
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A molecular dynamics simulation study for variant drug responses due to FMS-like tyrosine kinase 3 G697R mutation†
Chien-Cheng Leea,
Yu-Chung Chuangb,
Yu-Lin Liub and
Chia-Ning Yang *b
*b
aDepartment of Pharmacy, Zuoying Armed Forces General Hospital, Kaohsiung, Taiwan
bDepartment of Life Science, National University of Kaohsiung, No. 700, Kaohsiung University Road, Nan-Tzu District 811, Kaohsiung, Taiwan. E-mail: cnyang@nuk.edu.tw; Fax: +886-7-5919404; Tel: +886-7-5919717
First published on 8th June 2017
Abstract
FMS-like tyrosine kinase 3 (FLT3) is an attractive target for acute myeloid leukemia. Recent studies have suggested that the application of small-molecule kinase inhibitors is a promising treatment strategy for patients with primary activating mutations of FLT3; however, the development of secondary mutations, including those of A627T, N676D, F691I, and G697R, that confer acquired resistance to kinase inhibitors has become a severe problem. In this study, we conducted a series of molecular dynamics simulations on PKC412- and sorafenib-bound FLT3 kinases and different apo forms of the FLT3 kinase to explain the minor and severe G697R mutation-induced resistance to sorafenib and PKC412, respectively. Structural analysis on our simulation results revealed that the type II kinase inhibitor sorafenib (IC50 = 9 nM) assesses its binding site through either the adenine pocket entrance or the back pocket entrance, whereas the type I kinase inhibitor PKC412 (IC50 = 35 nM) intercalates to its binding site from the front pocket entrance. The G697 residue is located at the end of the FLT3 kinase hinge segment and is close to the front and adenine pockets. In G697R mutation where the substituted R697 residue affects both the front and adenine pocket entrances in different manners, sorafenib may approach its binding site through the back pocket entrance, whereas PKC412 is blocked by the FLT3 kinase. This observation rationalizes that the kinase activity of the G697R mutant does not decrease in the presence of PKC412 concentrations of up to 400 nM, whereas the IC50 value of sorafenib shifts to 200 nM in response to the same mutation.
Introduction
Protein tyrosine kinases (PTKs) are involved in various cellular signaling events such as cell growth and proliferation.1–3 Tyrosine kinase deregulation is related to tumorigenesis and cancer progression. PTKs can be classified into two categories: transmembrane receptor tyrosine kinases and nonreceptor tyrosine kinases.4,5 FMS-like tyrosine kinase 3 (FLT3) is a member of the class III receptor tyrosine kinase family.6 It contains an extracellular domain, a transmembrane domain, a juxtamembrane (JM) domain, and a tyrosine kinase domain interrupted by a short kinase insertion.5 In the FLT3 monomer, the kinase domain is autoinhibited by the JM segment interacting with both N- and C-terminal lobes of the kinase domain so that the αC helix orients away from the ATP binding pocket; therefore, the kinase conformation is stabilized in the inactive state. Upon FLT3 ligand binding in the extracellular domain, FLT3 dimerizes and undergoes a conformational change to remove the JM segment from the kinase domain, thus resulting in the rearrangement of some structural elements in the kinase domain into an active state to continue the kinase function.FLT3 is expressed in primitive hematopoietic precursors within bone marrow.6 Reports have indicated that two types of mutations of FLT3 resulting in ligand-independent activation are associated with acute myeloid leukemia (AML).7 The most common type, occurring in approximately 23% of patients with AML, is the FLT3-internal tandem duplication (ITD) mutation that involves the ITD of coding sequences within the JM domain by interrupting the autoinhibition function of the JM segment. The other type, occurring in 7% of patients with AML, is point mutations such as D835Y in the kinase domain. Accordingly, small-molecule inhibitor therapy targeting the FLT3 kinase domain has become an attractive strategy for AML.8–12 Related studies have proven that the inhibition of FLT3 kinase activity by using small-molecule inhibitors induces apoptosis in cell lines with FLT3-activating mutations and increases the survival rate of mice expressing mutant FLT3 in their bone marrow cells.13,14 Although such a therapeutic strategy is promising for patients with primary activating mutations of FLT3, the development of secondary resistance mutations such as A627T, N676D, F691I, and G697R is emerging as a new obstacle to the treatment.15–17 Among the secondary resistance mutations, G697R confers high-level resistance to various inhibitors including PKC412 (IC50 = 35 nM for FLT3ITD, no decrease in kinase activation at concentrations of up to 400 nM for FLT3ITD,G697R), SU5614, and K252a, but it remains sensitive to sorafenib (IC50 = 9 nM for FLT3ITD and 200 nM for FLT3ITD,G697R).16
As illustrated in Fig. 1(A), PKC412 is a staurosporine derivative that has indolocarbazole lactam rings to mimic the ATP adenine rings. PKC412 is classified as a type I kinase inhibitor that binds to the active kinase conformation where the DFG motif adopts a DFG-in orientation, the N- and C-terminal lobes form a cleft to accommodate ATP, and the activation loop flips in solvent to generate a protein substrate binding site for obtaining γ-phosphate transferred from ATP.18,19 Sorafenib is a type II kinase inhibitor that binds to kinase in the inactive conformation, featured by a DFG-out orientation where the DFG phenylalanine points toward the ATP phosphate group binding region and by the packing of the activation loop on the kinase surface to block the protein substrate binding.20,21 Sorafenib is a slender and flexible compound, as indicated in Fig. 1(B), with a signature urea linkage shared by several type II kinase inhibitors. In the DFG-out orientation, the flipping-outward DFG phenylalanine spares a back pocket. Type II kinase inhibitors are always wedged between the adenine pocket and the back pocket. Regardless of the type I or II classification, the portion of a kinase inhibitor occupying the adenine pocket is anchored through H-bond interaction formed by the backbone atoms of amino acid residues located on the hinge connecting the N- and C-terminal lobes of the kinase.22–24
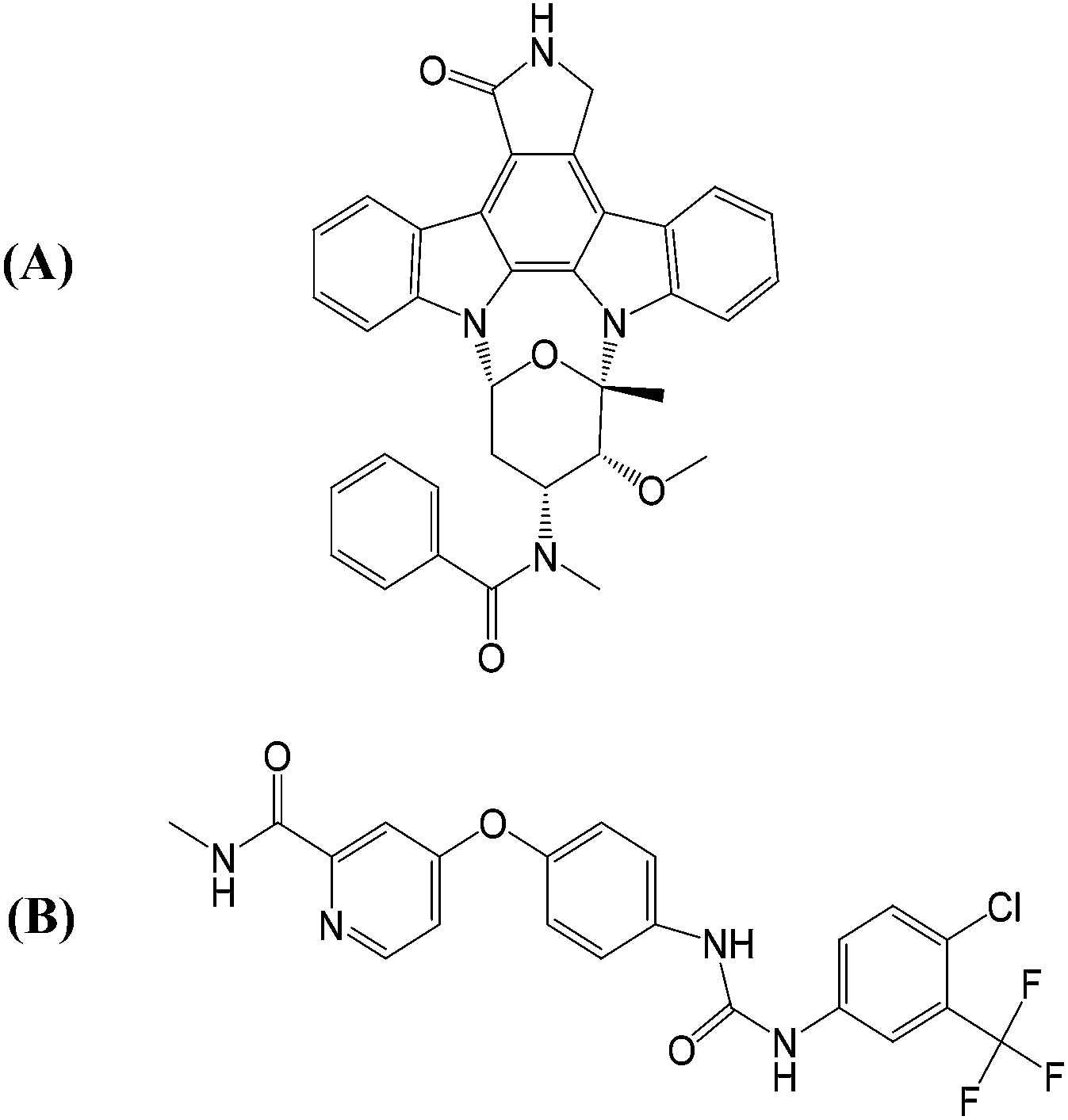 | ||
| Fig. 1 Structures of the two studied FLT3 kinase inhibitors: (A) PKC412 and (B) sorafenib. | ||
The above-mentioned features of FLT3 in active and inactive states are all collected in Fig. 2(A) and (B), respectively. It is also important to know that in active state there is an R-spine formed by four hydrophobic amino acid residues to strengthen the active conformation and maintain the kinase function.24,25 In FLT3 kinase, F691 (the gatekeeper residue), M665, F830 (in the DFG motif), and H809 are in close contact to form the R-spine, as indicated in Fig. 2(A). However, R-spine in the inactive state is disrupted, as shown in Fig. 2(B).
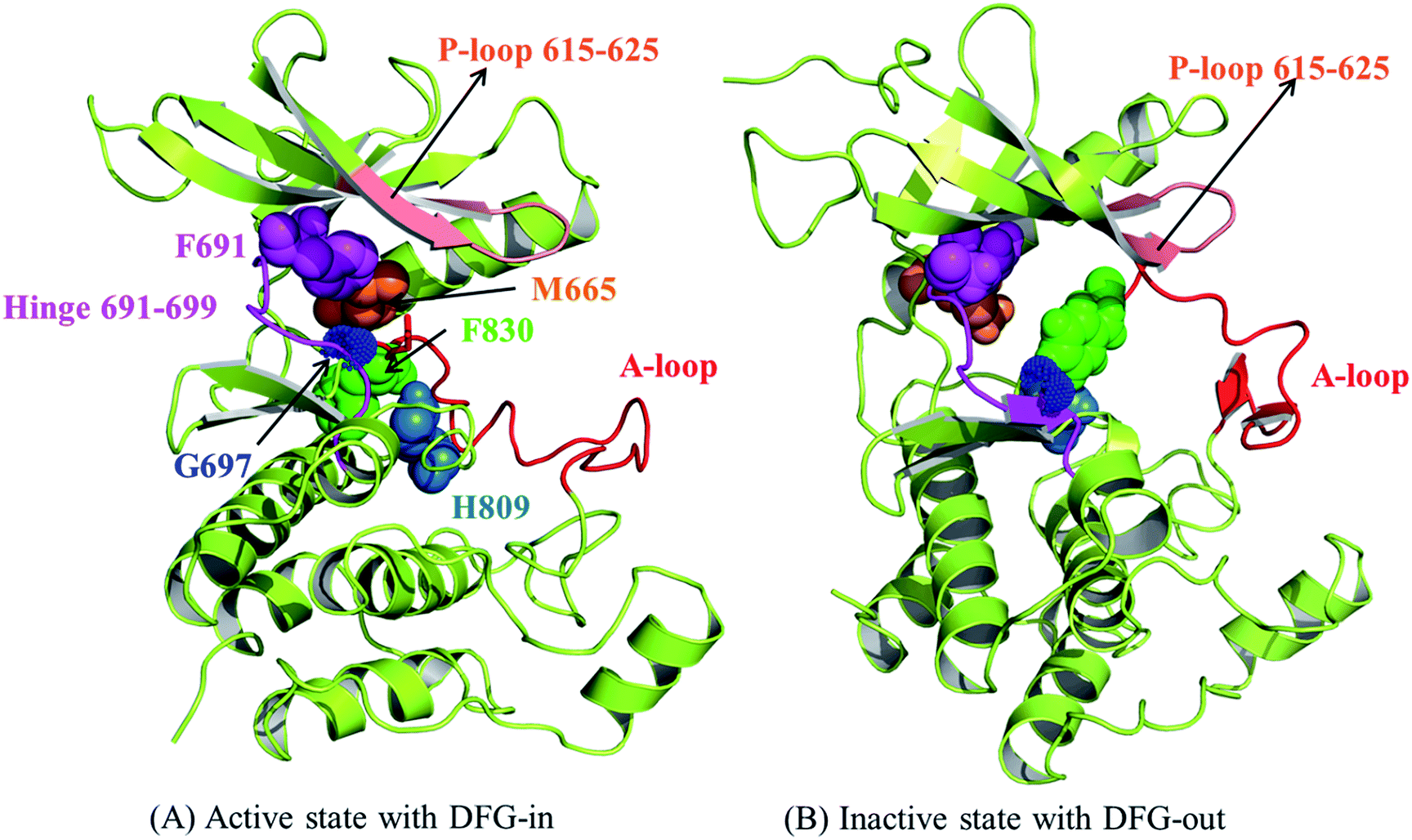 | ||
| Fig. 2 The structural features to differentiate the active and inactive states of the kinase domain of FLT3. (A) The active state and (B) the inactive state. | ||
As shown in Fig. 2, G697 in FLT3 kinase is located near the adenine pocket entrance. When G697 is mutated to a larger arginine residue, it is likely that the bulky R697 induces a steric clash to impede type I and II kinase inhibitors to access their binding site, but it does not interfere with ATP binding and kinase function. In this work, molecular dynamics (MD) simulations were applied to elucidate the different mechanisms underlying G697R mutation-induced resistance to PKC412 and sorafenib. We first evaluated the modeled structures of FLT3 kinase domain bound with PKC412 and sorafenib to obtain the binding site information. Meanwhile, four apo forms of FLT3 kinase domain in the active or inactive state, with G697 or R697, were constructed as well to differentiate the tolerance mechanisms by two types of kinase inhibitors toward the same point mutation. We believe this work provides useful structural information for the future development of FLT3 kinase inhibitors for a more effective treatment of diseases such as AML.
Materials and methods
Studied systems
MD simulations have long served as an efficient tool for evaluating the structural detail and dynamic behaviors of proteins, because the trajectory of individual atoms can be recorded and monitored over time. Numerous MD simulation-based studies have successfully provided insights into protein–protein and protein–small molecule interactions at the atomic level.26–30 In this study, we examined the different levels of drug resistance in response to G697R mutation on the FLT3 kinase domain carrying ITD, which implies that the JM segment flips away from the FLT3 kinase. Accordingly, we evaluated six systems including FLT3ITD:PKC412, FLT3ITD:sorafenib, FLT3ITD/DFGin, FLT3ITD,G697R/DFGin, FLT3ITD/DFGout, and FLT3ITD,G697R/DFGout.Construction of apo and inhibitor-bound FLT3 structures using homology
None of the aforementioned structures is available; therefore, we applied the following strategy to build the initial model structures for MD simulations. To build the FLT3ITD:sorafenib complex, we utilized the quizartinib-bound FLT3ITD complex structure (PDB ID: 4XUF, where quizartinib is also a type II kinase inhibitor bound to the inactive state of FLT3 kinase domain)25 as the target structure and sorafenib-bound hCDK8 kinase as the reference structure (PDB ID: 3RGF)21 to replace sorafenib into the related binding site of the FLT3 kinase after the structural alignment of the FLT3 and hCDK8 kinases with a root-mean-square deviation (RMSD) value of 1.427 Å. We selected the quizartinib-bound FT3 complex because sorafenib and quizartinib share a urea group in their scaffolds, and both compounds are type II kinase inhibitors recognizable by the inactive state conformation. To validate the resultant FLT3ITD:sorafenib complex structure, we monitored the presence of several signature interactions including H-bonds on the pyridine nitro atom, the amino hydrogen atom on the picolinamide moiety, and two hydrogen atoms in the urea linker. Additional details are provided in the Results and discussion section.To build the FLT3ITD/DFGout structure, we simply removed the bound ligand in quizartinib bound-FLT3ITD25 (PDB ID: 4XUF, where FLT3 is in the inactive state)25 and proceeded with structural optimization centered on the ligand binding site. Furthermore, we manually replaced the G697 residue with arginine in FLT3ITD/DFGout to generate FLT3ITD,G697R/DFGout.
PKC412 binds to kinase in the active state conformation with the DFG-in orientation.31 When we used the quizartinib-bound FLT3ITD complex structure (where FLT3ITD is in the inactive state conformation) as the target structure and PKC412-bound DYRK1A kinase (PDB ID: 4NCT) as the reference structure (where DYRK1A is in the active state conformation),31 we imposed the PCK412 binding mode into the FLT3ITD kinase domain and rearranged the coordinates of the activation loop, αC helix, and DFG motif to adjust FLT3ITD into the active state conformation. The resultant FLT3ITD:PKC412 complex structure possessing two H-bonds formed between the kinase hinge region and PKC412 (by using 3-pyrroline-2-one) was similar to the DYRK1A:PCK412 complex structure. The resultant FLT3ITD:PKC412 complex was further applied to create FLT3ITD/DFGin by removing the ligand PKC412. To generate the FLT3ITD,G697R/DFGin structure, we substituted the G697 residue with arginine and proceeded with local structural optimization centered at R697 and the ligand binding site.
Setup for molecular dynamics simulations
The N- and C-terminals of FLT3 were capped by acetyl and N-methyl groups, respectively. MD simulations were performed using the AMBER 12.0 software package32 with ff03.r1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 and ff99SB34 for four models. The force field parameters for the ligand were generated using the general AMBER force field by employing the Antechamber program. The partial atomic charges for the ligand atoms were assigned using the AM-BCC protocol35 after electrostatic potential calculations at the HF/6-31G* level. All hydrogen atoms of the FLT3 protein were assigned using the LEAP module under the consideration of ionizable residues set at their default protonation states at a neutral pH value. Each complex was immersed in a cubic box of the TIP3P water model.36 An appropriate size of the box was selected for the distance between the atoms in each system and the wall to be greater than 12 Å. The neighboring TIP3P water molecules within 2 Å of the complex were removed to prevent unnecessary H-bonds between the studied FLT3 system and the added solvent molecules at the beginning of the simulation. Each solvated system was energy minimized by considering three stages, each employing 500 steps of the steepest descent algorithm and 500 steps of the conjugate gradient algorithm with a non-bonded cutoff of 8.0 Å. At stage 1, the studied structure was restrained so that the added TIP3P water molecules reoriented appropriately in the system. At stage 2, the backbone of the studied structure was restrained so that the amino acid side chains could find appropriate ways to avoid neighboring species conflicts. At stage 3, the entire solvated structure was minimized without any restraint.
33 and ff99SB34 for four models. The force field parameters for the ligand were generated using the general AMBER force field by employing the Antechamber program. The partial atomic charges for the ligand atoms were assigned using the AM-BCC protocol35 after electrostatic potential calculations at the HF/6-31G* level. All hydrogen atoms of the FLT3 protein were assigned using the LEAP module under the consideration of ionizable residues set at their default protonation states at a neutral pH value. Each complex was immersed in a cubic box of the TIP3P water model.36 An appropriate size of the box was selected for the distance between the atoms in each system and the wall to be greater than 12 Å. The neighboring TIP3P water molecules within 2 Å of the complex were removed to prevent unnecessary H-bonds between the studied FLT3 system and the added solvent molecules at the beginning of the simulation. Each solvated system was energy minimized by considering three stages, each employing 500 steps of the steepest descent algorithm and 500 steps of the conjugate gradient algorithm with a non-bonded cutoff of 8.0 Å. At stage 1, the studied structure was restrained so that the added TIP3P water molecules reoriented appropriately in the system. At stage 2, the backbone of the studied structure was restrained so that the amino acid side chains could find appropriate ways to avoid neighboring species conflicts. At stage 3, the entire solvated structure was minimized without any restraint.
The MD simulations in this study were performed using a standard protocol, which entails gradual heating, density, equilibration, and production procedures in the isothermal isobaric ensemble (NPT, P = 1 atm and T = 300 K) MD method. A minimized solvated system was used as the starting structure for subsequent MD simulations. In the 500 ps heating procedure, the system was gradually heated from 0 to 300 K in 50 ps, followed by applying the density procedure at 300 K for 500 ps and constant equilibration at 300 K for 50 ps. After the equilibration procedure, the system underwent a 20 ns production procedure for determining the conformation. The time step was set at 2 fs. A snapshot was captured every 10 ps to record the conformation trajectory during the production procedure. An 8 Å cutoff was applied to treat nonbonding interactions, such as short-range electrostatic and van der Waals interactions, whereas the particle mesh Ewald method was applied to treat long-range electrostatic interactions.37 The SHAKE algorithm38 was used to constrain all bonds containing hydrogen atoms to their equilibrium lengths. To enlarge the data sampling pool, we submitted two independent simulation runs for each studied system. For structural analyses, we used the trajectory in the last 40 ns from each simulation run, covering 800 conformation snapshots, for each FLT3 system.
Results and discussion
MD stability of the studied systems
The Cα RMSD values for the six studied systems, each system with two 70 ns simulation runs, in the production duration as a function of time are plotted in Fig. 3 to evaluate the simulation trajectory quality and convergence. The curves in Fig. 3(A) and (B) represent the two inhibitor-bound complexes. Notably, the PKC412-bound curves fluctuate in a greater magnitude than do the sorafenib-bound curves. It is due to that FLT3 kinase in the FLT3ITD:sorafenib complex is in the inactive state with the activation loop packing against the C-terminal lobe (the red segment in Fig. 4) and the structural flexibility is reduced. By contrast, the higher oscillation in the PKC412-bound curves is attributed to the flexible activation loop (the red segment in Fig. 5) exposed in the solvent implying the FLT3 kinase is in the active state.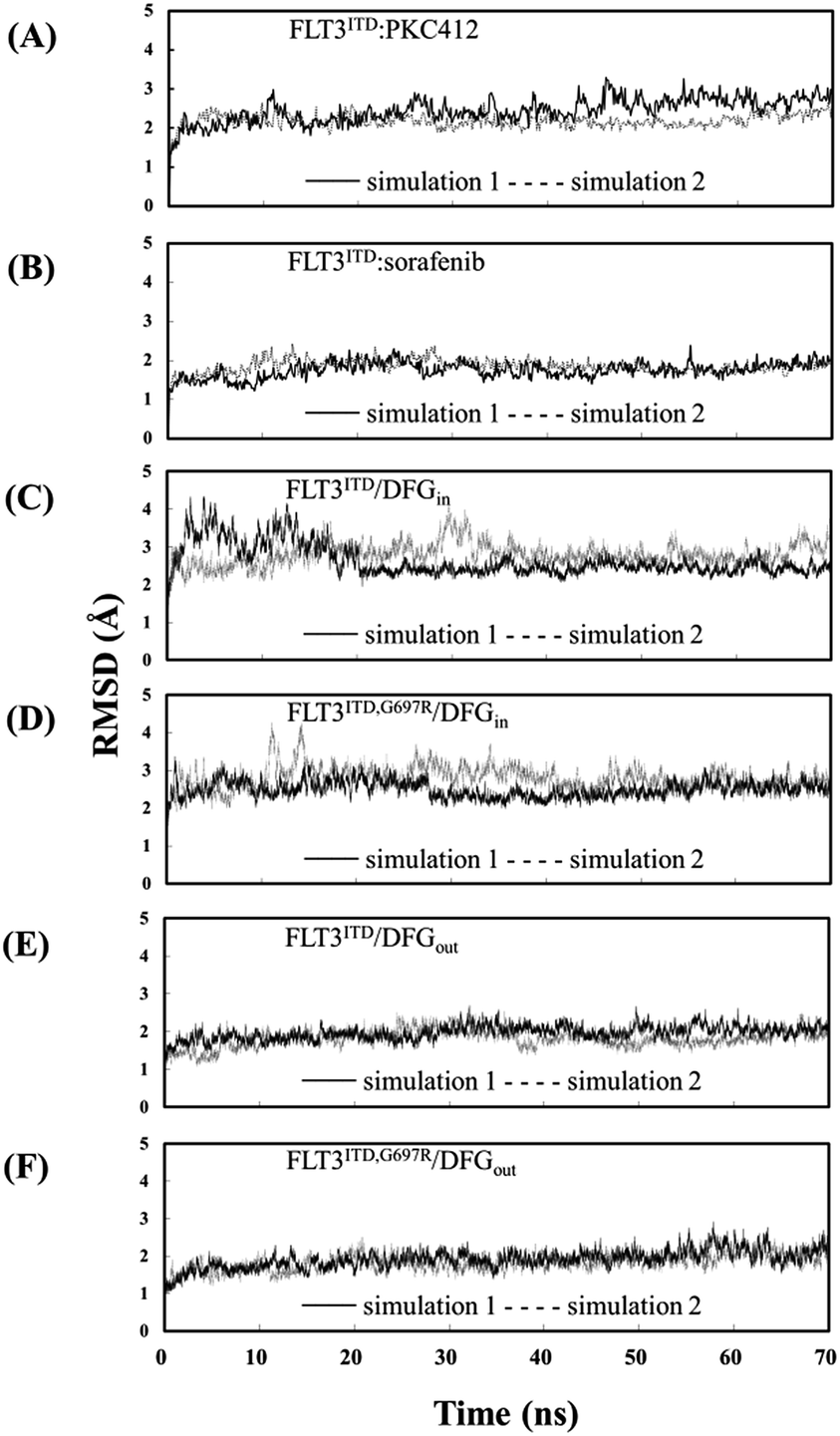 | ||
| Fig. 3 RMSDs of heavy atoms of (A) the PKC412-bound FLT3 kinase carrying G697; (B) the sorafenib-bound FLT3 kinase carrying G697; (C)–(F) the unbound FLT3 kinase carrying G697 or R697 and with different DFG motif orientations. | ||
 | ||
| Fig. 4 MD-predicted structure of the FLT3ITD:sorafenib complex. The red segment represents a relatively ordered activation loop of the FLT3 kinase in the inactive state, whereas the strand-loop-strand motif in salmon indicates the P-loop. Two close-up views of the adenine and back pockets show the interaction between the ligand and kinase. The four residues in pink, orange, green, and blue CPK colors indicate F691, M665, F830, and H809, respectively, in the interrupted R-spine. | ||
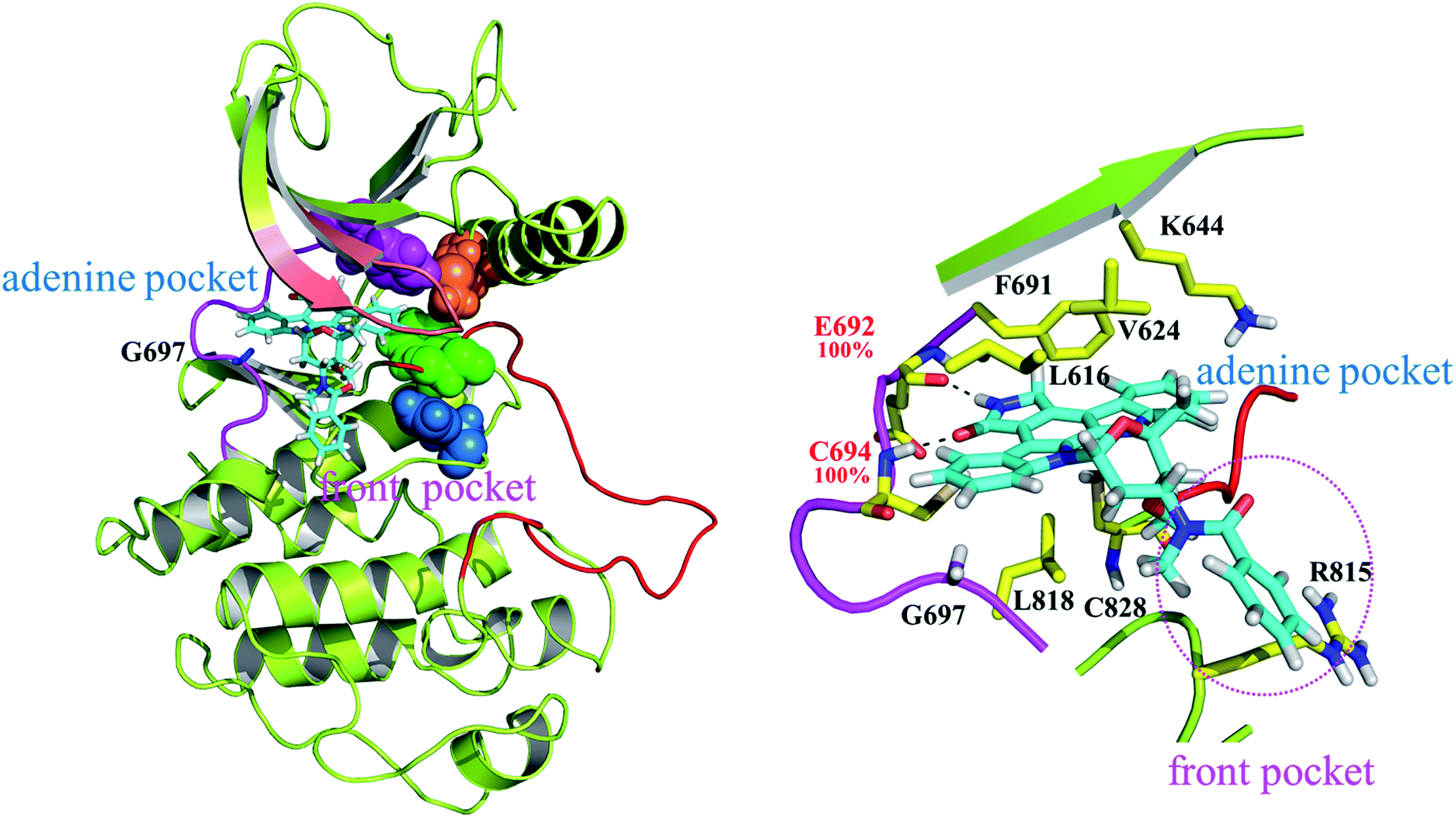 | ||
| Fig. 5 MD-predicted structure of the FLT3ITD:PKC412 complex. The red segment represents a relatively flexible activation loop of the FLT3 kinase in the active state, whereas the strand-loop-strand structure in salmon indicates the P-loop. A close-up view provides the detailed features of the adenine and front pockets. The four residues in pink, orange, green, and blue CPK indicate F691, M665, F830, and H809 lined up in the stable R-spine. | ||
The eight curves in Fig. 3(C)–(F) are responsible for the FLT3ITD/DFGin, FLT3ITD,G697R/DFGin, FLT3ITD/DFGout, and FLT3ITD,G697R/DFGout. These curves are relatively steady. As expected, the curves of the FLT3 kinase carrying DFG-out (i.e., in the inactive state with its activation loop packed on the C-terminal lobe surface, the red segment in Fig. 4 and 6) are less flexible than those of the FLT3 kinase carrying DFG-in (i.e., in the active state with the activation loop exposed in the solvent, the red segment in Fig. 5 and 7).
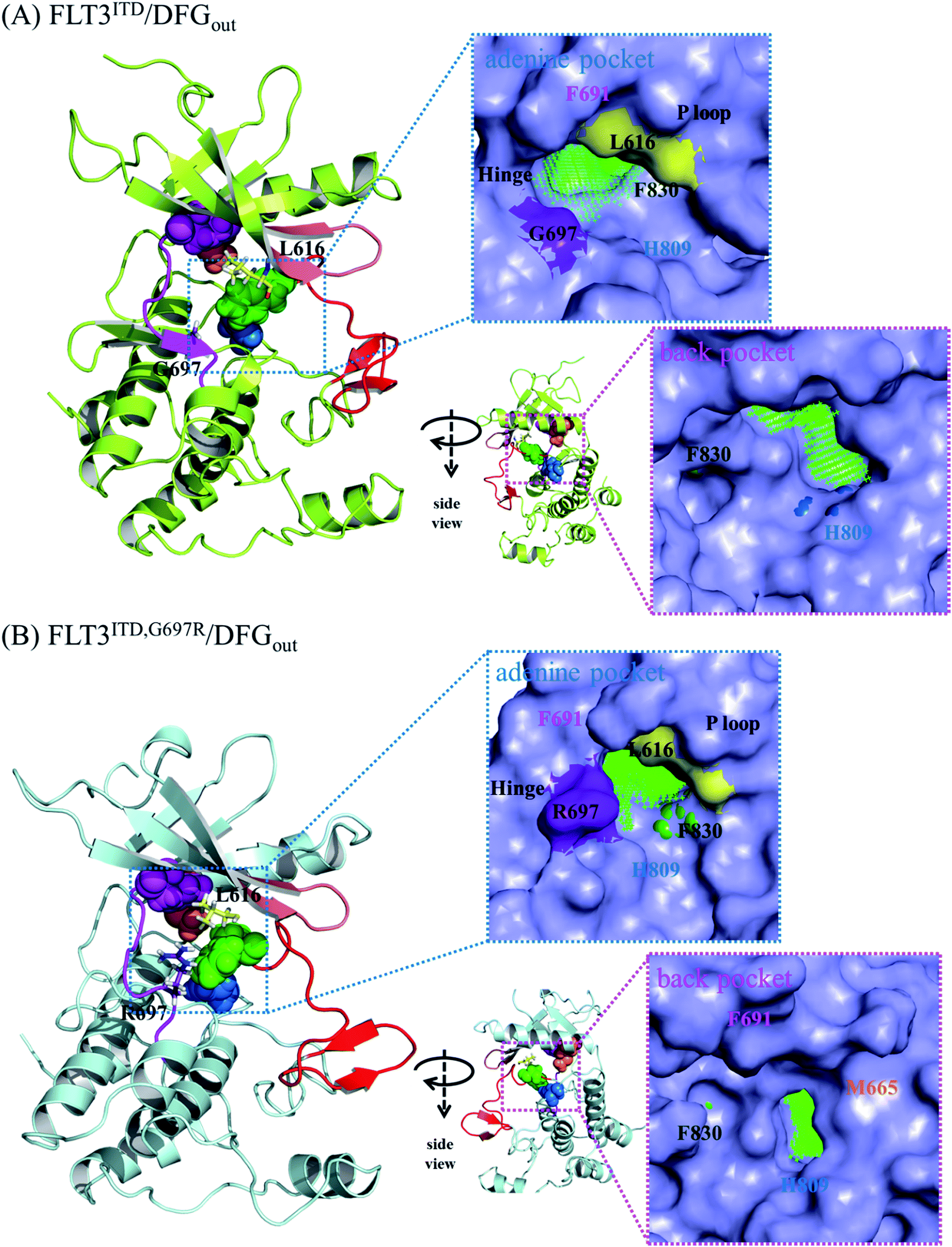 | ||
| Fig. 6 MD-predicted structures of (A) FLT3ITD/DFGout and (B) FLT3ITD,G697R/DFGout. The four residues in CPK show the interrupted R-spine in the inactive state conformation. | ||
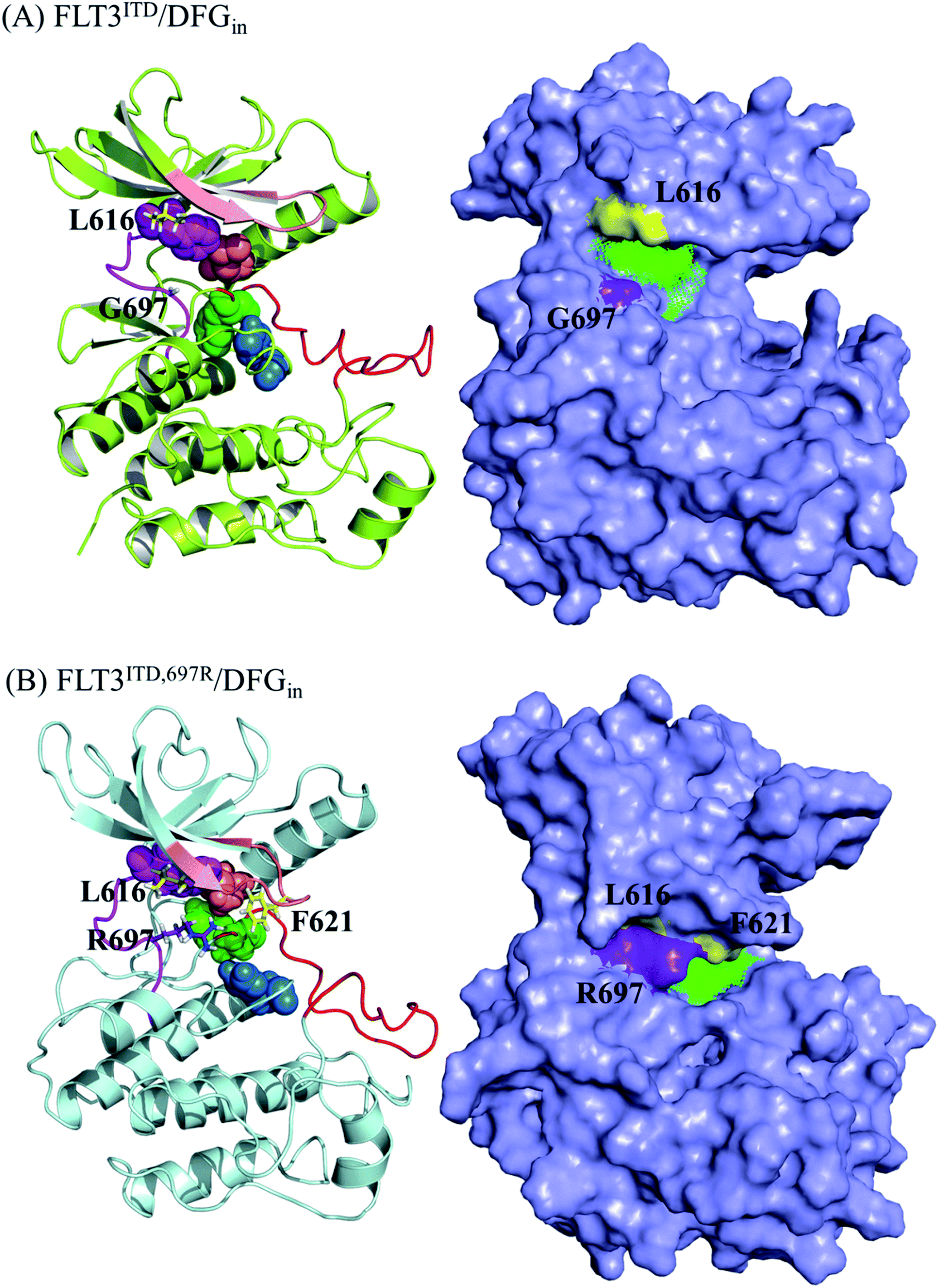 | ||
| Fig. 7 MD-predicted structure of (A) FLT3ITD/DFGin and (B) FLT3ITD,G697R/DFGin. The four residues in CPK define the intact R-spine. In FLT3ITD/DFGin, the surface representation structure indicates that the binding cavity for PKC412 is large and available since L616 and G697 point away from each other. However, in FLT3ITD,G697R/DFGin, the surface representation structure indicates that the binding cavity for PKC412 has shrunk owing to the interaction between L616 and R697, which indirectly drags down F621 to shield the front pocket entrance. | ||
Sorafenib binding mode
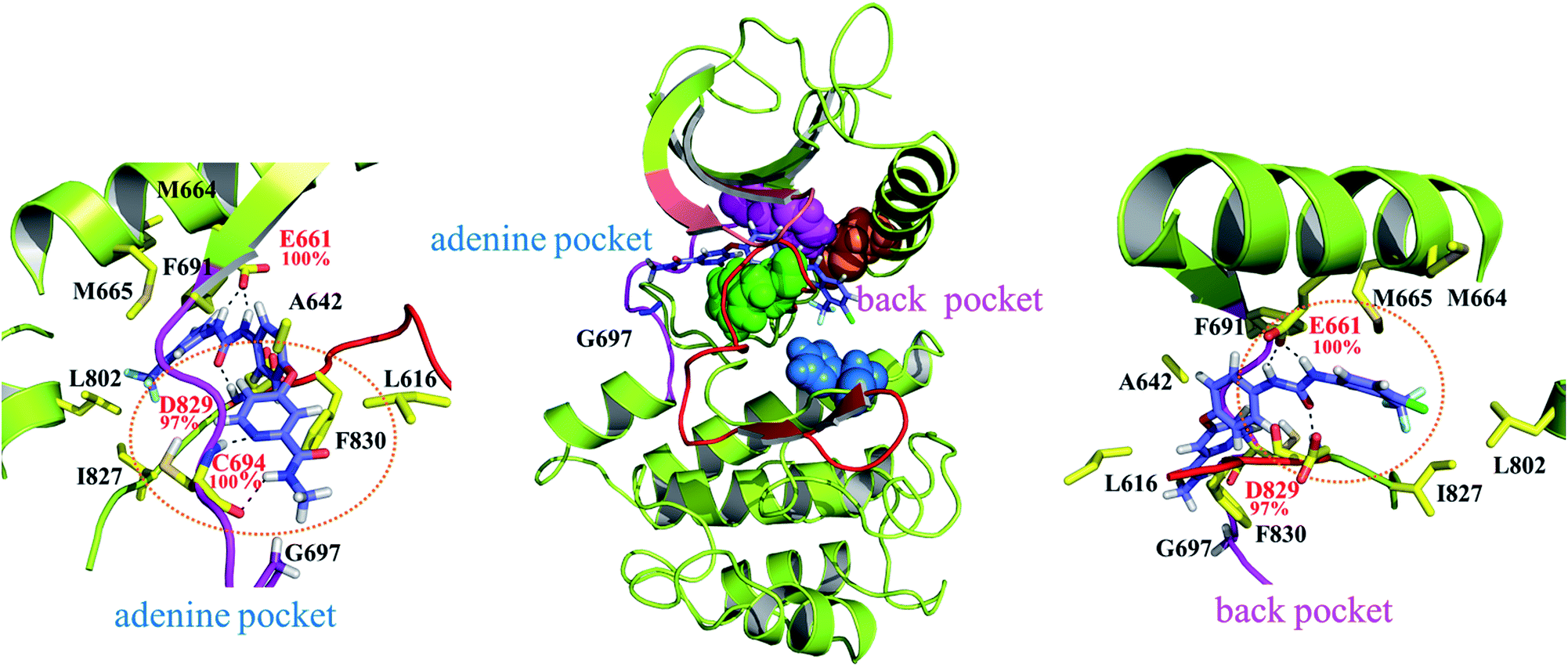
As indicated in Fig. 4 sorafenib stretches in the adenine and back pockets, where the back pocket is present exclusively with a DFG-out orientation rendered by F830 pointing outward. Two close-up panels centered on the adenine and back pockets are included for comprehensive binding detail. In the adenine pocket, C694 (the fourth amino acid residue on the FLT3 kinase hinge) forms two H-bonds through its backbone amino hydrogen atom and carbonyl oxygen atom to the pyridine nitro atom and amino hydrogen atom on the picolinamide moiety, respectively. In the back pocket, two amino hydrogen atoms in the urea linker are secured by E661 (on the αC helix), and the carbonyl oxygen atom in the urea linker forms a H-bond with the amino hydrogen atom of the D829 (of the DFG motif) backbone. C694, E661, and D829 fasten sorafenib in the FLT3 kinase by forming H-bonds whose occurrences are almost 100% among the 800 collected snapshots; these residues play similar roles in the CDK8 kinase (A100, E66, and D173)21 and p38α kinase (M109, E71, and D168)20 to interact with the bound sorafenib. In Fig. S1,† we summarized a structural comparison made among sorafenib-bound FLT3, CDK8, and p38α kinases to support our modeled FLT3ITD:sorafenib complex. In addition to these electrostatic interactions, sorafenib experiences van der Waal interactions by its surrounding hydrophobic residues. Specifically, the central phenoxy ring is clamped by the gatekeeper residue F691 and DFG motif's F830. The 4-chloro-3-(trifluoromethyl)phenyl moiety is surrounded by M664 and M665, on the αC helix, and by L802 and I827, on the C-terminal lobe; the pyridine ring is slightly surrounded by A642 and L616 on the N-terminal lobe. Notably, G697 is near the end of the hinge region and is too small to be involved in the sorafenib pocket. In G697R mutated FLT3, it is possible that the long side chain of R697 may provide hydrophobic interaction toward the methyl group of sorafenib, instead of occupying sorafenib binding site.In Fig. 4, the gatekeeper F691 (in pink CPK) and M665 (located on the αC helix, in orange CPK) cover the top region of the back pocket, whereas F830 (a member of the DFG motif, in green CPK) and H809 (a member of the HRD motif, in blue CPK) cover the bottom region of the back pocket. Conversely, these four residues in the active state of FLT3 form the so-called regulatory spine (R-spine), through their hydrophobic contact, to strengthen the active kinase conformation.39,40 Type II inhibitors such as sorafenib, imatinib, and quizartinib, which prefer to bind the inactive state of kinases, can fit their urea linker and an associated ring into the back pocket encompassed by the disrupted R-spine.24,25
PKC412 binding mode
Fig. 5 highlights the FLT3ITD:PKC412 complex. Unlike type II inhibitors wedged between the adenine and back pockets,22–24 PKC412 suits itself in the adenine pocket and a hydrophobic region adjacent to the end of the hinge,41 which is termed as the front pocket herein. Accordingly, the entire PKC412 binding site is relatively outward compared with the sorafenib binding site. As shown in the side panel, E692 and C694 (on the hinge) form two H-bonds with the amine and ketone groups in the 3-pyrroline-2-one moiety of PKC412. These two H-bond occurrences are nearly 100% among the 800 sampled snapshots. In addition, the binding interaction is mostly hydrophobic. Specifically, from the N-terminal lobe, L616, V624 (both on the P-loop), K644 (on β3, by using its long side chain), and F691 induce van der Waal interactions toward the upper face of PKC412, whereas from the C-terminal lobes, L818, C828, and R815 add some contact to the lower face of PKC412. Notably, except for the R815 residue interacting with the extended phenyl ring outside the ATP pocket, the other residues forming the hydrophobic pocket surround the indolocarbazol lactam ring moiety, which is the scaffold of staurosporine and related derivatives. Our predicted PKC412 binding mode in the FLT3 kinase follows the binding of PKC412 to DYRK1A.31 That is, F238 (the gatekeeper residue) and K118 (on the β3 strand) provide hydrophobic contacts toward the upper face of PKC412; E239 and L241 (the first and third hinge residues, respectively) connected with the 3-pyrroline-2-one region through two H-bonds; and the extended phenyl ring is in contact with E291 from the C-terminal lobe. Fig. S2† summarized a structural comparison made between PKC412-bound FLT3 and PKC412-bound DYRK1A with parallel ligand–receptor interaction to support our modeled FLT3ITD:PKC412 complex structure. As shown in a close-up panel, the rear side of the PKC412 binding pocket involves dense interactions among the amino acid residues, signifying that the ligand PKC412 can only bind to FLT3 from the front access. Moreover, the R-spine39,40 assembled by F691, M665, F830, and H809 (in pink, orange, green, and blue CPK, respectively) shows steady contact.Structural analysis on apo FLT3ITD and FLT3ITD,G697R
Because the DFG motif can switch between DFG-in (the active state, preferred by the PKC412 binding pocket) and DFG-out (the inactive state, preferred by the sorafenib binding pocket) orientations, we inspected four apo systems, namely FLT3ITD/DFGin, FLT3ITD,G697R/DFGin, FLT3ITD/DFGout, and FLT3ITD,G697R/DFGout, in our MD simulations to evaluate the binding site variance influenced by G697R mutation. Meanwhile, the activation loop and αC helix were also taken into consideration to follow the features in both active and inactive states. It is worth to mention that the mutation site G697 is near the C-terminal end of the hinge segment (F691-L699) which links the N- and C-lobes in FLT3 kinase. Our structural analysis shows that F691, E692, and Y693 are secured by amino acid residues on the N-lobe through H-bond formation whereas Y696, G697, D698, and L699 are tightened by amino acid residues on the C-lobe through H-bond formation as well. Because most of these H-bonds are formed between the backbone amino hydrogen atom and the backbone carbonyl oxygen atom, the mobility within the hinge segment is quite restricted. Such a layout allows C694 and C695, in the middle of the hinge, to be properly positioned to interact with the incoming ligand. On the other hand, this layout also implies that the flexibility of G697 is limited and when G697 is replaced to R697 the conformational influence is imported from the long side chain mobility.FLT3ITD/DFGout in Fig. 6(A) corresponds to the inactive state conformation that is preferred by sorafenib. F830 points outward and the R-spine39,40 is not formed, as evidenced by the four separated residues F691, M665, F830, and H809 (in pink, orange, green, and blue CPK, respectively). Structural analysis suggested that in the FLT3ITD/DFGout structure, the chamber to accommodate the slender sorafenib is a through tunnel with two openings on both the adenine and back pocket sides, as depicted in the two side panels in Fig. 6(A). As shown in the upper right panel of Fig. 6(A), the adenine pocket opening is surrounded by the hinge segment and P-loop,42 where the side chains of G697 and L616 point away from the opening to make the entrance more accessible. As shown in lower right panel of Fig. 6(A), the back pocket opening is furnished by the separated R-spine residues; these two openings afford two entrances for sorafenib to enter its binding site in the FLT3 kinase.
As shown in Fig. 6(B), the R-spine39,40 of the FLT3ITD,G697R/DFGout structure is also interrupted. Moreover, the long side chain of the substituted R697 residue can flip upward and downward, driven by the hydrophobic interactions with L616 in the N-terminal lobe and by the formation of a salt bridge with the nearby D698 in the C-terminal lobe, respectively. When R697 is in close contact with L616 through their long side chains, the adenine pocket entrance becomes narrow and consequently less reachable, thus hindering sorafenib from entering the binding site, as indicated in the upper right panel of Fig. 6(B). Under such a circumstance, the opening on the back pocket acts as an auxiliary entrance to accommodate sorafenib. Moreover, G697R mutation does not affect the back pocket entrance, as indicated in the lower right panel of Fig. 6(B). This could explain why replacing the small residue G696 with the bulky residue R697 does not induce severe drug resistance when IC50 = 9 nM for FLT3ITD and IC50 = 200 nM for FLT3ITD,G697R.
Fig. 7(A) shows the FLT3ITD/DFGin structure, with F830 adopting an inward orientation to point toward the back pocket; therefore, F691, M665, F830, and H809 line up to form a stable R-spine39,40 to ensure the active state conformation. Accordingly, type I kinase inhibitors such PKC412 can enter the binding pocket only from the front pocket entrance. The binding site shown in the surface presentation is wide open for PKC412 to intercalate in.
As shown in Fig. 7(B), for FLT3ITD,G697R/DFGin, R697 is located on the rim of the front entrance and flips upward and downward between L616 and D698 on the N- and C-terminal lobes, respectively. When R697 flips upward and interacted with L616, the P-loop (in pink) bends downward to cover the entrance for PKC412 binding. In particular, the downward-pointing F621 on the P-loop tip shields the front pocket entrance. Moreover, the rigidity of the PCK412 indolocarbazole lactam scaffold does not afford much flexibility for gaining access the binding site. To summarize, the mutation of G697R has some indirect effects on opposing PKC412 to enter its binding site.
Conclusion
Fig. 8 summarizes our analysis. Fig. 8(A) illustrates three pockets for the accommodation of type I and II kinase inhibitors based on the ATP-competitive mechanism.23 The adenine pocket usually takes in a ring system that mimics adenine rings and forms one to three H-bonds with the backbone amino hydrogen atom and the backbone carbonyl oxygen atom on the middle three amino acid residues in the FLT3 kinase hinge segment (E692, Y693, and C694). The back pocket is available exclusively in the inactive conformation when the DFG motif adopts the so-called DFG-out orientation that places the phenylalanine residue (F830 in the FLT3 kinase) outward. Type II kinase inhibitors such as sorafenib and quizartinib are slender; therefore, they stretch in both the adenine and back pockets, as shown in Fig. 8(B). By contrast, type I kinase inhibitors prefer to bind to the active state conformation of kinases with a DFG-in orientation, where the back pocket is filled with the DFG phenylalanine and is unavailable. Type I kinase inhibitors such as staurosporine and staurosporine-derivatives, including PKC412 and CEP701, use their indolocarbazole lactam rings to occupy the adenine pocket. Smaller type I inhibitors such as staurosporine and CEP70143,44 may only need the adenine pocket; however, larger type I kinase inhibitors such as PKC412 require both adenine and front pockets, as shown in Fig. 8(C). Regarding the restricted planarity of the PKC412 bulky fused ring system, structural analysis indicated that PKC412 accesses its binding site from the front pocket entrance. Notably, in the active state conformation, the back pocket entrance, blocked by the R-spine, is unavailable. Meanwhile, the adenine pocket entrance is too narrow to accommodate PKC412. However, because the DFG phenylalanine residue points outward, sorafenib cannot enter the binding site through the front pocket entrance; instead, it may manage to enter through the adenine or back pocket entrance. The back pocket entrance is available owing to the disrupted R-spine in the inactive state conformation,39,40 as indicated in Fig. 8(B). Furthermore, Fig. 8(B) shows that in FLT3ITD,G697R, the van der Waals interaction between R697 and L616 shrinks the adenine pocket entrance and therefore impedes sorafenib from entering through the adenine pocket entrance. However, using the back pocket entrance is an alternative; thus, G697R confers minor drug resistance to sorafenib. Fig. 8(C) illustrates the effects of G697R on PKC412 binding. R697 interacts with L616 and indirectly drags down the P-loop whose bulky F621 on the tip points downward to partially shield the front pocket entrance. Restricted by the rigidity of the planar indolocarbazole lactam rings, it is difficult for PKC412 to access the binding site; therefore, G697R causes severe drug resistance. We believe the proposed mechanism for PKC412 also explains the drug resistance of K-252a toward FLT3 carrying G697R, because K-252a is also a staurosporine derivative. On the basis of our simulation on both inhibitor-bound and apo forms of FLT3, we successfully distinguished the effects of G697R on PKC412 and sorafenib in two divergent mechanisms. We expect this rationale to be useful in investigating type I and II kinase inhibitors challenged by point mutations. The present study provided structural impact brought by G697R mutation toward two types of inhibitors, PKC412 and sorafenib. In the future, we may consider applying enhanced sampling simulations45,46 to compare the three pocket entrances.
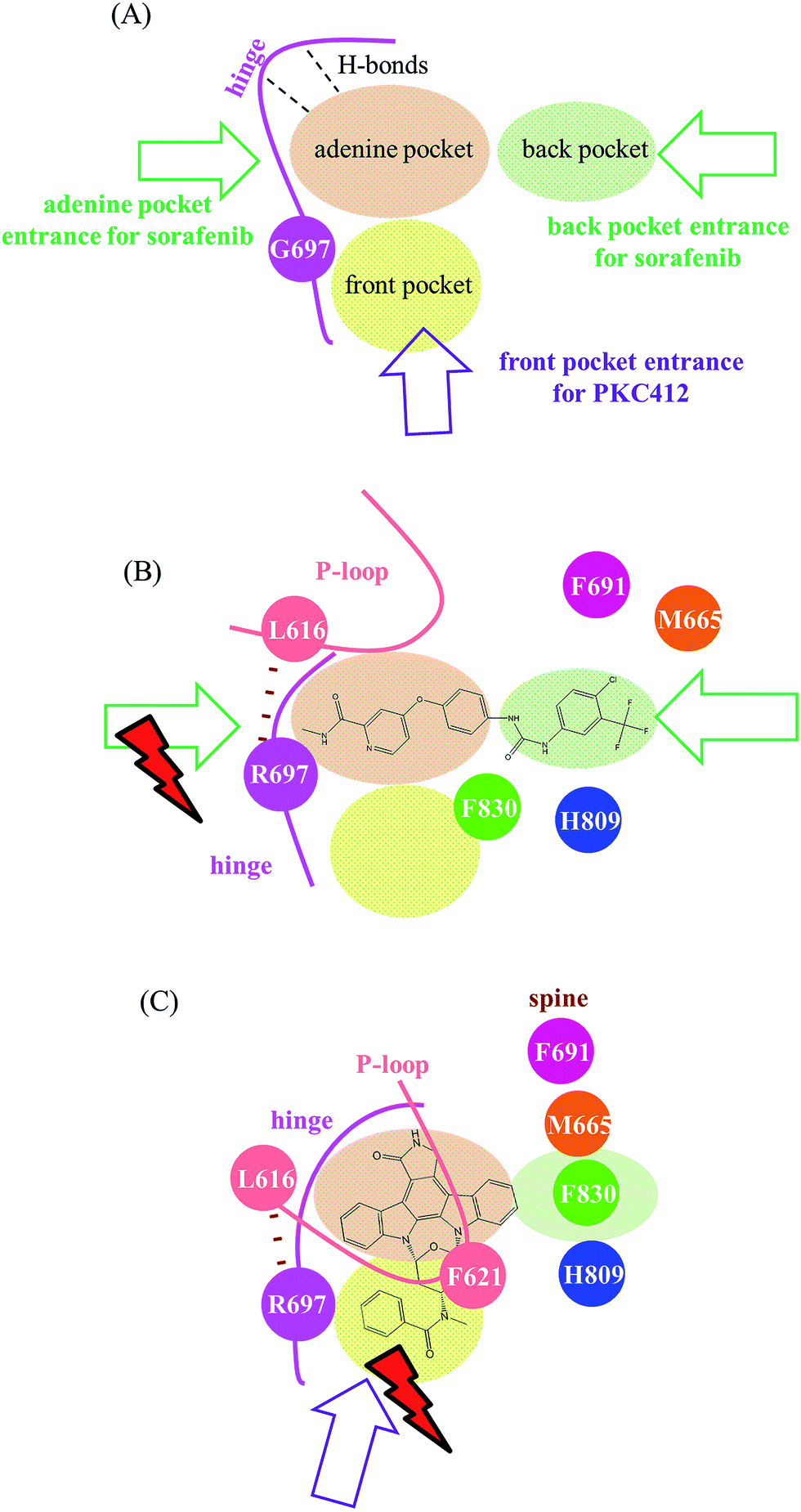 | ||
| Fig. 8 Summarized scheme distinguishing different mechanisms accounting for varying effects of G697R on sorafenib and PKC412. (A) Without G697R mutation, three possible entrances exist, where the adenine pocket entrance and the back pocket entrance are for sorafenib, and PKC412 would take the front pocket entrance. (B) In G697R, sorafenib is not allowed to enter through the adenine pocket entrance, but the back pocket entrance serves as an alternative to alleviate the effects caused by G697R. (C) In G697R, R697 in the studied mutant interacts with L616 and indirectly drags down F621, which therefore shields the front pocket entrance for PKC412 to intercalate into the binding site. | ||
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank Zuoying Armed Forces General Hospital (Kaohsiung, Taiwan) for providing the financial support for this study (Grant ZBH 102-25).Notes and references
- Y. Mizuguchi, S. Specht, K. Isse, E. Sasatomi, J. G. Lunz 3rd, T. Takizawa and A. J. Demetris, J. Hepatol., 2015, 63, 399–407 CrossRef CAS PubMed.
- R. Roskoski Jr, Pharmacol. Res., 2015, 94, 9–25 CrossRef PubMed.
- H. Zhang, A. Wang, S. Qi, S. Cheng, B. Yao and Y. Xu, Int. J. Mol. Sci., 2014, 15, 11665–11677 CrossRef CAS PubMed.
- M. K. Paul and A. K. Mukhopadhyay, Int. J. Med. Sci., 2004, 1, 101–115 CrossRef CAS PubMed.
- P. M. Chan, Protein Cell, 2011, 2, 108–115 CrossRef CAS PubMed.
- M. Bianchini, E. Ottaviani, T. Grafone, B. Giannini, S. Soverini, C. Terragna, M. Amabile, P. P. Piccaluga, M. Malagola, M. Rondoni, C. Bosi, M. Baccarani and G. Martinelli, Clin. Chem., 2003, 49, 1642–1650 CAS.
- M. R. Grunwald and M. J. Levis, Int. J. Hematol., 2013, 97, 683–694 CrossRef CAS PubMed.
- C. C. Smith, E. A. Lasater, K. C. Lin, Q. Wang, M. Q. McCreery, W. K. Stewart, L. E. Damon, A. E. Perl, G. R. Jeschke, M. Sugita, M. Carroll, S. C. Kogan, J. Kuriyan and N. P. Shah, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5319–5324 CrossRef CAS PubMed.
- Y. Y. Ke, V. K. Singh, M. S. Coumar, Y. C. Hsu, W. C. Wang, J. S. Song, C. H. Chen, W. H. Lin, S. H. Wu, J. T. A. Hsu, C. Shih and H. P. Hsieh, Sci. Rep., 2015, 5, 11702 CrossRef CAS PubMed.
- P. P. Zarrinkar, R. N. Gunawardane, M. D. Cramer, M. F. Gardner, D. Brigham, B. Belli, M. W. Karaman, K. W. Pratz, G. Pallares, Q. Chao, K. G. Sprankle, H. K. Patel, M. Levis, R. C. Armstrong, J. James and S. S. Bhagwat, Blood, 2009, 114, 2984–2992 CrossRef CAS PubMed.
- K. Spiekermann, R. J. Dirschinger, R. Schwab, F. Faber, C. Buske, S. Schnittger, L. M. Kelly, G. Gilliland and W. Hiddemann, Blood, 2002, 100, 34a CrossRef PubMed.
- S. A. Wander, M. J. Levis and A. T. Fathi, Ther. Adv. Hematol., 2014, 5, 65–77 CrossRef CAS PubMed.
- C. C. Smith, E. A. Lasater, X. Zhu, K. C. Lin, W. K. Stewart, L. E. Damon, S. Salerno and N. P. Shah, Blood, 2013, 121, 3165–3171 CrossRef CAS PubMed.
- C. C. Smith, Q. Wang, C. S. Chin, S. Salerno, L. E. Damon, M. J. Levis, A. E. Perl, K. J. Travers, S. Wang, J. P. Hunt, P. P. Zarrinkar, E. E. Schadt, A. Kasarskis, J. Kuriyan and N. P. Shah, Nature, 2012, 485, 260–263 CrossRef CAS PubMed.
- J. Cools, N. Mentens, P. Furet, D. Fabbro, J. J. Clark, J. D. Griffin, P. Marynen and D. G. Gilliland, Cancer Res., 2004, 64, 6385–6389 CrossRef CAS PubMed.
- E. Lierman, I. Lahortiga, H. Van Miegroet, N. Mentens, P. Marynen and J. Cools, Haematologica, 2007, 92, 27–34 CrossRef CAS PubMed.
- H. S. Ma, B. Nguyen, A. S. Duffield, L. Li, A. Galanis, A. B. Williams, P. A. Brown, M. J. Levis, D. J. Leahy and D. Small, Cancer Res., 2014, 74, 5206–5217 CrossRef CAS PubMed.
- A. A. Warkentin, M. S. Lopez, E. A. Lasater, K. Lin, B. L. He, A. Y. Leung, C. C. Smith, N. P. Shah and K. M. Shokat, eLife, 2014, 3, e03445 Search PubMed.
- E. Weisberg, C. Boulton, L. M. Kelly, P. Manley, D. Fabbro, T. Meyer, D. G. Gilliland and J. D. Griffin, Cancer Cell, 2002, 1, 433–443 CrossRef CAS PubMed.
- H. V. Namboodiri, M. Bukhtiyarova, J. Ramcharan, M. Karpusas, Y. Lee and E. B. Springman, Biochemistry, 2010, 49, 3611–3618 CrossRef CAS PubMed.
- E. V. Schneider, J. Bottcher, M. Blaesse, L. Neumann, R. Huber and K. Maskos, J. Mol. Biol., 2011, 412, 251–266 CrossRef CAS PubMed.
- D. K. Treiber and N. P. Shah, Chem. Biol., 2013, 20, 745–746 CrossRef CAS PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- Z. Zhao, H. Wu, L. Wang, Y. Liu, S. Knapp, Q. S. Liu and N. S. Gray, ACS Chem. Biol., 2014, 9, 1230–1241 CrossRef CAS PubMed.
- J. A. Zorn, Q. Wang, E. Fujimura, T. Barros and J. Kuriyan, PLoS One, 2015, 10, e0121177 Search PubMed.
- T. Agarwal, N. Annamalai, T. K. Maiti and H. Arsad, Gene, 2016, 580, 17–25 CrossRef CAS PubMed.
- J. Y. Zhan, J. L. Zhang, Y. Wang, Y. Li, H. X. Zhang and Q. C. Zheng, J. Biomol. Struct. Dyn., 2015, 34, 2351–2366 Search PubMed.
- H. Xie, Y. Li, F. Yu, X. Xie, K. Qiu and J. Fu, Int. J. Mol. Sci., 2015, 16, 27350–27361 CrossRef CAS PubMed.
- H. W. Chang, F. S. Chung and C. N. Yang, J. Chem. Inf. Model., 2013, 53, 1775–1786 CrossRef CAS PubMed.
- Y. C. Chuang, C. H. Chang, J. T. Lin and C. N. Yang, Mol. BioSyst., 2014, 11, 723–733 RSC.
- M. Alexeeva, E. Aberg, R. A. Engh and U. Rothweiler, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 1207–1215 CAS.
- T. A. D. D. A. Case, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. Wang and P. Kollman, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins: Struct., Funct., Genet., 2006, 65, 712–725 CrossRef CAS PubMed.
- A. Jakalian, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- M. Azam, M. A. Seeliger, N. S. Gray, J. Kuriyan and G. Q. Daley, Nat. Struct. Mol. Biol., 2008, 15, 1109–1118 CAS.
- A. P. Kornev, S. S. Taylor and L. F. Ten Eyck, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14377–14382 CrossRef CAS PubMed.
- D. R. Hauser, T. Scior, D. M. Domeyer, B. Kammerer and S. A. Laufer, J. Med. Chem., 2007, 50, 2060–2066 CrossRef CAS PubMed.
- G. Georghiou, R. E. Kleiner, M. Pulkoski-Gross, D. R. Liu and M. A. Seeliger, Nat. Chem. Biol., 2012, 8, 366–374 CrossRef CAS PubMed.
- T. J. Boggon, Y. Li, P. W. Manley and M. J. Eck, Blood, 2005, 106, 996–1002 CrossRef CAS PubMed.
- S. J. Miknyoczki, H. Chang, A. Klein-Szanto, C. A. Dionne and B. A. Ruggeri, Clin. Cancer Res., 1999, 5, 2205–2212 CAS.
- J. Kastner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 932–942 CrossRef.
- J. F. St-Pierre, M. Karttunen, N. Mousseau, T. Rog and A. Bunker, J. Chem. Theory Comput., 2011, 7, 1583–1594 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04099g |
| This journal is © The Royal Society of Chemistry 2017 |