DOI:
10.1039/C7RA03965D
(Paper)
RSC Adv., 2017,
7, 27530-27537
C-Quaternary alkynyl glycinols via the Ritter reaction of cobalt complexed alkynyl glycols†
Received
7th April 2017
, Accepted 12th May 2017
First published on 23rd May 2017
Abstract
A novel approach to C-quaternary alkynyl glycinols based on the Ritter reaction of acetonitrile with cobalt complexed alkynyl glycols is presented. The reaction is promoted by acids such as H2SO4 or BF3·Et2O to give oxazolines as the reaction products. These are subjected to cobalt complex cleavage in oxidative conditions and subsequent acidic hydrolysis providing amino alcohols. The substrates for the Ritter reaction can be easily assembled to introduce structural diversity at both variable positions. The Ritter reaction conditions for oxazoline formation is compatible with a range of substituents at the alkyne terminal position providing oxazolines in moderate to good yields. Methyl, hydroxymethyl and silyloxymethyl substituents at the reaction center of glycols are well tolerated, while a phenyl group in this position is detrimental to the reaction.
Introduction
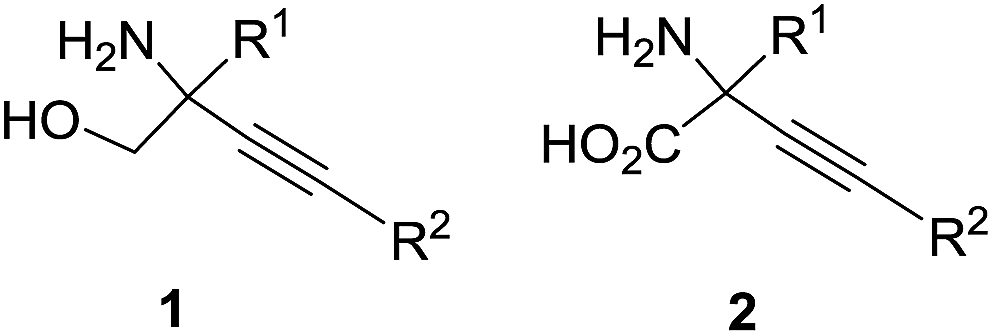
C-Quaternary alkynyl glycinols 1 and synthetically equivalent alkynyl glycine 2 derivatives (Fig. 1) are versatile building blocks for the construction of complex biologically active molecules.1–7 While there is a good arsenal of methods for the synthesis of C-quaternary alkynyl glycines 2,1,2 the direct access to C-quaternary alkynyl glycinols 1 is limited to few alternatives avoiding the reduction of carboxyl groups in glycines 2. The literature search revealed only the Seyferth–Gilbert homologation of a serinal derivative;8 aminolysis of alkynyl epoxides7,9–12 and the insertion of a nitrene into a propargylic C–H bond13 as synthetically useful approaches. Thus, a short synthesis of glycinol derivatives 1 from readily available variable building blocks is very desirable.
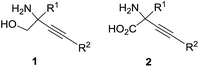 |
| | Fig. 1 C-Quaternary alkynyl glycinols 1 and glycines 2. | |
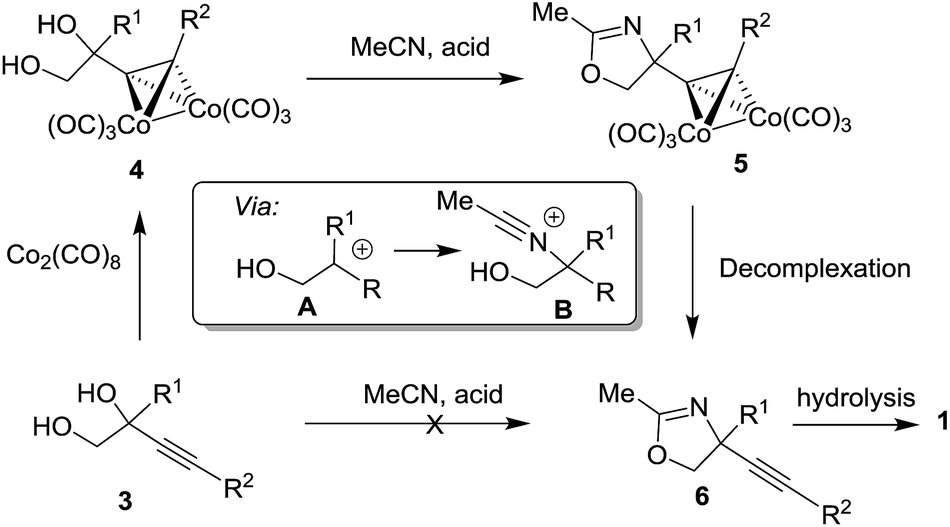
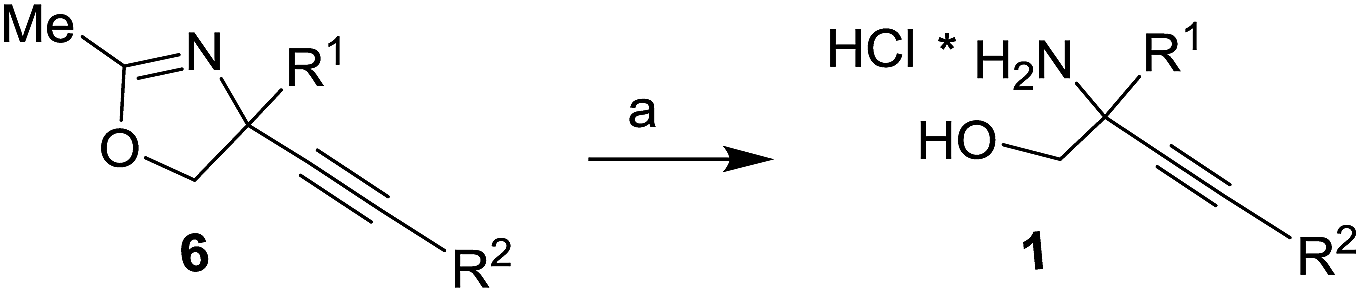
We have recently reported the synthesis of alkynyl glycinols 1 (R1 = H) via intramolecular propargylic amination of bis-trichloroacetimidates derived from alkynyl glycols.14 Our attempts to extend this approach for the synthesis of C-quaternary derivatives were not successful. As an alternative, we turned our attention to the Ritter reaction of 1,2-diols which is a known method for the synthesis oxazolines and oxazines involving carbenium ion A and nitrilium ion B intermediates.15–24 When alkynyl glycol 3 (R1 = Me, R2 = nPent) was directly subjected to the Ritter reaction conditions (MeCN, AcOH, H2SO4) the expected oxazoline 6 was obtained in a very low yield (<10%) (Scheme 1). This prompted us to explore the Ritter reaction of cobalt complexed alkynyl glycols 4 (ref. 25 and 26) which has better ability to stabilize the intermediate carbenium ion A27–29 providing oxazolines 5 as precursors of alkynyl glycinol derivatives 1. Such approach gave the expected results which are summarized in this article.
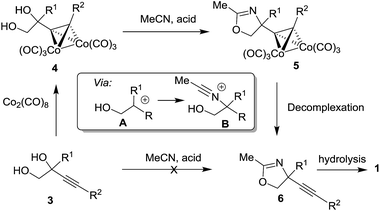 |
| | Scheme 1 The Ritter reaction for the synthesis of oxazolines 5 as precursors of amino alcohols 1. | |
Results and discussion
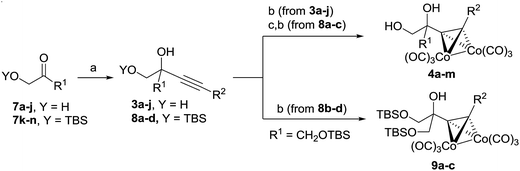
Cobalt complexed alkynyl glycols 4a–j were prepared starting from hydroxy ketone derivatives 7a–j. Addition of lithium acetylenides provided alkynyl diols 3a–j which were treated with Co2(CO8) (Table 1).
Table 1 Synthesis of cobalt complexed alkynyl diols 4 and 9b
|
| Entry |
R1 |
R2 |
Y |
3 or 8, yield% |
4, yield %a |
9, yield% |
| R1 = CH2OTBS in compounds 8 was transformed to R1 = CH2OH in compounds 4. Reagents and conditions: (a) alkyne, nBuLi, LiBr, THF, −40 °C-r.t.; (b) Co2(CO)8, CH2Cl2, r.t.; (c) TBAF, THF, 0 °C-r.t. |
| 1 |
Me |
nPent |
H |
3a, 98 |
4a, 98 |
— |
| 2 |
Me |
tBu |
H |
3b, 47 |
4b, 75 |
— |
| 3 |
Me |
TMS |
H |
3c, 86 |
4c, >99 |
— |
| 4 |
Me |
Ph |
H |
3d, >99 |
4d, 94 |
— |
| 5 |
Me |
2-ClPh |
H |
3e, 90 |
4e, 90 |
— |
| 6 |
Me |
4-MeOPh |
H |
3f, 60 |
4f, 83 |
— |
| 7 |
Me |
CH2OBn |
H |
3g, 39 |
4g, 70 |
— |
| 8 |
Me |
Me |
H |
3h, 47 |
4h, 70 |
— |
| 9 |
Ph |
nPent |
H |
3i, 96 |
4i, 56 |
— |
| 10 |
Ph |
Ph |
H |
3j, 97 |
4j, 82 |
— |
| 11 |
H |
nPent |
TBS |
8a, 82 |
4k, 40 |
|
| 12 |
CH2OTBS (CH2OH)a |
nPent |
TBS |
8b, 94 |
4l, 78 |
9a, 73 |
| 13 |
CH2OTBS (CH2OH)a |
Ph |
TBS |
8c, 95 |
4m, 73 |
9b, 79 |
| 14 |
CH2OTBS |
Me |
TBS |
8d, 75 |
— |
9c, 86 |
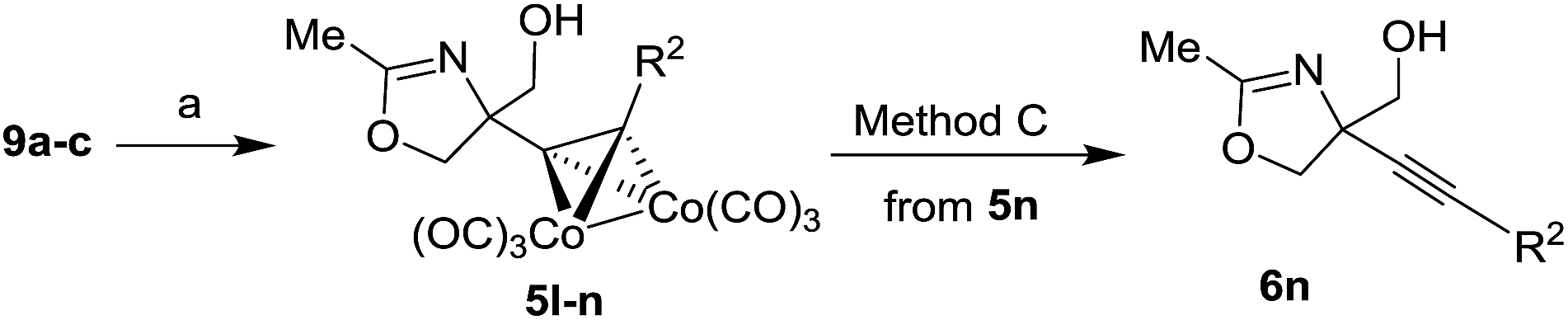
If O-TBS protected starting materials 7l–n were used, the corresponding addition products 8a–c were deprotected before the complex 4l–m formation. Several O-TBS protected alkynyl glycols 8b–d were transformed to the corresponding cobalt complexes 9a–c.
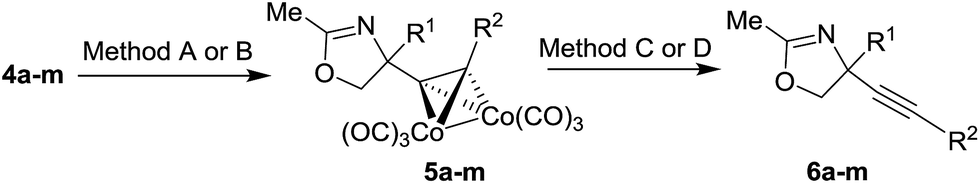
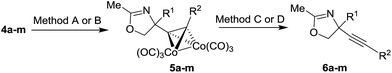
Cobalt complexed alkynyl glycols 4a–d gave the expected oxazolines 5a–d in the Ritter reaction with acetonitrile using both H2SO4 and BF3·EtO2 as acid promoters (Table 2, entries 1–4). Except for the substrate 4c, better yields were obtained under conditions involving BF3·Et2O. Using BF3·Et2O as acid, glycols 4e–h were transformed to oxazolines 5e–h (Table 2, entries 5–8). These results indicate that the Ritter reaction tolerates wide range of substituents at the terminal alkyne position in substrates 4. Diols 4i,j bearing Ph group at the reaction center were unsuitable substrates giving no yield of the expected oxazolines 5i,j (Table 2, entries 9 and 10). Secondary alcohol 4k could be successfully subjected to the Ritter reaction providing acetamide 5k (Table 2, entry 11). Hydroxymethyl substituent at the reaction center of the substrates 4l,m was tolerated to give the Ritter reaction products 5l,m in moderate and good yields (Table 2, entries 12 and 13).
Table 2 The Ritter reaction of cobalt complexed alkynyl glycols 4 and the cleavage of cobalt complexa
|
| Entry |
R1 |
R2 |
5, yield% (method) |
6, yield% (method) |
| Reagents and conditions: method A: MeCN, H2SO4, AcOH, 0 °C; method B: BF3·Et2O, MeCN, 0 °C; method C: DDQ, CH2Cl2, 0 °C. Method D: NMO CH2Cl2, 0 °C. |
| 1 |
Me |
nPent |
5a, 58 (A); 78 (B) |
6a, 84 (C), 42 (D) |
| 2 |
Me |
tBu |
5b, 75 (A); 82 (B) |
6b, 64 (C) |
| 3 |
Me |
TMS |
5c, 89 (A); 84 (B) |
6c, 88 (C) |
| 4 |
Me |
Ph |
5d, 57 (A); 86 (B) |
6d, 83 (C) |
| 5 |
Me |
2-ClPh |
5e, 61 (B) |
6e, 92 (C) |
| 6 |
Me |
4-MeOPh |
5f, 63 (B) |
6f, 85 (C) |
| 7 |
Me |
CH2OBn |
5g, 78 (B) |
6g, 82 (C) |
| 8 |
Me |
Me |
5h, 74 (B) |
6h, 46 (C) |
| 9 |
Ph |
nPent |
5i, dec. (B) |
— |
| 10 |
Ph |
Ph |
5j, dec. (B) |
— |
| 11 |
H |
nPent |
5k, 77 (B) |
6k, 78 (C) |
| 12 |
CH2OH |
nPent |
5l, 46 (B) |
6l, 61 (C); 65 (D) |
| 13 |
CH2OH |
Ph |
5m, 81 (B) |
6m, 26 (C); 65 (D) |
Several reaction conditions for the cleavage of cobalt complex 5a were investigated to obtain the uncomplexed oxazoline 6a (ethylenediamine, THF, 65 °C, yield of 6a, 28%; NMO, CH2Cl2, r.t. yield of 6a, 42%; DDQ, CH2Cl2, r.t. yield of 6a, 84%).30,31
The best yield of 6a was obtained in oxidative conditions with DDQ which to our knowledge has not yet been reported as the reagent for the decomplexation of alkyne cobalt complexes. Other cobalt complexes 5a–h,l–m were also cleaved with DDQ to give uncomplexed oxazolines 6a–h,l–m typically in good yields. The only exception was substrate 5m which gave product 6m in poor yield. For the cleavage of the complex 5m, NMO was better suited as oxidant to provide product 6m more efficiently.
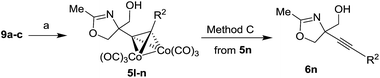
O-TBS protected alkynyl glycols 9a–c could also be used as substrates for the Ritter reaction (Table 3). The reaction proceeded with concomitant deprotection of O-TBS group to give oxazolines 5l–n. The cleavage of cobalt complex 5n was performed with DDQ to give uncomplexed oxazoline 6n (Table 3, entry 3).
Table 3 The Ritter reaction of cobalt complexed alkynyl diols 9a–c and the cleavage of cobalt complex in intermediate 5na
|
| Entry |
9, R2 |
5, yield% |
6, yield% |
| Reagents and conditions: (a) BF3·Et2O, MeCN, 0 °C; method C: DDQ, CH2Cl2, 0 °C. |
| 1 |
9a, nPent, |
5l, 37 |
See Table 2 |
| 2 |
9b, Ph |
5m, 78 |
See Table 2 |
| 3 |
9c, Me |
5n, 63 |
6n, 73 (C) |
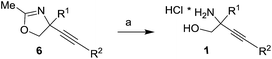
Selected oxazolines 6d,g,h,l,m were transformed to amino alcohols 1 by using acidic hydrolysis in mild conditions (Table 4). The hydrolysis proceed with good yields of product 1d,g,h,l,m formation which were purified by the trituration with EtOAc.
Table 4 Preparation of amino alcohols 1 via hydrolysis of oxazolinesa
|
| Entry |
R1 |
R2 |
1, yield% |
| Reagents and conditions: (a) aq. 20% HCl, MeOH, r.t. |
| 1 |
Me |
Ph |
1d, 96 |
| 2 |
Me |
CH2OBn |
1g, 64 |
| 3 |
Me |
Me |
1h, 62 |
| 4 |
CH2OH |
nPent |
1l, 82 |
| 5 |
CH2OH |
Ph |
1m, 77 |
Experimental
General information
Commercially available reagents were used without further purification. All air or moisture-sensitive reactions were carried out under an argon atmosphere using oven-dried glassware. Flash chromatography was carried out using Merck Kieselgel 60 (230–400 mesh). Thin layer chromatography was performed on silica gel and was visualized by staining with KMnO4. NMR spectra were recorded on a Varian Mercury spectrometer (400 MHz) and a Bruker Fourier spectrometer (300 MHz) with chemical shift values (δ) in ppm relative to TMS using the residual chloroform signal as an internal standard. Elemental analyses were performed using a Carlo-Erba EA1108 Elemental Analyser. HRMS were obtained using a Q-TOF micro high resolution mass spectrometer with ESI (ESI+/ESI−).
Preparation of diols/triols 3 and 8
Alcohols 3 and 8 were prepared according to the procedure described in the literature starting from the corresponding ketones 7.14
Alcohols 3a,32 3b,33 3c,34 3d,34 3g,34 3k,14 3i,j35 8a14 are known in literature.
4-(2-Chlorophenyl)-2-methylbut-3-yne-1,2-diol (3e). White powder. M.p. 63–65 °C. 1H NMR (400 MHz, CD3OD) δ 7.50 (dd, J = 7.3, 2.1 Hz, 1H, –C6H4Cl), 7.43 (dd, J = 7.7, 1.6 Hz, 1H, –C6H4Cl), 7.30 (td, J = 7.7, 2.0 Hz, 1H, –C6H4Cl), 7.26 (td, J = 7.5, 1.5 Hz, 1H, –C6H4Cl), 3.63 (d, J = 11.0 Hz, 1H, –CH2O–), 3.60 (d, J = 11.0 Hz, 1H, –CH2O–), 1.54 (s, 3H, –CH3). 13C NMR (100 MHz, CD3OD) δ 138.2, 135.9, 132.1, 131.7, 129.2, 125.4, 99.7, 82.5, 72.4, 71.1, 27.5. Anal. calcd for C12H14O3: C, 62.72%; H, 5.26%; found: C, 62.71%; H, 5.23%.
4-(4-Methoxyphenyl)-2-methylbut-3-yne-1,2-diol (3f). Off white powder. M.p. 74–77 °C. 1H NMR (400 MHz, CDCl3) δ 7.36 (d, J = 8.9 Hz, 2H, –C6H5), 6.83 (d, J = 8.9 Hz, 2H, –C6H5), 3.81 (s, 3H, –OCH3), 3.74 (dd, J = 11.0, 5.0 Hz, 1H, –CH2O–), 3.56 (dd, J = 11.0, 8.8 Hz, 1H, –CH2O–), 2.66 (s, 1H, –OH), 2.13 (dd, J = 8.8, 5.0 Hz, 1H, –OH), 1.55 (s, 3H, –CH3). 13C NMR (100 MHz, CDCl3) δ 159.9, 133.4, 114.3, 114.1, 89.0, 84.7, 71.0, 69.2, 55.4, 20.2. Anal. calcd for C12H14O3: C, 69.89%; H, 6.84%; found: C, 69.56%; H, 6.86%.
2-Methyl-pent-3-yne-1,2-diol (3h). Colourless oil. 1H NMR (300 MHz, CDCl3) δ 2.76 (d, J = 10.9 Hz, 1H, –CH2OH), 2.61 (d, J = 10.9 Hz, 1H, –CH2OH), 1.72 (s, 1H, –OH), 1.28 (s, 1H, –OH), 1.00 (s, 3H, –CH3), 0.58 (s, 3H, –CH3). 13C NMR (100 MHz, CDCl3) δ 109.9, 80.8, 70.9, 68.6, 25.5, 3.5. In HRMS conditions no signal observed. GC-MS (EI): m/z: 83 [M − CH2OH]+.
6-(Hept-1-yn-1-yl)-2,2,3,3,9,9,10,10-octamethyl-4,8-dioxa-3,9-disilaundecan-6-ol (8b). Colourless oil. 1H NMR (300 MHz, CDCl3) δ 3.61 (d, J = 9.5 Hz, 2H, –CH2O–), 3.51 (d, J = 9.5 Hz, 2H, –CH2O–), 2.80 (s, 1H, –OH), 2.11 (t, J = 7.1 Hz, 2H, –CH2(CH2)3CH3), 1.52–1.37 (m, 2H, –CH2CH2(CH2)2CH3), 1.29–1.14 (m, 4H, –CH2CH2(CH2)2CH3), 0.82 (s, 18H, –SiC(CH3)3), 0.81–0.77 (m, 3H, –CH2(CH2)3CH3), 0.00 (d, J = 1.1 Hz, 12H, –Si(CH3)2). 13C NMR (100 MHz, CDCl3) δ 85.6, 79.8, 71.1, 65.9, 31.0, 28.2, 25.8, 22.2, 18.7, 18.3, 13.9, −5.4. In HRMS conditions no signal observed. GC-MS (EI): m/z: 357 [M − tBu]+.
Deprotection of silyl groups gave 2-(hept-1-yn-1-yl)propane-1,2,3-triol (3k). Colourless oil. 1H NMR (400 MHz, CDCl3) δ 3.70 (s, 4H, –CH2OH), 2.18 (ddt, J = 9.2, 7.1, 3.7 Hz, 2H, –CH2(CH2)3CH3), 1.59–1.44 (m, 2H, –CH2CH2(CH2)2CH3), 1.30 (qd, J = 3.6, 3.1, 1.5 Hz, 4H, –CH2CH2(CH2)2CH3), 0.96–0.81 (m, 3H, –CH2(CH2)3CH3). 13C NMR (100 MHz, CDCl3) δ 87.9, 78.3, 71.5, 67.4, 31.0, 28.2, 22.1, 18.6, 13.9. In HRMS conditions no signal observed. GC-MS (EI): m/z: 155 [M − CH2OH]+.
2,2,3,3,9,9,10,10-Octamethyl-6-(phenylethynyl)-4,8-dioxa-3,9-disilaundecan-6-ol (8c). Colourless oil. 1H NMR (400 MHz, CDCl3) δ 7.44–7.39 (m, 2H, –C6H5), 7.30–7.26 (m, 3H, m, –C6H5), 3.80 (d, J = 9.5 Hz, 2H, –CH2O–), 3.72 (d, J = 9.5 Hz, 2H, –CH2O–), 3.04 (s, 1H, –OH), 0.90 (s, 18H, –SiC(CH3)3), 0.09 (d, J = 0.9 Hz, 12H, –Si(CH3)2). 13C NMR (100 MHz, CDCl3) δ 131.7, 128.1, 122.7, 89.3, 84.8, 71.6, 65.9, 25.8, 18.3, −5.4. HR-MS (ESI-TOF) m/z: calcd for C23H40O3Si2Na 443.2399; found [M + Na]+ 443.2414.
Deprotection of silyl groups gave 2-(phenylethynyl)propane-1,2,3-triol (3l). Colourless oil. 1H NMR (300 MHz, CDCl3) δ 7.52–7.41 (m, 2H, o-C6H5–), 7.39–7.31 (m, 3H, m,p-C6H5–), 3.92–3.82 (m, 4H, –CH2OH), 3.08 (s, 1H, –OH), 2.18 (dd, J = 8.6, 4.8 Hz, 2H, –CH2OH). 13C NMR (100 MHz, CDCl3) δ 131.3, 127.9, 127.9, 122.6, 88.9, 84.5, 71.5, 65.3. In HRMS conditions no signal observed. GC-MS (EI): m/z: 161 [M − –CH2OH]+.
2,2,3,3,9,9,10,10-Octamethyl-6-(prop-1-yn-1-yl)-4,8-dioxa-3,9-disilaundecan-6-ol (8d). Colourless oil. 1H NMR (400 MHz, CDCl3) δ 3.67 (d, J = 9.5 Hz, 2H, –CH2O–), 3.58 (d, J = 9.5 Hz, 2H, –CH2O–), 2.85 (s, 1H, –OH), 1.81 (s, 3H, –CH3), 0.89 (s, 18H, –SiC(CH3)3), 0.06 (d, J = 1.7 Hz, 12H, Si(CH3)2). 13C NMR (100 MHz, CDCl3) δ 81.1, 79.2, 71.1, 65.8, 25.8, 18.3, 3.5, −5.3, −5.4. HR-MS (ESI-TOF) m/z: calcd for C18H48O3Si2Na 381.2247; found [M + Na]+ 381.2257.
General procedure for the preparation of cobalt-complexed propargyl alcohols 4 and 9
To a solution of alkyne (1 mmol) in CH2Cl2 (5 mL), Co2(CO)8 (1.1 mmol) was added under argon atmosphere. The solution was stirred at room temperature until no evolution of CO2 was observed (TLC showed the formation of the complex to be completed). The solvent was removed in vacuo and the residue was purified by column chromatography on silica gel eluting with a mixture of ethyl acetate and petroleum ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30–1:4) to yield the Co2(CO)6-alkyne complex.
:30–1:4) to yield the Co2(CO)6-alkyne complex.
13C-NMR for compounds 4 and 9 was not possible to record due to Co induced line broadening. Typically compounds 4 and 9 were not stable under conditions used for HRMS.
Hexacarbonyl[μ-η4-(2-methylnon-3-yne-1,2-diol)]dicobalt (4a). Red powder. 1H NMR (300 MHz, CDCl3) δ 3.70 (d, J = 4.7 Hz, 2H, –CH2O–), 2.90–2.73 (m, 2H, –CH2(CH2)3CH3), 2.26 (s, 1H, –OH), 2.06–1.95 (m, 1H, –OH), 1.73–1.31 (m, 9H, –CH3, –CH2CH2(CH2)2CH3, –(CH2)2(CH2)2CH3), 0.93 (t, J = 6.2 Hz, 3H, –(CH2)4CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(2,5,5-trimethylhex-3-yne-1,2-diol)]dicobalt (4b). Red powder. 1H NMR (300 MHz, CDCl3) δ 3.72 (d, J = 5.3 Hz, 2H, –CH2O–), 2.25 (s, 1H, –OH), 2.15–2.02 (m, 1H, –OH), 1.62 (s, 3H, –CH3), 1.35 (s, 9H, –C(CH3)3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(2-methyl-4-(trimethylsilyl)but-3-yne-1,2-diol)]dicobalt (4c). Red powder. 1H NMR (300 MHz, CDCl3) δ 3.66 (d, J = 5.9 Hz, 2H, –CH2O–), 2.04 (s, 1H, –OH), 2.04 (t, J = 5.9 Hz, 1H, –OH), 1.57 (s, 3H, –CH3), 0.33 (s, 9H, –Si(CH3)3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(2-methyl-4-phenylbut-3-yne-1,2-diol)]dicobalt (4d). Red powder. 1H NMR (400 MHz, CDCl3) δ 7.69–7.56 (m, J = 6.8 Hz, 2H, o-C6H5–), 7.40–7.29 (m, 3H, p,m-C6H5–), 3.89–3.74 (br, 2H, –CH2O–), 2.58 (s, 1H, –OH), 2.08–1.99 (br, 1H, –OH), 1.67 (s, 3H,–CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(4-(2-chlorophenyl)-2-methylbut-3-yne-1,2-diol)]dicobalt (4e). Red powder. 1H NMR (300 MHz, CDCl3) δ 8.01–7.94 (m, 1H, C6H4Cl–), 7.45–7.37 (m, 1H), 7.32–7.26 (m, 2H, C6H4Cl–, overlapping with CHCl3 signal), 3.82 (d, J = 5.8 Hz, 2H, –CH2O–), 2.87 (s, 1H, –OH), 2.05 (t, J = 5.8 Hz, 1H, –OH), 1.67 (s, 3H, –CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(4-(4-methoxyphenyl)-2-methylbut-3-yne-1,2-diol)]dicobalt (4f). Red powder.1H NMR (300 MHz, CDCl3) δ 7.61 (s, 2H, m-MeO–C6H4–), 6.90 (s, 2H, o-MeO–C6H4–), 4.14 (s, 1H, –CH2OH), 3.85 (s, 4H, CH3O–C6H4– (3H), and overlapping –CH2OH (1H)), 2.59 (s, 1H, –OH), 2.18 (s, 1H, –OH), 1.69 (s, 3H, –CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(5-(benzyloxy)-2-methylpent-3-yne-1,2-diol)]dicobalt (4g). Red powder.1H NMR (300 MHz, CDCl3) δ 7.37 (m, 5H, C6H5–), 4.76 (d, J = 8.4 Hz, 4H, –CH2O–CH2–), 3.65 (d, J = 5.2 Hz, 1H, –CH2OH), 3.53 (s, 1H, –CH2OH), 3.10 (s, 1H, –OH), 2.19 (s, 1H, –OH), 1.53 (s, 3H, –CH3). HR-MS (ESI-TOF) m/z: calcd for C19H16O9Co2504.9382; found 504.9380.
Hexacarbonyl[μ-η4-(2-methyl-pent-3-yne-1,2-diol)]dicobalt (4h). Red powder. 1H NMR (300 MHz, CDCl3) δ 3.72 (s, 2H, –CH2OH), 2.72 (s, 3H, –CH3), 2.31 (s, 1H, –OH), 2.06 (s, 3H, –CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(2,4-diphenylbut-3-yne-1,2-diol)]dicobalt (4i). 1H NMR (300 MHz, CDCl3) δ 7.59 (d, J = 7.5 Hz, 2H, o-C6H4–), 7.41 (t, J = 7.5 Hz, 2H, m-C6H4–), 7.33 (d, J = 7.5 Hz, 1H, p-C6H4–), 4.36 (dd, J = 10.0, 4.5 Hz, 1H, –CH2OH), 3.95 (t, J = 10.0 Hz, 1H, –CH2OH), 3.10 (s, 1H, –OH), 2.75 (m, 2H, –CH2(CH2)3CH3), 2.19 (s, 1H, –OH), 1.69–1.58 (m, 2H, –CH2CH2(CH2)2CH3), 1.52–1.30 (m, 4H, –CH2CH2(CH2)2CH3), 0.95 (t, J = 6.9 Hz, 3H, –CH2(CH2)3CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(6-(hept-1-yn-1-yl)-2,2,3,3,9,9,10,10-octamethyl-4,8-dioxa-3,9-disilaundecan-6-ol)]dicobalt (4j). 1H NMR (300 MHz, CDCl3) δ 7.56 (d, J = 7.2 Hz, 4H, o-C6H4–), 7.42–7.29 (m, 6H, m,p-C6H4–), 4.60–4.43 (m, 1H, –CH2OH), 4.25–4.03 (m, 1H, –CH2OH), 3.39 (s, 1H, –OH), 1.84 (s, 1H, –OH). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(non-3-yne-1,2-diol)]dicobalt (4k). Red powder. 1H NMR (300 MHz, CDCl3) δ 4.39 (s, 1H, –CH(OH)-), 3.62 (s, 2H, –CH2OH), 2.21–2.09 (m, 2H, –CH2(CH2)3CH3), 1.44 (t, J = 7.2 Hz, 2H, –CH2CH2(CH2)2CH3), 1.35–1.13 (m, 4H, –CH2CH2(CH2)2CH3), 0.89–0.77 (m, 3H, –CH2(CH2)3CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(2-(hept-1-yn-1-yl)propane-1,2,3-triol)]dicobalt (4l). 1H NMR (300 MHz, CDCl3) δ 3.62 (s, 4H, –CH2OH), 2.11 (t, J = 7.2 Hz, 2H, –CH2(CH2)3CH3), 1.99 (s, 1H, –OH), 1.51–1.35 (m, 2H, –CH2CH2(CH2)2CH3), 1.29–1.19 (m, 4H, –CH2CH2(CH2)2CH3), 0.88–0.72 (m, 3H, –CH2(CH2)3CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(2-(phenylethynyl)propane-1,2,3-triol)]dicobalt (4m). 1H NMR (300 MHz, CDCl3) δ 7.46 (dd, J = 7.4, 2.2 Hz, 2H, o-C6H4–), 7.37–7.32 (m, 3H, m,p-C6H4–), 3.87 (dd, J = 6.3, 3.5 Hz, 4H, –CH2OH), 3.08 (s, 1H, –OH), 1.57 (s, 1H, –OH). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(6-(hept-1-yn-1-yl)-2,2,3,3,9,9,10,10-octamethyl-4,8-dioxa-3,9-disilaundecan-6-ol)]dicobalt (9a). Red powder. 1H NMR (300 MHz, CDCl3) δ 3.63 (dd, J = 24.5, 9.5 Hz, 4H, –CH2OTBS), 2.80 (s, 1H, –OH), 2.75–2.65 (m, 2H, –CH2–), 1.66–1.37 (m, 6H, (–CH2–)3), 1.31 (d, J = 3.0 Hz, 3H, –CH3), 0.83 (s, 18H, –SiC(CH3)3), 0.00 (d, J = 3.1 Hz, 12H, –Si(CH3)2). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(2,2,3,3,9,9,10,10-octamethyl-6-(phenylethynyl)-4,8-dioxa-3,9-disilaundecan-6-ol)]dicobalt (9b). Red powder. 1H NMR (300 MHz, CDCl3) δ 7.62 (dd, J = 7.7, 1.7 Hz, 2H, o-C6H5–), 7.30–7.23 (m, 3H, p,m-C6H5–), 3.89 (d, J = 9.6 Hz, 2H, –CH2OTBS), 3.76 (d, J = 9.6 Hz, 2H, –CH2OTBS), 3.20 (s, 1H, –OH), 0.85 (s, 18H, –SiC(CH3)3), 0.02 (d, J = 9.8 Hz, 12H, –Si(CH3)2). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(2,2,3,3,9,9,10,10-octamethyl-6-(prop-1-yn-1-yl)-4,8-dioxa-3,9-disilaundecan-6-ol)]dicobalt (9c). Red powder. 1H NMR (300 MHz, CDCl3) δ 3.70 (dd, J = 21.3, 9.5 Hz, 4H, –CH2OTBS), 2.85 (s, 1H, –OH), 2.65 (s, 3H, –CH3), 0.91 (s, 18H, –SiC(CH3)3), 0.08 (d, J = 2.8 Hz, 12H, –Si(CH3)2). Not stable under HR-MS conditions.
Method A for the Ritter reaction
A solution of the cobalt complex of diol 4 (2.2 mmol) in CH3CN (54 eq., 118.8 mmol, 6.2 mL) was cooled to 0–3 °C (ice/water bath) and AcOH (8 eq., 17.6 mmol, 1.0 mL) was added followed by dropwise addition of H2SO4 (9 eq., 19.9 mmol, 1.0 mL). The reaction mixture was allowed to stir at this temperature until complete conversion of the starting material was observed (TLC control, usually 8 min). The reaction mixture was diluted with Et2O (30 mL) and poured into water (15 mL). The organic phase was separated and the aqueous phase was extracted with Et2O (30 mL). The combined organic phase was washed with aq. NaHCO3, dried over Na2SO4, filtered and evaporated. The crude residue was purified by chromatography on silica gel eluting with a mixture of ethyl acetate and petroleum ether (1:20–1:10) to afford oxazoline cobalt complex.
Method B for the Ritter reaction
A solution of the cobalt complex 4 or 9 (0.3 mmol) in MeCN (3 mL) was cooled to 0–3 °C (ice/water bath) and BF3 Et2O (0.38 mL, 10 eq., 2.96 mmol) was added dropwise. The reaction mixture was allowed to stir at this temperature until complete conversion of the starting material (TLC control, usually 5–10 min). The reaction mixture was diluted with DCM (15 mL) and aq. NaHCO3 (7 mL) was added. The organic phase was separated and washed with brine (1 × 7 mL), dried over Na2SO4, filtered and evaporated. The crude residue was purified by chromatography on silica gel eluting with a mixture of ethyl acetate and petroleum ether (1:20–1:3) to afford oxazoline cobalt complex.
13C-NMR for compounds 5 was not possible to record due to Co induced line broadening. Typically compounds 5 were not stable under conditions used for HRMS.
Hexacarbonyl[μ-η4-(4-(hept-1-yn-1-yl)-2,4-dimethyloxazoline)]dicobalt (5a). Viscous colorless oil. 1H NMR (400 MHz, CDCl3) δ 4.28 (d, J = 8.4 Hz, 1H, –CH2O–), 4.12 (d, J = 8.4 Hz, 2H, CDCl3), 2.89–2.76 (m, 2H, –CH2(CH2)3CH3), 1.97 (s, 3H, –CH3), 1.74–1.58 (m, 5H, –CH3, –CH2CH2(CH2)2CH3), 1.43 (qd, J = 15.2, 7.4 Hz, 4H, –(CH2)2(CH2)2CH3), 0.93 (t, J = 7.1 Hz, 3H, –(CH2)4CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(4-(3,3-dimethylbut-1-yn-1-yl)-2,4-dimethyloxazoline)]dicobalt (5b). Viscous colorless oil with tendency to crystalize. 1H NMR (300 MHz, CDCl3) δ 4.28 (d, J = 8.4 Hz, 1H, –CH2O–), 4.14 (d, J = 8.4 Hz, 1H, –CH2O–), 1.97 (s, 3H, –CH3), 1.68 (s, 3H, –CH3), 1.35 (s, 9H, –C(CH3)3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(2,4-dimethyl-4-((trimethylsilyl)ethynyl) oxazoline)]dicobalt (5c). Viscous colorless oil. 1H NMR (300 MHz, CDCl3) δ 4.21 (d, J = 8.4 Hz, 1H, –CH2O–), 4.15 (d, J = 8.4 Hz, 1H, –CH2O–), 1.98 (s, 3H, –CH3), 1.65 (s, 3H, –CH3), 0.32 (s, 9H, –Si(CH3)3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(2,4-dimethyl-4-(phenylethynyl)oxazoline)] dicobalt (5d). Viscous colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.74–7.68 (m, Hz, 2H, –C6H5), 7.41–7.27 (m, 3H, –C6H5), 4.40 (d, J = 8.4 Hz, 1H, –CH2O–), 4.21 (d, J = 8.4 Hz, 1H, –CH2O–), 2.02 (s, 3H, –CH3), 1.70 (s, 3H, –CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(4-((2-chlorophenyl)ethynyl)-2,4-dimethyl oxazoline)]dicobalt (5e). Viscous colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.31–8.09 (br, 1H, –C6H4Cl), 7.53–7.28 (br, 3H, –C6H4Cl overlapping with CDCl3), 4.50 (d, J = 8.2 Hz, 1H, –CH2O–), 4.28 (d, J = 8.2 Hz, 1H, –CH2O–), 2.02 (s, 3H, –CH3), 1.68 (s, 3H, –CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(4-((4-methoxyphenyl)ethynyl)-2,4-dimethyl-oxazoline)]dicobalt (5f). Red oil. 1H NMR (300 MHz, CDCl3) δ 7.69 (2H, –C6H4–), 6.92 (2H, C6H4–), 4.42 (1H, –CH2O–), 4.24 (1H, –CH2O–), 3.85 (3H, –OCH3), 2.03 (3H, –CH3), 1.71 (3H, –CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(4-(3-(benzyloxy)prop-1-yn-1-yl)-2,4-dimethyl-oxazoline)]dicobalt (5g). Red oil. 1H NMR (300 MHz, CDCl3) δ 7.45–7.29 (m, 5H, C6H5–), 4.70 (t, J = 14.3 Hz, 4H, –CH2O–CH2–), 4.31 (d, J = 8.5 Hz, 1H, –CH2O–), 4.13 (d, J = 8.5 Hz, 1H, –CH2O–), 1.96 (s, 3H, –CH3), 1.63 (s, 3H, –CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(2,4-dimethyl-4-(prop-1-yn-1-yl)-oxazoline)]dicobalt 5h. Red oil. 1H NMR (300 MHz, CDCl3) δ 4.28 (1H, –CH2O–), 4.15 (1H, –CH2O–), 2.72 (s, 3H, –CH3), 1.99 (s, 3H, –CH3), 1.65 (s, 3H, –CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-(4-(hept-1-yn-1-yl)-2-methyl-oxazoline)]dicobalt (5k). Red oil. 1H NMR (300 MHz, CDCl3) δ 5.32 (dd, J = 8.7, 6.4 Hz, 1H, –CHN–), 4.58–4.45 (m, 1H, –CH2O–), 4.18 (dd, J = 8.5, 5.6 Hz, 1H, –CH2O–), 2.92–2.83 (m, 2H, –CH2–), 2.01 (d, J = 1.0 Hz, 3H, –CH3), 1.68 (dd, J = 15.6, 8.0 Hz, 2H, –CH2–), 1.55–1.33 (m, 4H, –CH2–CH2–), 0.93 (dd, J = 13.7, 6.5 Hz, 3H, –CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-((4-(hept-1-yn-1-yl)-2-methyl-4,5-dihydrooxazol-4-yl)methanol)]dicobalt (5l). Red oil. 1H NMR (300 MHz, CDCl3) δ 4.46 (d, J = 8.4 Hz, 1H, –CH2O–), 4.15 (d, J = 8.4 Hz, 1H, –CH2O–), 3.84 (dd, J = 10.8, 4.4 Hz, 1H, –CH2OH), 3.54 (dd, J = 20.4, 10.8 Hz, 1H, –CH2OH), 3.09 (dd, J = 8.9, 4.4 Hz, 1H, –OH), 2.81–2.68 (m, 2H, –CH2(CH2)3CH3), 1.93 (s, 3H, –CH3), 1.66–1.49 (m, 2H, –CH2CH2(CH2)2CH3), 1.43–1.25 (m, 4H, –(CH2)2(CH2)2CH3), 0.85 (t, J = 7.1 Hz, 3H, –CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-((2-methyl-4-(phenylethynyl)-4,5-dihydrooxazol-4-yl)methanol)]dicobalt (5m). Red oil. 1H NMR (300 MHz, CDCl3) δ 7.45 (d, J = 6.5 Hz, 2H, o-C6H5–), 7.13 (d, J = 7.4 Hz, 3H, m,p-C6H5), 4.42 (d, J = 8.4 Hz, 1H, –CH2O–), 4.12 (d, J = 8.4 Hz, 1H, –CH2O–), 3.82–3.69 (m, 1H, –CH2OH), 3.42 (t, J = 10.4 Hz, 1H, –CH2OH), 2.36 (d, J = 6.1 Hz, 1H, –OH), 1.84 (s, 3H, –CH3). Not stable under HR-MS conditions.
Hexacarbonyl[μ-η4-((2-methyl-4-(prop-1-yn-1-yl)-4,5-dihydrooxazol-4-yl)methanol)]dicobalt (5n). Red oil. 1H NMR (300 MHz, CDCl3) δ 4.50 (d, J = 8.2 Hz, 1H, –CH2O–), 4.24 (d, J = 8.2 Hz, 1H, –CH2O–), 4.00–3.84 (m, 2H, –CH2OH), 3.76–3.60 (m, 1H, –OH), 2.72 (s, 3H, –CH3), 2.06 (s, 3H, –CH3). Not stable under HR-MS conditions.
General procedure for the cleavage of cobalt complexes 5, method C
DDQ (3 eq., 1.23 mmol) was added in portions to a solution of cobalt complexed oxazoline 5 (1 eq., 0.41 mmol) in CH2Cl2 (4 mL) at 0 °C (ice/water bath). The reaction mixture was stirred until complete conversion of the starting material (TLC control, 30 min – 2 h). The reaction mixture was diluted with CH2Cl2 (30 mL) and aq. NaHCO3 (10 mL) was added. The organic phase was separated and washed with H2O (1 × 10 mL). Organic phase was dried over Na2SO4, filtered and evaporated. The crude residue was purified by chromatography on silica gel eluting with a mixture of ethyl acetate and petroleum ether 1:4–1:1 to afford oxazoline 6.
General procedure for the cleavage of cobalt complexes 5, method D
N-Methylmorpholine N-oxide (NMO) (10 eq., 4.1 mmol) was added in portions to a solution of cobalt complexed oxazoline 5 (1 eq., 0.41 mmol) in CH2Cl2 (4 mL) at 0 °C (ice/water bath). The reaction mixture was stirred until complete conversion of the starting material (TLC control, usually 30 min). The reaction was quenched with aq. NaHCO3 (10 mL) and extracted with ethyl acetate (2 × 8 mL). The organic phase was washed with brine (1 × 10 mL), dried over Na2SO4, filtered and evaporated. The crude residue was purified by chromatography on silica gel eluting with a mixture of ethyl acetate and petroleum ether (1:4–1:1) to afford oxazoline 6.
4-(Hept-1-yn-1-yl)-2,4-dimethyloxazoline (6a). Colorless oil. 1H NMR (400 MHz, CDCl3) δ 4.28 (d, J = 8.1 Hz, 1H, –CH2O–), 4.02 (d, J = 8.1 Hz, 1H, –CH2O–), 2.17 (t, J = 7.1 Hz, 2H, –CH2(CH2)3CH3), 1.98 (s, 3H, –CH3), 1.54–1.44 (m, 5H, –CH3, –CH2CH2(CH2)2CH3), 1.37–1.24 (m, 4H, –(CH2)2(CH2)2CH3), 0.89 (t, J = 7.1 Hz, 3H, –(CH2)4CH3). 13C NMR (100 MHz, CDCl3) δ 165.0, 84.2, 82.8, 79.6, 64.4, 31.2, 29.3, 28.5, 22.3, 18.8, 14.2, 14.1. HR-MS (ESI-TOF) m/z: calcd for C12H20NO 194.1545; found [M + H]+ 194.1548.
4-(3,3-Dimethylbut-1-yn-1-yl)-2,4-dimethyloxazoline (6b). Colorless oil. 1H NMR (400 MHz, CDCl3) δ 4.23 (d, J = 8.0 Hz, 1H, –CH2O–), 4.02 (d, J = 8.0 Hz, 1H, –CH2O–), 1.97 (s, 3H, –CH3), 1.45 (s, 3H, –CH3), 1.18 (s, 9H, –C(CH3)3). 13C NMR (100 MHz, CDCl3) δ 164.7, 92.1, 81.2, 79.8, 64.3, 31.2, 29.6, 27.4, 14.2. HR-MS (ESI-TOF) m/z: calcd for C11H18NO 180.1388; found 180.1389 [M + H]+.
2,4-Dimethyl-4-((trimethylsilyl)ethynyl)oxazoline (6c). Colorless oil. 1H NMR (400 MHz, CDCl3) δ 4.32 (d, J = 8.2 Hz, 1H, –CH2O–), 4.03 (d, J = 8.2 Hz, 1H, –CH2O–), 1.98 (s, 3H, –CH3), 1.49 (s, 3H, –CH3), 0.14 (s, 9H, –Si(CH3)3). 13C NMR (100 MHz, CDCl3) δ 164.4, 107.1, 86.6, 78.4, 63.8, 28.1, 13.2, −0.9. HR-MS (ESI-TOF) m/z: calcd for C10H18NOSi 196.1158; found 196.1156 [M + H]+.
2,4-Dimethyl-4-(phenylethynyl)oxazoline (6d). Colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.42–7.36 (m, 2H, –C6H5), 7.28–7.22 (m, 3H, –C6H5), 4.41 (d, J = 8.2 Hz, 1H, –CH2O–), 4.09 (d, J = 8.2 Hz, 1H, –CH2O–), 1.99 (s, 3H, –CH3), 1.58 (s, 3H, –CH3). 13C NMR (100 MHz, CDCl3) δ 165.5, 131.8, 128.4, 128.3, 122.9, 91.7, 83.5, 79.5, 64.9, 29.1, 14.2. HR-MS (ESI-TOF) m/z: calcd for C13H14NO 200.1075; found 200.1075 [M + H]+.
4-((2-Chlorophenyl)ethynyl)-2,4-dimethyloxazoline (6e). Colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.45 (dd, J = 7.4, 1.9 Hz, 1H, –C6H4Cl), 7.37 (dd, J = 7.9, 1.3 Hz, 1H, –C6H4Cl), 7.22 (td, J = 7.7, 1.9 Hz, 1H, –C6H4Cl), 7.17 (td, J = 7.5, 1.4 Hz, 1H, –C6H4Cl), 4.49 (d, J = 8.2 Hz, 1H, –CH2O–), 4.14 (d, J = 8.2 Hz, 1H, –CH2O–), 2.02 (s, 3H, –CH3), 1.63 (s, 3H, –CH3). 13C NMR (100 MHz, CDCl3) δ 165.7, 136.2, 133.4, 129.4, 129.3, 126.4, 122.8, 96.9, 80.4, 79.4, 65.0, 28.9, 14.2. HR-MS (ESI-TOF) m/z: calcd for C13H13NOCl 234.0686; found 234.0684 [M + H]+.
4-((4-Methoxyphenyl)ethynyl)-2,4-dimethyl-oxazoline (6f). Colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.33 (d, J = 8.9 Hz, 2H, –C6H4OMe), 6.79 (d, J = 8.9 Hz, 2H, –C6H4OMe), 4.41 (d, J = 8.1 Hz, 1H, –CH2O–), 4.09 (d, J = 8.1 Hz, 1H, –CH2O–), 3.78 (s, 3H, –OCH3), 2.00 (s, 3H, –CH3), 1.58 (s, 3H, –CH3). 13C NMR (100 MHz, CDCl3) δ 165.2, 159.5, 133.1, 133.1, 114.8, 113.7, 90.1, 83.2, 79.4, 64.7, 55.2, 28.9, 14.1. HR-MS (ESI-TOF) m/z: calcd for C14H15NO2 229.1181; found 230.1178 [M + H]+.
4-(3-(Benzyloxy)prop-1-yn-1-yl)-2,4-dimethyloxazoline (6g). Brownish oil. 1H NMR (300 MHz, CDCl3) δ 7.30–7.21 (m, 5H, C6H5–), 4.51 (s, 2H, –OCH2Ph), 4.28 (d, J = 8.2 Hz, 1H, –CH2O–), 4.13 (s, 2H, –CH2OBn), 3.98 (d, J = 8.2 Hz, 1H, –CH2O–), 1.93 (s, 3H, –CH3), 1.46 (s, 3H, –CH3). 13C NMR (100 MHz, CDCl3) δ 165.5, 137.4, 128.4, 128, 127.8, 88.9, 79.3, 79.1, 71.6, 64.2, 57.5, 28.8, 13.9. HR-MS (ESI-TOF) m/z: calcd for C15H17NO2 243.1338; found 244.1335 [M + H]+.
2,4-Dimethyl-4-(prop-1-yn-1-yl)-oxazoline (6h). Colorless oil. 1H NMR (400 MHz, CDCl3) δ 4.26 (d, J = 8.1 Hz, 1H, –CH2O–), 3.98 (d, J = 8.1 Hz, 1H, –CH2O–), 1.95 (s, 3H, –CH3), 1.79 (s, 3H, –CH3), 1.44 (s, 3H, –CH3). 13C NMR (100 MHz, CDCl3) δ 164.9, 81.8, 79.4, 79.3, 64.2, 29.0, 13.9, 3.6. HR-MS (ESI-TOF) m/z: calcd for C8H11NO 137.0918; found 138.0919 [M + H]+.
4-(Hept-1-yn-1-yl)-2-methyl-oxazoline (6k). Colorless oil. 1H NMR (300 MHz, CDCl3) δ 4.68 (d, J = 8.0 Hz, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NCH–), 4.33 (dd, J = 10.0, 8.0 Hz, 1H, –CH2O–), 4.11–3.99 (m, 1H, –CH2O–), 2.12 (td, J = 7.1, 2.0 Hz, 2H, –CH2–), 1.93 (s, 3H, –CH3), 1.49–1.37 (m, 2H, –CH2–), 1.30–1.22 (m, 4H, –CH2CH2–), 0.82 (t, J = 7.1 Hz, 3H, –CH3). 13C NMR (100 MHz, CDCl3) δ 171.1, 84.9, 67.9, 60.4, 53.4, 31.0, 28.2, 22.1, 18.7, 14.2, 13.9. HR-MS (ESI-TOF) m/z: calcd for C11H17NO 179.1388; found 180.1384 [M + H]+.
NCH–), 4.33 (dd, J = 10.0, 8.0 Hz, 1H, –CH2O–), 4.11–3.99 (m, 1H, –CH2O–), 2.12 (td, J = 7.1, 2.0 Hz, 2H, –CH2–), 1.93 (s, 3H, –CH3), 1.49–1.37 (m, 2H, –CH2–), 1.30–1.22 (m, 4H, –CH2CH2–), 0.82 (t, J = 7.1 Hz, 3H, –CH3). 13C NMR (100 MHz, CDCl3) δ 171.1, 84.9, 67.9, 60.4, 53.4, 31.0, 28.2, 22.1, 18.7, 14.2, 13.9. HR-MS (ESI-TOF) m/z: calcd for C11H17NO 179.1388; found 180.1384 [M + H]+.
(4-(Hept-1-yn-1-yl)-2-methyl-4,5-dihydrooxazol-4-yl)methanol (6l). Colorless oil. 1H NMR (300 MHz, CDCl3) δ 4.30 (d, J = 8.2 Hz, 1H, –CH2OH), 4.19 (d, J = 8.2 Hz, 1H, –CH2OH), 3.66 (d, J = 11.3 Hz, 1H, –CH2O–), 3.46 (d, J = 11.3 Hz, 1H, –CH2O–), 2.13 (t, J = 7.2 Hz, 2H, –CH2–), 1.95 (s, 3H, –CH3), 1.43 (t, J = 7.2 Hz, 2H, –CH2–), 1.33–1.16 (m, 4H, –CH2CH2–), 0.82 (t, J = 6.9 Hz, 3H, –CH3). 13C NMR (100 MHz, CDCl3) δ 167.4, 86.5, 79.2, 75.2, 69.4, 67.4, 31.0, 28.2, 22.1, 18.7, 14.0, 13.9. HR-MS (ESI-TOF) m/z: calcd for C12H19NO2 209.1494; found [M + H]+ 210.1492.
(2-Methyl-4-(phenylethynyl)-4,5-dihydrooxazol-4-yl)methanol (6m). Colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.45–7.37 (m, 2H, –C6H5), 7.32–7.24 (m, 3H, –C6H5), 4.46 (d, J = 8.3 Hz, 1H, –CH2OH), 4.40 (d, J = 8.3 Hz, 1H, –CH2OH), 3.87 (d, J = 11.3 Hz, 1H, –CH2O–), 3.64 (d, J = 11.3 Hz, 1H, –CH2O–), 2.04 (s, 3H, –CH3). 13C NMR (100 MHz, CDCl3) δ 167.9, 131.8, 128.5, 128.2, 122.2, 88.1, 85.5, 75.0, 69.8, 67.1, 14.0. HR-MS (ESI-TOF) m/z: calcd for C13H13NO2 215.1022; found [M + H]+ 216.1025.
(2-Methyl-4-(prop-1-yn-1-yl)-4,5-dihydrooxazol-4-yl)methanol (6n). Colorless oil. 1H NMR (300 MHz, CDCl3) δ 4.28 (d, J = 8.2 Hz, 1H, –CH2OH), 4.20 (d, J = 8.2 Hz, 1H, –CH2OH), 3.66 (d, J = 11.3 Hz, 1H, –CH2O–), 3.47 (d, J = 11.3 Hz, 1H, –CH2O–), 1.95 (s, 3H, –CH3), 1.78 (s, 3H, –CH3). 13C NMR (100 MHz, CDCl3) δ 170.2, 81.2, 76.3, 67.3, 66.8, 56.9, 14.2, 3.7. HR-MS (ESI-TOF) m/z: calcd for C8H11NO2 153.0864; found [M + H]+ 154.0868.
General procedure for the synthesis of alkynyl glycinols 1
Aqueous 6 M HCl (1 mL) was added dropwise to a solution of oxazoline 6 (0.15 mmol) in MeOH (1.5 mL) at room temperature. The reaction mixture was stirred at room temperature for 2 h and the solvent was evaporated. Toluene (1 mL) was added to the mixture and evaporated. This procedure was repeated one more time. The residue was suspended in EtOAc and filtered to give amino alcohol hydrochloride salt 1.
1-Hydroxy-2-methyl-4-phenylbut-3-yn-2-aminium chloride (1c). Amorphous compound. 1H NMR (400 MHz, methanol-d4) δ 7.50–7.43 (m, 2H, C6H5–), 7.43–7.26 (m, 3H, C6H5–), 3.83 (d, J = 11.5 Hz, 1H, –CH2OH), 3.70 (d, J = 11.5 Hz, 1H, –CH2OH), 1.64 (s, 3H, –CH3). 13C NMR (100 MHz, CD3OD) δ 131.4, 129.1, 128.2, 121.1, 86.4, 84.7, 66.3, 52.7, 21.5. HR-MS (ESI-TOF) m/z: calcd for C11H13NO 175.23; found 159.0810 [M − OH]+.
5-(Benzyloxy)-1-hydroxy-2-methylpent-3-yn-2-aminium chloride (1g). Amorphous compound. 1H NMR (300 MHz, CD3OD) δ 7.35–7.15 (m, 5H, C6H5–), 4.49 (s, 2H, –OCH2Ph), 4.16 (s, 2H, –CH2OBn), 3.66 (d, J = 11.4 Hz, 1H, –CH2OH), 3.54 (d, J = 11.4 Hz, 1H, –CH2OH), 1.48 (s, 3H, –CH3). 13C NMR (100 MHz, CD3OD) δ 137.3, 128.0, 127.7, 127.6, 83.0, 82.5, 71.5, 66.2, 56.5, 54.4, 21.4. HR-MS (ESI-TOF) m/z: calcd for C13H18NO2 220.1336; found 220.1338 [M + H]+.
1-Hydroxy-2-methylpent-3-yn-2-aminium chloride (1h). Amorphous compound. 1H NMR (400 MHz, methanol-d4) δ 3.67 (d, J = 11.5 Hz, 1H, –CH2OH), 3.55 (d, J = 11.5 Hz, 1H, –CH2OH), 1.85 (s, 3H, –CH3), 1.49 (s, 3H, –CH3), 1.36 (dt, J = 7.4, 3.9 Hz, 1H, –OH). 13C NMR (101 MHz, CD3OD) δ 83.4, 75.3, 66.4, 52.2, 21.6, 1.6. HR-MS (ESI-TOF) m/z: calcd for C12H12NO 114.0919; found 114.0922 [M + H]+.
1-Hydroxy-2-(hydroxymethyl)non-3-yn-2-aminium chloride (1l). Amorphous compound. 1H NMR (400 MHz, CD3OD) δ 3.74 (d, J = 11.3 Hz, 2H, –CH2OH), 3.67 (d, J = 11.3 Hz, 2H, –CH2OH), 2.24 (t, J = 7.1 Hz, 2H, –CH2(CH2)3CH3), 1.61–1.46 (m, 2H, –CH2CH2(CH2)2CH3), 1.44–1.27 (m, 4H, –CH2CH2(CH2)2CH3), 0.89 (t, J = 7.1 Hz, 3H, –CH2(CH2)3CH3). 13C NMR (101 MHz, CD3OD) δ 88.9, 74.1, 62.9, 56.9, 30.6, 27.6, 21.8, 17.8, 12.8. HR-MS (ESI-TOF) m/z: calcd for C10H20NO2 186.1494; found. 186.1494 [M + H]+.
1-Hydroxy-2-(hydroxymethyl)-4-phenylbut-3-yn-2-aminium chloride (1m). Amorphous compound. 1H NMR (400 MHz, CD3OD) δ 7.53–7.43 (m, 2H, o-C6H5–), 7.43–7.33 (m, 3H, p,m-C6H5–), 3.87 (d, J = 11.4 Hz, 2H, –CH2OH), 3.83 (d, J = 11.4 Hz, 2H, –CH2OH). 13C NMR (101 MHz, CD3OD) δ 131.5, 129.1, 128.2, 121.1, 87.5, 82.8, 62.7, 57.4. HR-MS (ESI-TOF) m/z: calcd for C11H14NO2 192.10; found 175.0759 [M − OH]+.
Conclusions
In summary, we have developed a novel approach to C-quaternary alkynyl glycinols. This is based on the Ritter reaction of acetonitrile with cobalt complexed alkynyl glycols to give oxazolines. The substrates can be easy assembled to introduce the structural diversity at both variable positions. The Ritter reaction is compatible with a range of substituents at the alkyne terminal position providing oxazolines in moderate to good yields. Hydroxymethyl substituent at the reaction center in both unprotected or O-TBS protected form was well tolerated. The Ritter reaction proceeds also with bis-O-TBS protected alkynyl glycerols with concomitant cleavage of the TBS groups. However, the phenyl group at the reaction center of glycols was detrimental inducing low or no yield of the product formation. Cobalt alkyne complexes in the oxazolines produced by the Ritter reaction can be cleaved in oxidative conditions using DDQ, or NMO as reagents. Hydrolysis of oxazoline ring in mild acidic conditions efficiently provides amino alcohols. We believe that method presented in this paper will find an application for the synthesis of complex amino alcohol derivatives. A version based on catalytic amount of cobalt additive or a protocol for efficient cobalt recovery needs to be developed in the future. This would enable the use of the method for economic and eco-friendly manufacturing processes.
Acknowledgements
Financial support from the EU H2020 Marie Curie Skłodowska Curie ETN program, project INTEGRATE (Contract No. 642620), is gratefully acknowledged.
Notes and references
- J. Bolsakova and A. Jirgensons, Eur. J. Org. Chem., 2016, 4591 CrossRef CAS.
- T. Boibessot, D. Bénimélis, P. Meffre and Z. Benfodda, Amino Acids, 2016, 48, 2081 CrossRef CAS PubMed.
- H. Fukumoto, K. Takahashi, J. Ishihara and S. Hatakeyama, Angew. Chem., Int. Ed., 2006, 45, 2731 CrossRef CAS PubMed.
- S. N. Osipov, P. Tsouker, L. Hennig and K. Burger, Tetrahedron, 2004, 60, 271 CrossRef CAS.
- K. Morisaki, M. Sawa, J. Nomaguchi, H. Morimoto, Y. Takeuchi, K. Mashima and T. Ohshima, Chem.–Eur. J., 2013, 19, 8417 CrossRef CAS PubMed.
- V. M. Girijavallabhan, L. Chen, C. Dai, R. J. Feltz, L. Firmansjah, D. Li, S. H. Kim, J. A. Kozlowski, B. J. Lavey and A. Kosinski, et al., Bioorg. Med. Chem. Lett., 2010, 20, 7283 CrossRef CAS PubMed.
- G. Pattenden and G. Rescourio, Org. Biomol. Chem., 2008, 6, 3428 CAS.
- Z. Benfodda, D. Bénimélis, M. Jean, J.-V. Naubron, V. Rolland and P. Meffre, Amino Acids, 2015, 47, 899 CrossRef CAS PubMed.
- G. Hattori, A. Yoshida, Y. Miyake and Y. Nishibayashi, J. Org. Chem., 2009, 74, 7603 CrossRef CAS PubMed.
- U. Schmidt, M. Respondek, A. Lieberknecht, J. Werner and P. Fischer, Synthesis, 1989, 256 CrossRef CAS.
- S. Hatakeyama, H. Matsumoto, H. Fukuyama, Y. Mukugi and H. Irie, J. Org. Chem., 1997, 62, 2275 CrossRef CAS PubMed.
- C. J. Brennan, G. Pattenden and G. Rescourio, Tetrahedron Lett., 2003, 44, 8757 CrossRef CAS.
- R. D. Grigg, J. W. Rigoli, S. D. Pearce and J. M. Schomaker, Org. Lett., 2012, 14, 280 CrossRef CAS PubMed.
- J. Sirotkina, L. Grigorjeva and A. Jirgensons, Eur. J. Org. Chem., 2015, 6900–6908 CrossRef CAS.
- R. Bishop, in Compr. Org. Synth. II, Elsevier, Amsterdam, 2nd edn, 2014, pp. 239–295 Search PubMed.
- I. R. Morgan, A. Yazici, S. G. Pyne and B. W. Skelton, J. Org. Chem., 2008, 73, 2943 CrossRef CAS PubMed.
- M. Vangala and G. P. Shinde, Beilstein J. Org. Chem., 2015, 11, 2289 CrossRef CAS PubMed.
- J. L. Jiménez Blanco, E. M. Rubio, C. Ortiz Mellet and J. M. García Fernández, Synlett, 2004, 2230 Search PubMed.
- D. Noort, G. A. van der Marel, G. J. Mulder and J. H. van Boom, Synlett, 1992, 224 CrossRef CAS.
- D. M. Gordon and S. J. Danishefsky, J. Org. Chem., 1991, 56, 3713 CrossRef CAS.
- I. W. Davies, C. H. Senanayake, R. D. Larsen, T. R. Verhoeven and P. J. Reider, Tetrahedron Lett., 1996, 37, 813 CrossRef CAS.
- C. H. Senanayake, L. M. DiMichele, J. Liu, L. E. Fredenburgh, K. M. Ryan, F. E. Roberts, R. D. Larsen, T. R. Verhoeven and P. J. Reider, Tetrahedron Lett., 1995, 36, 7615 CrossRef CAS.
- E.-J. Tillmanns and J. Ritter, J. Org. Chem., 1957, 22, 839 CrossRef CAS.
- A. Toshimitsu, C. Hirosawa and K. Tamao, Tetrahedron, 1994, 50, 8997 CrossRef CAS.
- S. Top and G. Jaouen, J. Chem. Soc., Chem. Commun., 1979, 224 RSC.
- S. Top and G. Jaouen, J. Org. Chem., 1981, 46, 78 CrossRef CAS.
- R. F. Lockwood and K. M. Nicholas, Tetrahedron Lett., 1977, 18, 4163 CrossRef.
- K. M. Nicholas, Acc. Chem. Res., 1987, 20, 207 CrossRef CAS.
- B. J. Teobald, Tetrahedron, 2002, 58, 4133 CrossRef CAS.
- G. B. Jones, J. M. Wright, T. M. Rush, G. W. Plourde, T. F. Kelton, J. E. Mathews, R. S. Huber and J. P. Davidson, J. Org. Chem., 1997, 62, 9379 CrossRef CAS.
- T. Sugihara, H. Ban and M. Yamaguchi, J. Organomet. Chem., 1998, 554, 163 CrossRef CAS.
- D. Kalaitzakis, T. Montagnon, I. Alexopoulou and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2012, 51, 8868 CrossRef CAS PubMed.
- B. Gabriele, R. Mancuso, V. Maltese, L. Veltri and G. Salerno, J. Org. Chem., 2012, 77, 8657 CrossRef CAS PubMed.
- S.-T. Chen and J.-M. Fang, J. Org. Chem., 1997, 62, 4349 CrossRef CAS PubMed.
- R. Spina, E. Colacino, J. Martinez and F. Lamaty, Chem.–Eur. J., 2013, 19, 3817 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03965d |
| ‡ These authors provided an equal contribution to the publication. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*