
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation of label-free and viable circulating tumour cells (CTCs) from blood samples of cancer patients through a two-step process: negative selection-type immunomagnetic beads and spheroid cell culture-based cell isolation
Chia-Jung Liao†
a,
Chia-Hsun Hsieh†bc,
Hung-Ming Wang†b,
Wen-Pin Choua,
Tzu-Keng Chiuc,
Jyun-Huan Changa,
A.-Ching Chao*de and
Min-Hsien Wu *ab
*ab
aGraduate Institute of Biochemical and Biomedical Engineering, Chang Gung University, Taoyuan City, Taiwan, Republic of China. E-mail: mhwu@mail.cgu.edu.tw; Fax: +886-3-2118668; Tel: +886-3-2118800 ext. 3599
bDivision of Haematology/Oncology, Department of Internal Medicine, Chang Gung Memorial Hospital, Taoyuan City, Taiwan, Republic of China
cDepartment of Chemical and Materials Engineering, Chang Gung University, Taoyuan City, Taiwan, Republic of China
dDepartment of Neurology, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan, Republic of China. E-mail: achch@cc.kmu.edu.tw; Fax: +886-7-3162158; Tel: +886-7-3121101 ext. 6833
eDepartment of Neurology, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan, Republic of China
First published on 6th June 2017
Abstract
Isolation of high-purity, label-free, and viable circulating tumour cells (CTCs) from cancer patients is crucial for subsequent analyses. To address this issue, a two-step CTC isolation scheme was proposed, wherein a spheroid cell culture was used to further purify viable CTCs after conventional negative selection-based CTC isolation methods. Our results from a cancer cell line model revealed that the survival of leukocytes in spheroid cell cultures was significantly decreased with time, whereas OECM-1 cells maintained viability and proliferated. Therefore, such a cell culture operation was expected to increase cancer cell purity in the cell spheroids. This assumption was confirmed by our results, which showed that cancer cell purities were 10.6 to 80.3-fold increased after spheroid cell culture for 8 days. In the following clinical tests, CTC-related cells were observed in 6 of 13 blood samples. Furthermore, the average purity of CTC-related cells was 34.8 ± 14.0%. By utilizing a second-step spheroid cell culture operation, the purity of CTC-related cells was greatly improved when compared with that (less than 10%) achievable by conventional negative selection-based CTC isolation. Overall, this study proposed a two-step process for the isolation of high-purity, label-free, and viable CTCs.
Introduction
Cancer metastasis is a leading cause of cancer-derived death.1 Circulating tumour cells (CTCs) are cells shed from primary tumours into adjacent vasculature and subsequently present in blood circulation.2 Growing evidence has suggested that the existence of CTCs in blood circulation is associated with cancer metastasis or relapse.1,3,4 Therefore, fundamental studies on CTCs have great potential for determining the mechanisms underlying cancer metastasis, which could facilitate the development of therapeutic solutions for cancer care. Moreover, several emerging studies have proposed that CTCs act as a real-time tumour biopsy to be utilized in the selection of therapeutic regimens for each unique cancer patient.5 For this clinical utility, the responses of CTCs to anti-cancer drugs (e.g., through cell-based chemosensitivity assays) or CTC gene expression analyses can be used to guide personalized cancer chemotherapy and serve as a clinically important indicator for monitoring long-term therapeutic efficacy.6 This can provide predictive information for the adjustment of therapeutic schemes throughout the stages of cancer care.To achieve these goals, the isolation of high-purity, label-free, viable, and clinically meaningful CTCs from blood samples of cancer patients is crucial. Recent progress in cell isolation techniques has allowed for the isolation of CTCs through various strategies, which can be broadly categorized into physical and biochemical-based schemes.7 The isolation of CTCs in a blood sample based on physical differences (e.g., size8 and density9) between the CTCs and surrounding mononuclear cells is generally regarded as easy, label-free, and possible isolated viable cells but not as specific as biochemical-based methods.10,11 Biochemical techniques commonly utilize magnetic beads (e.g., the FDA-approved CellSearch system12) or specific surfaces (e.g., Isoflux,13 CTC chip,5,14 and MagSweeper15) coupled with CTC surface antigen-specific antibodies to recognize and selective capture of CTCs. In these CTC isolation schemes, the targeted surface antigens on CTCs are either tumour-specific markers or epithelial-specific markers. For the latter, the most common used biomarkers are epithelial cell adhesion molecule (EpCAM) and cytokeratins (CKs).16 These two surface antigens are expressed by cancer cells of epithelial origin and are normally absent in normal blood cells. The magnetic beads or surface-bound CTCs are then separated from the leukocyte background by a magnetic field or buffer solution flow. CTC isolations based on this strategy are usually referred to as positive selections of CTCs and are considered mainstream methods in CTC isolation.
Although the positive selection-based CTC isolation schemes have been widely shown to be effective in isolating CTCs with high cell purities (e.g., enrichment factor of CellSearch system and CTC chip are 4.0 × 104, and 7.1 × 104, respectively17–19), a few important biological issues should be addressed. First, EpCAM and CKs are not expressed in all tumours;20 thus, such cell isolation strategies may not be suitable for some types of CTCs. Second, CTCs, particularly those with metastatic natures, may undergo the so-called epithelial-to-mesenchymal transition (EMT).21 These CTCs subsequently reduce the expressions of EpCAM and CKs.20 This phenomenon could prevent positive selection-based CTC isolation schemes from isolating clinically meaningful CTCs associated with cancer metastases. Moreover, CTCs harvested via these methods are typically labeled with magnetic beads or immobilized on a surface. This could greatly hamper their applications for subsequent cell-based assays (e.g., CTCs-based chemosensitivity assays). To address these issues, a few recent studies have proposed negative selection-based strategies for CTC isolation, wherein only blood cells are targeted for depletion using standard immunomagnetic beads-based cell isolation methods.22–24 This would leave all possible and label-free CTCs in the remaining cells. For example, Balasubramanian P. et al. utilized a negative selection strategy for CTC purification from head and neck cancer patients and demonstrated that putative CTCs are multitudinous phenotypes, both biological and physical. They identified putative CTCs, negative for EpCAM but positive for CK, epidermal growth factor receptor (EGFR), vimentin, CD44, from patients.24 The negative selection methods are no prone to selection bias as positive selection strategies. However, most of these CTC isolation methods suffer from low CTC purity (e.g., enrichment factor of density gradient separation is 4.5 × 102),18,25 which could hinder the subsequent utilization of CTCs for specific applications (e.g., gene expression analyses26).
To address these technical hurdles in positive or negative selection-based CTC isolation methods, this study proposed the combination of a three-dimensional (3-D) cell culture technique and a negative selection-based CTC isolation method. With recent advances in cell culture techniques, there is growing evidence that 3-D cell culture models provide more physiologically meaningful and biomimetic culture conditions for cultured cells in comparison with conventional 2-D monolayer cell culture models.27 In general, 3-D cell cultures cultivate cells within 3-D scaffolding biomaterials or in cell aggregate spheroids.28 3-D cell techniques have been utilized in a wide variety of research areas, such as tissue engineering,29 drug testing models,28 and various life science-related studies.27 Recent reports have demonstrated that CTCs can be cultured in 3-D cell culture models (e.g., scaffolding biomaterials30 or spheroid6 based cell cultures). In our preliminary tests it was found that human leukocytes gradually die out in 3-D cell cultures within a few days. Based on these findings, a two-step CTC isolation process was proposed, wherein a 3-D cell culture was utilized to further purify CTCs after a negative selection-based CTC isolation process. One of the technical advantages of this hybrid cell isolation protocol is its ability to isolate label-free, high-purity and, most importantly, all possible CTCs in a blood sample without encountering issues related to the complexity of surface antigens on CTCs, as has been experienced by conventional positive selection-based CTC isolation methods.20 Furthermore, recent reports have indicated that most CTCs in vivo die out soon after entering blood circulation, leaving few viable CTCs. These physiologically unique, viable CTCs may be associated with subsequent cancer metastases.31 Recently, a magnetic negative depletion following by the EPISPOT assay was proposed to detect viable CTCs.32 Ramirez J. M. et al. demonstrated that concentrations of viable CTC (secretion of EpCAM and/or CK19 proteins) serve as an independent prognostic factor for metastatic breast cancer.33 Thus, another key advantageous feature of the proposed two-step CTC isolation protocol is its ability to harvest viable CTCs with physiological and clinically meaningful characteristics.
In this study, we quantified the main cell populations (i.e., erythrocytes, CD45pos leukocytes, and CD45neg peripheral blood mononuclear cells (PBMCs), which may contain all possible CTCs) in blood samples, of healthy donors and head-and-neck cancer patients after conventional negative selection-based CTC isolation methods. We further tested the feasibility of using 3-D cell culture models for CTC isolation and purification after the negative selection-based CTC isolation processes. In the feasibility tests, we evaluated the survival of leukocytes isolated from the blood samples of healthy donors and cancer patients and the survival of OECM-1 cells (a cancer cell line model) in 3-D cell culture models (e.g., agarose hydrogel and spheroid-based cell cultures). Based on these experiments, the performance of the proposed two-step CTC isolation process was evaluated. Furthermore, the utilization of such a cell isolation process for the isolation of CTCs from the blood samples of head-and-neck cancer patients was demonstrated. Our results revealed that the major cell population in cell samples subjected to negative selection-based CTC isolation was CD45pos leucocytes (85.4–90.7%), demonstrating the need for further CTC isolation and purification. Additionally, our results showed that the numbers of CD45neg PBMCs in the blood samples of cancer patients were significantly higher than those of healthy blood donors. For the 3-D cell culture CTC isolation feasibility test, our results showed that the 8 day spheroid cell culture model improved cancer cell purity by 10.6 to 80.3-fold. In clinical tests, the proposed two-step CTC isolation protocol was able to isolate viable CTCs at an improved purity (34.8 ± 14.0%) compared with CTC purity values (<10%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25) obtained through a conventional negative selection-based CTC isolation process. Overall, this study proposed a two-step process for the isolation of high-purity, label-free and viable CTCs.
25) obtained through a conventional negative selection-based CTC isolation process. Overall, this study proposed a two-step process for the isolation of high-purity, label-free and viable CTCs.
Materials and methods
Quantification of erythrocytes, CD45pos leukocytes, and CD45neg PBMCs in blood samples of healthy donors and head-and-neck cancer patients after conventional negative selection-based CTC isolation
We compared the ratio differences of the main cell populations (i.e., erythrocytes, CD45pos leukocytes, and the CD45neg PBMCs, which may contain all possible CTCs) in the blood samples of healthy donors and head-and-neck cancer patients after negative selection-based CTC isolation. This study was performed in strict accordance with the Taiwan Ministry of Health and Welfare guidelines for the care and use of cancer cell lines and human samples and was approved by the Institutional Review Board of Chang Gung Memorial Hospital at Linkou, Taiwan (approval ID: 104-7249B). Informed consent was obtained from all blood sample donors. Advanced head-and-neck cancer patients with histopathologically confirmed squamous cell carcinoma and healthy blood donors were enrolled in a single medical centre, the Chang Gung Memorial Hospital in Linkou, Taoyuan, Taiwan. All methods were carried out in accordance with relevant guidelines. Briefly, peripheral blood samples (8 ml each) were obtained from healthy blood donors (n = 6), and head-and-neck cancer patients (n = 4). The samples were kept at 4 °C and processed within 24 h. PBMCs were first isolated from a whole blood sample using a commonly used density gradient-based separation method (Ficoll-Paque Premium, 1.077 g ml−1; GE Healthcare, Piscataway, NJ).34 In the subsequent negative selection-based CTC isolation, CD45 magnetic beads (EasySep Human CD45 Depletion Kit; StemCell Technologies, Vancouver, BC, Canada) were used to deplete leukocytes from PBMCs per manufacturer instructions. The leukocyte depletion rate was evaluated by counting the numbers of PBMCs before and after depletion treatment and calculated using the following equation: depletion rate = [(PBMCsoriginal − PBMCsremaining)/PBMCsoriginal] × 100%.After leukocyte depletion, the ratio of the erythrocytes, the CD45pos leukocytes, and the CD45neg PBMCs in the remaining cell sample were quantified microscopically with the aid of immunofluorescent staining as previously described.35 Donkey anti-mouse Alexa Fluor 594 or 488 secondary antibody (1:1000 dilution; Thermo Fisher Scientific Inc. Waltham, MA), and Hoechst 33342 (5 μg ml−1, Molecular Probes, Carlsbad, CA) were used to recognize the remaining CD45pos cells and all nucleated cells, respectively. Briefly, the remaining cell samples were prepared in phosphate buffered saline (PBS; 1 × 106 cells per 100 μl). The abovementioned immunofluorescent reagents were added to the prepared cell suspension and incubated for 30 min at room temperature. The treated cells were then washed twice with PBS. Afterwards, half of each sample was observed microscopically to quantify the number of cells and the ratios of erythrocytes, CD45pos leukocytes, and CD45neg PBMCs. The CD45neg/Hoechstneg, CD45pos/Hoechstpos, and CD45neg/Hoechstpos populations were defined as erythrocytes, leukocytes, and CD45neg PBMCs, respectively. The other half of each sample was analyzed using flow cytometry (FACSAria II; BD Bioscience, San Jose, CA) to quantify the number of CD45neg PBMCs.
Feasibility evaluation of using 3-D cell culture for further CTC isolation and purification after negative selection-based CTC isolation process: cell line model
A two-step process was proposed for CTC isolation, wherein a 3-D cell culture model was utilized to further purify CTCs after a negative selection-based CTC isolation. The process was based on our initial hypothesis that leukocytes may gradually die in a 3-D cell culture model, whereas CTCs would survive. To test this hypothesis, experiments were carried out. The survival of leukocytes and OECM-1 cells (an oral cavity cancer cell line model) in the 3-D cell culture models (i.e., agarose hydrogel and spheroid-based cell cultures) were evaluated. Briefly, the PBMCs were first isolated from the blood samples of healthy donors and head-and-neck cancer patients as described earlier. The agarose hydrogel-based 3-D cell cultures were carried out using a CytoSelect Clonogenic Tumour Cell Isolation Kit (Cell Biolabs, Inc., San Diego, CA), and prepared per manufacturer instructions. Briefly, the wells of a 96-well microplate were coated with base agar matrix and incubated at 4 °C for 30 min for gelation. PBMC (2 × 104 cells per well), and OECM-1 (1 × 103 cells per well) cell suspensions were prepared and individually mixed with the pre-melted agar matrix. Subsequently, these mixes were loaded onto the base agar matrix layer of each well. After 20 min of incubation at 4 °C, 100 μl of CTC culture medium (RPMI1640 medium (Gibco Invitrogen, Carlsbad, CA) supplemented with epidermal growth factor (EGF, 10 ng ml−1; Gibco Invitrogen), basic fibroblast growth factor (bFGF, 10 ng ml−1; Gibco Invitrogen) and B-27 supplements (Gibco Invitrogen)) was added to each well.The spheroid-based cell culture model was performed based on a previously described method.36 Briefly, PBMCs (2 × 104 cells per well) and OECM-1 (1 × 103 cells per well) suspensions were prepared in CTC culture medium and seeded in the wells of a 48-well microplate pre-coated with 2% agarose. In addition to the aforementioned cell culture preparations, OECM-1 cells (5.0%) were also co-cultured with PBMCs in the two 3-D cell culture models as described earlier. To determine the cancer cell purity before and after the spheroid-based cell culture, the OECM-1 cells capable of stably expressing green fluorescence protein (GFP), referred to as OECM-1-GFP cells, were spiked into PBMCs (2 × 104 cells) at 5.0%, 1.0%, and 0.2% and cultured in the spheroid-based cell culture model. Culture media were replaced every 3 days.
PMBC survival in the two 3-D cell culture models was assayed using trypan blue staining and microscopic observations37 at days 0, 4, 8, and 12. The data is presented as survival ratio [(the number of live cells at a particular time point/the number of live cells at day 0) × 100%]. For the OECM-1 cells, the commonly used methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay38 was used to quantify viable OECM-1 cells at days 0, 2, 4, and 8. Additionally, to determine cell viability, cells were stained with calcein AM viability dye (1:1000 dilution, Invitrogen, Carlsbad, CA) and Hoechst 33342 (5 μg ml−1, Molecular Probes, Carlsbad, CA) and subsequently observed by fluorescence microscopy. For the OECM-1-GFP and PBMC co-cultures in the spheroid-based cell culture model, fluorescent images of OECM-1-GFP cells were observed at days 0 and 8. Cells at days 0, 4, and 8 were harvested and stained with DRAQ5 nuclear dye (5 μM; eBioscience, San Diego, CA) and assayed cancer cell purity by flow cytometry. Cancer cell purity (%) was calculated: (number of GFPpos cells/number of all nucleated cells) × 100%.
Demonstration of using the two-step cell isolation process for CTC isolation: clinical sample tests
To demonstrate the feasibility of using the proposed two-step cell isolation for CTC isolation, the following clinical tests were carried out. Blood samples (8 ml each; n = 13) were obtained from patients diagnosed with head-and-neck cancer. PBMCs were isolated and subjected to a negative selection-based CTC isolation as described earlier. After these, a quarter of each cell sample was analysed to quantify the EpCAMpos CTC number based on a previously described method.34 Each remaining sample was cultured in a spheroid-based cell culture model as described earlier. After 8 days of culture, cells in the cell spheroid were stained with calcein AM viability dye and other immunofluorescent dyes to label viable CTC-related cells observed by fluorescence microscopy. For immunofluorescent assays, the cultured cells were pre-treated according to previously described methods.35 Cell samples were incubated in primary antibodies for 2 h at room temperature. After washing with PBST (0.05% Triton X-100 in PBS) twice, the cells were incubated in secondary antibodies and Hoechst 33342 dye for 30 min at room temperature. Fluorescent images were then acquired using a fluorescent microscope. The antibodies used were anti-CD45-PE conjugated antibody (1:100 dilution; clone 5B1, Miltenyi Biotec, GmbH, Germany), anti-wide spectrum cytokeratin antibody (1:100 dilution; Abcam, Inc., Cambridge, MA), anti-vimentin antibody (1:500 dilution; GeneTex, San Antonio, TX), Alexa Fluor 488 conjugated goat anti-rabbit secondary antibody (1:500 dilution; Thermo Fisher Scientific), and Alexa Fluor 594 conjugated donkey anti-mouse secondary antibody (1:500 dilution; Thermo Fisher Scientific). CD45neg/CKpos/Hoechstpos and/or CD45neg/vimentinpos/Hoechstpos cells were considered CTC-related cells in the samples.
Results and discussion
Quantification of erythrocytes, CD45pos leukocytes, and CD45neg PBMCs in blood samples of healthy donors and head-and-neck cancer patients after negative selection-based CTC isolation
Growing evidence have shown that CTCs have heterogeneous biological features.39 Therefore, the isolation or identification of CTCs with single or few biomarkers (i.e., via conventional positive selection-based CTC isolation schemes) is technically insufficient.20 To address this issue, a two-step, label-free, high-purity, and physiologically meaningful CTC isolation process was presented in this study. A density-gradient separation and negative selection-type immunomagnetic beads-based method was carried out in a first step, followed by CTC purification using a 3-D cell culture technique. In this study, the capture efficiency of OECM-1 cells was evaluated by performing a cancer cell spiking test. Results showed that around 53% and 84% of the spiked OECM-1 cells were recovered by the density-gradient separation and negative depletion processes, respectively. These performances were comparable with previously reported.34,40 We quantified the differences in the main cell populations in blood samples of healthy donors and head-and-neck cancer patients after negative selection-based CTC isolation. PBMCs were isolated from whole blood samples (average PBMCs per ml blood: 1.4 ± 0.6 × 106 cells per ml; n = 10). In the subsequent immunomagnetic beads-based CTC isolation, the average leukocyte depletion rate was found to be 98.8 ± 0.9%, indicating that most of the leukocytes in a treated sample were removed. The remaining cell populations in the cell samples were quantified via microscopy (Fig. 1A) and flow cytometry (Fig. 1B and C). The results (Fig. 1A) revealed that the major cell population in the remaining cell samples was CD45pos leukocytes (average percentage for healthy blood donors and cancer patient cases were 90.7 ± 11.9% and 85.4 ± 2.4%, respectively). CD45neg PBMCs, which may contain CTCs (e.g., EpCAMpos or EpCAMneg cancer cells41,42), were detected in all blood samples (Fig. 1A). More importantly, the CD45neg PBMCs were more prevalent in the blood samples of cancer patients than in those of healthy donors (the average percentage for healthy blood donors and cancer patients were 1.9 ± 1.4% and 4.5 ± 2.7%, respectively; mean ± S.D., P = 0.27, Mann-Whitney U test). Consistent results were also reported via flow cytometry (Fig. 1B and C). These results (Fig. 1C) showed that the numbers of CD45neg PBMCs per milliliter of blood were 46 ± 23 and 196 ± 162 (mean ± S.D., P = 0.019, Mann-Whitney U test) for healthy blood donors and cancer patients, respectively. In the blood samples of healthy donors, CD45neg PBMCs were detected based on the above results. These cells could be immature blasting myeloid cells, neutrophils, and myeloid-derived suppressor cells as reported previously.42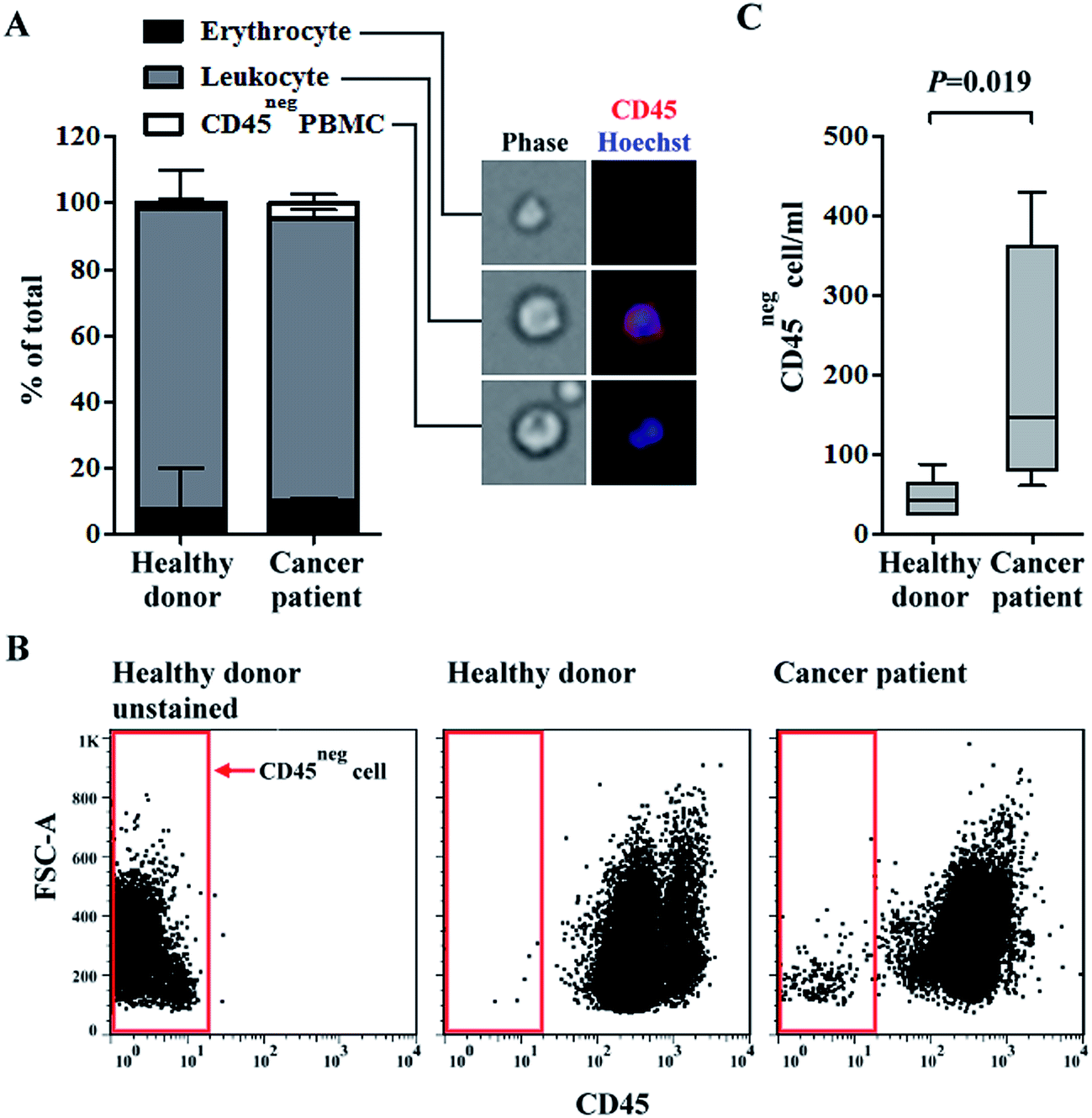 | ||
| Fig. 1 Quantification of main cell populations in blood samples after negative selection-based CTC isolation. (A) The ratios of erythrocytes, CD45pos leukocytes, and CD45neg PBMCs in blood samples of healthy donors and head-and-neck cancer patients after negative selection-based CTC isolation were quantified via immunofluorescent staining and microscopy (red: CD45; blue: Hoechst). (B) Cell samples after negative selection were analysed using flow cytometry. The FSC-A-CD45 dot plots were shown (left, unstained cells of healthy donor serves as negative control; middle, cell sample of healthy donor stained with anti-CD45 antibody and Hoechst dye; right, cell sample of cancer patient stained with anti-CD45 antibody and Hoechst dye). The red frame circumscribes the signals of CD45neg PBMCs. The quantification results (the CD45neg cells per ml blood) were shown in panel (C) and the statistic differences between groups were compared using Mann-Whitney U test. | ||
Similar results have been previously reported, wherein the numbers of EpCAMpos/CD45neg and EpCAMneg/CD45neg cells were significantly higher in blood samples of head-and-neck cancer patients than in those of healthy blood donors.43 Additionally, the number of EpCAMneg/CD45neg cells were significantly higher in blood samples of metastatic breast cancer patients, and the number of EpCAMneg/CD45neg/CKpos subpopulation cells was significantly associated with poorer overall survival.44 Based on previous studies using CellSearch detection system, CTC (EpCAMpos/CKpos/CD45neg/DAPIpos) detection rates were 12.5–40.0% in patients diagnosed with advanced head-and-neck cancer.45 This implied that more than half of advanced head-and-neck cancer cases were not properly diagnosed by conventional positive selection strategies. Taken together, the key technical advantage of exploiting a negative selection strategy for CTC isolation is its ability to harvest EpCAMneg/CD45neg cancer cells, which are generally ignored by positive selection. However, the majority of cell species (85.4 ± 2.4% for cancer patient cases) in cell samples obtained after negative selection-based CTC isolations was CD45pos leukocytes (Fig. 1A) even through 98.8% of the original leukocyte population were removed by negative selection. The low CTC purity is a major shortcoming of negative selection-based CTC isolation methods.25 Such a low CTC purity may cause problems when harvested CTCs are used for subsequent analyses (e.g., gene expression analyses26). To address this issue, a 3-D cell culture technique was proposed to further purify CTCs.
Use of 3-D cell culture for further CTC isolation and purification after negative selection-based CTC isolation process: feasibility evaluation based on cell line model
There are approximately 3–4 × 106 CTCs in blood circulation per one gram of tumour tissue.46 However, only 0.01% of CTCs survive and are capable of forming micro-metastases in distant tissues.31 Thus, only small cell populations within CTCs are considered responsible for cancer metastases.31 Analyses of these biologically and clinically meaningful CTCs would likely provide useful information on mechanisms underlying cancer metastases or on suitable therapeutic drugs.47 To achieve this goal, the isolation of CTCs in a high-purity and a biologically meaningful manner is crucial. However, this is not achievable using conventional positive or negative selection-based cell isolation schemes.19To address this issue, we utilized a 3-D cell culture method to further improve the cell purity of isolated CTCs and to selectively harvest viable CTCs after a negative selection-based CTC isolation process. Recent progress in cell culture techniques, especially the culture of CTCs from blood samples of breast,6 prostate,30 colon,48 and lung49 cancer patients, has been successfully demonstrated. In these studies, spheroid cell culture models (i.e., a 3-D cell aggregate culture) were used due to a number of advantages. First, sphere formation is a biological feature of stem cells.50 A few types of cancer cells have been recognized to possess characteristic properties of stem cells; these cells are often referred to as cancer stem cells. With similar stem cell properties, CTCs could more effectively survive the challenges of blood circulation and establish cancer metastases.51–54 Second, a recent report disclosed that CTC clusters (i.e., CTC aggregates) provide survival advantages in the bloodstream55 and thus have higher metastatic ability than single CTCs.56 These advantages of utilizing a spheroid cell culture model for CTC culture would likely preserve viable CTCs.
In this work, we assumed that CTCs in cell samples obtained after negative selection-based CTC isolation survived spheroidal cell culture, whereas leukocytes, which comprise the major cell population (Fig. 1A), gradually die in such cell culture conditions. Leukocytes are naturally mobile cells in blood circulation and thus, would unlikely adapt to a cell aggregate-based culture format. In addition to a spheroid cell culture model, we also explored the suitability of using a 3-D hydrogel-based cell culture, wherein cultured cells are encapsulated within a 3-D hydrogel matrix. Results (Fig. 2A) revealed that the survival ratio of leukocytes from healthy blood donors and cancer patients in the two 3-D cell culture models significantly decreased with culture time. For the 3-D hydrogel-based cell culture, the survival ratio of leukocytes from healthy blood donors and cancer patients at days 8 and 12 were 16.5 ± 6.0% and 7.2 ± 1.7%, and 19.7 ± 5.6% and 12.1 ± 1.4%, respectively. Regarding the spheroid cell culture, the survival ratio were 24.5 ± 3.1% and 15.3 ± 6.4%, and 23.6 ± 1.2% and 18.7 ± 1.7%. No significant differences were observed among the survival ratios of leukocytes at each time point tested (Fig. 2A). These results indicated that the origin of leukocytes and the different 3-D cell culture models did not affect leukocyte cell viability within the investigated conditions.
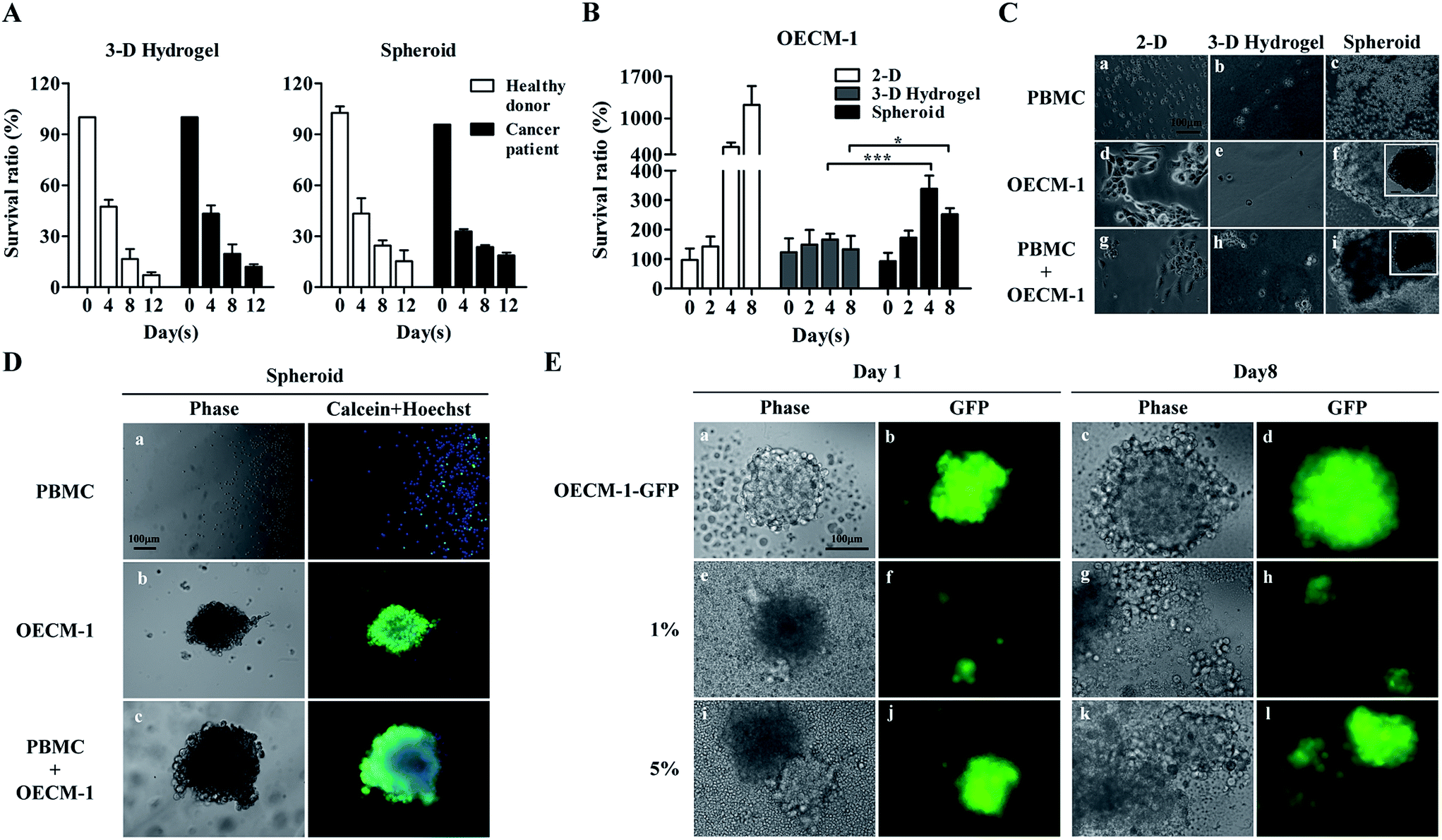 | ||
| Fig. 2 Feasibility evaluation of using a 3-D cell culture for further CTC purification. (A) Survival ratios of the leucocytes from healthy blood donors and cancer patients in 3-D hydrogel and spheroid-based culture models were assayed using trypan blue staining. (B) Survival ratios of OECM-1 cells in 2-D monolayer-, 3-D hydrogel, and spheroid-based culture models were analysed using MTT assay. (C) The light microscopy images of cells (i.e., PBMCs, OECM-1 cells, and their co-culture) cultured in the 3 cell culture models. (D) Viabilities of cells (i.e., PBMCs, OECM-1 cells, and their co-culture) in spheroid cultures were assayed via immunofluorescent staining and microscopy (green, calcein; blue, Hoechst). (E) Light and fluorescent microscopy images of pure OECM-1-GFP cells (upper row) and OECM-1-GFP cells co-culture with PBMCs (middle row, OECM-1-GFP cells spike-in at 1.0%; lower row, OECM-1-GFP cells spike-in at 5.0%) at days 1 and 8. | ||
OECM-1 cultures were also examined using two 3-D culture models. A conventional 2-D monolayer cell culture model was also prepared for comparison. As expected, OECM-1 cells maintained viability and significantly proliferated in the monolayer model (Fig. 2B). Conversely, while the OECM-1 cultures in the 3-D hydrogel-based cell culture maintained viability (survival ratio: 133.3–166.7%), they proliferated less significantly when compared with the cultures in the monolayer model. These outcomes were consistent with previous findings showing that cells cultured in monolayers proliferated more rapidly than those in 3-D environments.57 For the spheroid cell culture model (Fig. 2B), the OECM-1 cells maintained viability (survival ratio: 172.2–338.9%) and proliferated more significantly than those in the 3-D hydrogel-based culture, particularly after day 4. Fig. 2C shows light microscopy images of cells (i.e., PBMCs, OECM-1 cells, and their co-culture) cultured in the three cell culture models. The PBMCs did not adhere on 2-D surfaces (Fig. 2C(a)) or form cell spheroids in the two 3-D cell cultures (Fig. 2C(b) and (c)) after 7 days of culture. This could be attributed to their inherent non-anchoring nature. For the OECM-1 cells, they adhered and spread on 2-D surfaces (Fig. 2C(d)) as expected, maintained spherical morphologies in the 3-D hydrogel-based culture (Fig. 2C(e)), and formed spheroids in the spheroid culture model (Fig. 2C(f)). In the co-cultures of PBMCs and OECM-1 cells, the results (Fig. 2C(g)–(i)) showed that the existence of PBMCs in a cell sample did not affect the formation of cancer cell spheroids (Fig. 2C(i)). However, the results described above were based on a cancer cell line model. Reverse result was reported in previous publication58 showing that primary cancer cells proliferated less in a 2-D monolayer cell culture model than they in a 3-D cell culture model. As discussed earlier, 3-D spheroid culture model was more commonly adopted for CTC culture in recent publications.6,48 Taken together, the spheroid cell culture model was selected for CTC isolation in this study.
To determine the cell viability of the cultures in the spheroid cell culture model, cell samples were stained with fluorescent dyes and observed microscopically. The results (Fig. 2D(a)) showed that most of the PBMCs in the spheroid cell culture models were dead (calceinneg/Hoechstpos) after 7 days of culture, which correlated with the results shown in Fig. 2A. Additionally, the OECM-1 cells cultured in the spheroid model maintained high cell viability as seen in Fig. 2D(b) (calceinpos/Hoechstpos). In the co-culture microscopy images (Fig. 2D(c)) revealed that cancer cell aggregates had formed, and high cell viability was maintained. To evaluate the performance of the proposed spheroid-based cell culture for improving the CTC purity, OECM-1-GFP cells were spiked into PBMCs at the 5.0%, 1.0% and 0.2%, to mimic the CTC purity range normally achieved by conventional negative selection-based CTC isolation.25 After 8 days of co-culture of OECM-1-GFP cells and PBMCs, fluorescent images of the cell aggregate spheroids were obtained. The results (Fig. 2E) showed that the pure OECM-1-GFP cells formed cell aggregate spheroids at day 1 (Fig. 2E(a) and (b)). The spheroid slightly expanded due to cell growth by day 8 (Fig. 2E(c) and (d)). Similar results were also observed for the cell samples containing 1.0% and 5.0% OECM-1-GFP cells (Fig. 2E(e) to (l)). Based on the proliferation of cancer cells (Fig. 2B and E) and the death of leukocytes (Fig. 2A) in cell spheroids, such a cell culture operation was expected to increase cancer cell purity in the spheroid sample. To quantitatively analyse cancer cell purity, cell aggregate spheroids formed at days 0, 4, and 8 were harvested and assayed using flow cytometry. The results (Table 1) showed that cancer cell purities were 4.2–10.3 and 10.6–80.3-fold higher after spheroid cell culture for 4 and 8 days, respectively. Therefore, an 8 day spheroid cell culture was used for clinical tests to improve cancer cell purity to 49.6%, 29.5%, and 6.4% from original cancer cell purities of 4.7%, 1.1%, and 0.08%, respectively (Table 1).
Demonstration of the two-step cell isolation process for CTC isolation: clinical tests
The spheroid cell culture model was selected for CTC isolation based on the results shown in Fig. 2A–E. In the following clinical tests, 13 blood samples obtained from patients diagnosed with head-and-neck cancer were first subjected to negative selection-based CTC isolation processes followed by EpCAMpos cell enumeration using a published protocol.34 Finally, the treated cell samples were cultured in the spheroid culture model. Immunofluorescent staining and fluorescent microscopy were used to determine whether a cell spheroid sample contained CTC-related cells. If positive, the CTC culture was considered a cultivable case. From microscopy, the CD45neg/CKpos/Hoechstpos and/or CD45neg/vimentinpos/Hoechstpos cells were considered CTC-related cells. CKs are surface antigens expressed by epithelial cancer cells, and are normally absent in normal blood cells.16 Vimentin is the intermediate filament cytoskeleton and which is expressed by the cancer cells that have undergone the epithelial-to-mesenchymal transition (EMT).59 For the later, cancer patients with vimentinpos CTCs showed poor prognoses than those without vimentinpos CTCs.60,61 Based on these two surface markers, all possible CTCs in a cell sample can be observed. As shown in our results (Fig. 3A), cell spheroids were formed after 8 days of culture, wherein most of the viable cells were CD45neg/Hoechstpos PBMCs. Furthermore, in the immunofluorescent images (Fig. 3B), some cells in the cell spheroids were CD45neg/CKpos/Hoechstpos and/or CD45neg/vimentinpos/Hoechstpos cells, indicating the existence of CTC-related cells. Due to the equipment limitation, the CD45neg/CKpos and CD45neg/vimentinpos cells were unable to be separately identified in this work. However, clinical significances of vimentinpos CTCs have been demonstrated recently,60,61 identification of these CTCs in cultures would be a valuable issue to explore. | ||
| Fig. 3 Using two-step cell isolation process for CTC isolation from blood samples of cancer patients. (A) Light and fluorescent microscopy images of cells harvested from spheroid cultures (red, CD45; green, calcein; blue, Hoechst). Several viable CD45neg PBMCs were noticed in the cell spheroid (the green dots). (B) Microscopy images (upper row, cell spheroid sample 1; the lower row, cell spheroid sample 2) of CTC-related cells (CD45neg/CKpos/Hoechstpos and/or CD45neg/vimentinpos/Hoechstpos cells) found in spheroid cultures (red: CD45; green: CK or vimentin; blue: Hoechst). The white arrows pointed out the CTC-related cells. | ||
Table 2 shows the results of the clinical tests. CTC-related cells were observed in 6 of 13 blood samples. Moreover, the number of EpCAMpos cells was significantly correlated with successful CTC culture, with a P value of 0.045 (Fisher's exact test), i.e., blood samples with higher numbers of EpCAMpos cells contributed to successful CTC culture. Based on these results, conventional EpCAMpos cell enumeration would be required to determine the suitability of using the proposed two-step process for CTC isolation. In this study, EpCAMpos CTCs were detected in all tested samples (except for no. 5 sample, we failed to acquire the enumeration data). However, CTC-related cells were only observed in 6 of 13 cell culture samples (Table 2). The phenomenon could be explained by the fact that most CTCs could die soon after entering into bloodstream and only a small portion of them could survive in such condition.31 This could again point out the key advantage of utilizing an additional in vitro cell culture operation is its ability to further purify the live and clinically meaningful cancer cells. Within the boundaries of our experimental conditions, other pathological characteristics, such as lymph node involvement, tumour grade, and distant metastases, did not seem to correlate with the successful CTC culture (Table 2). Furthermore, the average purity of CTC-related cells (CTC-related cells/nucleated cells × 100%) in the tested samples were 34.8 ± 14.0%. By utilizing a second-step spheroid cell culture, the purity of CTC-related cells was greatly improved when compared with the CTC purity range (less than 10%) achievable by conventional negative selection-based CTC isolation processes. To our knowledge, only one study reporting successful short term culture of head-and-neck CTCs was published last year.62 Arutha Kulasinghe, et al. demonstrated that blood samples with higher number of EpCAMpos CTCs could lead to higher successful rate of culture. The consistent results were also demonstrated in this study. Nevertheless, some different findings were also found in these two studies. First, after 8 days of spheroid cell culture, some CD45pos leukocytes were still fund in cultures in our study. However, this phenomenon was not observed in the work done by Arutha Kulasinghe, et al. further investigations would be required to compare the differences of culture conditions used in these two studies and their influence on the survival of CD45pos leukocytes. Second, comparing to the works published by Arutha Kulasinghe, et al. (CK and EGFR were used to identify CTCs), CK and vimentin were used in our study to comprehensively identify the epithelial and mesenchymal type CTC-related cells. Third, in the study done by Arutha Kulasinghe, et al., the CTCs were successfully cultured in 7/25 (28%) samples. Among them, 3 cases (12%) were cultured using 3-D cell culture model. In our study, the CTC-related cells (CKpos and/or vimentinpos) were successfully cultured in 6/13 (46%) samples in 3-D cell culture model.
| No. | Tumor site | Tumor stage | CTCsa/ml | Cultivable CTC-related cellsb |
|---|---|---|---|---|
| a The number of EpCAMpos cell per ml blood.b CTC-related cell = CD45neg/CKpos/Hoechstpos and/or CD45neg/vimentinpos/Hoechstpos cell. | ||||
| 1 | Paranasal sinus | T4bN1M1 | 140 | Yes |
| 2 | Hopypharynx | T4bN2bM0 | 5 | No |
| 3 | Hopypharynx | T4aN2bM0 | 42 | No |
| 4 | Larynx | T4aN2bM0 | 48 | No |
| 5 | Oral cavity | T2N2bM0 | Yes | |
| 6 | Oropharynx | T4aN2bM0 | 94 | Yes |
| 7 | Oropharynx | T2N2bM0 | 37 | No |
| 8 | Oropharynx | T4bN1M0 | 18 | No |
| 9 | Oral cavity | T4bN0M0 | 15 | Yes |
| 10 | Oral cavity | T4bN0M0 | 47 | No |
| 11 | Oral cavity | T4bN2cM1 | 57 | No |
| 12 | Hopypharynx | T4aN2cM0 | 129 | Yes |
| 13 | Hopypharynx | T3N2bM0 | 30 | Yes |
Conclusion
In this study, we proposed a two-step CTC isolation and purification scheme. A 3-D cell culture model was used to further purify viable, label-free, and high purity CTCs after conventional negative selection-based isolation process. Our results revealed that the majority cell species in cell sample obtained after negative selection was CD45pos leukocytes (85.4–90.7%) even though 98.8% of the original leukocytes were removed by negative depletion processes. Therefore, strategies to further purify CTC are required. Additionally, our results showed that the CD45neg PBMCs were more prevalent in the blood samples of cancer patients than in those of healthy donors. These results highlight the key technical advantage of exploiting a negative selection strategy for CTC isolation is its ability to harvest EpCAMneg/CD45neg CTCs, which are generally ignored by positive selection. Based on the proliferation of cancer cells and the death of leukocytes in cell spheroids, we proposed to utilize the spheroid-based cell culture to increase cancer cell purity in the cell sample after negative selection. Our results demonstrated that such a cell culture operation could improve the purities of cancer cell by 10.6 to 80.3-fold after 8 days of culture. In the following clinical tests, cultivable CTC-related cells were observed in approximately half of samples (6/13) after 8 days of culture. The average purity of CTC-related cells was 34.8 ± 14.0%, which was greatly improved when compared with those achievable by conventional negative selection-based CTC isolation methods. Overall, this study proposed a two-step process, negative selection-based CTC isolation followed by 3-D spheroid culture, for the isolation of high-purity, label-free, and viable CTCs.Acknowledgements
This work was sponsored by the Ministry of Science and Technology, R.O.C. (MOST: 104-2628-E-182-002-MY3, 105-2314-B-182A-030 and MOST 104-2314-B-182-031-MY3), and Chang Gung Memorial Hospital (CMRPD2E0011-13, CMRPG3F0131, CMRPG3G0591-3, CMRPG3E0512, and CMRPG3E1633). The authors would like to thank for the technical supports from the Core Instrument Center, Chang Gung University, Taoyuan, Taiwan.References
- P. Mehlen and A. Puisieux, Nat. Rev. Cancer, 2006, 6, 449–458 CrossRef CAS PubMed.
- M. G. Krebs, R. L. Metcalf, L. Carter, G. Brady, F. H. Blackhall and C. Dive, Nat. Rev. Clin. Oncol., 2014, 11, 129–144 CrossRef CAS PubMed.
- M. G. Krebs, J. M. Hou, T. H. Ward, F. H. Blackhall and C. Dive, Ther. Adv. Med. Oncol., 2010, 2, 351–365 CrossRef PubMed.
- A. Toss, Z. Mu, S. Fernandez and M. Cristofanilli, Ann. Transl. Med., 2014, 2, 108 Search PubMed.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- M. Yu, A. Bardia, N. Aceto, F. Bersani, M. W. Madden, M. C. Donaldson, R. Desai, H. Zhu, V. Comaills, Z. Zheng, B. S. Wittner, P. Stojanov, E. Brachtel, D. Sgroi, R. Kapur, T. Shioda, D. T. Ting, S. Ramaswamy, G. Getz, A. J. Iafrate, C. Benes, M. Toner, S. Maheswaran and D. A. Haber, Science, 2014, 345, 216–220 CrossRef CAS PubMed.
- S. A. Joosse, T. M. Gorges and K. Pantel, EMBO Mol. Med., 2015, 7, 1–11 CrossRef CAS PubMed.
- G. Vona, L. Estepa, C. Beroud, D. Damotte, F. Capron, B. Nalpas, A. Mineur, D. Franco, B. Lacour, S. Pol, C. Brechot and P. Paterlini-Brechot, Hepatology, 2004, 39, 792–797 CrossRef PubMed.
- D. Marrinucci, K. Bethel, A. Kolatkar, M. S. Luttgen, M. Malchiodi, F. Baehring, K. Voigt, D. Lazar, J. Nieva, L. Bazhenova, A. H. Ko, W. M. Korn, E. Schram, M. Coward, X. Yang, T. Metzner, R. Lamy, M. Honnatti, C. Yoshioka, J. Kunken, Y. Petrova, D. Sok, D. Nelson and P. Kuhn, Phys. Biol., 2012, 9, 016003 CrossRef PubMed.
- M. Yu, S. Stott, M. Toner, S. Maheswaran and D. A. Haber, J. Cell Biol., 2011, 192, 373–382 CrossRef CAS PubMed.
- J. Zhang, K. Chen and Z. H. Fan, Adv. Clin. Chem., 2016, 75, 1–31 Search PubMed.
- W. J. Allard, J. Matera, M. C. Miller, M. Repollet, M. C. Connelly, C. Rao, A. G. Tibbe, J. W. Uhr and L. W. Terstappen, Clin. Cancer Res., 2004, 10, 6897–6904 CrossRef PubMed.
- W. Harb, A. Fan, T. Tran, D. C. Danila, D. Keys, M. Schwartz and C. Ionescu-Zanetti, Transl. Oncol., 2013, 6, 528–538 CrossRef PubMed.
- S. L. Stott, C. H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd Jr, A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS PubMed.
- A. H. Talasaz, A. A. Powell, D. E. Huber, J. G. Berbee, K. H. Roh, W. Yu, W. Xiao, M. M. Davis, R. F. Pease, M. N. Mindrinos, S. S. Jeffrey and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3970–3975 CrossRef CAS PubMed.
- G. Deng, M. Herrler, D. Burgess, E. Manna, D. Krag and J. F. Burke, Breast Cancer Res., 2008, 10, R69 CrossRef PubMed.
- B. Hong and Y. Zu, Theranostics, 2013, 3, 377–394 CrossRef CAS PubMed.
- R. L. Eifler, J. Lind, D. Falkenhagen, V. Weber, M. B. Fischer and R. Zeillinger, Cytometry, Part B, 2011, 80, 100–111 CrossRef PubMed.
- M. M. Ferreira, V. C. Ramani and S. S. Jeffrey, Mol. Oncol., 2016, 10, 374–394 CrossRef CAS PubMed.
- C. Raimondi, C. Nicolazzo and A. Gradilone, Chin. J. Cancer Res., 2015, 27, 461–470 CAS.
- J. P. Thiery, Nat. Rev. Cancer, 2002, 2, 442–454 CrossRef CAS PubMed.
- L. Dieguez, M. A. Winter, K. J. Pocock, K. E. Bremmell and B. Thierry, Analyst, 2015, 140, 3565–3572 RSC.
- Y. Lu, H. Liang, T. Yu, J. Xie, S. Chen, H. Dong, P. J. Sinko, S. Lian, J. Xu, J. Wang, S. Yu, J. Shao, B. Yuan, L. Wang and L. Jia, Cancer, 2015, 121, 3036–3045 CrossRef CAS PubMed.
- P. Balasubramanian, J. C. Lang, K. R. Jatana, B. Miller, E. Ozer, M. Old, D. E. Schuller, A. Agrawal, T. N. Teknos, T. A. Summers Jr, M. B. Lustberg, M. Zborowski and J. J. Chalmers, PLoS One, 2012, 7, e42048 CAS.
- S. B. Huang, M. H. Wu, Y. H. Lin, C. H. Hsieh, C. L. Yang, H. C. Lin, C. P. Tseng and G. B. Lee, Lab Chip, 2013, 13, 1371–1383 RSC.
- T. K. Chiu, W. P. Chou, S. B. Huang, H. M. Wang, Y. C. Lin, C. H. Hsieh and M. H. Wu, Sci. Rep., 2016, 6, 32851 CrossRef CAS PubMed.
- D. Antoni, H. Burckel, E. Josset and G. Noel, Int. J. Mol. Sci., 2015, 16, 5517–5527 CrossRef CAS PubMed.
- R. Edmondson, J. J. Broglie, A. F. Adcock and L. Yang, Assay Drug Dev. Technol., 2014, 12, 207–218 CrossRef CAS PubMed.
- E. Carletti, A. Motta and C. Migliaresi, Methods Mol. Biol., 2011, 695, 17–39 CAS.
- D. Gao, I. Vela, A. Sboner, P. J. Iaquinta, W. R. Karthaus, A. Gopalan, C. Dowling, J. N. Wanjala, E. A. Undvall, V. K. Arora, J. Wongvipat, M. Kossai, S. Ramazanoglu, L. P. Barboza, W. Di, Z. Cao, Q. F. Zhang, I. Sirota, L. Ran, T. Y. MacDonald, H. Beltran, J. M. Mosquera, K. A. Touijer, P. T. Scardino, V. P. Laudone, K. R. Curtis, D. E. Rathkopf, M. J. Morris, D. C. Danila, S. F. Slovin, S. B. Solomon, J. A. Eastham, P. Chi, B. Carver, M. A. Rubin, H. I. Scher, H. Clevers, C. L. Sawyers and Y. Chen, Cell, 2014, 159, 176–187 CrossRef CAS PubMed.
- K. J. Luzzi, I. C. MacDonald, E. E. Schmidt, N. Kerkvliet, V. L. Morris, A. F. Chambers and A. C. Groom, Am. J. Pathol., 1998, 153, 865–873 CrossRef CAS PubMed.
- C. Alix-Panabieres, J. P. Brouillet, M. Fabbro, H. Yssel, T. Rousset, T. Maudelonde, G. Choquet-Kastylevsky and J. P. Vendrell, J. Immunol. Methods, 2005, 299, 177–188 CrossRef CAS PubMed.
- J. M. Ramirez, T. Fehm, M. Orsini, L. Cayrefourcq, T. Maudelonde, K. Pantel and C. Alix-Panabieres, Clin. Chem., 2014, 60, 214–221 CAS.
- P. J. Su, M. H. Wu, H. M. Wang, C. L. Lee, W. K. Huang, C. E. Wu, H. K. Chang, Y. K. Chao, C. K. Tseng, T. K. Chiu, N. M. Lin, S. R. Ye, J. Y. Lee and C. H. Hsieh, Sci. Rep., 2016, 6, 31423 CrossRef CAS PubMed.
- J. He, E. Sun, M. V. Bujny, D. Kim, M. W. Davidson and X. Zhuang, PLoS Pathog., 2013, 9, e1003701 Search PubMed.
- S. F. Chen, Y. C. Chang, S. Nieh, C. L. Liu, C. Y. Yang and Y. S. Lin, PLoS One, 2012, 7, e31864 CAS.
- S. Sjostedt and E. Bezak, Acta Oncol., 2012, 51, 1086–1094 CrossRef PubMed.
- D. Grebenova, H. Cajthamlova, J. Bartosova, J. Marinov, H. Klamova, O. Fuchs and Z. Hrkal, J. Photochem. Photobiol., B, 1998, 47, 74–81 CrossRef CAS.
- X. Guan, F. Ma, S. Liu, S. Wu, R. Xiao, L. Yuan, X. Sun, Z. Yi, H. Yang and B. Xu, Oncotarget, 2016, 7, 65993–66002 Search PubMed.
- W. He, S. A. Kularatne, K. R. Kalli, F. G. Prendergast, R. J. Amato, G. G. Klee, L. C. Hartmann and P. S. Low, Int. J. Cancer, 2008, 123, 1968–1973 CrossRef CAS PubMed.
- S. D. Mikolajczyk, L. S. Millar, P. Tsinberg, S. M. Coutts, M. Zomorrodi, T. Pham, F. Z. Bischoff and T. J. Pircher, J. Oncol., 2011, 2011, 252361 Search PubMed.
- J. L. Schehr, Z. D. Schultz, J. W. Warrick, D. J. Guckenberger, H. M. Pezzi, J. M. Sperger, E. Heninger, A. Saeed, T. Leal, K. Mattox, A. M. Traynor, T. C. Campbell, S. M. Berry, D. J. Beebe and J. M. Lang, PLoS One, 2016, 11, e0159397 Search PubMed.
- H. C. Lin, H. C. Hsu, C. H. Hsieh, H. M. Wang, C. Y. Huang, M. H. Wu and C. P. Tseng, Clin. Chim. Acta, 2013, 419, 77–84 CrossRef CAS PubMed.
- M. B. Lustberg, P. Balasubramanian, B. Miller, A. Garcia-Villa, C. Deighan, Y. Wu, S. Carothers, M. Berger, B. Ramaswamy, E. R. Macrae, R. Wesolowski, R. M. Layman, E. Mrozek, X. Pan, T. A. Summers, C. L. Shapiro and J. J. Chalmers, Breast Cancer Res., 2014, 16, R23 CrossRef PubMed.
- A. Kulasinghe, C. Perry, L. Jovanovic, C. Nelson and C. Punyadeera, Int. J. Cancer, 2015, 136, 2515–2523 CrossRef CAS PubMed.
- T. P. Butler and P. M. Gullino, Cancer Res., 1975, 35, 512–516 CAS.
- J. Mateo, M. Gerlinger, D. N. Rodrigues and J. S. de Bono, Genome Biol., 2014, 15, 448 CrossRef PubMed.
- L. Cayrefourcq, T. Mazard, S. Joosse, J. Solassol, J. Ramos, E. Assenat, U. Schumacher, V. Costes, T. Maudelonde, K. Pantel and C. Alix-Panabieres, Cancer Res., 2015, 75, 892–901 CrossRef CAS PubMed.
- G. Hamilton, O. Burghuber and R. Zeillinger, Lung, 2015, 193, 451–452 CrossRef PubMed.
- E. Pastrana, V. Silva-Vargas and F. Doetsch, Cell Stem Cell, 2011, 8, 486–498 CrossRef CAS PubMed.
- B. Aktas, M. Tewes, T. Fehm, S. Hauch, R. Kimmig and S. Kasimir-Bauer, Breast Cancer Res., 2009, 11, R46 CrossRef PubMed.
- P. A. Theodoropoulos, H. Polioudaki, S. Agelaki, G. Kallergi, Z. Saridaki, D. Mavroudis and V. Georgoulias, Cancer Lett., 2010, 288, 99–106 CrossRef CAS PubMed.
- I. Baccelli, A. Schneeweiss, S. Riethdorf, A. Stenzinger, A. Schillert, V. Vogel, C. Klein, M. Saini, T. Bauerle, M. Wallwiener, T. Holland-Letz, T. Hofner, M. Sprick, M. Scharpff, F. Marme, H. P. Sinn, K. Pantel, W. Weichert and A. Trumpp, Nat. Biotechnol., 2013, 31, 539–544 CrossRef CAS PubMed.
- C. L. Hodgkinson, C. J. Morrow, Y. Li, R. L. Metcalf, D. G. Rothwell, F. Trapani, R. Polanski, D. J. Burt, K. L. Simpson, K. Morris, S. D. Pepper, D. Nonaka, A. Greystoke, P. Kelly, B. Bola, M. G. Krebs, J. Antonello, M. Ayub, S. Faulkner, L. Priest, L. Carter, C. Tate, C. J. Miller, F. Blackhall, G. Brady and C. Dive, Nat. Med., 2014, 20, 897–903 CrossRef CAS PubMed.
- A. Fabisiewicz and E. Grzybowska, Med. Oncol., 2017, 34, 12 CrossRef PubMed.
- N. Aceto, A. Bardia, D. T. Miyamoto, M. C. Donaldson, B. S. Wittner, J. A. Spencer, M. Yu, A. Pely, A. Engstrom, H. Zhu, B. W. Brannigan, R. Kapur, S. L. Stott, T. Shioda, S. Ramaswamy, D. T. Ting, C. P. Lin, M. Toner, D. A. Haber and S. Maheswaran, Cell, 2014, 158, 1110–1122 CrossRef CAS PubMed.
- S. B. Huang, S. S. Wang, C. H. Hsieh, Y. C. Lin, C. S. Lai and M. H. Wu, Lab Chip, 2013, 13, 1133–1143 RSC.
- K. F. Lei, M. H. Wu, C. W. Hsu and Y. D. Chen, Int. J. Electrochem. Sci., 2012, 7, 12817–12828 CAS.
- M. G. Mendez, S. Kojima and R. D. Goldman, FASEB J., 2010, 24, 1838–1851 CrossRef CAS PubMed.
- C. R. Lindsay, S. Le Moulec, F. Billiot, Y. Loriot, M. Ngo-Camus, P. Vielh, K. Fizazi, C. Massard and F. Farace, BMC Cancer, 2016, 16, 168 CrossRef CAS PubMed.
- A. Satelli, A. Mitra, Z. Brownlee, X. Xia, S. Bellister, M. J. Overman, S. Kopetz, L. M. Ellis, Q. H. Meng and S. Li, Clin. Cancer Res., 2015, 21, 899–906 CrossRef CAS PubMed.
- A. Kulasinghe, C. Perry, M. E. Warkiani, T. Blick, A. Davies, K. O'Byrne, E. W. Thompson, C. C. Nelson, I. Vela and C. Punyadeera, Oncotarget, 2016, 7, 60101–60109 Search PubMed.
Footnote |
| † Liao, Hsieh, and Wang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |