healing biomaterials with adhesion strength over 150 kPa; 10053595, Development of functionalized hydrogel scaffold based on medical grade biomaterials with 30% or less of molecular weight reduction]. This study was financially supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2016M3A9E9941743).References
- M. Mehdizadeh and J. Yang, Macromol. Biosci., 2013, 13, 271–288 CrossRef CAS PubMed.
- P. J. M. Bouten, M. Zonjee, J. Bender, S. T. K. Yauw, H. V. Goor, J. C. M. V. Hest and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1375–1405 CrossRef CAS.
- L. P. Bré, Y. Zheng, A. P. Pêgo and W. Wang, Biomater. Sci., 2013, 1, 239–253 RSC.
- J. S. Boateng, K. H. Matthews, H. N. Stevens and G. M. Eccleston, J. Pharm. Sci., 2008, 97, 2892–2923 CrossRef CAS PubMed.
- J. A. Hubbell, J. Controlled Release, 1996, 39, 305–313 CrossRef CAS.
- E. Lih, J. S. Lee, K. M. Park and K. D. Park, Acta Biomater., 2012, 8, 3261–3269 CrossRef CAS PubMed.
- Y. Lee, J. W. Bae, D. H. Oh, K. M. Park, Y. W. Chun, H.-J. Sung and K. D. Park, J. Mater. Chem. B, 2013, 1, 2407–2414 RSC.
- M. C. Giano, Z. Ibrahim, S. H. Medina, K. A. Sarhane, J. M. Christensen, Y. Yamada, G. Brandacher and J. P. Schneider, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- O. Jeon, J. E. Samorezov and E. Alsberg, Acta Biomater., 2014, 10, 47–55 CrossRef CAS PubMed.
- N. Artzi, T. Shazly, C. Crespo, A. B. Ramos, H. K. Chenault and E. R. Edelman, Macromol. Biosci., 2009, 9, 754–765 CrossRef CAS PubMed.
- S. H. Hyon, N. Nakajima, H. Sugai and K. Matsumura, J. Biomed. Mater. Res., Part A, 2013, 102, 2511–2520 CrossRef PubMed.
- C. Fan, J. Fu, W. Zhu and D. A. Wang, Acta Biomater., 2016, 33, 51–63 CrossRef CAS PubMed.
- C. Ghobril and M. W. Grinstaff, Chem. Soc. Rev., 2015, 44, 1820–1835 RSC.
- E. M. M. Del Valle, Process Biochem., 2004, 39, 1033–1046 CrossRef CAS.
- V. J. Stella and Q. He, Toxicol. Pathol., 2008, 36, 30–42 CrossRef CAS PubMed.
- O. Jeon, D. S. Alt, S. M. Ahmed and E. Alsberg, Biomaterials, 2012, 33, 3503–3514 CrossRef CAS PubMed.
- S. Lu, C. Gao, X. Xu, X. Bai, H. Duan, N. Gao, C. Feng, Y. Xiong and M. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 13029–13037 CAS.
- C. G. Gomez, M. Rinaudo and M. A. Villar, Carbohydr. Polym., 2007, 67, 296–304 CrossRef CAS.
- K. M. Park, Y. Lee, J. Y. Son, J. W. Bae and K. D. Park, Bioconjugate Chem., 2012, 23, 2042–2050 CrossRef CAS PubMed.
- K. M. Park, Y. Lee, J. Y. Son, D. H. Oh, J. S. Lee and K. D. Park, Biomacromolecules, 2012, 13, 604–611 CrossRef CAS PubMed.
- R. Jin, C. Hiemstra, Z. Zhong and J. Feijen, Biomaterials, 2007, 28, 2791–2800 CrossRef CAS PubMed.
- E. S. Dragan, Chem. Eng. J., 2014, 243, 572–590 CrossRef CAS.
- J.-A. Yang, J. Yeom, B. W. Hwang, A. S. Hoffman and S. K. Hahn, Prog. Polym. Sci., 2014, 39, 1973–1986 CrossRef CAS.
- P. J. M. Bouten, M. Zonjee, J. Bender, S. T. K. Yauw, H. van Goor, J. C. M. van Hest and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1375–1405 CrossRef CAS.
- F. A. E. Curotto, Anal. Chem., 1993, 211, 240–241 Search PubMed.
- L. Cui, J. Jia, Y. Guo, Y. Liu and P. Zhu, Carbohydr. Polym., 2014, 99, 31–38 CrossRef CAS PubMed.
- A. M. Azevedo, V. C. Martins, D. M. F. Prazeres, V. Vojinović, J. M. S. Cabral and L. P. Fonseca, Biotechnol. Annu. Rev., 2003, 9, 199–247 CAS.
- H. N. Sven-olov Molin, L. Dolonius and H.-a. Hansson, J. Histochem. Cytochem., 1978, 26, 1053–1056 CrossRef PubMed.
- Y. Ogushi, S. Sakai and K. Kawakami, Macromol. Biosci., 2009, 9, 262–267 CrossRef CAS PubMed.
- K. M. Park, K. S. Ko, Y. K. Joung, H. Shin and K. D. Park, J. Mater. Chem., 2011, 21, 13180–13187 RSC.
- L. Schöne and H. Herrmann, Atmos. Chem. Phys., 2014, 14, 4503–4514 Search PubMed.
- A. P. Duarte, J. F. Coelho, J. C. Bordado, M. T. Cidade and M. H. Gil, Prog. Polym. Sci., 2012, 37, 1031–1050 CrossRef CAS.
- M. E. Davis and M. E. Brewster, Nat. Rev. Drug Discovery, 2004, 3, 1023–1035 CrossRef CAS PubMed.
- S. Jia, P. Xie, S. J. Hong, R. Galiano, A. Singer, R. A. Clark and T. A. Mustoe, Wound Repair Regen., 2014, 22, 730–739 CrossRef PubMed.
- V. Kant, A. Gopal, N. N. Pathak, P. Kumar, S. K. Tandan and D. Kumar, Int. Immunopharmacol., 2014, 20, 322–330 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Ninhydrin assay, synthetic scheme for GTA and oβ-CD, 1H NMR and IR spectra of oβ-CD, elastic modulus of GTA5–oβ-CD1 hydrogels as a function of H2O2 concentration. See DOI: 10.1039/c7ra04137c |
| ‡ Thai Thanh Hoang Thi and Yunki Lee are co-first authors. |
| This journal is © The Royal Society of Chemistry 2017 |