
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation on the effect of known potent S. aureus NorA efflux pump inhibitors on the staphylococcal biofilm formation
Stefano Sabatini *a,
Miranda Piccionib,
Tommaso Felicettia,
Stefania De Marcob,
Giuseppe Manfronia,
Rita Pagiottib,
Morena Nocchettia,
Violetta Cecchettia and
Donatella Pietrella*b
*a,
Miranda Piccionib,
Tommaso Felicettia,
Stefania De Marcob,
Giuseppe Manfronia,
Rita Pagiottib,
Morena Nocchettia,
Violetta Cecchettia and
Donatella Pietrella*b
aDepartment of Pharmaceutical Sciences, Chemistry and Technology of the Drug Section, University of Perugia, Via del Liceo, 1, 06123, Perugia, Italy. E-mail: stefano.sabatini@unipg.it; Tel: +39-075-585-5130
bDepartment of Pharmaceutical Sciences, Biochemical Sciences and Health Section, University of Perugia, Via del Giochetto, 5, 06122, Perugia, Italy. E-mail: donatella.pietrella@unipg.it; Tel: +39-075-585-7208
First published on 25th July 2017
Abstract
The emergence of multidrug resistant microorganisms has triggered the impending need of developing effective antibacterial strategies. Staphylococcus epidermidis and the more virulent S. aureus are able to colonize and form biofilms on implanted medical devices causing clinical acute and chronic infections. The aim of the present work was to assess the effect of NorA efflux pump inhibitors (EPIs) on S. aureus and S. epidermidis biofilm formation by using known synthetic NorA EPIs and thus giving support to the hypothesis that efflux pumps could play an intriguing role in Staphylococcus biofilm formation. In particular, three 2-phenylquinolines (1a–c) and a pyrazolobenzothiazine (2), our previously reported NorA EPIs, at concentrations lower than MIC showed a good ability in reducing biofilm formation in S. aureus (ATCC 29213) and S. epidermidis (ATCC 35984). Then, to assess if biofilm formation inhibitors could be combined with an antibacterial to limit biofilm production, the two best compounds 1c and 2 were tested with ciprofloxacin (CPX) and the results showed an excellent synergistic effect against Staphylococcus strains with a biofilm formation inhibition over 60% at low concentrations. Further, to confirm the involvement of the NorA efflux pump in the biofilm production, NorA EPIs 1c and 2 were tested against a modified S. aureus strain overexpressing the NorA efflux pump (SA-1199B) and results highlighted a strong decrease in biofilm mass. In conclusion, cytotoxicity assays on HeLa and HepG2 cells showed that concentrations useful to inhibit biofilm formation in Staphylococcus strains in combination with CPX were lower than the toxic concentrations for human cells.
Introduction
To date, most of the research concerning the discovery of new antibacterial agents has been based on the spreading of bacteria within liquid media, and not considering that this method is useful for the prediction of antimicrobial activity against acute infections only characterized by planktonic bacteria.1 Unfortunately, less than 0.1% of the total microbes spreads in the planktonic mode of growth, whereas most of the microbial species are attached to a living or abiotic surface and organized in aggregates, surrounded by an extracellular matrix, that are termed biofilms.2 Most microorganisms can grow in biofilms, including bacteria (Gram-positive and Gram-negative; aerobic and anaerobic),2 mycobacteria3 and fungi (mould and yeast).4,5 Due to the physical and biological properties of the biofilm, microorganisms can establish persistent infections with an extreme resistance to the antimicrobial agents (about 100–1000 times),6 as well as an exceptional ability to evade the host defence.7–10 Indeed, the hyper-mutability of the biofilm growing bacterial cells during chronic infection and the high tolerance to the common antimicrobial agents make treatment very difficult if not impossible.11,12 In particular, for infections related to contaminated medical implants with a biofilm, the surgical removal of the prosthesis is often the only recommended practice.13 Thus, preventive approaches should be undertaken but, unfortunately, early biofilm formation generates very little inflammation, making it difficult to detect the initial infection.2Antimicrobial tolerance of bacteria growing in biofilms is multifactorial and concerns several resistance mechanisms: (i) a lower diffusion of antimicrobial agents through the biofilm polysaccharide matrix;14 (ii) physiological and phenotypic changes of the cell population due to the different nutrient availability within the layers of the biofilm;15 (iii) communication between bacteria of the same or different species, also called quorum sensing (QS);16 and (iv) expression of efflux pumps (EPs).17
Bacterial EPs are transmembrane proteins, grouped in five different families,18 present in almost all bacterial species; their function is to extrude structurally unrelated noxious agents before they reach their target.19–23 In detail, a basal expression and, especially, an overexpression of EPs in bacteria lead to a rapid efflux of antibacterials from the microbial cells, resulting in a reduced concentration at the target site. In turn, this EP-mediated sub-inhibitory concentration of antimicrobials is the reason for an increasing rate of new mutations generating further resistance mechanisms.24 Interestingly, in recent years, EPs have been described to be involved in several different resistance mechanisms, but their role is not yet clearly understood. Among these results, of particular interest is their involvement in the biofilm formation of different microbes (Escherichia coli, Pseudomonas putida, Klebsiella pneuomoniae, and Staphylococcus aureus), indirectly shown by inhibiting EPs with known EP inhibitors (i.e. phenylalanine-arginine beta-naphthylamide – PAβN, thioridazine – TZ, carbonyl cyanide 3-chlorophenylhydrazone – CCCP, and 1-(1-naphthylmethyl)-piperazine – NMP). Unfortunately, their activity is thought to be related with nonspecific mechanisms of efflux inhibition and often also coupled with a high toxicity.25–27 Further, EPs involvement in biofilm formation was clearly proved through genetic studies in Gram-negative bacteria by the demonstration that deletions of EP genes impaired the production of biofilm.25,26
In S. aureus, a well-known pathogen growing in biofilm and causing tremendous chronic infections in patients with prostheses or cystic fibrosis,28 the most expressed EP is NorA encoded by the norA gene.29 The NorA pump is a cytoplasmic membrane protein belonging to the Major Facilitator Superfamily and it uses a proton motive force to energize the transport of antimicrobial agents (e.g. ciprofloxacin – CPX) across the cell membrane via an H+/drug antiporter mechanism.30 The inhibition of NorA is a promising strategy aimed to block drug extrusion from the bacterial cells, but the lack of a crystal structure of NorA makes difficult the medicinal chemistry efforts to find efficient NorA efflux pump inhibitors (EPIs). Furthermore, the few known NorA EPIs differ in terms of shape and structure and to date their mechanism of action has not been fully clarified. Anyway, given our experience in the design and synthesis of NorA EPIs,31–34 in this work we investigated the capability of previously reported potent NorA EPIs32–34 to inhibit biofilm formation in different Staphylococcus strains. To the best of our knowledge, few examples of biofilm formation inhibition by EPIs in S. aureus have been reported so far, and, in any case, they concern the use of known and toxic compounds such as CCCP and TZ.26,27
In addition, because of the presence of a norA gene codifying for a NorA-like efflux pump in Staphylococcus epidermidis,35 our study was also enlarged to S. epidermidis, which is less virulent than S. aureus, but also able to colonize and to form biofilms on implanted medical devices and cause several clinical infections. Anyway, the ability of both Staphylococcus spp to form biofilms on an abiotic surface or damaged host tissue is an important virulence factor that represents one of the main survival mechanisms for the bacteria.36
Thus, with the aim to find dual EP-biofilm formation inhibitors, we selected four of the in house most potent NorA EPIs (three 2-phenylquinolines 1a–c32,33 and a pyrazolobenzothiazine 2 34 Fig. 1), in order to assess their ability to inhibit biofilm formation and/or induce biofilm dispersal on S. aureus (ATCC 29213) and S. epidermidis (ATCC 35984).
 | ||
| Fig. 1 In house NorA EPIs (2-phenylquinolines 1a–c and pyrazolobenzothiazine 2) selected to investigate their biofilm inhibition activity. | ||
The obtained encouraging results prompted us to assess two out of the four compounds in combination with CPX in order to demonstrate their ability, in synergism with CPX, in preventing biofilm formation with Staphylococcus strains.
Then, once we have proved that synthetic EPIs can synergize with CPX in reducing biofilm formation, looking to future medicinal chemistry efforts aimed to identify compounds endowed with both EPI and biofilm inhibition activity, we have shown that a S. aureus (SA-1199B overexpressing norA gene) strain,37 commonly used to identify new NorA EPIs in preliminary screenings, produced a greater biofilm mass than its parent wild-type strain SA-1199.37 In parallel, the same two strains were grown in the presence of the two best NorA EPIs in an attempt to prove that, by blocking EPs, it is possible to significantly reduce biofilm mass in the overexpressing EP strain SA-1199B.37
Experimental
NorA EPIs
2-Phenylquinolines (1a,32 1b33 and 1c33) and pyrazolobenzothiazine 2 34 (Fig. 1) were selected from our in house library of NorA EPI compounds and tested to assess their inhibition activity on biofilm formation. All the stock solutions of the compounds were prepared during the day of the experiment in DMSO (10 mg mL−1), with the exceptions of CPX, which was dissolved with sterile water. Further dilutions were set up with the medium.Microbial strains and growth conditions
The microbial strains used in this study were S. aureus (ATCC 29213), S. aureus SA-1199B (overexpressing norA and also possessing an A116E GrlA substitution),37 its isogenic parental strain SA-1199 (norA wt),37 and S. epidermidis (ATCC 35984). The bacterial cultures were maintained in tryptic soy agar (TSA). The day before the test, one single colony was inoculated in Muller Hinton Broth (MHB) and incubated for 24 h at 37 °C. Microbial cells were harvested by centrifugation, washed, counted by spectrophotometric analysis (OD600) and resuspended to the desired concentration in the appropriate culture medium (MHB). S. aureus (ATCC 29213) and S. epidermidis (ATCC 35984) strains were obtained from LGC standards (Milan, Italy) while S. aureus SA-1199B and its isogenic parental strain SA-1199 were a kind gift from Prof. Glenn W. Kaatz (John D. Dingell Department of Veterans Affairs Medical Center and the Department of Internal Medicine, Division of Infectious Diseases, School of Medicine, Wayne State University, Detroit, Michigan 48201, United States).MIC assay
MIC was determined by a micro-broth dilution method according to the Clinical and Laboratory Standards Institute/National Committee for Clinical Laboratory Standards (CLSI/NCCLS) Approved Standard.38 Briefly, to determine the MIC: all the NorA EPIs (1a–c32,33 and 2 34), TZ, and CPX, were diluted in the MHB. The dilutions, ranging from 0.11 to 250 μg mL−1 of the compounds, were prepared in U bottom 96 well plates. The inoculum size of bacteria in the MHB was about 103 to 104 bacteria per well. The plates were incubated at 37 °C for 24 h. The MIC of each compound was defined as the lowest concentration that inhibited visible growth of the microorganisms and bacterial growth was also determined by spectrophotometer analysis (600 nm). MIC assays were performed in duplicate and each experiment was repeated at least twice.Effect of NorA EPIs on biofilm formation
The in vitro static biofilm assay was performed using a 96-well microtiter plate as previously described, but with some modifications.39,40 To grow biofilms, overnight cultures of S. aureus or S. epidermidis were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 (corresponding to 1–2 × 106 CFU mL−1) in TSB supplemented with 2% sucrose. Bacteria were incubated in the presence or absence of different NorA EPIs (1a–c32,33 and 2 34) at concentrations of 250, 125, 62.5, 25, and 2.5 μg mL−1; TZ (at 250, 125, 62.5, 25, and 2.5 μg mL−1) and CPX at 0.45 μg mL−1 were used as the positive controls. Cultures were incubated at 37 °C for 24 h in static conditions. After incubation, the biofilm developed in each well was washed twice with 200 μL of distilled water. In each well, 100 μL of 0.4% crystal violet were added for 30–45 min. After this procedure, the wells were washed four times with distilled water, immediately discoloured with 200 μL of 95% ethanol. After 45 minutes, 100 μL of discoloured solution was transferred to a well of a new plate and the crystal violet measured at 570 nm in a microplate reader (TECAN). The amount of biofilm formed was measured by comparing the absorbance values of the compound-treated wells versus untreated control wells. Biofilm assays were performed in triplicate in at least three individual experiments for each concentration.
:100 (corresponding to 1–2 × 106 CFU mL−1) in TSB supplemented with 2% sucrose. Bacteria were incubated in the presence or absence of different NorA EPIs (1a–c32,33 and 2 34) at concentrations of 250, 125, 62.5, 25, and 2.5 μg mL−1; TZ (at 250, 125, 62.5, 25, and 2.5 μg mL−1) and CPX at 0.45 μg mL−1 were used as the positive controls. Cultures were incubated at 37 °C for 24 h in static conditions. After incubation, the biofilm developed in each well was washed twice with 200 μL of distilled water. In each well, 100 μL of 0.4% crystal violet were added for 30–45 min. After this procedure, the wells were washed four times with distilled water, immediately discoloured with 200 μL of 95% ethanol. After 45 minutes, 100 μL of discoloured solution was transferred to a well of a new plate and the crystal violet measured at 570 nm in a microplate reader (TECAN). The amount of biofilm formed was measured by comparing the absorbance values of the compound-treated wells versus untreated control wells. Biofilm assays were performed in triplicate in at least three individual experiments for each concentration.
Effect of NorA EPIs on biofilm dispersion
To test the effect of NorA EPIs on biofilm dispersion, biofilms, grown on the inside surface of 96-well microtiter plates for 24 h as described above, were treated with three different concentrations (1 × MIC, 2 × MIC, and 4 × MIC; when MIC values were >250 μg mL−1, the used MIC value was 250 μg mL−1) of NorA EPIs (1a–c 32,33 and 2 34) at 37 °C for further 24 h. Diluent of NorA EPIs was used as a control. The positive controls were TZ and CPX. Afterward, the biofilm mass was quantified by crystal violet assay. Biofilm dispersal assays were performed in triplicates in at least three individual experiments for each concentration.Checkerboard assays
The synergism of the NorA EPIs (1c33 and 2 34) with CPX was evaluated by a checkerboard titration assay in microplates as recommended by the NCCLS and expressed as the sum of the fractional inhibitory concentration (FIC) index for each agent. In brief, checkerboards were set up with double dilutions of CPX (0–1.8 μg mL−1) in the horizontal wells and NorA EPIs (0–250 μg mL−1) in the vertical wells. Then, a 100 μL suspension of S. aureus or S. epidermidis adjusted to 105/104 CFU mL−1 was added to each well and cultured at 37 °C for 24 h in MHB.After incubation, plates were visually inspected for turbidity to determine growth. Visual readings of the MIC were then performed and the OD600 nm values were measured. Experiments were performed in triplicate on different days. Each checkerboard test generates many different combinations and, by convention, the FIC value of the most effective combination was used in calculating the fractional inhibitory concentration index (FICI). FICI was calculated by adding both FICs:
| FICI = FICA + FICB = CcombA/MICaloneA + CcombB/MICaloneB |
Cell lines
HeLa cell line (ATCC CCL-2), a human epithelial carcinoma cell line derived from cervical cancer, and HepG2 (ATCC HB-8065), a human liver cancer cell line, were maintained in RPMI 1640. The culture medium consisted of RPMI 1640 with 2 mM glutamine, 10% fetal bovine serum (FBS), 10000 penicillin units and streptomycin 10 μg mL−1, referred to as cRPMI. Confluent cultures were split using 0.25% trypsin/EDTA. Monolayers were incubated at room temperature for 5–10 min until cell detachment. Fresh medium (cRPMI) was added to disperse the cells, suspensions were then centrifuged and resuspended in culture medium at the desired concentration. Human cervical cancer HeLa (ATCC CCL-2) and human hepatocellular carcinoma HepG2 (ATCC HB-8065) cell lines were obtained from LGC standards (Milan, Italy).
Cytotoxicity assay
The cytotoxicity of NorA EPIs (1a–c 32,33 and 2 34), TZ, and CPX was tested by the determination of the cell ATP level by a ViaLight® Plus Kit (Lonza). The method is based upon the bioluminescent measurement of ATP that is present in all metabolically active cells. The bioluminescent method utilizes an enzyme, luciferase, which catalyses the formation of light from ATP and luciferin. The emitted light intensity is linearly related to the ATP concentration and is measured using a luminometer. To perform cytotoxicity tests, cells were recovered, counted and adjusted to the concentration 2 × 105/mL, seeded in a flat bottom 96-well culture plate and incubated until monolayer formation. The examinations were carried out for the test compounds and the control (cells not treated). Various 1:2 dilutions of the above mentioned NorA EPIs were diluted 1:2 in cRPMI in order to achieve final concentrations in the wells from 250 to 0.11 μg mL−1. Each concentration was tested in triplicate. In parallel, the cytotoxicity of CPX-1c (0.056–3.9 and 0.014–31.25 μg mL−1, respectively) and CPX-2 (0.00356–15.625 and 0.028–7.8 μg mL−1, respectively) combinations were tested on HepG2 cells. After adding the test compounds onto cell monolayers, plates were incubated for 24 h at 37 °C. After incubation, plates were left to cool at room temperature for 10 minutes and then the Cell Lysis Reagent was added to each well to extract ATP from the cells. Next, after 10 minutes the AMR Plus (ATP Monitoring Reagent Plus) was added and after 2 more minutes the luminescence was read using a microplate luminometer (TECAN). The results are expressed as 50% cytotoxic concentration (CC50), defined as the concentration required to reduce the live cell number by 50% compared to the untreated controls. Cytotoxicity values referred to the combination of CPX plus 1c33 or 2 34 assessed at their FICI values and were expressed as percentage of live cells.
Statistical analysis
All the data were expressed as mean ± SD. Differences between groups were compared using the Student's t-test (two-tailed). A P-value of <0.05 was considered significant.Results & discussion
Antimicrobial activity of NorA EPIs
The MICs of the four NorA EPIs (the 2-phenylquinolines 1a–c32,33 and the pyrazolobenzothiazine 2 34 Fig. 1) and TZ were firstly determined on S. aureus (ATCC 29213) and S. epidermidis (ATCC 35984). Data shown in Table 1 illustrate how the selected compounds had generally no intrinsic antibacterial activity with MIC values equal to or higher than 250 μg mL−1, with the exception of 1b33 that showed a MIC value of 62.5 μg mL−1 when tested against S. epidermidis. On the contrary, both the strains were susceptible to CPX, according to CLSI breakpoints. CPX was included in the test as a positive control.| Compd. | Staphylococcus aureus | Staphylococcus epidermidis |
|---|---|---|
| MIC (μg mL−1) | MIC (μg mL−1) | |
| 1a | 250 | >250 |
| 1b | 250 | 62.5 |
| 1c | >250 | >250 |
| 2 | 250 | >250 |
| TZ | 250 | 250 |
| CPX | 0.45 | 0.45 |
Since resistance develops only for compounds endowed with antibacterial activity, high MIC values for NorA EPIs were considered favourably.
Anti-biofilm activity
To evaluate if the NorA efflux pump inhibition could influence the biofilm formation of S. aureus and S. epidermidis, biofilms were grown in the absence or presence of NorA EPIs (1a–c32,33 and 2)34 and TZ at different concentrations (250, 125, 62.5, 25 and 2.5 μg mL−1) and the biofilm mass was analysed by Cristal Violet staining. The results in Fig. 2 show a similar trend of activity for NorA EPIs on both the strains tested.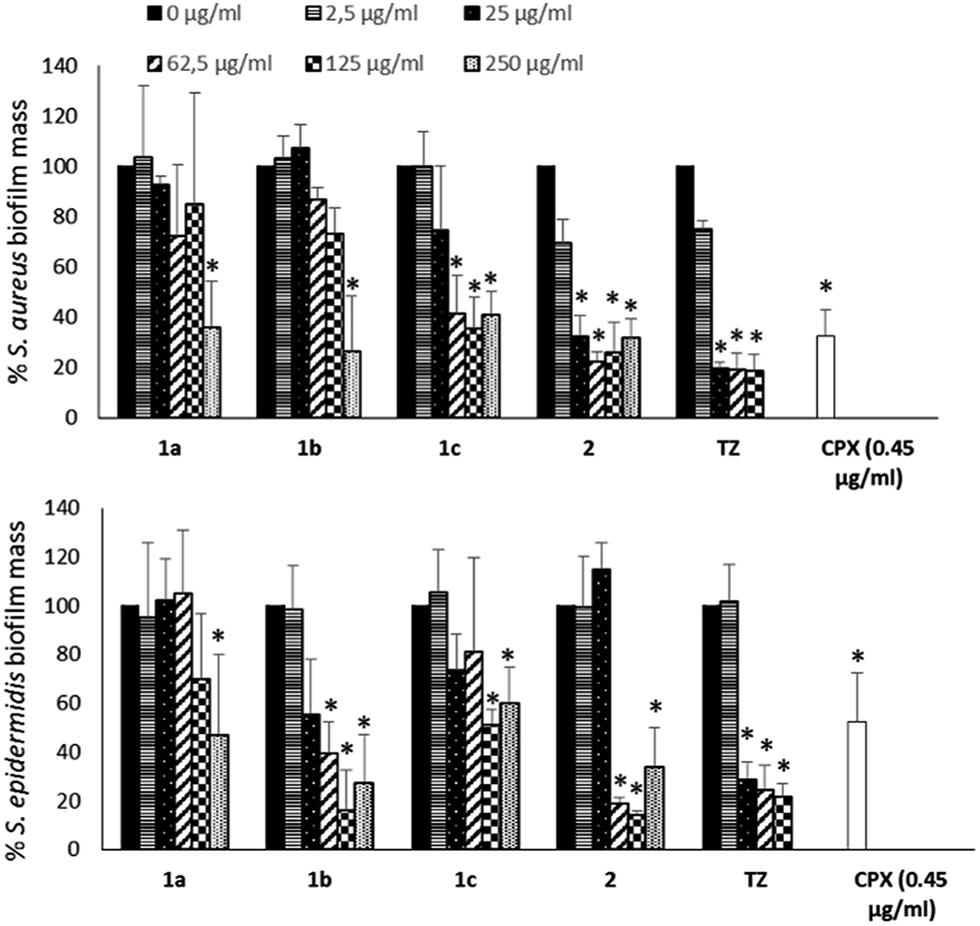 | ||
| Fig. 2 Effect of NorA EPIs (1a–c and 2) on the biofilm formation of S. aureus (ATCC 29213) and S. epidermidis (ATCC 35984). TZ was used as a positive control. CPX was used at its MIC to evaluate its ability to inhibit biofilm. Data represent the mean ± SD of two independent experiments performed in triplicate. *P < 0.05 (NorA EPI-treated or CPX-treated microorganisms vs. untreated bacteria). | ||
In particular, with respect to S. aureus, compounds 1a32 and 1b33 were able to reduce the biofilm by 50% only at the concentrations corresponding to MIC. On the contrary, of considerable interest, compounds 1c33 and 2 34 at concentrations corresponding to 1/2 and 1/4 MICs showed a biofilm mass decrease greater than 60%, comparable to that observed for TZ. Then, at lower concentrations, only compound 2 34 was again able to reduce biofilm formation, by 60% at 25 μg mL−1 (1/10 MIC) up to 30% at 2.5 μg mL−1 (1/100 MIC), the same as the positive control TZ. Similar results were observed on S. epidermidis, where 2 34 showed an ability to reduce the biofilm mass up to 20% at 1/4 MIC, an inhibition higher than that observed for TZ. Compound 1b33 showed a good biofilm inhibition activity against S. epidermidis only when tested at 1×, 2×, and 4× MIC. Noteworthy, biofilm inhibition data concerning CPX showed that the antibacterial alone at its MIC is only partially able to reduce biofilm formation (Fig. 2) and consequently, once the biofilm is formed, CPX antibacterial activity will be hampered; therefore, a synergistic action of CPX with EPIs could increase the antibacterial drug efficacy.
In parallel, the ability of NorA EPIs, TZ, and CPX to disperse preformed biofilm was assayed on the same S. aureus and S. epidermidis strains, grown in biofilms, for 24 h (Fig. 3), by using 1× MIC concentrations for all the tested compounds.
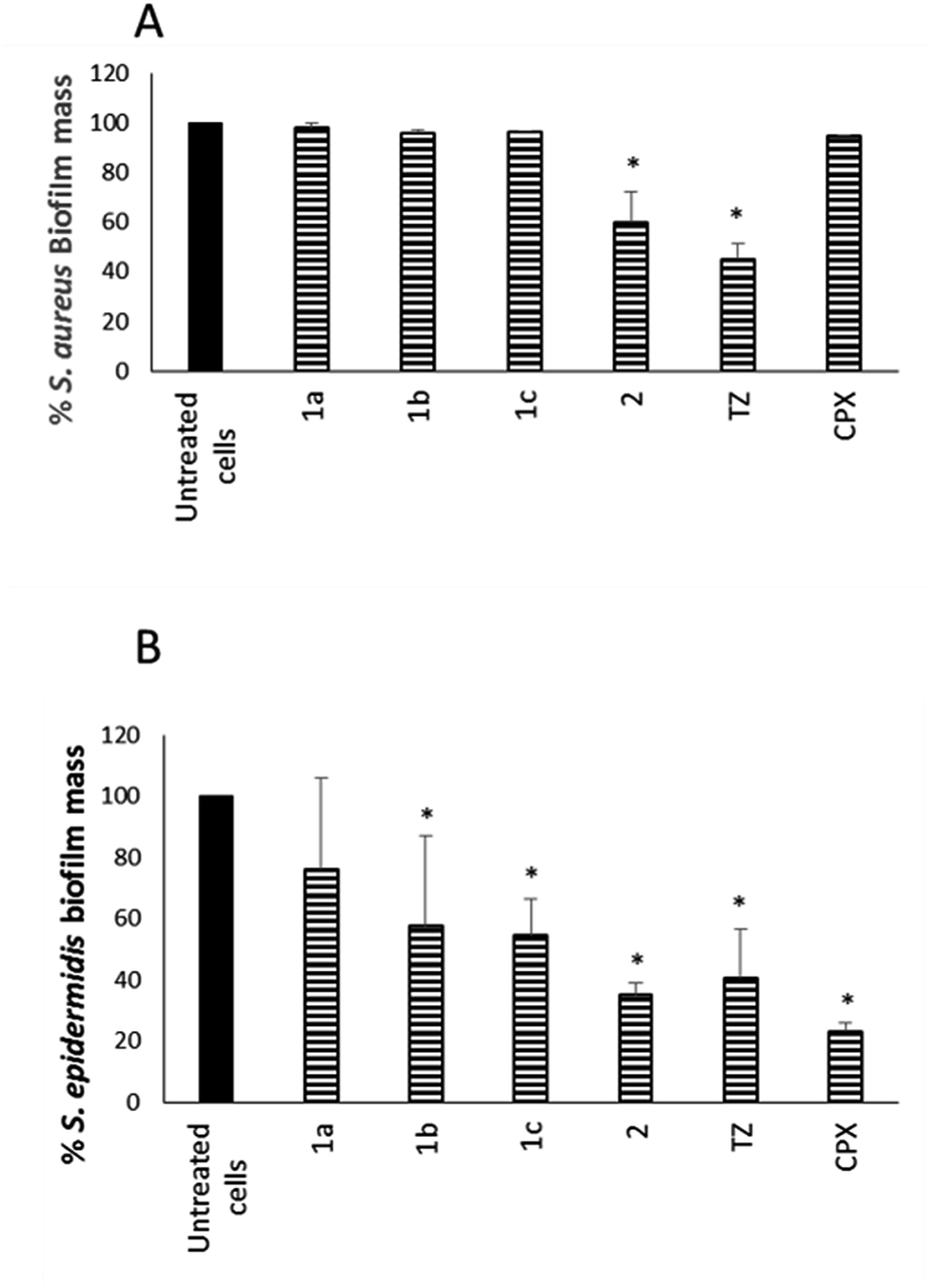 | ||
| Fig. 3 Effect of NorA EPIs 1a–c and 2 on the dispersal of a biofilm of S. aureus (ATCC 29213) (A) and S. epidermidis (ATCC 35984) (B). Data represent the mean ± SD of two independent experiments performed in triplicate. *P < 0.05 (NorA EPI-treated microorganisms vs. untreated cells), #P < 0.05 (CPX-treated microorganisms vs. untreated cells). | ||
The results in Fig. 3A show that compound 2 34 exhibited a good dispersal activity (50% mass reduction) at 1 × MIC, comparable to that observed for the reference TZ and much better than that of antibacterial CPX. Compounds 1a–c32,33 were able to reduce a S. aureus preformed biofilm by 48.26, 34.37 and 50.70%, respectively, only at a very high concentration (4 × MIC – data not shown). Considering S. epidermidis, EPI compounds 1b,33 1c33 and 2 34 showed a good dispersal activity while compound 1a32 was the worst of the four with the percentage of mass biofilm reduction corresponding to only 20% (Fig. 3B). In detail, 1b33 and 1c33 exhibited an activity comparable to that observed for TZ with a 50% reduction of preformed biofilm mass and compound 2 34 showed the best activity comparable to that of the antibacterial CPX alone with a 65% reduction of the biofilm mass.
Synergistic antibiofilm activity of NorA EPIs with CPX
With the aim to consider a feasible medical application, two of the four EPIs (1c33 and 2 34) having the best biofilm inhibition activity were tested in synergism with CPX on S. aureus ATCC 29213 and S. epidermidis ATCC 35984 forming biofilms in order to evaluate their ability to improve the anti-biofilm effect of CPX.Calculations of FIC and FICI (always less than 0.5) obtained by checkerboard assays on S. aureus and S. epidermidis showed significant synergistic effects of NorA EPIs (1c33 and 2 34) with CPX (Table 2).
| MIC alone (μg mL−1) | MIC combination (μg mL−1) | FIC (MICcomb/MICalone) | FICI | Biofilm inhibitiona (%) | ||
|---|---|---|---|---|---|---|
| a Percentage of biofilm inhibition was calculated at concentrations two-fold lower than the FICI values. | ||||||
| S. epidermidis (ATCC 35984) | CPX | 0.45 | 0.056 | 0.124 | 0.1396 | 64.32 |
| 1c | 250 | 3.9 | 0.0156 | |||
| CPX | 0.45 | 0.0035 | 0.0078 | 0.0703 | 60.73 | |
| 2 | 250 | 15.625 | 0.0625 | |||
| S. aureus (ATCC 29213) | CPX | 0.45 | 0.014 | 0.031 | 0.156 | 59.82 |
| 1c | 250 | 31.25 | 0.125 | |||
| CPX | 0.45 | 0.028 | 0.062 | 0.093 | 74.26 | |
| 2 | 250 | 7.8 | 0.0312 | |||
Since a basal expression of EPs contributes to the CPX MIC increase and according to the previously reported EPI activity of compounds 1c33 and 2,34 the data shown in Table 2 highlight that, for both strains, EPI compounds completely synergize with CPX in a dose-dependent manner.
Data were collected by using strains enabled to grow in biofilm in order to check the concentrations at which compounds did not possess an antimicrobial effect (1/2 FICI values). Indeed, bactericidal concentrations do not allow monitoring of the combined EPI-CPX ability to inhibit biofilm formation because, of course, bacteria are killed when still in the planktonic mode. Therefore, at concentrations two-fold lower than FICI values, the planktonic cells were removed from the wells and the production of biofilm on the bottom of the well was evaluated. The biofilm reduction was calculated as a percentage in comparison with untreated bacterial cells. Compound 1c33 in combination with CPX showed an inhibition of the biofilm equal to 60% against both Staphylococcus strains, while compound 2 34 plus CPX highlighted a 60% biofilm inhibition against S. epidermidis and an even greater (74%) biofilm inhibition against S. aureus, highlighting that at very low concentrations NorA EPIs in combination with CPX are able to inhibit biofilm formation in Staphylococcus strains. Therefore, it is evident that known NorA inhibitors could be used with a dual effect: (i) to avoid the extrusion of CPX from S. aureus cells, as previously reported, and (ii) to abolish biofilm formation thereby allowing CPX to carry out the bactericidal effect. In this way, all of the NorA EPIs could be thought as putative S. aureus biofilm inhibitors. Moreover, worthy of note is the prophylactic purpose that low doses of CPX plus a NorA EPI can prevent the biofilm formation that, otherwise, once formed, is almost impossible to eradicate.
Biofilm formation evaluation in different S. aureus strains (SA-1199/SA-1199B)
Driven by the encouraging results obtained, we decided to monitor the biofilm production in the absence or presence of the two best compounds (1c33 and 2 34) against SA-1199B,37 a S. aureus strain commonly used by microbiologists to demonstrate the inhibition of the NorA pump. Indeed, due to the overexpression of the norA gene and mutation in A116E GrlA,37 the SA-1199B strain is commonly used (i) to evaluate the NorA EPI activity by inhibition of the ethidium bromide efflux, a dye NorA substrate, and (ii) to prove the synergistic effect in restoring antimicrobials MIC through checkerboard assays with antimicrobial agents substrates of the NorA pump.32–34 Therefore, we employed this strain to verify the inhibition of the biofilm formation in correlation with the NorA EP inhibition and indirectly show the correlation between NorA overexpression and biofilm formation. Thus, biofilm production of SA-1199B37 was compared to that of its parental strain SA-1199 norA wild-type37 and the results show that SA-1199B37 was able to form the biofilm with a mass significantly higher (P < 0.01) than its parental wild-type strain SA-1199 37 (Fig. 4).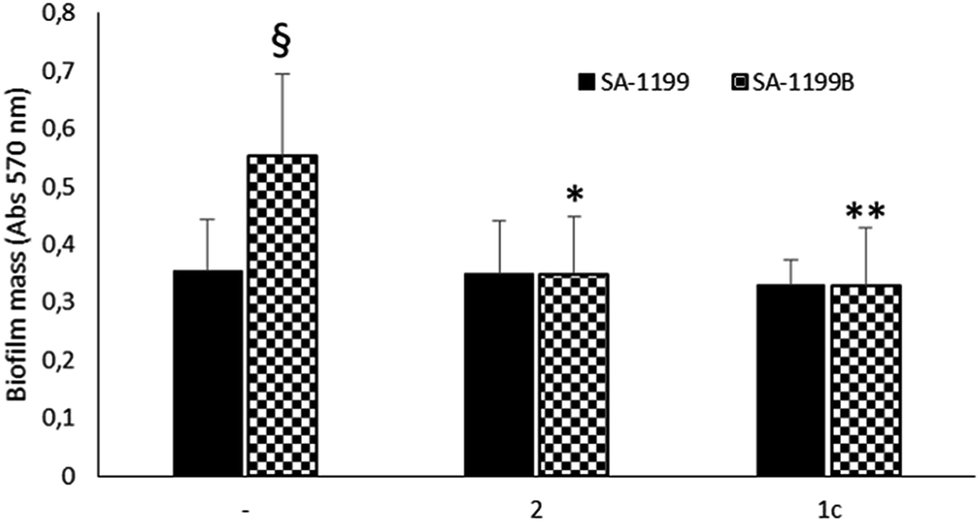 | ||
| Fig. 4 Effect of NorA EPIs (1c and 2) on biofilm formation of SA-1199 and SA-1199B strains. Data represent the mean ± SD of two independent experiments performed in triplicate. §P < 0.01 (SA-1199B vs. SA-1199); *P < 0.05, **P < 0.01 (NorA EPI-treated microorganisms vs. related untreated cells). | ||
Moreover, when the biofilm production was monitored in the presence of NorA EPIs, 1c33 and 2,34 at 25 μg mL−1 (the MIC values of compounds 1c33 and 2 34 were >100 μg mL−1 and 50 μg mL−1, respectively),33,34 SA-1199B37 showed a significant reduction of the biofilm mass (Fig. 4). As a consequence, the presence of EPIs reduced the biofilm formation of the SA-1199B37 strain to the same level as SA-1199,37 highlighting that EP overexpression takes part in the mechanism involved in biofilm formation. Moreover, from these data it is evident that inhibiting the NorA EP activity with EPIs could be a promising strategy to decrease the biofilm formation of S. aureus strains.
Cytotoxicity evaluation
For all the EPIs reported in this study, cytotoxic activity on both human epithelial (HeLa) and human liver (HepG2) cancer cells was evaluated by measurement of the ATP levels (Table 3). Considering the CPX CC50 values (86.02 μg mL−1 on HeLa and 138.58 μg mL−1 on HepG2), cytotoxic activity data obtained for the 2-phenylquinoline derivatives (1a,32 1b,33 and 1c33) were considered too high: 5.44, 9.02, and 8.72 μg mL−1, respectively, on HeLa cells and 4.91, 9.08, and 21.74 μg mL−1, respectively, on HepG2 cells. Conversely, in respect to CC50 values of the TZ (3.47 μg mL−1 on HeLa and 3.67 μg mL−1 on HepG2), a known on-the-market antipsychotic drug, 2-phenylquinoline CC50 values were significantly more acceptable. In addition, the CC50 value of compound 234 (34.68 μg mL−1 on HeLa and 62.84 μg mL−1 on HepG2) was half of the CPX CC50 value, and, definitely, ten times higher than that of TZ.
| Compd. | HeLa cells | HepG2 cells |
|---|---|---|
| CC50 (μg mL−1) | CC50 (μg mL−1) | |
| 1a | 5.44 | 4.91 |
| 1b | 8.72 | 9.08 |
| 1c | 9.02 | 21.74 |
| 2 | 34.68 | 62.84 |
| TZ | 3.47 | 3.67 |
| CPX | 86.02 | 138.58 |
In detail, by comparing the cytotoxicity values with the concentrations to which the dual EP-biofilm formation inhibitory activities synergize with CPX (Tables 2 and 4), the CC50 values of compound 2 34 are higher than the concentration used in combination with CPX against both Staphylococcus strains. In the same way, the CC50 values of compound 1c33 were higher than the concentration used in combination with CPX against S. epidermidis but, unfortunately, not against the S. aureus strain.
| Compd. | FICI values | HepG2 cells |
|---|---|---|
| μg mL−1 | % live cells | |
| a FICI values are referred to combinations against S. epidermidis (ATCC 35984).b FICI values are referred to combinations against S. aureus (ATCC 29213). | ||
| CPX + 1c | 0.056 + 3.9a | 100% |
| CPX + 2 | 0.0035 + 15.625a | 100% |
| CPX + 1c | 0.014 + 31.25b | 11% |
| CPX + 2 | 0.028 + 7.8b | 100% |
Thus, in order to confirm the safety profile of these combinations, the cytotoxicity of CPX plus 1c33 or 2 34 at the concentrations corresponding to the FICI values resulting from checkerboard assays were evaluated on HepG2 cells. With the exception of the combination CPX-1c against S. aureus, all the combinations CPX-EPI showed a 100% of cell vitality (Table 4). Therefore, it can be concluded that compound 2 34 can be used in combination with antimicrobials at sub-toxic concentrations for human cells to prevent biofilm formation.
Conclusions
In this paper, we show that the biofilm formation of S. aureus and S. epidermidis strains can be affected by using synthetic NorA EPIs. Further, in order to collect data about the possible involvement of NorA EP in S. aureus biofilm formation, we show that potent NorA EPIs possess the ability to decrease biofilm mass in a S. aureus strain overexpressing norA (SA-1199B).37 Although the mechanism is still unclear, these data indirectly confirm that the NorA EP is involved in the formation of biofilm, highlighting its role as virulence factor.Finally, data collected in this study suggest that potent EPIs, used in synergism with CPX, increase the efficiency of the antibacterial in preventing biofilm formation in Staphylococcus strains, presumably through EP inhibition. Thus, a starting point for an innovative research field addressed to restore the antibacterial activity of old ineffective drugs has been identified. Moreover, this method to combine dual EP/biofilm inhibitors with antimicrobial agents should be considered for the production of film to spread on materials requiring sterility with the aim to prevent biofilm formation.
Funding
This work was supported by Università degli Studi di Perugia (Fondo per il sostegno della Ricerca di Base 2014).Conflict of interest
No potential conflict of interest was reported by the authors.Abbreviations
| AMR | ATP Monitoring Reagent |
| CC50 | Half Cytotoxicity Concentration |
| CCCP | Carbonyl Cyanide 3-Chlorophenylhydrazone |
| CPX | Ciprofloxacin |
| EP | Efflux Pump |
| EPI | Efflux Pump Inhibitor |
| FBS | Fetal Bovine Serum |
| FIC | Fractional Inhibitory Concentration |
| FICI | FIC index |
| MHB | Muller Hinton Broth |
| MIC | Minimum Inhibitory Concentration |
| NMP | 1-(1-Naphthylmethyl)-piperazine |
| PAβN | Phenyl-arginine-beta-naphthylamide |
| QS | Quorum Sensing |
| TSA | Tryptic Soy Agar |
| TZ | Thioridazine |
Acknowledgements
The authors are grateful to Prof. G. W. Kaatz for his kind gift of the S. aureus strains SA-1199B and SA-1199.References
- C. J. Murray and A. D. Lopez, Lancet, 1997, 349, 1498–1504 CrossRef CAS.
- T. Bjarnsholt, O. Ciofu, S. Molin, M. Givskov and N. Hoiby, Nat. Rev. Drug Discovery, 2013, 12, 791–808 CrossRef CAS PubMed.
- M. S. Islam, J. P. Richards and A. K. Ojha, Expert Rev. Anti-Infect. Ther., 2012, 10, 1055–1066 CrossRef CAS PubMed.
- M. Cuellar-Cruz, E. Lopez-Romero, J. C. Villagomez-Castro and E. Ruiz-Baca, Future Microbiol., 2012, 7, 755–771 CrossRef CAS PubMed.
- F. M. Muller, M. Seidler and A. Beauvais, Med. Mycol., 2011, 49, S96–S100 CrossRef PubMed.
- N. Hoiby, O. Ciofu, H. K. Johansen, Z. J. Song, C. Moser, P. O. Jensen, S. Molin, M. Givskov, T. Tolker-Nielsen and T. Bjarnsholt, Int. J. Oral Sci., 2011, 3, 55–65 CrossRef PubMed.
- T. Bjarnsholt, K. Kirketerp-Moller, P. O. Jensen, K. G. Madsen, R. Phipps, K. Krogfelt, N. Hoiby and M. Givskov, Wound Repair Regen., 2008, 16, 2–10 CrossRef PubMed.
- C. A. Fux, J. W. Costerton, P. S. Stewart and P. Stoodley, Trends Microbiol., 2005, 13, 34–40 CrossRef CAS PubMed.
- P. S. Stewart, Lancet, 2003, 361, 97 CrossRef.
- P. S. Stewart and J. W. Costerton, Lancet, 2001, 358, 135–138 CrossRef CAS.
- O. Ciofu, B. Riis, T. Pressler, H. E. Poulsen and N. Hoiby, Antimicrob. Agents Chemother., 2005, 49, 2276–2282 CrossRef CAS PubMed.
- K. Driffield, K. Miller, J. M. Bostock, A. J. O'Neill and I. Chopra, J. Antimicrob. Chemother., 2008, 61, 1053–1056 CrossRef CAS PubMed.
- H. Wu, C. Moser, H. Z. Wang, N. Hoiby and Z. J. Song, Int. J. Oral Sci., 2015, 7, 1–7 CrossRef CAS PubMed.
- J. N. Anderl, M. J. Franklin and P. S. Stewart, Antimicrob. Agents Chemother., 2000, 44, 1818–1824 CrossRef CAS PubMed.
- J. D. Oliver, FEMS Microbiol. Rev., 2010, 34, 415–425 CrossRef CAS PubMed.
- A. Brooun, S. Liu and K. Lewis, Antimicrob. Agents Chemother., 2000, 44, 640–646 CrossRef CAS PubMed.
- P. Gilbert, D. G. Allison and A. J. McBain, J. Appl. Microbiol., 2002, 92, 98s–110s CrossRef PubMed.
- L. J. Piddock, Nat. Rev. Microbiol., 2006, 4, 629–636 CrossRef CAS PubMed.
- J. M. Pages, L. Amaral and S. Fanning, Curr. Med. Chem., 2011, 18, 2969–2980 CrossRef CAS PubMed.
- J. M. Pages and L. Amaral, Biochim. Biophys. Acta, 2009, 1794, 826–833 CrossRef CAS PubMed.
- L. Amaral, G. Spengler, M. Viveiros, L. Rodrigues, A. Martins, I. Couto, M. Martins, S. Fanning, J. M. Pages and J. Molnar, Anticancer Res., 2008, 28, 3193–3194 Search PubMed.
- L. Amaral, S. Fanning and J. M. Pages, in Adv Enzymol Relat Areas Mol Biol, ed. E. J. Toone, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011, vol. 77, pp. 61–108 Search PubMed.
- L. Amaral, A. Martins, J. Molnar, J. E. Kristiansen, M. Martins, M. Viveiros, L. Rodrigues, G. Spengler, I. Couto, J. Ramos, S. Dastidar, S. Fanning, M. McCusker and J. M. Pages, In Vivo, 2010, 24, 409–424 CAS.
- J. Sun, Z. Deng and A. Yan, Biochem. Biophys. Res. Commun., 2014, 453, 254–267 CrossRef CAS PubMed.
- S. Baugh, A. S. Ekanayaka, L. J. Piddock and M. A. Webber, J. Antimicrob. Chemother., 2012, 67, 2409–2417 CrossRef CAS PubMed.
- S. Baugh, C. R. Phillips, A. S. Ekanayaka, L. J. Piddock and M. A. Webber, J. Antimicrob. Chemother., 2014, 69, 673–681 CrossRef CAS PubMed.
- M. Kvist, V. Hancock and P. Klemm, Appl. Environ. Microbiol., 2008, 74, 7376–7382 CrossRef CAS PubMed.
- A. Jenkins, B. A. Diep, T. T. Mai, N. H. Vo, P. Warrener, J. Suzich, C. K. Stover and B. R. Sellman, mBio, 2015, 6, e02272-14 CrossRef PubMed.
- D. Patel, C. Kosmidis, S. M. Seo and G. W. Kaatz, Antimicrob. Agents Chemother., 2010, 54, 5070–5073 CrossRef CAS PubMed.
- S. M. Soto, Virulence, 2013, 4, 223–229 CrossRef PubMed.
- S. Sabatini, G. W. Kaatz, G. M. Rossolini, D. Brandini and A. Fravolini, J. Med. Chem., 2008, 51, 4321–4330 CrossRef CAS PubMed.
- S. Sabatini, F. Gosetto, G. Manfroni, O. Tabarrini, G. W. Kaatz, D. Patel and V. Cecchetti, J. Med. Chem., 2011, 54, 5722–5736 CrossRef CAS PubMed.
- S. Sabatini, F. Gosetto, N. Iraci, M. L. Barreca, S. Massari, L. Sancineto, G. Manfroni, O. Tabarrini, M. Dimovska, G. W. Kaatz and V. Cecchetti, J. Med. Chem., 2013, 56, 4975–4989 CrossRef CAS PubMed.
- S. Sabatini, F. Gosetto, S. Serritella, G. Manfroni, O. Tabarrini, N. Iraci, J. P. Brincat, E. Carosati, M. Villarini, G. W. Kaatz and V. Cecchetti, J. Med. Chem., 2012, 55, 3568–3572 CrossRef CAS PubMed.
- M. A. Juarez-Verdayes, B. Parra-Ortega, C. Hernandez-Rodriguez, G. Betanzos-Cabrera, S. Rodriguez-Martinez, M. E. Cancino-Diaz and J. C. Cancino-Diaz, Microb. Pathog., 2012, 52, 318–325 CrossRef CAS PubMed.
- H. McCarthy, J. K. Rudkin, N. S. Black, L. Gallagher, E. O'Neill and J. P. O'Gara, Front. Cell. Infect. Microbiol., 2015, 5, 1 Search PubMed.
- G. W. Kaatz and S. M. Seo, Antimicrob. Agents Chemother., 1997, 41, 2733–2737 CAS.
- CLSI, Performance Standards for Antimicrobial Susceptibility Testing; Twenty-First Informational Supplement. CLSI document M100–S21, Clinical and Laboratory Standards Institute, Wayne, PA, 2011 Search PubMed.
- T. Iwase, Y. Uehara, H. Shinji, A. Tajima, H. Seo, K. Takada, T. Agata and Y. Mizunoe, Nature, 2010, 465, 346–349 CrossRef CAS PubMed.
- L. Sancineto, M. Piccioni, S. De Marco, R. Pagiotti, V. Nascimento, A. L. Braga, C. Santi and D. Pietrella, BMC Microbiol., 2016, 16, 220 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |