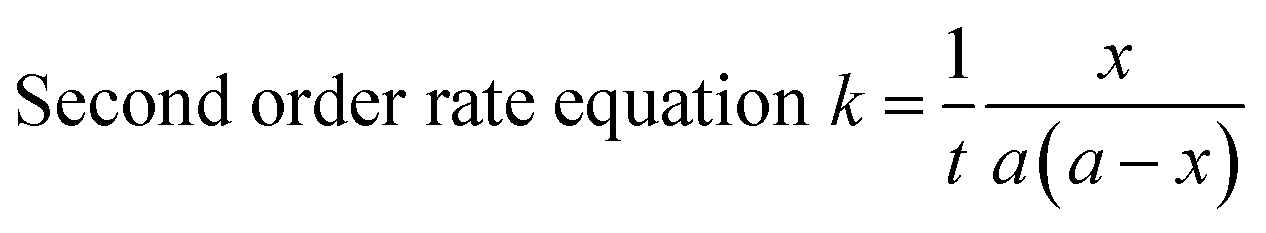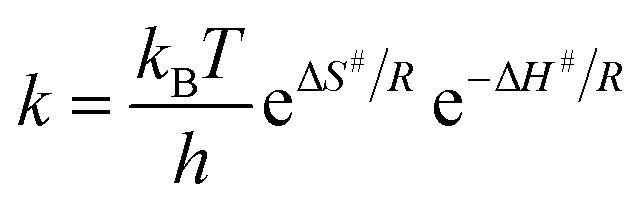DOI:
10.1039/C7RA03734A
(Paper)
RSC Adv., 2017,
7, 34149-34159
Forward and reverse reactions of N-methylaniline-blocked polyisocyanates: a clear step into double Arrhenius plots and equilibrium temperature of thermally reversible reactions†
Received
1st April 2017
, Accepted 29th June 2017
First published on 7th July 2017
Abstract
Blocked polyisocyanates are important building blocks mainly used in a variety of polyurethane coatings. In this study, the synthesis, kinetics of blocking and deblocking reactions and deblocking temperatures of a series of N-methylaniline-blocked polyisocyanates were studied in detail using a hot-stage FT-IR spectrophotometer adopting neat conditions. The results were compared with an aim to resolve the complex questions on the relationship between the forward and reverse reaction parameters. As a result, double Arrhenius plots for these thermally reversible reactions were proposed which paved the way for the prediction of equilibrium temperatures, i.e., the temperature at which both the forward and reverse reactions coexist. From these plots, the equilibrium temperature and the equilibrium rate constants for the forward and reverse reactions were determined. It was found that the electronic and steric effects of the substituents were reflected uniformly in the deblocking temperature, equilibrium temperature and rate constants of the reverse reactions. Among the six N-methylanilines studied, the one having a chloro substituent at a para position was found to be equally reactive compared to the unsubstituted N-methylaniline in the forward reaction and at the same time it was easily cleaved off in the reverse reaction. On the other hand the N-methylaniline possessing a methyl ester substituent at a para position was found to be slow in the forward reaction and cleaved off with highest rate in the reverse reaction. The data such as time required for 50% conversion into product and the equilibrium temperature in combination with deblocking temperature reported in this work will increase the prospects of these blocked polyisocyanates in the polyurethane industry.
Introduction
Thermal equilibrium is said to exist when the rates of the forward and reverse reactions proceed at the same rate in thermally reversible reactions. The equilibrium state achieved in these chemical reactions raises the way to find out the equilibrium temperature which determines the end- and onset-temperatures of the forward and reverse reaction respectively. This temperature boundary is a very useful parameter for industries producing and using the blocked polyisocyanate raw materials. Thermodynamics plays a crucial role in the determination of equilibrium states of reactions, energy changes on reactions, building a relationship between temperature, concentration and time. In this study, we are able to predict the existence of the equilibrium state when a double Arrhenius plot is constructed for the blocking (forward) and the deblocking (reverse) reactions, which showed the equilibrium temperature when the plot was extrapolated to temperature scale. This method is more authentic and allows direct determination of equilibrium temperature using kinetic data of thermally reversible reactions.
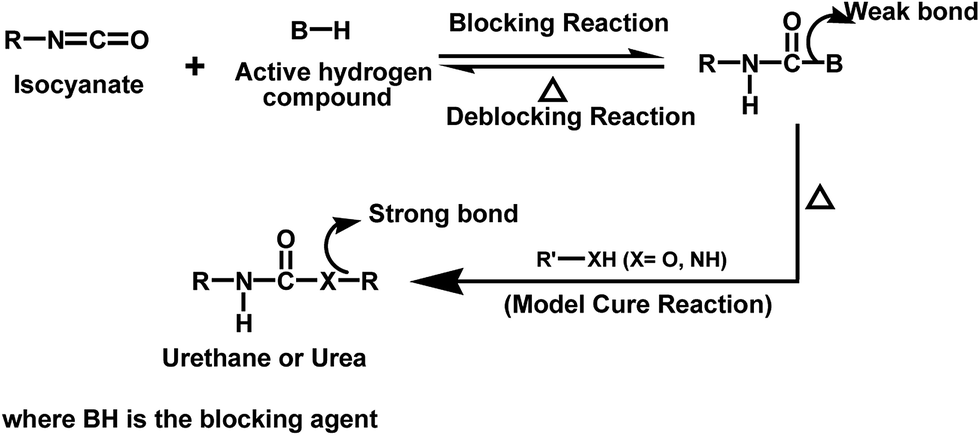
Blocked isocyanates are ideal organic systems suitable for the study of forward and reverse reactions of a thermally reversible process in the temperature range of ambient to 100 °C.1 They are adducts containing a comparatively weak bond formed by the reaction between isocyanates and compounds containing an active hydrogen atom. At elevated temperatures, the reaction tends to proceed in such a way to regenerate the isocyanate and the blocking agent. The regenerated isocyanate can react with a co-reactant containing a nucleophile (alcohol or amine) to form a urethane or urea with thermally more stable bonds. The concept of blocked isocyanates used to make polyurethane heat-curable products is described in Fig. 1.
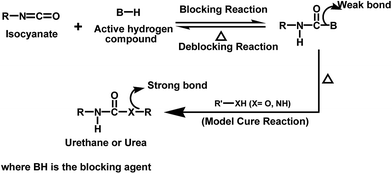 |
| | Fig. 1 Concept of blocked isocyanate. | |
Blocked isocyanates have been the subject of thorough and well-up dated reviews.2–7 Several compounds, namely phenols,8–11 oximes,12,13 amides,14 imides,14 imidazoles,15,16 amidines,17 lactams,18–21 mercaptans,22 aromatic secondary amines,23–25 alcohols,26–28 sodium bisulphite,29–32 pyrazoles,33–35 1,2,4-triazoles,36 hydroxamic acid esters,37 formyl acrylate,38 N-hydroxypthalimide,39 N,N-dimethylacrylamide,40 and diethylene glycol monobutyl ether41 have been reported as blocking agents. Among these blocking agents, phenols are most studied blocking agent because of the possibilities of introducing a number of substituents on the benzene ring. A similar trend of substituent effect is expected in the N-methylaniline (aromatic secondary amines) blocked isocyanates also. Thus it is intended to study the synthesis of a series of N-methylaniline-blocked polyisocyanates, their blocking (forward) and deblocking (reverse) kinetics, their deblocking temperature and the determination of their equilibrium temperature from the double Arrhenius plot.
Chemistry of polyurethane formation reactions has been studied extensively.42–44 However, investigations dealing both blocking and deblocking reactions in a comparative manner are yet to be found in the literature. Recently we studied phenol-blocked polyisocyanates1 with an intension to resolve the complex question on the relationship between kinetic and activation parameters of the forward and reverse reactions. As a result double Arrhenius plots for thermally reversible reactions were proposed for the first time from which equilibrium temperature range and most probable equilibrium temperature were assessed. In this line of work, herein we report new findings on N-methylaniline-blocked polyisocyanates with better accuracy in the results. The results reported in this paper are indeed necessary to optimize the conditions for the developments of new products and their application.
Experimental
Materials
N-Methylaniline (Lancaster), N-methyl-o-toluidine (Aldrich), N-methyl-o-anisidine (Aldrich), 2-chloro N-methylaniline (Aldrich), 4-chloro N-methylaniline (Aldrich), methyl 4-(methylamino)benzoate, poly(tetrahydrofuran) (Terathane, Mn = 2000) (Aldrich) and dibutylamine (Fluka) were used as received. The TDI (TCI) used for the preparation of blocked polyisocyanates was a mixture of isomers containing 98% 2,4-isomer and 2% 2,6-isomer. The solvents, tetrahydrofuran (Merck), toluene (SRL, India) and methanol (SRL, India) were purified by standard procedures.
Measurements
FT-IR spectra of polyurethane prepolymer (polyisocyanate) and blocked polyisocyanates were recorded on a PerkinElmer Spectrum Two model (FT-IR C101375) spectrophotometer by the neat thin film method.
General procedure for the synthesis of N-methylaniline-blocked polyisocyanates (2–7)
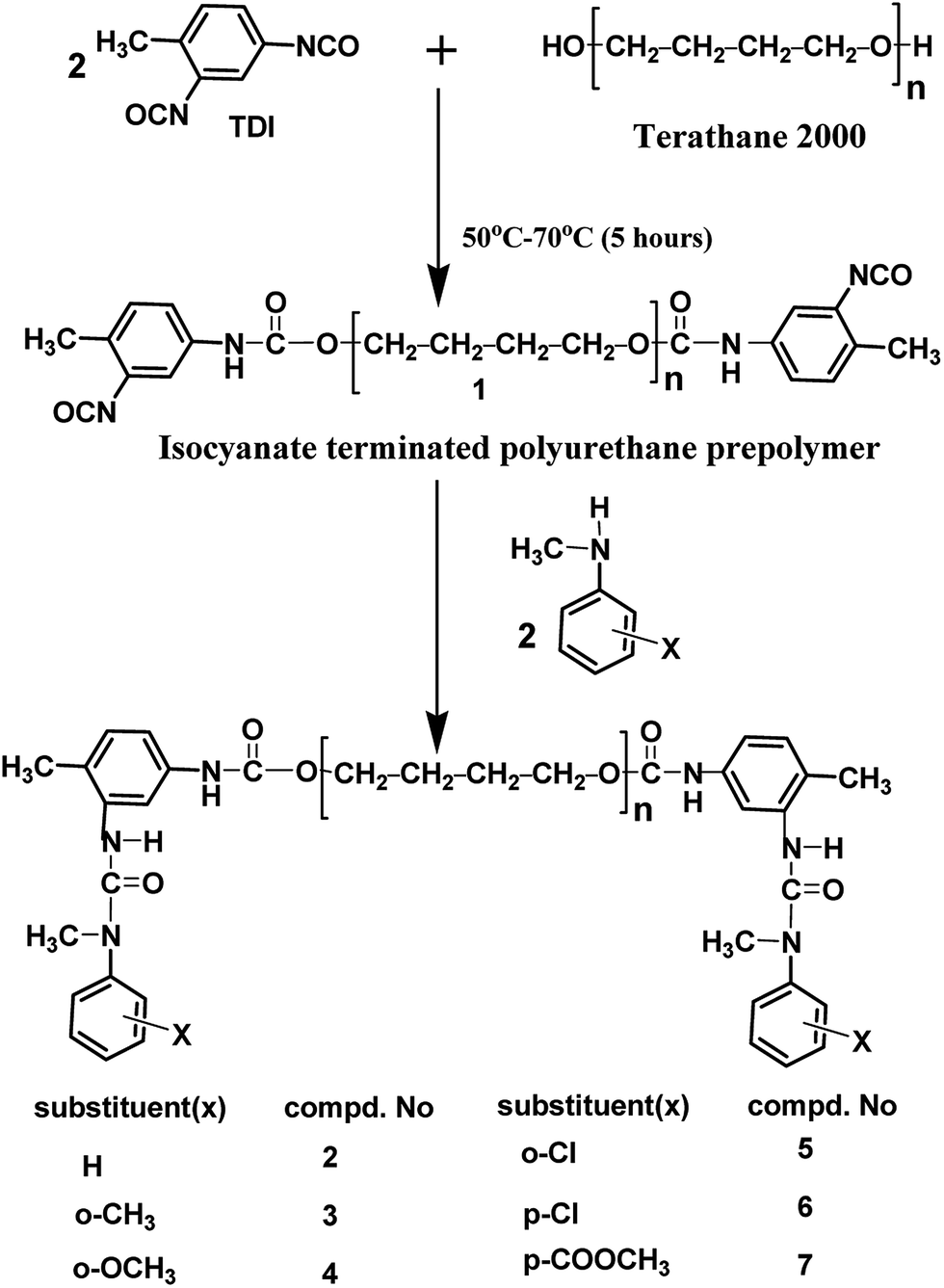
Blocked polyisocyanates were prepared in a two-step process. In the first step, isocyanate-terminated polyurethane prepolymer (1) was prepared. Subsequently in the second step, the terminal –NCO groups of the prepolymer were blocked with N-methylaniline. The following general procedure was adopted for the preparation of blocked polyisocyanates. In a typical synthesis, 1.69 g (9.712 mmol) of TDI was taken in a three-neck round bottomed flask fitted with a mechanical stirrer and a nitrogen inlet. 9.7 g (4.85 mmol) of polyol, [poly(tetrahydrofuran): Mn = 2000] was added drop wise into the flask using an addition funnel with stirring by mechanical means. The experimental setup was maintained in a temperature controlled oil bath. The addition rate was such that it took one hour for the complete transfer. The reaction time was 5 hours; for the first 2 hours the temperature of oil bath was maintained at 50 °C and for the next 3 hours it was maintained at 70 °C. After 5 hours, the reaction temperature was reduced to 40 °C; the –NCO content was determined by the dibutylamine method45 and found to be close to the expected theoretical value. This –NCO terminated prepolymer was then blocked with an equivalent amount (i.e., 4.85 mmol) of N-methylaniline. After thorough mixing, a very small quantity of the sample was taken with the help of a glass-rod for FT-IR measurements towards kinetic analysis and the reaction in the flask was allowed to continue until the complete disappearance of –NCO absorption in the FT-IR spectrum. The structures of the blocked polyurethane prepolymers (blocked polyisocyanates) were confirmed by FT-IR spectroscopic method.
Determination of blocking kinetics parameters
The blocking kinetics of –NCO terminated prepolymer with a series of N-methylaniline was followed by a hot-stage FT-IR spectrophotometer. The experiments were carried out isothermally at any three temperatures suitable for a particular system between the temperature ranges of 30–70 °C. In a typical experiment, a thin film of the reaction mixture of N-methylaniline and –NCO terminated prepolymer was cast on two NaCl discs and they covered each other separated 0.5 mm by lead spacer. Then, the NaCl discs with a lead spacer were placed in a heating transmission cell (HT-32, model 0019-200, ThermoElectron Corp., Madison, WI, USA, connected to a microprocessor-based programmable temperature controller, Omega CT-3251) maintained at desired temperature. Eight scans at a resolution of 4 cm−1 were co-added to produce a single spectrum in a single run. The spectrum at time-zero was recorded within two minutes, and then was recorded at regular time intervals. As the time increased, the absorption by the –NCO group of isocyanate-terminated polyurethane prepolymer at 2270 cm−1 decreased due to the blocking of the –NCO group by N-methylaniline. The peak area under the absorption by the –NCO group in each spectrum was calculated using PerkinElmer Spectrum Two software (Version 10.4.4) and considered as equivalent to the concentration of the polyisocyanate at time ‘t’. The obtained data were treated according to a second-order rate equation in which the two reactants have an equal initial concentration because it gave a linear fit.
where ‘a’ is the initial concentration of the polyisocyanate at time t = 0 and (a − x) is the concentration of the polyisocyanate at time ‘t’. The activation energy (Ea) of the blocking reactions of polyisocyanate was calculated from the slope of the Arrhenius plot (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k versus T−1) using the equation Ea = 2.303 × R × slope. The entropy of activation (ΔS#) was calculated from the well-known Eyring equation,
k versus T−1) using the equation Ea = 2.303 × R × slope. The entropy of activation (ΔS#) was calculated from the well-known Eyring equation,
where kB is the Boltzmann constant (1.3806 × 10−23 J K−1), h is Planck's constant (6.626 × 10−34 J s), R is the gas constant (8.314 J K−1 mol−1), T is the reaction temperature in kelvin, k is the rate constant at T and ΔH# is the enthalpy of activation. The ΔH# in the above equation was obtained from the energy of activation using the relation ΔH = Ea − RT.
Determination of deblocking temperatures of blocked polyisocyanates
In a typical experiment, a thin film of a blocked isocyanate was cast on a NaCl disc and was covered with an uncoated disc. Both the discs were placed on a heating transmission cell connected with a microprocessor based programmable temperature controller. The temperature of the cell was increased from ambient to 170 °C with a heating rate of 5 °C min−1. Eight scans at a resolution of 4 cm−1 were co-added to produce a single spectrum in a single run. The lowest temperature at which a detectable absorption in the 2270 cm−1 range due to regeneration of the –NCO group was noted as the minimum deblocking temperature.
Determination of deblocking kinetics parameters
Deblocking kinetics of the blocked polyisocyanate was followed using FT-IR spectrophotometer attached with hot-stage accessories described in the preceding section. The experiments were carried out isothermally at any four suitable temperatures for a particular system between the temperature ranges of 100–160 °C. Here, the time set to reach the desired temperature from ambient was 7 minutes. The program was made in the heating device in such a way that once the desired temperature was reached, the experiment proceeded isothermally.
In a typical experiment, 7.5 μl of 5.0 × 10−4 molar solution of un-blocked polyisocyanate (i.e., –NCO terminated prepolymer) in dry THF was placed on the center of one of the NaCl discs. The solvent was evaporated under the IR lamp and then the discs were assembled and fitted with the heating accessory. The spectrum was recorded immediately. The peak area under the –NCO absorption was calculated and considered as ‘a’ appearing in the first-order rate equation (here ‘a’ must be considered as equal to amount of blocked isocyanate). Then, the solution of the same concentration of a blocked polyisocyanate was used for deblocking kinetic studies. The spectrum was recorded for zero-time immediately when the sample reached the desired temperature and then the reaction was followed at regular time intervals. At zero-time, in all the cases, there was no isocyanate absorption at 2270 cm−1 and as the time increased, the absorption by the –NCO group at 2270 cm−1 increased due to the deblocking of blocked polyisocyanates. The peak area of the –NCO group in each spectrum was calculated using the PerkinElmer Spectrum Two software and considered as ‘x’ (amount of isocyanate regenerated) appearing in the first order rate equation. From the ‘a’ and ‘x’, a − x was calculated and the data obtained were treated according to a first order rate equation since it gave a linear fit.
The energy of activation (Ea), enthalpy of activation (ΔH#), free energy of activation (ΔG#) and entropy of activation (ΔS#) of the deblocking reactions were calculated as described in the preceding section.
Results and discussion
Synthesis of blocked polyisocyanates (2–7)
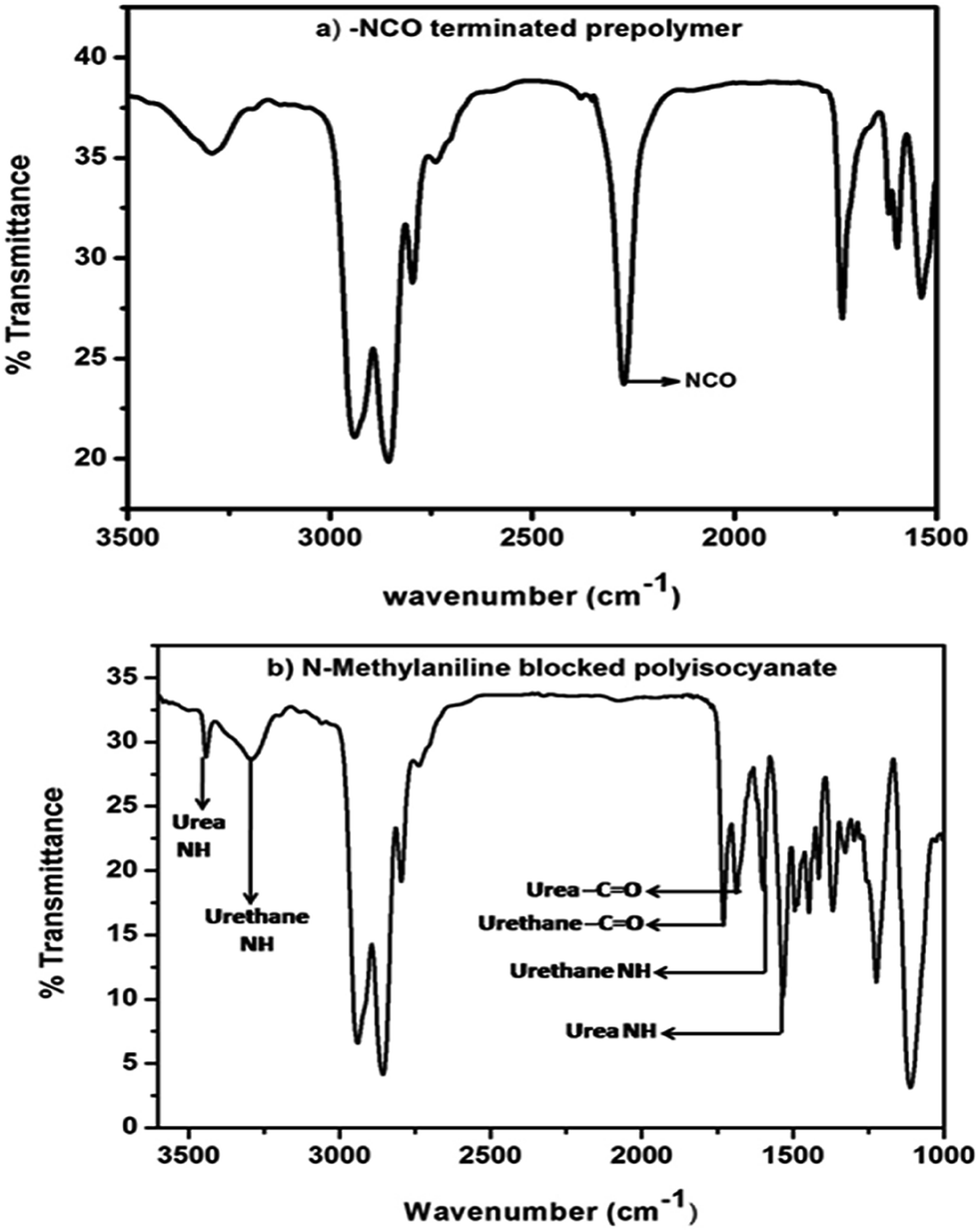
The polyisocyanate was prepared separately for each entry given in Table 1 by the reaction of two equivalents of TDI with one equivalent of poly(tetrahydrofuran)diol (Scheme 1). The reaction was monitored using a FT-IR spectrophotometer. To ensure the completion of the reaction, the –NCO content of the polyisocyanate was determined by dibutylamine method45 and the value was found to be in good agreement with the theoretically calculated value of 6.33% (w/w). The strong absorption band appeared at 2270 cm−1 range in the FT-IR spectrum (Fig. 2a) indicated the presence of terminal –NCO groups in the polyisocyanate. To block the terminal –NCO group, several substituted N-methylanilines – aromatic secondary amines – were used as blocking agents. The blocking reactions were not catalysed since secondary amines are highly reactive nucleophiles for isocyanates.23 The blocking reactions were monitored using FT-IR spectrophotometer and the reactions were stopped upon complete disappearance of –NCO absorption at 2270 cm−1 range in the FT-IR spectra; time required for 50% completion of the reactions in the IR cell are given in Table 1.
Table 1 Synthesis of N-methylaniline-blocked polyisocyanates (2–7)
| Compd no. |
Blocking agents |
Time (min) required for 50% completion of blocking reaction at 50 °C |
| 2 |
N-Methylaniline |
11 |
| 3 |
N-Methyl-o-toluidine |
80 |
| 4 |
N-Methyl-o-anisidine |
21 |
| 5 |
2-Chloro N-methylaniline |
130 (12%) |
| 6 |
4-Chloro N-methylaniline |
15 |
| 7 |
Methyl 4-(methylamino)benzoate |
180 |
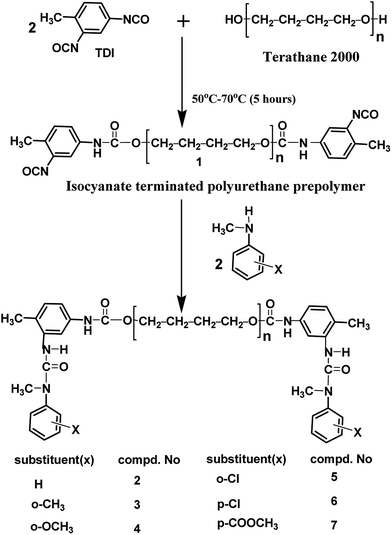 |
| | Scheme 1 Synthesis of N-methylaniline-blocked polyisocyanates (2–7). | |
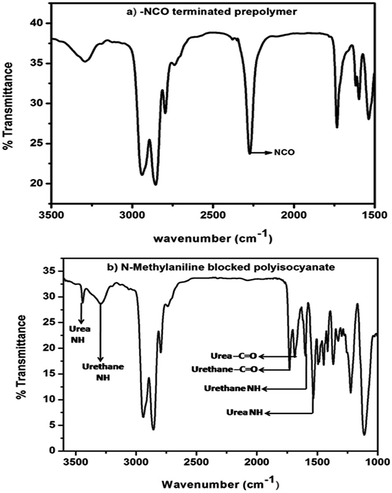 |
| | Fig. 2 FT-IR spectrum of (a) isocyanate-terminated polyurethane prepolymer and (b) N-methylaniline-blocked polyisocyanate. | |
For this work, it was decided to use N-methylaniline containing electron releasing substituents like methyl and methoxy groups and electron withdrawing substituents like chloro and ester substituents at ortho and para positions as blocking agents. Accordingly, all the N-methylanilines were used for the preparation of blocked polyisocyanates. Blocking reaction with N-methyl-p-toluidine and N-methyl-p-anisidine were found to be very fast; more than 50% of the reaction was completed within two minutes, thus the blocking kinetics of these substituted N-methylanilines were not able to follow using FT-IR spectrophotometer. On the other hand, the blocking reaction with methyl 2-(methylamino)benzoate (N-methylaniline containing methyl ester substituent at ortho position) was found to be extremely slow; no detectable changes in the intensity of –NCO peak was observed for 120 minutes at 50 °C, thus the blocking kinetics of this reaction was also not carried out. In view of these facts, deblocking kinetic data of the blocked polyisocyanates based on these three blocking agents were unable to include in this work, because both forward and reverse reaction rate constants are together required to construct double Arrhenius plots; construction of double Arrhenius plots, obtaining the equilibrium temperature thereof and comparison of forward and reverse reaction parameters of thermally reversible reactions are the objectives of this work.
The very high reactivity of N-methyl-p-toluidine and N-methyl-p-anisidine (no data given) is due to the electron donating methyl and methoxy substituents which render the nitrogen atom of N-methylaniline more basic for its easy attack on the partially positive carbon atom of –NCO group during the blocking reaction. The slow blocking reaction of N-methylaniline substituted with electron donating substituents at ortho position is due to the steric factor (Table 1). Here, when compare the reactivity of N-methyl-o-anisidine to N-methyl-o-toluidine, the former blocks the isocyanate quickly even though its substituent is more bulky compared to methyl substituent of the later. This indicates that, within these two cases, electronic effect overcomes the steric effect of the methoxy substituent. Electron withdrawing chloro and ester substituents at ortho and para position of N-methylaniline require long time, high temperatures and in some cases (2-chloro N-methylaniline and methyl 4-(methylamino)benzoate) use of excess blocking agent (10 mol%) was inevitable for the completion of the reaction. The electron withdrawing substituents decrease the electron density at the nitrogen atom and make it comparatively less basic than that of unsubstituted N-methylaniline, thus the reactivity of these blocking agents is less towards –NCO group. Extremely high reaction time and requirement of temperatures more than 75 °C for the reactions involving 2-chloro N-methylaniline indicates presence of intramolecular hydrogen bonding in these blocking agents which leads to the non-availability of the hydrogen atom for the addition reaction with the isocyanate group.
The FT-IR spectra of all the N-methylaniline blocked polyisocyanates were essentially identical and showed urea –NH stretching absorption at 3425 cm−1, urea –NH bending absorption at 1504 cm−1, urea –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching absorption at 1685 cm−1, urethane –NH stretching absorption at 3303 cm−1, urethane –NH bending absorption at 1521 cm−1 and urethane –CO stretching absorption at 1735 cm−1. The absence of absorption at 2270 cm−1 indicated that the isocyanate groups were completely blocked with N-methylaniline. Typical example spectrum is given in Fig. 2b. This confirms the formation of amine-blocked polyisocyanates without any ambiguity.
O stretching absorption at 1685 cm−1, urethane –NH stretching absorption at 3303 cm−1, urethane –NH bending absorption at 1521 cm−1 and urethane –CO stretching absorption at 1735 cm−1. The absence of absorption at 2270 cm−1 indicated that the isocyanate groups were completely blocked with N-methylaniline. Typical example spectrum is given in Fig. 2b. This confirms the formation of amine-blocked polyisocyanates without any ambiguity.
Blocking kinetics
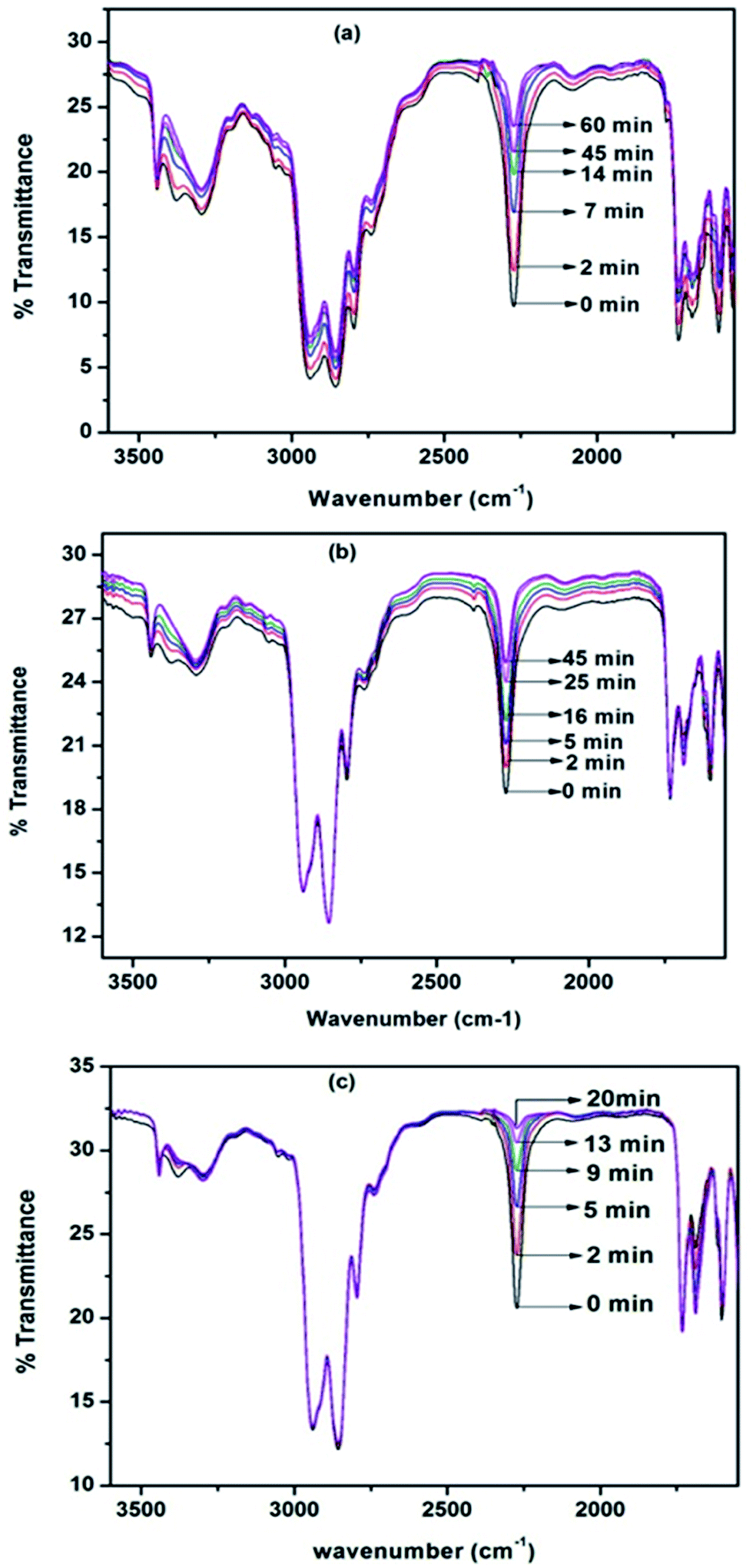
Hot stage FT-IR spectrophotometer is the best analytical tool to study the neat blocking kinetics of polyisocyanate with N-methylaniline as this tool directly measures the disappearance of the isocyanate (–NCO) functional group of the polyisocyanate during the course of the blocking reaction. With the intention of calculating the kinetic and activation parameters, isothermal experiments were carried out at three different temperatures. The distinct changes observed in the FT-IR spectrum of each blocking reaction mixture were the steady intensity decrease of the absorption peak at 2270 cm−1 corresponding to the disappearance of –NCO groups, and the steady increase at 1730 cm−1 corresponding to the formation of urea carbonyl group with respect to time. We followed the disappearance of the isocyanate absorption at 2270 cm−1 for the blocking kinetics. Typical FT-IR spectra recorded for different time intervals at three different temperatures for the blocking reaction are given in Fig. 3.
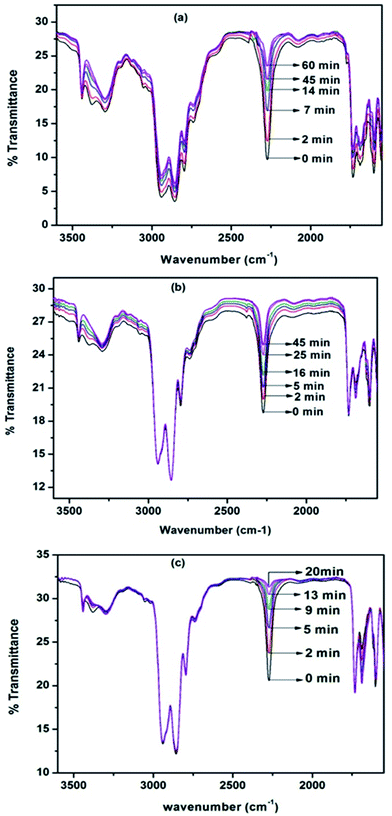 |
| | Fig. 3 FT-IR spectra recorded for different time intervals under isothermal condition for the blocking reaction of polyisocyanate with N-Methylaniline: (a) 40 °C (b) 50 °C and (c) 60 °C. | |
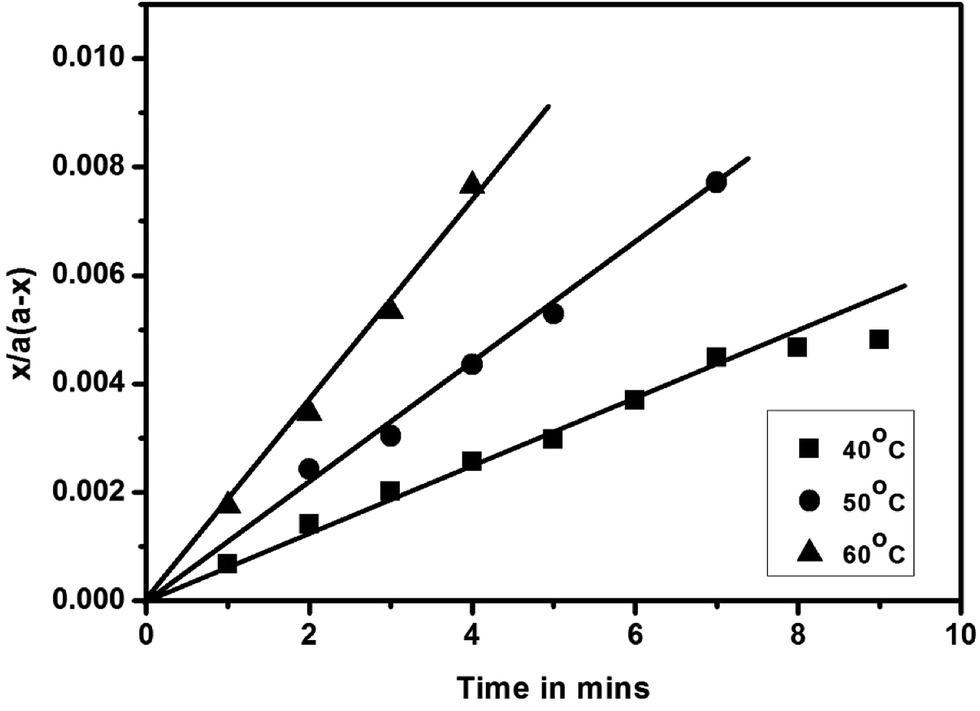
While the blocked isocyanate formation via isocyanate–phenol reaction that follow second order kinetics have been thoroughly studied,46,47 the formation of the same via isocyanate – secondary amine has not been reported yet. In this study, the plots of x/a (a − x) versus time were found to be linear, passes through the origin and confirm that the blocking reactions of polyisocyanate with a series of N-methylanilines also follow second order kinetics of the type A + B → products, in which the two reactants have the same initial concentration. Typical second order rate plots for these reactions carried out at three different temperatures are given in Fig. 4 and the second order rate constants for 50% conversion along with activation parameters for all the reactions studied are given in Table 2. The rate constants of all the substituted N-methylanilines are found low compared to unsubstituted N-methylaniline. This observation and the trend present in these results are absolutely in agreement with the preceding discussion and consistent with the time required for 50% conversion of the reaction given in Table 1. The activation energy (Ea) values of the reactions involving all the substituted N-methyanilines are found to be high compared to unsubstituted N-methylaniline except for the case of N-methyl-o-anisidine; the high Ea values are not proportional but consistent with low rate constant values. The high negative entropy of activation indicates that these blocking reactions proceed through rigid intermediate at transition state.
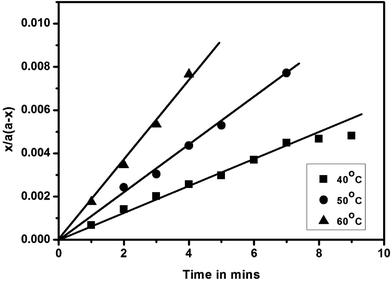 |
| | Fig. 4 Second-order kinetic plots of blocking reaction of polyisocyanate with N-methylaniline. | |
Table 2 Second-order rate constants and activation parameters for blocking reactions of polyisocyanates with N-methylaniline
| Compd no. |
Blocking agents |
Second order rate constant k × 102 (mol−1 min−1) |
Ea (kJ mol−1) |
ΔH# (kJ mol−1) |
ΔG# (kJ mol−1) |
ΔS# (J K−1 mol−1) |
| 30 °C |
40 °C |
50 °C |
60 °C |
70 °C |
| 2 |
N-Methylaniline |
|
5.74 |
10.21 |
17.38 |
|
56.22 |
53.53 |
135.7 |
−254.7 |
| 3 |
N-Methyl-o-toluidine |
|
0.92 |
2.03 |
5.19 |
|
74.73 |
72.04 |
158.5 |
−267.8 |
| 4 |
N-Methyl-o-anisidine |
3.75 |
6.43 |
9.08 |
|
|
35.99 |
33.31 |
117.2 |
−259.9 |
| 5 |
2-Chloro N-methylaniline |
|
0.05 |
0.19 |
0.46 |
|
95.48 |
92.8 |
185.6 |
−287.2 |
| 6 |
4-Chloro N-methylaniline |
3.09 |
7.43 |
9.76 |
|
|
52.26 |
49.58 |
131.6 |
−255.0 |
| 7 |
Methyl 4-(methylamino)benzoate |
|
|
0.53 |
1.54 |
1.83 |
147.7 |
144.6 |
234.6 |
−278.8 |
Deblocking temperatures of N-methylaniline-blocked polyisocyanates
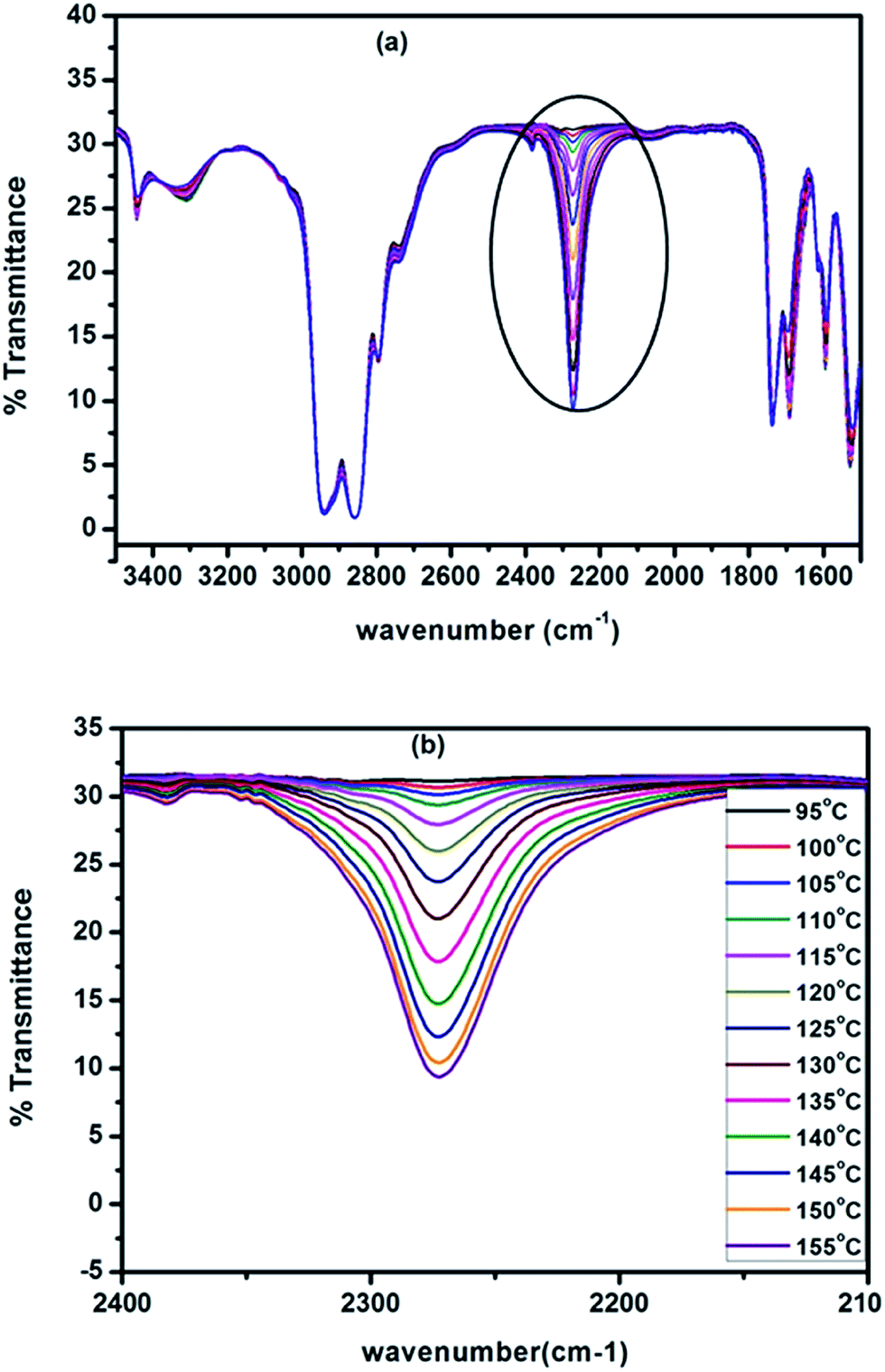
As mentioned in the preceding section, hot-stage FT-IR spectrophotometer is the best analytical tool to study the deblocking reaction of blocked isocyanates as this tool directly measures the regeneration of the –NCO functional group and the disappearance of the carbonyl group of the blocked –NCO group. All the blocked polyisocyanates were subjected to FT-IR analyses under dynamic condition for the purpose of assessment of deblocking temperatures. The spectra recorded from room temperature to 170 °C for a typical N-methylaniline-blocked polyisocyanate are given in Fig. 5, in which some spectra have been removed for the sake of clarity and the minimum deblocking temperature determined from these spectra are given in Table 3.
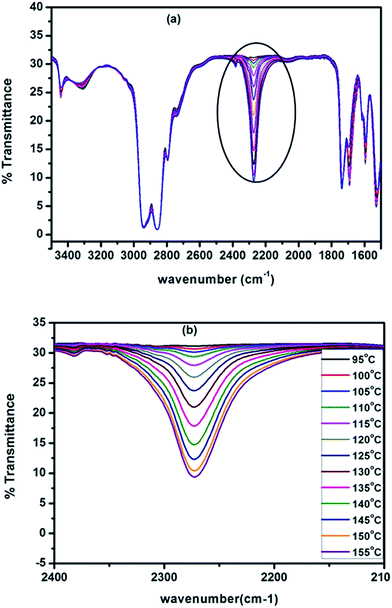 |
| | Fig. 5 FT-IR spectra of N-methylaniline-blocked polyisocyanate recorded at (a) different temperatures (b) zoomed range of isocyanate absorption region. | |
Table 3 Deblocking temperatures of N-methylaniline-blocked polyisocyanates determined using hot-stage FT-IR spectrophotometer
| Compd no. |
Blocking agents |
Deblocking temp. (°C) |
| 2 |
N-Methylaniline |
115 |
| 3 |
N-Methyl-o-toluidine |
130 |
| 4 |
N-Methyl-o-anisidine |
140 |
| 5 |
2-Chloro N-methylaniline |
125 |
| 6 |
4-Chloro N-methylaniline |
110 |
| 7 |
Methyl 4-(methylamino)benzoate |
105 |
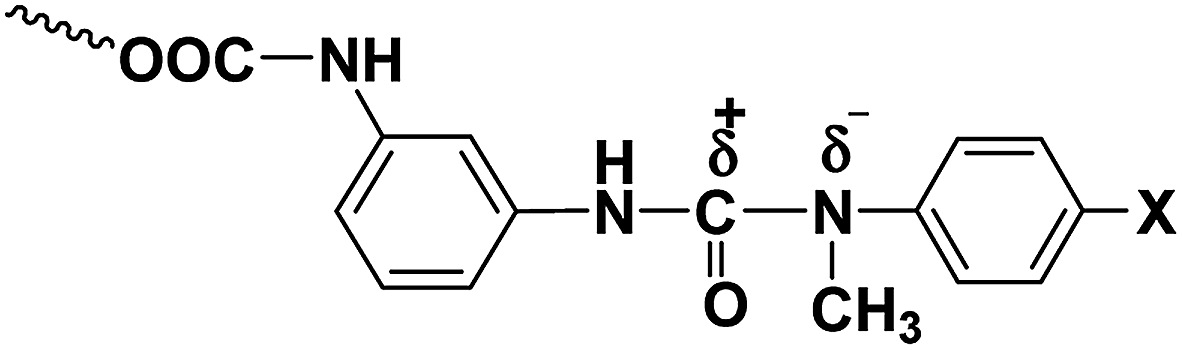
The ease with which a blocked isocyanate will dissociate into isocyanate and the blocking agent depend upon the magnitude of charge separation between the carbonyl carbon of isocyanate moiety and the nitrogen atom of the blocking agent (Fig. 6). Thus, electron-donating substituents in the blocking agent will strengthen the labile bond and electron-withdrawing substituents in the blocking agent will weaken the bond, resulting in a low deblocking temperature. In accordance with this argument, the deblocking temperature obtained was consistent with the electronic effects, i.e., the blocking agent with electron-releasing substituents, like methyl and methoxy groups, deblock at higher temperatures whereas the blocking agent with electron-withdrawing substituents, like chlorine and ester groups, deblock at lower temperatures compared to N-methylaniline (Table 3). The high deblocking temperature of 2-chloro-N-methylaniline need explanation and this is given in the forthcoming section.
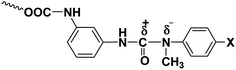 |
| | Fig. 6 Polarised structure of N-methylaniline-blocked polyisocyanate. | |
Deblocking kinetics (a comparison with the blocking reaction)
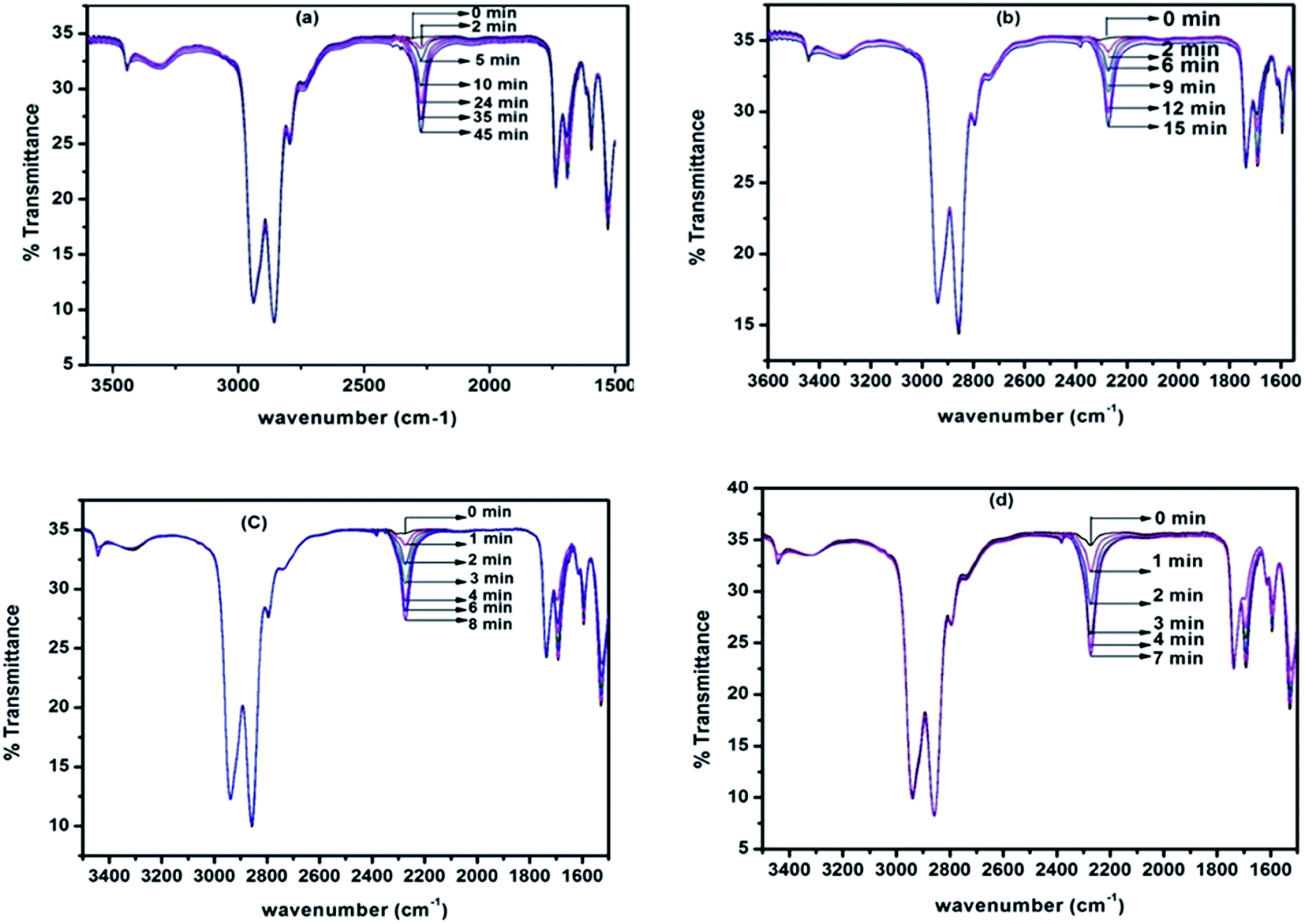
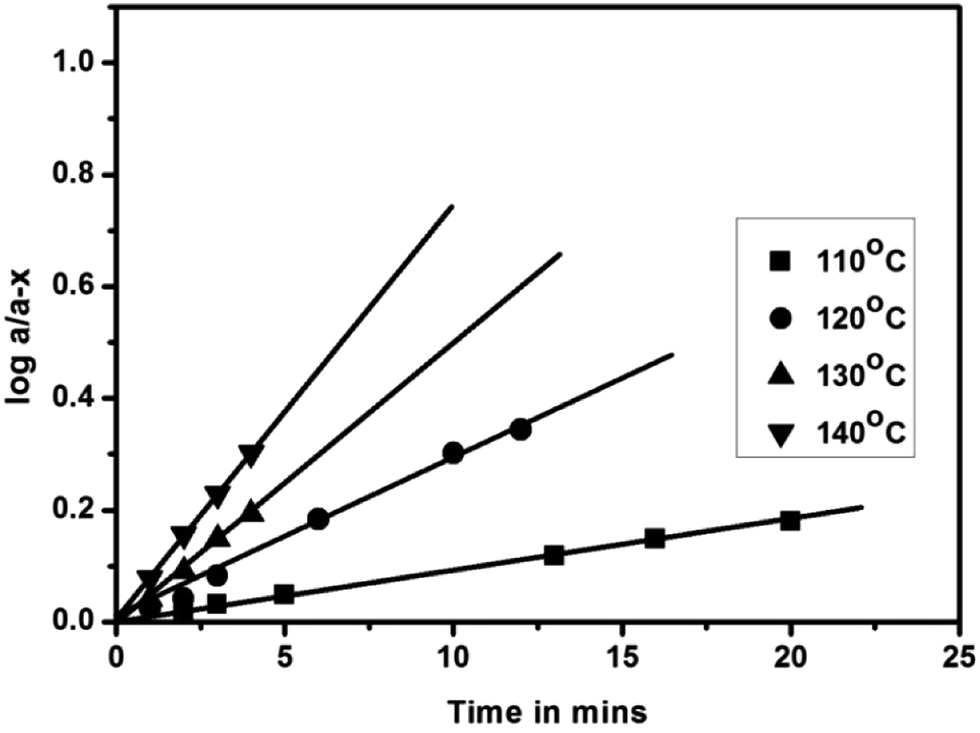
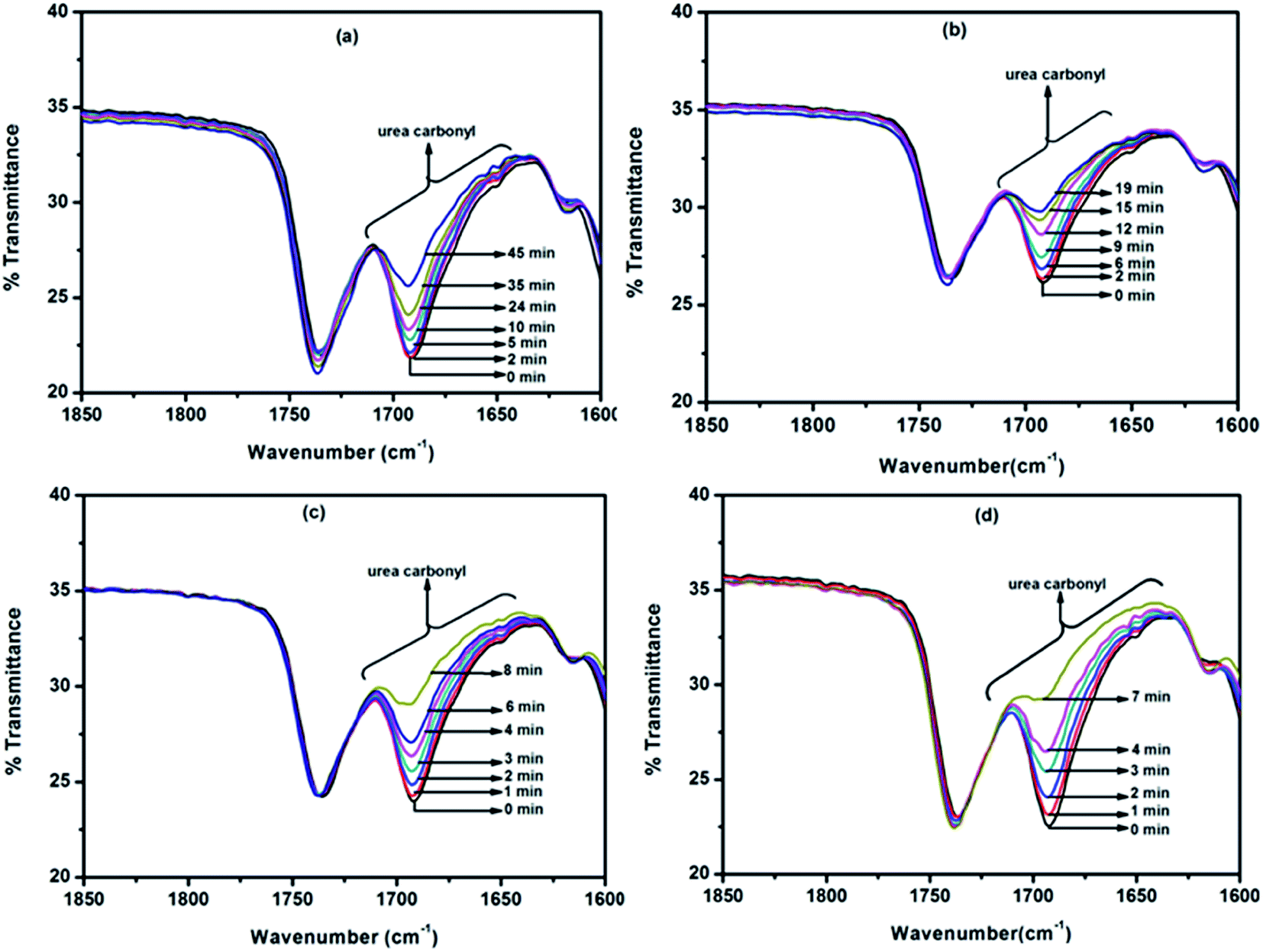
Deblocking kinetics of each N-methylaniline-blocked polyisocyanate was carried out substantially above its deblocking temperature using a hot stage FT-IR spectrophotometer. Three changes were observed in the FT-IR spectra recorded for different time intervals. The first change observed was an increase in the intensity of the –NCO absorption at 2270 cm−1 with respect to time, and then it becomes constant for some time, after which it decreased slowly due to the reaction between regenerated isocyanate and the –NH group of remaining urethane moieties leading to the formation of allophanate groups. The second change observed was due to the absorption by the urea carbonyl at 1732 cm−1 that was decreased due to the cleavage of blocked isocyanate groups. This change was convenient to follow only in the case of uncatalysed, 2-methyl, 2-methoxy and 4-chloro substituted N-methylaniline-blocked isocyanates. In the case of 2-chloro-N-methylaniline-blocked isocyanate, the urea carbonyl was not well separated from the urethane carbonyl and in the case of N-methylaniline with ester substituent the urea carbonyl was merged with substituent's carbonyl group. The third change observed was the decrease in absorption intensity of –N–H at 3300 cm−1 due to the cleavage of blocked isocyanate groups. Here, the urethane and urea N–H groups present in the blocked polyisocyanate absorb in the same region. Hence, the absorption peak of the –NCO group at 2270 cm−1 was conveniently followed for deblocking kinetics as it absorbs strongly and exclusively in that region. Typical FT-IR spectra recorded for different time intervals at different temperatures for the deblocking kinetic analysis are given in Fig. 7. The plots of loga/(a − x) versus time for all the reactions were found to be linear and confirm that the deblocking reaction of all the N-methylaniline-blocked polyisocyanates follow first order kinetics. Typical example plots drawn for four different temperatures are given in Fig. 8. The first order rate constants and activation parameters calculated for all the blocked polyisocyanates are given in Table 4.
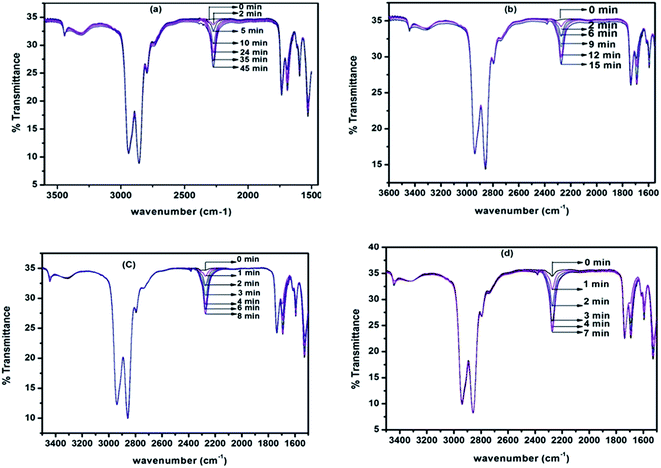 |
| | Fig. 7 FT-IR spectra recorded for different time intervals under isothermal condition for the deblocking reaction of N-methylaniline-blocked polyisocyanate: (a) 110 °C, (b) 120 °C, (c) 130 °C and (d) 140 °C. | |
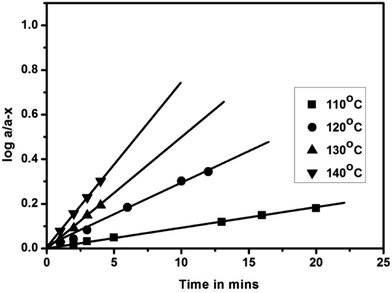 |
| | Fig. 8 First-order kinetic plots of the deblocking reaction of N-methylaniline-blocked polyisocyanate. | |
Table 4 First-order rate constants and activation parameters for deblocking reactions of N-methylaniline-blocked-polyisocyanates
| Compd no. |
Blocking agents |
First order rate constant k × 102 (min−1) |
Ea (kJ mol−1) |
ΔH# (kJ mol−1) |
ΔG# (kJ mol−1) |
ΔS# (J K−1 mol−1) |
| 100 °C |
110 °C |
120 °C |
130 °C |
140 °C |
150 °C |
160 °C |
| 2 |
N-Methylaniline |
|
2.2 |
4.7 |
10.3 |
16.6 |
|
|
89.95 |
86.6 |
173.7 |
−216.2 |
| 3 |
N-Methyl-o-toluidine |
|
|
|
2.2 |
3.6 |
5 |
6.2 |
51.74 |
48.38 |
140.6 |
−228.9 |
| 4 |
N-Methyl-o-anisidine |
|
|
|
5.3 |
9.3 |
13.2 |
19.6 |
61.61 |
58.25 |
106.9 |
−221.7 |
| 5 |
2-Chloro N-methylaniline |
|
|
3.4 |
5.3 |
9.3 |
13.6 |
|
127.5 |
124.1 |
205.6 |
−202.5 |
| 6 |
4-Chloro N-methylaniline |
|
4.1 |
8.0 |
12.0 |
23.4 |
|
|
106.3 |
102.9 |
189.5 |
−214.9 |
| 7 |
Methyl 4-(methylamino) benzoate |
2.6 |
5.8 |
9.7 |
15.2 |
|
|
|
73.06 |
69.71 |
155.5 |
−213.0 |
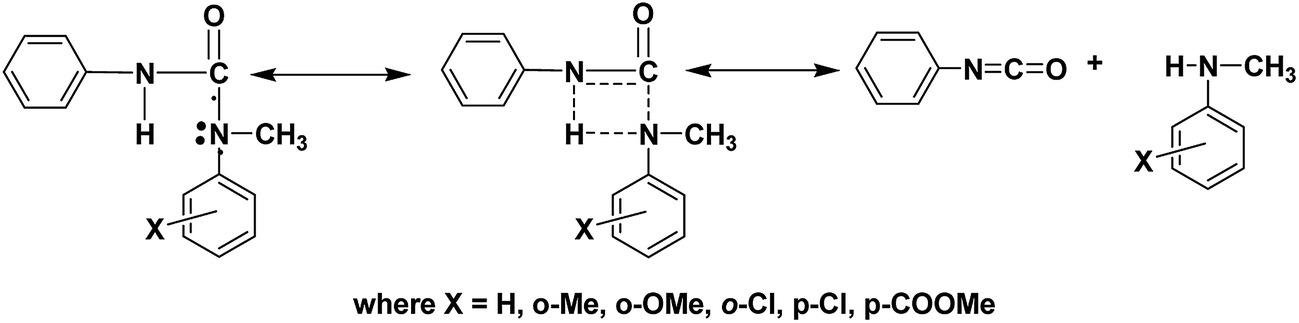
Analysis of the rate constants (k) of all the deblocking reactions studied clearly reveals that like in the blocking reactions, both electronic and steric effects determine the rate of the deblocking reaction and the results are consistent with the deblocking temperatures. In contrast to blocking reactions, no trend was observed between Ea and k values of deblocking reactions. Effect of steric constrain on the dissociation of labile bond present in the blocked isocyanate is able to realize only when comparing the rate of N-methyl-o-anisidine blocked polyisocyanate with that of N-methyl-o-toluidine blocked polyisocyanate. The high negative entropy value indicates that the deblocking occurs through the formation of rigid intermediate (Fig. 9). The formation of four membered rigid ring structure through the association of urea hydrogen atom with nitrogen atom of the blocking agent leading to auto-catalysis has been already proven using variable temperature 1H-NMR spectroscopy.23 According to the structure of the transition state proposed in the Fig. 9, it is obvious that once such structure is formed, then the labile bond present between carbonyl carbon and nitrogen of blocking agent become susceptible against the nature of the substituent present in the blocking agent and this is reflected in the deblocking kinetic results. The high deblocking temperature and low deblocking rate constant of 2-chloro-N-methylaniline blocked polyisocyanate can be explained based on the structure of transition state proposed in the Fig. 9. While all the N-methylanilines form the four centered transition state, the 2-chloro-N-methylaniline blocked isocyanate will form five membered ring structure through the association of urea hydrogen with chlorine substituent of the blocking agent. Thus, the autocatalytic activity of the blocking agent moiety is suppressed here and for the same reason, the labile bond will not be susceptible against the chlorine substituent. Hence, this blocking agent cleaves at relatively high temperature with slow rate compared to the expectation.
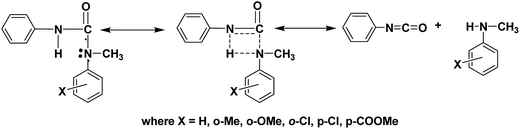 |
| | Fig. 9 Transition state proposed for deblocking of N-methylaniline-blocked isocyanate. | |
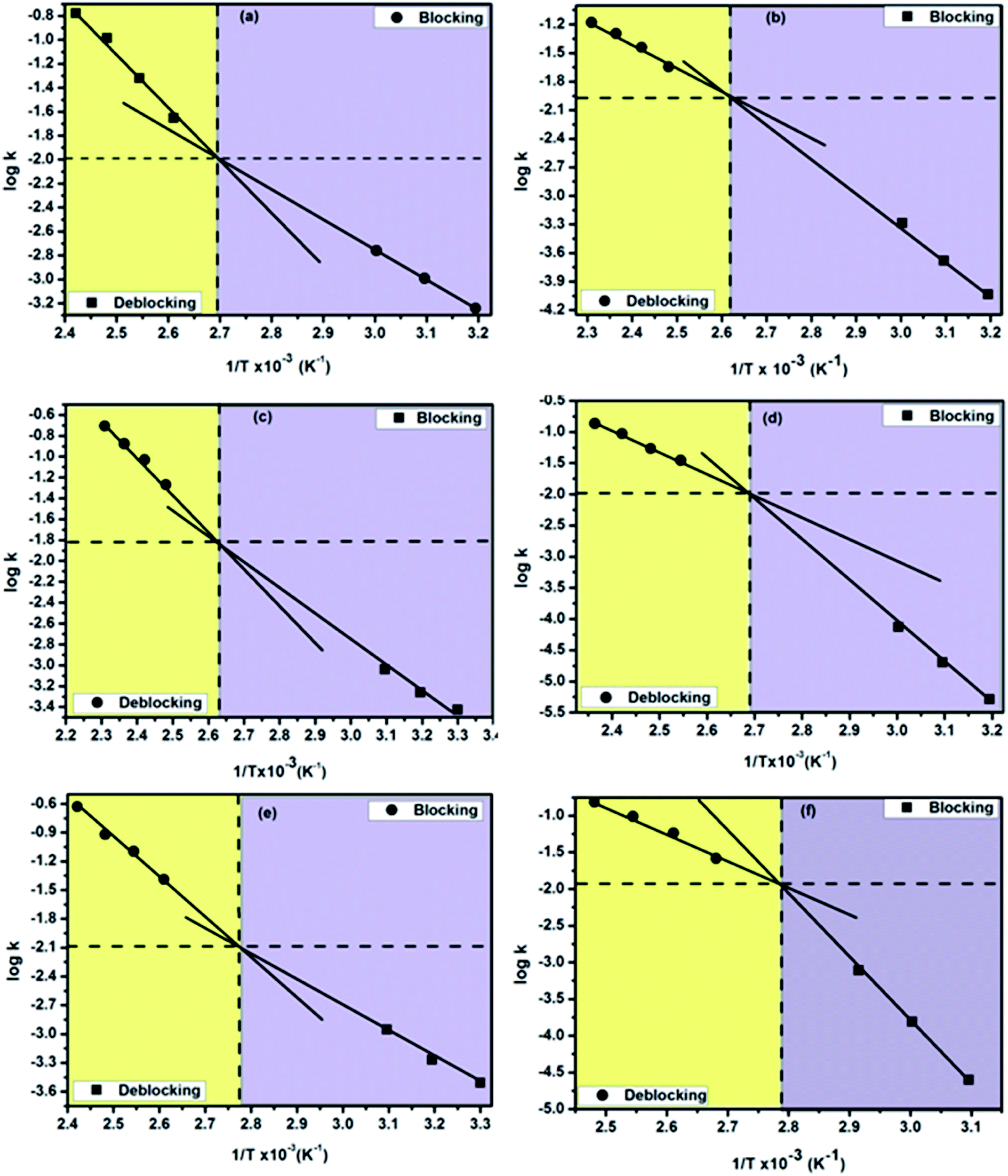
Double Arrhenius plots of thermally reversible blocking and deblocking reactions of amine-blocked polyisocyanates
Construction of double Arrhenius plots for thermally reversible blocking and deblocking reaction of a blocked isocyanate in a temperature range on which both the reaction proceed at a maximum rate is indeed a novel idea because such plots will pave the way for an insight into equilibrium state of the reaction. Very recently, we reported the outcome of such attempt for a variety of phenol-blocked polyisocyanates.1 Compared to phenols, N-methylanilines are functional group wise different and are recognized as blocking agents owing to their autocatalytic activity which could not be expected from phenolic-type blocking agents. The double Arrhenius plots of all the thermally reversible reactions studied in this work are shown in Fig. 10a–f and the data such as equilibrium temperature and equilibrium rate constants obtained from the plots are given in Table 5. It is very surprising to observe that all the Arrhenius plots of blocking and deblocking reactions of the present study found intersect each other, as a result, all the reactions showed their equilibrium temperature as a single value. This observation is interesting and new compared to phenol-blocked polyisocyanates, where among the nine mono substituted phenols studied, none of them showed distinct equilibrium temperature, instead of that all the reactions showed this temperature as a range, thereby we reported the most probable equilibrium temperature for phenol-blocked polyisocyanates. The trend observed in the equilibrium temperature given in the Table 5 is in agreement with the electronic effect of the substituents and the secondary bond force present in a blocking agent discussed for the deblocking temperature and deblocking kinetics. In principle, below or above this equilibrium temperature only the forward or reverse reaction respectively will take place. Based on the equilibrium temperatures of blocked polyisocyanates, a temperature range for their production and a temperature range for their application can be suggested, thus this parameter in general is industrially important for blocked polyisocyanates concerned. As for as equilibrium rate constants concerned, this parameter could not be related to each other, because the equilibrium temperature is different for different reaction.
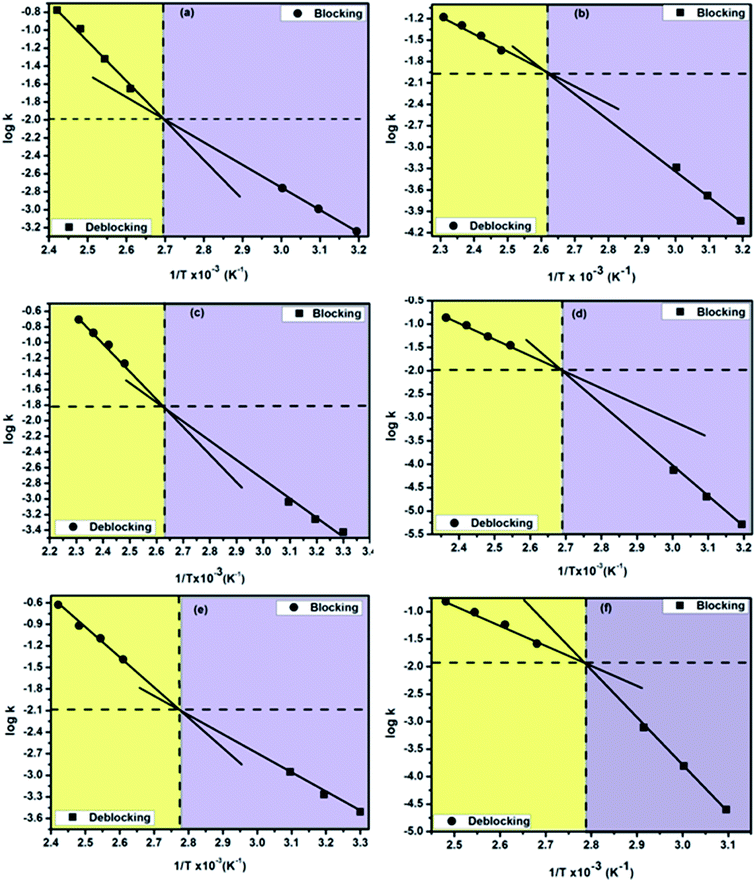 |
| | Fig. 10 Double Arrhenius plots of the forward and reverse reactions of amine-blocked polyisocyanates. Blocking agent: (a) N-methylaniline, (b) N-methyl-o-toluidine, (c) N-methyl-o-anisidine, (d) 2-chloro N-methylaniline, (e) 4-chloro N-methylaniline and (f) methyl 4-(methylamino)benzoate. | |
Table 5 Thermal equilibrium data of N-methylaniline-blocked polyisocyanates determined from the double Arrhenius plots of forward and reverse reactions
| Compd no. |
Blocking agents |
Equilibrium temperature for forward and reverse reaction (°C) |
Equilibrium rate constant k × 102 |
| 2 |
N-Methylaniline |
98 |
1.01 |
| 3 |
N-Methyl-o-toluidine |
111 |
1.09 |
| 4 |
N-Methyl-o-anisidine |
102 |
1.60 |
| 5 |
2-Chloro N-methylaniline |
100 |
0.97 |
| 6 |
4-Chloro N-methylaniline |
87 |
0.75 |
| 7 |
Methyl 4-(methylamino) benzoate |
87 |
1.15 |
To authenticate the double Arrhenius plots and the virtually single equilibrium temperature obtained thereof, the disappearance of carbonyl absorption was followed (Fig. 11) to calculate the kinetic parameters and these kinetic data were used for the construction of Arrhenius plots for all the reactions except the reaction involved 2-chloro- and methyl ester substituted N-methylanilines. The advantage of following the disappearance of carbonyl peak is that all the concentration terms appearing in the rate equation could be obtained from the same spectrum and this will give more accurate results. The pattern of the double Arrhenius plots and the equilibrium temperature obtained by this method were found absolutely similar to that obtained following regeneration of isocyanate peak for kinetic analysis. Thus, this method can be considered as authentic and direct determination of equilibrium temperature for thermally reversible reactions.
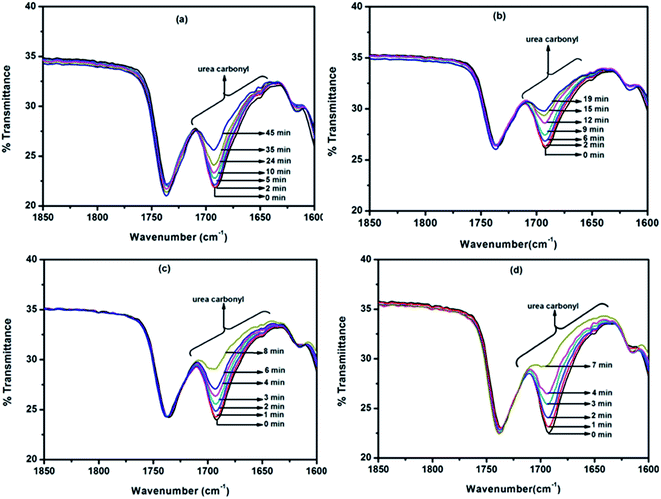 |
| | Fig. 11 FT-IR spectra recorded for different time intervals under isothermal condition for the deblocking reaction of N-methylaniline-blocked polyisocyanate: (a) 110 °C, (b) 120 °C, (c) 130 °C and (d) 140 °C. | |
Conclusion
Blocked polyisocyanates are main raw material used to produce a variety of heat curable polyurethane products. In this article we report synthesis and forward and reverse reaction parameters generated under identical neat experimental conditions for a series of N-methylaniline-blocked polyisocyanates. Using the forward and reverse reaction rate constants, double Arrhenius plots were constructed and equilibrium temperatures and equilibrium rate constants were determined directly from the plots. This method of determination of equilibrium temperature in general, can be applied for any thermally reversible reaction. Equilibrium temperature reported in this article is an important parameter in view of the production of blocked isocyanates and this temperature in combination with deblocking temperature of a blocked polyisocyanate is important in view of its application.
Acknowledgements
One of the authors (G. L.) thank the University Grants Commission, India for the award of Teacher Fellowship under Faculty Development Programme of UGC-XII Plan to carry out this work. The authors also thank CSIR for providing FT-IR with hot stage accessories under a scheme No. 02(0100)/12/EMR-II. Dt 31.10.2012.
References
- A. Sultan Nasar and S. Kalaimani, RSC Adv., 2016, 6, 76802–76812 RSC.
- Z. W. Wicks Jr, Prog. Org. Coat., 1975, 3, 73–99 CrossRef.
- Z. W. Wicks Jr, Prog. Org. Coat., 1981, 9, 3–28 CrossRef.
- D. A. Wicks and Z. W. Wicks Jr, Prog. Org. Coat., 1999, 36, 148–172 CrossRef CAS.
- D. A. Wicks and Z. W. Wicks Jr, Prog. Org. Coat., 2001, 41, 1–83 CrossRef CAS.
- D. A. Wicks and Z. W. Wicks Jr, Prog. Org. Coat., 2001, 43, 131–140 CrossRef CAS.
- E. Delebecq, J. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- S. Guo, J. He, W. Luo and F. Liu, Materials, 2016, 9, 100–110 CrossRef.
- M. S. Fedoseev, O. A. Noskova and L. F. Derzhavinskaya, Polym. Sci., Ser. D, 2016, 9, 199–204 CrossRef CAS.
- C. Lou and M. Di, J. Adhes. Sci. Technol., 2013, 27, 2340–2351 CrossRef CAS.
- A. Sultan Nasar, V. Shrinivas, T. Shanmugam and A. Raghavan, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4047–4050 CrossRef.
- T. Shen, M. Lu and L. Liang, Macromol. Res., 2012, 20, 827–834 CrossRef CAS.
- P. E. Engonga, V. Marchetti, P. Gerardin, P. Tekely and B. Loubinoux, J. Fluorine Chem., 2000, 101, 19–25 CrossRef CAS.
- I. Muramatsu, Y. Tanimoto, M. Kase and N. Okoshi, Prog. Org. Coat., 1993, 22, 279–286 CrossRef CAS.
- A. Sultan Nasar, S. N. Jaisankar, S. Subramani and G. Radhakrishnan, J. Macromol. Sci., Part A: Pure Appl. Chem., 1997, 34, 1237–1247 CrossRef.
- A. Li, G. Fan, H. Chen and Q. Zhao, Res. Chem. Intermed., 2013, 39, 3565–3577 CrossRef CAS.
- A. Wenning and F. Schmitt, Ger. Offen., DE 19 627 951, 1998.
- T. Shen, D. Zhou, L. Liang, J. Zheng, Y. Lan and M. Lu, J. Appl. Polym. Sci., 2011, 122, 748–757 CrossRef CAS.
- L. Yin, Y. Liu, Z. Ke and J. Yin, Eur. Polym. J., 2009, 45, 191–198 CrossRef CAS.
- M. Bertoldo, C. Cappelli, S. Catanorchi, V. Liuzzo and S. Bronco, Macromolecules, 2005, 38, 1385–1394 CrossRef CAS.
- L. van Ravenstein, W. Ming, R. D. van de Grampel, R. van derLinde, G. de With, T. Loontjens, P. C. Thune and J. W. Niemantsverdriet, Macromolecules, 2004, 37, 408–413 CrossRef CAS.
- J. H. Macmillan, E. R. Bertozzi and B. E. Streeter, US Pat., 4430 489, 1984.
- G. Sankar and A. Sultan Nasar, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1557–1570 CrossRef CAS.
- A. S. Nasar, S. Subramani and G. Radhakrishnan, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1815–1821 CrossRef CAS.
- G. Sankar and A. Sultan Nasar, J. Appl. Polym. Sci., 2008, 109, 1168–1176 CrossRef CAS.
- A. M. Issam and G. Shankar, Polym. Sci., Ser. B, 2011, 53, 52–56 CrossRef CAS.
- I. Ahmad, J. H. Zaidi, R. Hussain and A. Munir, Polym. Int., 2007, 56, 1521–1529 CrossRef CAS.
- H. Yeganeh and M. A. Shamekhi, Polym. Int., 2005, 54, 754–763 CrossRef CAS.
- Y. Zhang, F. Cao, H. Tan and F. Gu, Pigm. Resin Technol., 2014, 43/4, 194–200 CrossRef.
- Z. Zhou, H. Lv, X. Wang, F. Ren and W. Xu, J. Appl. Polym. Sci., 2013, 597–599 CrossRef CAS.
- Y. Zuo, J. Gu, Y. Zhang, H. Tan, P. Li and M. Di, J. Adhes. Sci. Technol., 2012, 26, 1685–1698 CAS.
- Y. Zhang, F. Gu, X. Fiang, L. Zhu and H. Tan, Pigm. Resin Technol., 2011, 40, 379–385 CrossRef CAS.
- A. Muehlebach, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 753–765 CrossRef CAS.
- S. Petrak, V. Shadurka and W. H. Binder, Prog. Org. Coat., 2009, 66, 296–305 CrossRef CAS.
- S. Subramani, Y. J. Park, Y. S. Lee and J. H. Kim, Prog. Org. Coat., 2003, 48, 71–79 CrossRef CAS.
- J. M. Lee, S. Subramani, Y. S. Lee and J. H. Kim, Macromol. Res., 2005, 13, 427–434 CrossRef CAS.
- E. Querat, L. Tighzert and J.-P. Pascault, Angew. Makromol. Chem., 1994, 219, 185–203 CrossRef CAS.
- X. Tassel, D. Barbry and L. Tighzert, Eur. Polym. J., 2000, 36, 1745–1751 CrossRef CAS.
- W. J. Wei, Z. R. Guo, Y. F. Zhang and E. L. Pan, J. Appl. Polym. Sci., 2002, 84, 1346–1352 CrossRef CAS.
- M. Vigano, M. Levi, S. Turri, M. Chiari and F. Damin, Polymer, 2007, 48, 4055–4062 CrossRef CAS.
- Z. Ranjbar, S. H. Montazeri, M. M. R. Nayini and A. Jannesari, Prog. Org. Coat., 2010, 69, 426 CrossRef CAS.
- M. Szycher, Scygher's handbook of polyurethanes, CRC Press, Taylor and Francis group, New York, 2nd edn, 2013 Search PubMed.
- E. Delebecq, J. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113(1), 80–113 CrossRef CAS PubMed.
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- D. J. David, Analytical Chemistry of the polyurethanes, Wiley-Interscience, New York, 1969, part III, vol. 16, p. 86 Search PubMed.
- H. Kothandaraman and A. Sultan Nasar, J. Indian Chem. Soc., 1992, 69, 281–282 CAS.
- H. Kothandaraman and A. Sultan Nasar, J. Indian Chem. Soc., 1992, 69, 269–270 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03734a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  * and
G. Libni
* and
G. Libni