
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-quality conjugated polymers via one-pot Suzuki–Miyaura homopolymerization†
Difei Zhou a,
Nutifafa Y. Doumonb,
Mustapha Abdu-Aguyeb,
Davide Bartesaghib,
Maria A. Loib,
L. Jan Anton Kosterb,
Ryan C. Chiechiab and
Jan C. Hummelen*ab
a,
Nutifafa Y. Doumonb,
Mustapha Abdu-Aguyeb,
Davide Bartesaghib,
Maria A. Loib,
L. Jan Anton Kosterb,
Ryan C. Chiechiab and
Jan C. Hummelen*ab
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: j.c.hummelen@rug.nl
bZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands
First published on 24th May 2017
Abstract
This paper describes a one-pot Suzuki–Miyaura homopolymerization that involves in situ borylation/cross coupling of dibrominated donor–acceptor conjugated macromonomers, in contrast to the standard Stille copolymerization of thienosilole and isoindigo monomers. Reaction kinetics investigation reveals that bis(pinacolato)diboron promotes an efficient polymerization. The homopolymer showed blue-shifted absorption compared to the Stille copolymer, which is rationalized by quantum chemical calculations of a series of oligomers containing various donor–acceptor configurations. The calculations suggest that the homopolymerization of asymmetrical macromonomers likely introduced both acceptor–acceptor and donor–donor segments into the backbone. The acceptor–acceptor segment is found to contribute mostly to the blue-shift of maximum absorption wavelength. Furthermore, detailed analysis of MALDI-TOF (matrix-assisted laser-desorption ionization-time of flight) spectra of these two polymers indicated that while the homopolymer is well defined, the Stille copolymer is end-capped mostly with the thienosilole moieties and/or methyl groups, implicating that destannylation and methyl transfer are the most-likely chain-termination pathways that limit high molecular weight. This is in sharp contrast to the homopolymerization, where chain-terminators are required to control the molecular weight for obtaining soluble material. The photovoltaic performances of bulk-heterojunction solar cells based on these polymers are compared.
Introduction
The Stille cross-coupling copolymerization1 has been a method of choice for the development of donor–acceptor conjugated alternating copolymers for a host of applications such as polymer solar cells,2–4 polymer light-emitting diodes,5 thin-film transistors6 and even polymer-based sensors.7 Such popularity is largely due to its remarkable tolerance for many functional groups, which enables the design and preparation of a variety of functional polymers. Yet the convenience of Stille cross-coupled polymerization comes at a cost in terms of the unavoidable toxicity and the difficulty of purifying the organostannanes that are necessary to achieve high molecular weights, as well as possible synthetic complications due to tin–tin homocouplings when certain catalyst/organostannane combinations are used.8 These drawbacks may pose limitations on the molecular weights and structural integrity of the resulting polymers. In this sense, a number of research groups have set out to prepare better copolymers from Stille copolymerization.9–12 Results suggest that high molecular weight, low polydispersity and better structural integrity can contribute to enhanced light absorption and improved morphology, which translate to better overall device performance.Along with the popularity of Stille cross-coupled copolymerization, Suzuki cross-coupling has been widely used to synthesize high molecular weight alternating copolymers. However the monomer scope of the Suzuki copolymerization is primarily limited to mildly electron-rich building blocks such as carbazoles,13,14 and fluorenes,15,16 which is probably due to the tendency towards deborylation of the boronic acid/esters of such moieties. These challenges highlight the significance of developing one-pot reactions that eliminate the necessity to acquire purified boronic acids/esters. To this end, a handful of research groups have investigated the scope of one-pot synthesis of biaryls in terms of catalyst, base and aryl halides.17–22 Early discoveries have shown that bis(pinacolato)diboron could be used as a condensation reagent for the one-pot synthesis of low to moderate molecular weight poly(arylene)s,23 and that certain bases such as K2CO3 may lead to oligomer formation in biaryl systems.24 In 2009, Reynolds et al. revisited this synthetic pathway and specifically studied the effect of CsF on the homopolymerization of fluorenes.25 Furthermore, in one of our earlier contributions, it has been shown that with symmetrical model systems, in situ borylation/cross coupling demonstrates a general superiority over the Stille copolymerization in terms of end-group control.26
In this work, we further develop the chemistry of one-pot borylation/cross-coupling polymerization. Instead of employing simple, head-to-tail symmetrical, relatively electron-neutral monomers, we constructed a head-to-tail asymmetrical donor–acceptor conjugated macromonomer that was subjected to the homopolymerization. Particularly, we compare the as-obtained homopolymer with a copolymerized counterpart synthesized from the standard Stille cross coupling, in terms of the optoelectronic properties and structural integrity.
Experimental section
General
All reagents and solvents were purchased from commercial sources and were used without further purification unless indicated otherwise. UV/vis measurements were carried out on a Perkin Elmer Lambda 9000 spectrometer in 1 cm quartz cuvettes with concentrations of 0.03–0.1 mg mL−1 in CHCl3. MALDI-TOF spectra were taken on a Biosystems Voyager apparatus. Samples were prepared by mixing the matrix (10 mg mL−1 in THF) and polymer sample (1 mg mL−1 in THF or CHCl3) at room temperature in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio. All measurements were performed in positive ion mode. NMR spectra were measured using a Varian AMX400 (400 MHz) instrument at 25 °C. FT-IR spectra were recorded on a Nicolet Nexus FT-IR fitted with a Thermo Scientific Smart iTR sampler. GPC measurements were done on a Spectra Physics AS 1000 series machine equipped with a Viskotek H-502 viscometer and a Shodex RI-71 refractive index detector. The columns (PLGel 5 m mixed-C) (Polymer Laboratories) were calibrated using narrow disperse polystyrene standards (Polymer Laboratories). Samples were made in CHCl3 at a concentration of 2–3 mg mL−1.
:1 volume ratio. All measurements were performed in positive ion mode. NMR spectra were measured using a Varian AMX400 (400 MHz) instrument at 25 °C. FT-IR spectra were recorded on a Nicolet Nexus FT-IR fitted with a Thermo Scientific Smart iTR sampler. GPC measurements were done on a Spectra Physics AS 1000 series machine equipped with a Viskotek H-502 viscometer and a Shodex RI-71 refractive index detector. The columns (PLGel 5 m mixed-C) (Polymer Laboratories) were calibrated using narrow disperse polystyrene standards (Polymer Laboratories). Samples were made in CHCl3 at a concentration of 2–3 mg mL−1.
Synthesis
Compounds 1,27 2 and 3 (ref. 28 and 29) were synthesized according to literature methods. The thienosilole precursors for 2 and 3 were synthesized with a modified procedure based on literature method.29 The synthetic steps of the two polymers are shown in Scheme 1.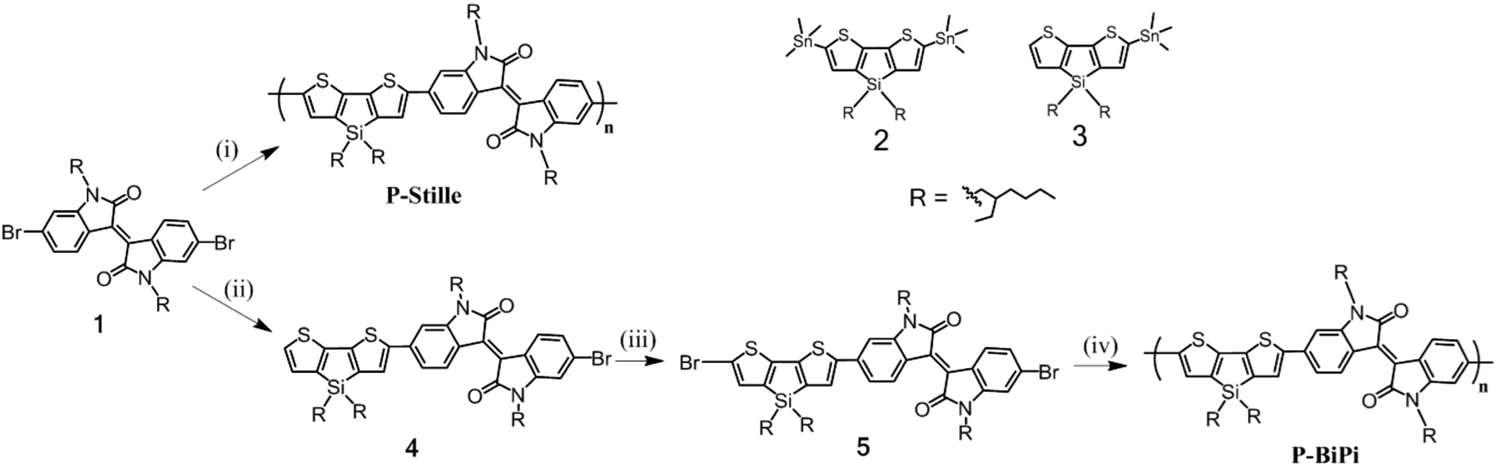 | ||
| Scheme 1 Synthesis of polymers. Reagents: (i) 2, Pd2(dba)3 (CHCl3 adduct, recrystallized from acetone), P(o-tol)3, toluene; (ii) 3, Pd2(dba)3 (CHCl3 adduct), P(o-tol)3, toluene; (iii) NBS, THF; (iv) bis(pinacolato)diboron, K3PO4, Pd(dppf)Cl2 (CH2Cl2 adduct), toluene/DMF (v/v = 4/1). P-Stille refers to the polymer obtained from a standard Stille copolymerization, while P-BiPi is the polymer synthesized with the BiPi promoted polycondensation. BiPi means bis(pinacolato)diboron. | ||
P-Stille
A 25 mL, flame-dried two neck flask, filled with dry N2, was charged with 1 (100 mg, 0.155 mmol) and 2 (118 mg, 0.16 mmol), followed by the addition of Pd2(dba)3 (CHCl3 adduct, recrystallized from acetone) (4 mg), P(o-tol)3 (16 mg). Degassed dry toluene was then introduced into the reaction flask via a cannula. The reaction was then set at 100 °C for 24 h. Afterwards, the reaction mixture was allowed to cool to room temperature, and added dropwise into 500 mL of methanol. The precipitate was collected with a cellulose thimble, which was then subjected to Soxhlet extraction with methanol for 24 h, acetone for 7 h, hexane for 5 h, and CHCl3 for 3 h, subsequently. The chloroform fraction was obtained as a purple-blue solid (128 mg, 85%). 1H NMR (400 MHz, CDCl3) δ 9.08 (2H, br), 7.54 (1H, br), 7.43 (1H, br), 6.51 (1H, br), 3.74 (4H, br), 2.94 (1H, br), 1.22 (34H, br), 0.79 (30H, br). IR (cm−1): 2954, 2920, 2853, 1689, 1608, 1453, 1376, 1352, 1170, 1102, 1073, 1013, 874, 817, 739.Compound 4
A 50 mL, flame-dried three neck flask, filled with dry N2, was charged with of 1 (500 mg, 0.776 mmol) and of 3 (480 mg, 0.83 mmol), followed by the addition of Pd(PPh3)4 (45 mg). Degassed dry toluene was then introduced into the reaction flask via a cannula. The reaction was then stirred at 100 °C overnight. After cooling to room temperature, most of the solvent of the resulting mixture was removed under reduced pressure. The resulting purple oil was subjected to silica gel chromatography with toluene/hexanes (v/v = 1/3) as the eluent. Compound 4 was obtained as a purple oil (450 mg, 60% yield). 1H NMR (400 MHz, CDCl3) δ 9.13 (d, J = 8 Hz, 1H), 9.02 (d, J = 8 Hz, 1H), 7.40 (s, 1H), 7.27 (dd, J = 8 Hz, 1H), 7.17 (dd, J = 8 Hz, 1H), 7.08 (d, J = 4 Hz, 1H), 6.92 (d, 1H), 6.86 (d, 1H), 3.62 (m, 4H), 1.84 (m, 2H), 1.35 (m, 20H), 1.18 (m, 10H), 0.93 (m, 20H), 0.80 (m, 12H). 13C NMR (101 MHz, CDCl3) δ 168.59, 168.20, 149.84, 148.59, 145.94, 145.79, 144.51, 144.38, 143.31, 138.71, 133.19, 130.61, 130.55, 130.44, 130.01, 127.38, 126.12, 125.82, 124.85, 120.65, 120.35, 118.83, 111.30, 104.62, 44.30, 44.10, 37.77, 37.45, 35.92, 35.64, 30.81, 30.59, 28.87, 28.60, 24.28, 24.00, 22.97, 17.63, 14.17, 10.82.Compound 5
A 50 mL round-bottom flask was charged with of 4 (320 mg, 0.33 mmol) and THF (20 mL). NBS (70.5 mg, 0.396 mmol) was added all at once. After 2 h, the product mixture was extracted with diethyl ether, and the organic phase was collected and dried under reduced pressure. The resulting purple oil was subjected to silica gel chromatography with toluene/hexanes (v/v = 1/3) as the eluent, yielding 245 mg of 5 (70% yield). 1H NMR (400 MHz, CDCl3) δ 9.14 (d, J = 8 Hz, 1H), 9.03 (d, J = 8 Hz, 1H), 7.37 (s, 1H), 7.26 (dd, J = 8 Hz, 1H), 7.16 (dd, J = 8 Hz, 1H), 7.03 (s, 1H), 6.93 (d, 1H), 6.89 (d, 1H), 3.64 (m, 4H), 1.85 (m, 2H), 1.37 (m, 24H), 1.17 (m, 10H), 0.96 (m, 16H), 0.79 (m, 12H).Reaction for P-BiPi kinetics
A 50 mL, flame-dried two neck flask, filled with dry N2, was charged with 5 (80 mg, 0.0755 mmol) and then Pd(dppf)Cl2 (CH2Cl2 adduct) (5 mg), K3PO4 (64 mg) and bis(pinacolato)diboron (19 mg, 0.0775 mmol). Degassed toluene/DMF (v/v = 4/1) (15 mL) was introduced into the reaction flask via a cannula. The reaction was set at 80 °C. A sample of 1 mL was taken from the reaction flask at given time spot for UV-vis and GPC measurements.P-BiPi
A 25 mL, flame-dried two neck flask, filled with dry N2, was charged with 4 (11 mg, 0.0113 mmol), 5 (100 mg, 0.0943 mmol) and then Pd(dppf)Cl2 (CH2Cl2 adduct) (4 mg), K3PO4 (80 mg) and bis(pinacolato)diboron (25 mg). Degassed toluene/DMF (v/v = 4/1) (10 mL) was introduced into the reaction flask via a cannula. The reaction was set at 80 °C overnight. Afterwards, the reaction mixture is cooled room temperature, and added dropwise into 500 mL of methanol. The precipitate was collected with a cellulose thimble, which was subjected to Soxhlet extraction with methanol for 24 h, acetone for 7 h, hexane for 5 h, and CHCl3 for 5 h, respectively. Production of the chloroform fraction appears as a purple solid (70 mg, 70%). 1H NMR (400 MHz, CDCl3) δ 9.22 (1H, br), 9.19 (1H, br), 7.41 (1H, br), 7.31 (2H, br), 7.03 (1H, br), 7.00 (2H, br), 3.74 (4H, br), 1.93 (2H, br), 1.25 (34H, br), 0.83 (28H, br). IR (cm−1): 2954, 2920, 2853, 1689, 1608, 1453, 1376, 1352, 1170, 1102, 1073, 874, 817, 739.Density functional theory calculations
Geometries of dimer (DAAD, DADA and ADDA) and tetramers (DADADADA and ADDAADDA) were optimized using Density Functional Theory (DFT) (B3LYP/6-311G*) with ORCA30 without any symmetry constraints. Subsequently, the vertical excitation energies were calculated using time-dependent DFT (TD-DFT) (B3LYPg/6-311G*). The lowest 10 excited states were calculated for the tetramers.Photoluminescence measurements
Photoluminescence measurements were carried out on solutions contained in quartz cuvettes (2 mm path length) in transmission mode. The excitation source was the second harmonic (approximately 400 nm) of a mode-locked Ti:Sapphire laser (Mira 900, Coherent) delivering 150 ps pulses at a repetition rate of 76 MHz. The laser power was adjusted using neutral density filters; and the excitation beam was spatially limited by an iris. The beam was focused with a 150 mm focal length lens onto a spot of approximately 50 μM. Steady state spectra were collected into a spectrometer with a grating of 50 lines per mm and recorded with an em-CCD array (Hamamatsu, ImagEM) sensitive between 400 nm and 900 μm. For time resolved measurements, the same pulsed excitation source was used. Spectra were collected on a Hamamatsu streak camera (Hamamatsu, Japan) working in Synchroscan mode (time resolution 2 ps) with a cathode sensitive in the visible. All spectra were corrected for the spectral response of the setup using a calibrated lamp.Solar cell device fabrications and measurements
Indium tin oxide (ITO) coated glass substrates were precleaned with deionized water, CMOS grade acetone, and then isopropanol, each for 15 min. Possible organic residues were removed by UV-ozone cleaning for 20 min. A layer of PEDOT:PSS (poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate), VP AI4083, H. C. Stark) (thickness 60 nm) was spin-cast on top at 1500 rpm for 50 s. After being baked at 140 °C for 10 min, the substrates were transferred into a nitrogen-filled glovebox (<0.1 ppm of O2 and H2O). Atop that, active layers of polymer:[60]PCBM (99.5%, Solenne B.V.) (D/A weight ratio = 1:4) were spin-coated in a N2-filled glovebox from o-DCB solution with 3% (v/v) 1,8-diiodooctane (DIO, Sigma Aldrich) at 600 rpm for 5 s and 400 rpm for 120 s. The donor-to-acceptor total concentration was 30 mg mL−1. The thickness of the photoactive layers was about 100 nm. Finally, LiF/Al (1 nm and 100 nm, respectively) were thermally evaporated at a pressure of <10−6 Torr on top of the organic layer to make the sandwiched structure ITO/PEDOT:PSS/active layer/LiF/Al. The active area of the device was 0.04 cm2. The current–voltage characteristics of all solar cell devices were measured with a Keithley 2400 source meter and were conducted in a nitrogen-filled glovebox. An AM 1.5 G solar simulator (AAA grade, XES-70S1) was used as the light source. The light intensity of the solar simulator was calibrated to be 100 mW cm−2 (at the position of sample) using a standard silicon reference solar cell (area 20 × 20 mm2, the certification of the reference cell is accredited by NIST to the ISO-17025 standard). Hole-only devices were fabricated on bare glass substrates which were carefully cleaned by washing with detergent solution and ultrasonication in acetone and isopropyl alcohol, followed by UV-ozone treatment. A Cr(1 nm)/Au(30 nm) anode was then thermally evaporated onto the substrate, followed by the coating of a 60 nm thick film of PEDOT-PSS. The film was then baked at 120 °C for 10 min. Subsequently, polymer:fullerene blends were deposited by spin-coating from ortho-dichlorobenzene solutions under N2 atmosphere. The hole-only devices were finished by thermal evaporation of a Pd(20 nm)/Au(80 nm) top contact.
Results and discussion
Synthesis of polymers
Scheme 1 presents the synthesis of the two polymers P-Stille and P-BiPi, where P-Stille is the copolymerization result of the dibrominated compound 1 and the distannyl thienosilole 2 and P-BiPi is the polymer obtained by the homopolymerization method. The obtainable molecular weight of the P-Stille product is maximized by gently refluxing the polymerization mixture for 24 h, giving a Mn and Mw of 11 and 16 kDa (Đ = 1.45), respectively. On the other hand, after a successful synthesis of the dibrominated macromonomer 5, an initial attempt to homopolymerize this macromonomer 5 overnight at 100 °C led to a 97% yield of intractable black solid, which could be either be due to cross-linking or a very high molecular weight. The possibility of cross-linking under the given conditions was readily excluded with the high purity of 5 determined from its HPLC absorption trace (Fig. S1†), excluding significant over-bromination of compound 4. Furthermore, infrared spectroscopy measurements of 5 and the insoluble material showed overlapping spectra (Fig. S2†), unambiguously indicating that the intractable material comprises ultra-high molecular weight polymer chains, based on macromonmer 5. These observations underscore the capacity of the in situ borylation/cross coupling route to yield high molecular weight polymeric materials.To better control the molecular weight, we further studied the kinetics of this specific homopolymerization by monitoring the molecular weight and light absorption changes over time, as shown in Fig. 1(a). It can be seen that the polymerization was notably fast, with the absorption saturating 4 h after the reaction was started at room temperature. The consecutively red-shifting absorptions suggest a continuous growth of the polymer chains. This observation is further correlated with the GPC results shown in Fig. 1(b), which indicate that polymers with Mn = 9.3 kDa are obtained after a reaction period of 4.5 h. Further prolonging the reaction time quickly led to insoluble product. Moreover, Fig. 1(b) shows a strong attenuation in the growth of the molecular weight over time (and over the conversion of monomers). Additionally, the fact that P-BiPi becomes completely intractable in a small time window after the initial polymerization period hints to cross coupling of long oligomers, leading to long and insoluble polymer chains that precipitate. The kinetic study suggests that with the specific macromonomer, the one-pot SM polymerization yields molecular weights comparable to that of the standard Stille copolymerization. Since molecular weight is not our pursuit within the current chemical investigation, no further optimization on molecular weights was conducted.
 | ||
| Fig. 1 (a) Normalized UV-vis absorptions and (b) molecular weight changes over time of the homopolymerization. The black bar in (b) means the reaction product has already become intractable. | ||
Note that the above analysis does not provide direct insight into the polymerization mechanism (step-growth or chain-growth), but a highly efficient polymerization of dibrominated donor–acceptor macromonomers was indeed observed. In terms of controlling the molecular weight, an efficient way is to pre-mix chain-terminators with the starting materials at the very beginning, such that part of the bifunctional monomers will be partially deactivated towards further reaction. Indeed, when using compound 4 as the chain-terminator for reaction (iv), we were able to isolate CHCl3-soluble polymers with a yield of 70%. Compound 4 was chosen as the chain-terminator because it shows the same reactivity as the macromonomer, ensuring a proper and efficient chain termination. The molecular weight of the polymer is limited by the chain terminator, which is reflected by increased product solubility in our case. An initial attempt of adding 5% of chain terminator (molar percentage relative to macromonomer) did not allow a satisfactory isolation of CHCl3-soluble polymers, even when the reaction time was limited. Isolation of CHCl3-soluble polymers was possible when the chain terminator percentage was increased to 10%.
Optoelectronic properties
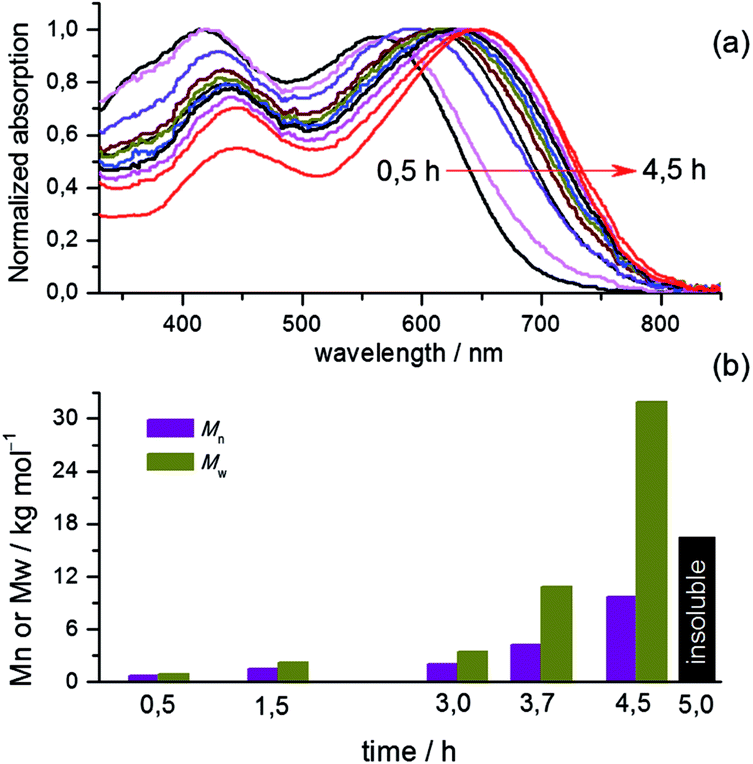
With the insights obtained from the kinetic study, we were able to synthesize soluble polymeric materials for characterization. We first measured the UV-vis absorption spectra of polymers P-Stille and P-BiPi in chloroform, as shown in Fig. 2. Interestingly, while the cut-off wavelengths of the two absorptions are close, there is a 75 nm blue shift of the maximum absorption wavelength from P-Stille (737 nm) to P-BiPi (662 nm). P-BiPi also showed apparently enhanced absorption of high energy photons (an absorption band peaking around 410 nm) compared to P-Stille. These observations indicate a significant structural difference between P-Stille and P-BiPi. Considering the fact that both the Miyarau and Suzuki catalytic cycles involve oxidative insertion as the first step, which preferably takes place on the isoindigo end due to its electron deficiency, this structural difference is most likely related to the cross coupling of two isoindigo ends (forming an A–A segment). The resulting oligomers ending with donor moieties may then lead to donor–donor cross couplings (forming a D–D segment), in addition to the normal donor–acceptor cross coupling (forming a D–A segment).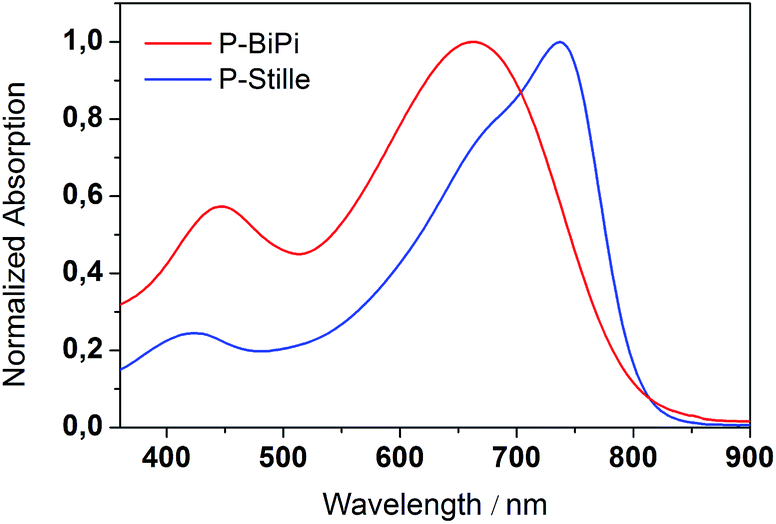 | ||
| Fig. 2 UV-vis absorption spectra of the Stille copolymer P-Stille and Suzuki–Miyarau homopolymer P-BiPi. | ||
To understand the structural origin of these absorption features, we considered a series of oligomers containing different conjugation segments, and calculated their absorption characteristics by time-dependent DFT (TD-DFT). We first calculated the absorption bands of DADA, DAAD and ADDA segments. These results are shown in Fig. S3.† It is clear that an ADDA fragment enables the absorption of low-energy photons, while a DAAD fragment absorbs the higher energy photons more amongst these three oligomers. We note that these three dimers were minimized in a coplanar conformation. In this sense, the calculated absorptions of these dimers are fairly qualitative. Nevertheless, the assessment between theory and measurement suggests that the existence of DAAD segments in the backbone will promote the absorption of high-energy photons, which is in accord with the observation shown in Fig. 1. This phenomenon is also present in the calculated absorption bands of DADADADA and ADDAADDA DA-tetramers, the latter of which contains both AA and DD segments (Fig. 2). On the basis of these observations, we further calculated the electronic transitions of the ADDAADDA DA-tetramer, for which an absorption spectrum was calculated as shown in Fig. 3(B). Despite the differences, it is clear that the calculated spectrum tends to replicate the absorption peaks of the experimental counterpart. This result provides indirect evidence that the as-presented in situ borylation/cross coupling polymerization has introduced DD, AA and DA segments into the backbone, probably in a random pattern.
 | ||
| Fig. 3 (A) Calculated absorption bands of two planarized DA-tetramers, DADADADA and ADDAADDA. (B) A comparison of the calculated absorption spectrum of an ADDAADDA tetramer, and the measured absorption spectra of the P-BiPi. | ||
These random donor–acceptor configurational patterns introduce structural disorder in the backbone of homopolymerized P-BiPi, which is further reflected in several optoelectronic properties of the material. For example, as shown in Fig. 4(a), while the Stille copolymer P-Stille shows a Stokes shift of 0.18 eV in chloroform solution, the Stokes shift of the homopolymer P-BiPi is notably increased to 0.35 eV. As noted by several authors, this might indicate that the structural disorder gives rise to a broader density of states, with emission occurring after relaxation towards low energy sites.31 Moreover, as seen in Fig. 4(b), while P-Stille clearly showed a mono-exponential photoluminescence decay with a fitted excitonic lifetime of 313 ps, the photoluminescence decay of P-BiPi is bimodal exponential, suggesting a multi-pathway decay with lifetimes of 41 ps and 5 ps.
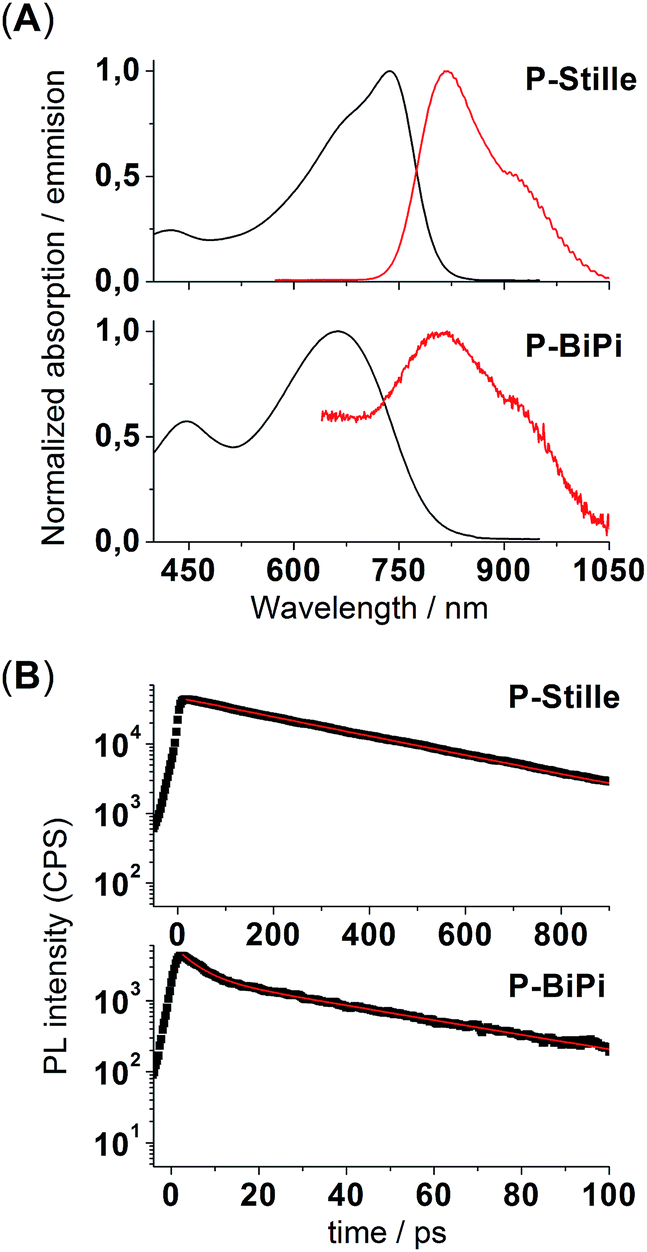 | ||
| Fig. 4 (A) Normalized absorption (black lines) and emission (red lines) spectra of P-Stille and P-BiPi measured in chloroform. (B) Photoluminescence spectra of P-Stille, hexane and chloroform fractions of P-BiPi. The red lines are mono-exponential (P-Stille) or bimodal-exponential (P-BiPi) fits. | ||
Although the detailed mechanism of this phenomenon is beyond the scope of this work, these observations seem to suggest that the existence of AA and/or DD segments in conjugated polymers adversely affects exciton lifetimes, and may also lead to significant fluorescence quenching as shown by the photoluminescence intensities in Fig. 4(B).
Mass spectrometry analysis
MALDI-TOF spectrometry has been widely applied to polymers, both with saturated backbones and with conjugated backbones, to infer structural information.32–34 Due to the soft-ionization nature of MALDI-TOF, the chemical composition of the polymers remains largely intact (i.e. absence of fragmentation). Therefore, given preliminary structural information and proper sample preparations, the analysis of MALDI-TOF spectra can be straightforward and reliable, although it does not give adequate quantitative information regarding the average molecular weight of the polymer sample. The MALDI-TOF spectra of P-Stille and P-BiPi are shown in Fig. 5. Note that due to the relatively poor solubility of P-BiPi in CHCl3, the hexane fraction instead of CHCl3 fraction of the homopolymerization product was used to maintain a sufficient concentration at room temperature. As seen in Fig. 5(A), the in situ borylation/cross coupling polymerization yielded samples showing a clean repetition of peaks, with a repetition interval of 900 Da, in accord with the mass of a D–A repeating unit. Additionally, no obvious residual Br or boronic ester end groups were observed in the case of P-BiPi. This is most likely due to the fact that a H-capped chain terminator (compound 4 in Scheme 1) was used. There are some side-peaks which are of slightly higher m/z values following the major peaks, as shown in Fig. 5(B). One of them might be a polymer chain with hydroxyl groups transferred from the dithranol matrix. The structural origin of the other lower peaks is not clear.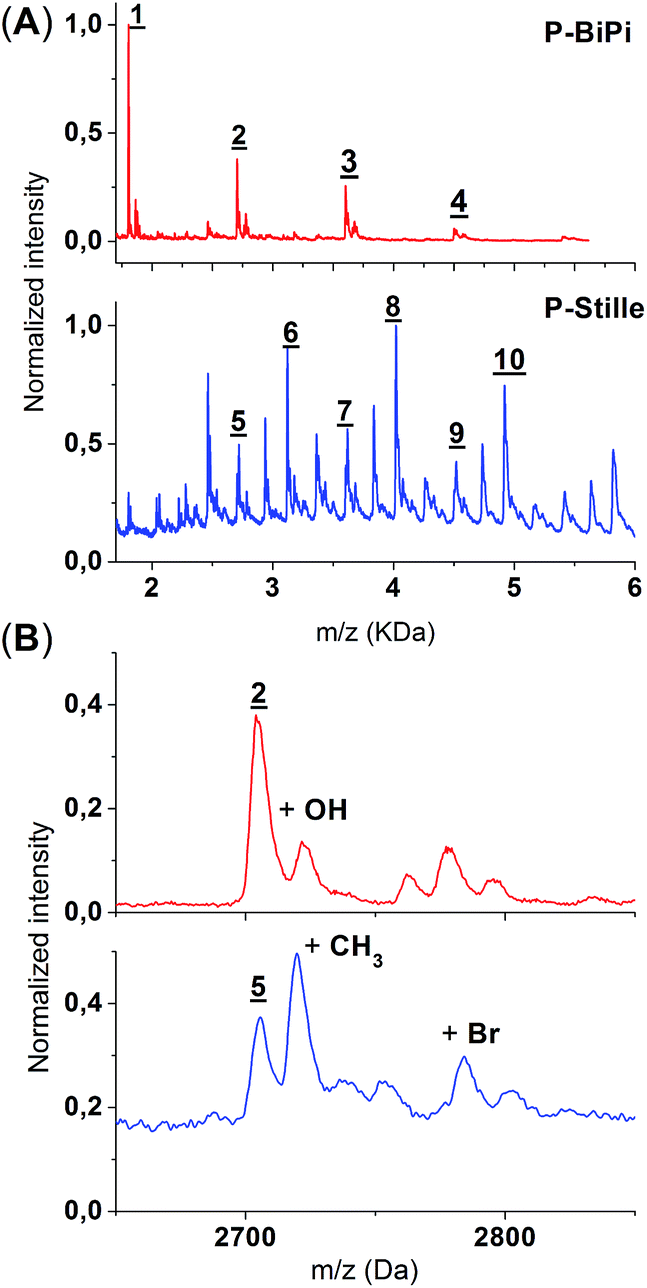 | ||
| Fig. 5 (A) Comparison of the MALDI-TOF spectra of P-Stille and P-BiPi. Peaks of interest are marked with numbers. Peaks 1–4 (peak-to-peak interval: 900 Da) of polymer P-BiPi correspond to dimer to pentamer with hydrogen end groups as the homopolymerization result. Peaks 5, 7, 9 of polymer P-Stille correspond to peaks 2–4 of P-BiPi. The peak-to-peak interval of 5 → 6, 7 → 8 and 9 → 10 is 416, which indicates the presence of an extra donor moiety in the backbone. Other peaks are not convincingly assignable. Dithranol is used as the matrix for both spectra. (B) A zoom-in of peaks 2 and 5. The zoom-in reveals that in the case of the Stille copolymerization, there are peaks representing polymers + methyl group. This is mostly likely due to methyl transfer from the trimethyltin monomer. Residual bromo end groups are visible. | ||
In contrast to the clean and simple mass spectrum of P-BiPi, a very different scenario was recorded for the Stille-copolymerized P-Stille. Although well-defined repetitions are found for all major peaks, only some of them can be sensibly assigned to corresponding structures. As shown in Fig. 5(A), oligomeric molecules of the same chemical composition can be found for P-Stille and P-BiPi. Yet it is interesting to notice that compared to the dihydro-terminated oligomers based on the same number of donor and acceptor moieties (as defined by the P-BiPi spectrum of Fig. 5(A)), all peaks with the highest intensity in the P-Stille spectrum are 416 Da (mass of a repeating donor moiety) higher (5 → 6, 7 → 8, and 9 → 10). This indicates two possibilities: (i) a majority of the resulted polymerized molecules are ended with a donor moiety; (ii) this extra donor moiety is incorporated into the backbone through tin–tin homocoupling. Since the spectrum is clearly free of polymer chains with trimethyltin end groups, the first possibility further implies that destannylation behaves as a major pathway for chain termination, impeding further chain growth. The origin of destannylation was not investigated specifically, but we hypothesize that destannylation is more likely to occur when electron-rich heteroarylstannanes are involved.35 Destannylation as a side reaction is expected to imbalance the stoichiometry of the original monomer ratio, leading to polymer chains with residual reactive Br end groups, which are clearly visible in the MALDI-TOF spectrum. Although homocoupling of organostannanes in conjugated polymer formation has been highlighted elsewhere,12 we did not directly observe it in our case. We note that homocoupling of organostannanes should not be a noticeable problem in this case, primarily owing to the usage of a Pd(0) instead of a Pd(II) catalyst. Upon zooming in around peaks 2 and 5, methyl transfer from trimethyltin is evidenced with a clear peak,12,36 as shown in Fig. 5(B). This highlights the potential drawbacks of routine Stille copolymerizations that potentially hinder the achievement of high quality conjugated polymers. Future synthetic effort should be dedicated to diminish these structural flaws.
Photovoltaic characterization
The photovoltaic performance of these two polymers was compared by fabricating bulk heterojunction solar cells under the same conditions, with a device structure of ITO/PEDOT:PSS/polymer:[60]PCBM/LiF/Al. AFM pictures (Fig. S4†) of the active layer indicate that both polymers can be mixed with [60]PCBM without noticeable phase separation, though P-Stille seems to be slightly aggregated in the blend. Despite the better chemical integrity of the homopolymerized P-BiPi, the random donor–acceptor configuration has notably reduced the absorption of the incoming light spectrum, which is indicated by the drastically lower molar extinction coefficient per repeating unit of P-BiPi than P-Stille, from 550 nm on (Fig. S5†). We believe this is largely responsible for its lower photocurrent of 1.08 mA cm−2, compared to the 3.34 mA cm−2 of the P-Stille device, as shown in Fig. 6. The random donor–acceptor configurations in P-BiPi likely impair effective intermolecular packing, resulting in a lower hole mobility of 5 × 10−8 cm2 V−1 s−1, compared to that of 6 × 10−6 cm2 V−1 s−1 in the P-Stille case as measured by space charge limited current measurements (Fig. S6†). Note that the studied polymers were not optimized for high mobility, as it is well-known that a variety of structural variations may pose notable effect on this physical parameter. While the present research demonstrates the capacity of one-pot homopolymerization in producing polymers of enhanced chemical integrity, its translation into improved physical characteristics shall be achieved in conjunction with dedicated consideration on physical aspects. Overall, with a low photovoltage of 0.6 V and fill factor of 29%, the P-BiPi-based device showed a power conversion efficiency of 0.99%, while P-Stille generated a power conversion efficiency of 1.66% with a photovoltage of 0.79 V and a fill factor of 44%.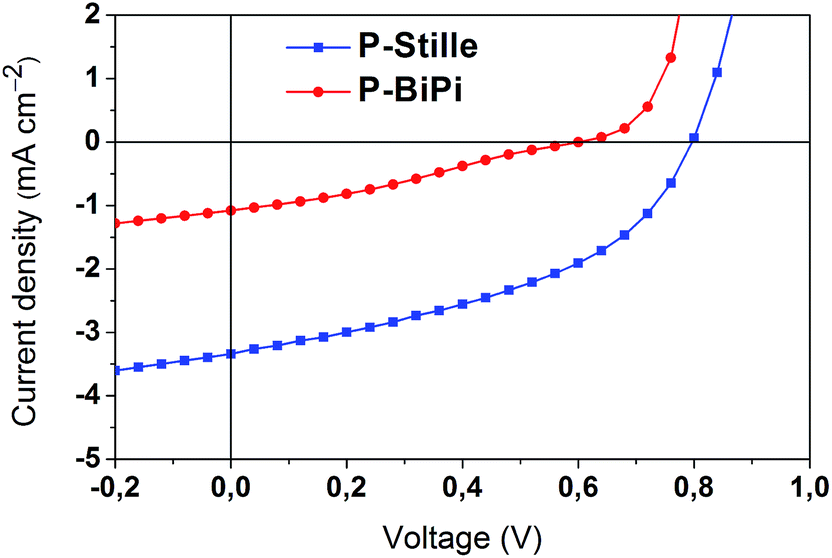 | ||
| Fig. 6 Current–Voltage curves of the P-Stille and P-BiPi-based bulk heterojunction solar cells with DIO additive, under AM1.5 solar illuminations. | ||
Conclusions
In conclusion, we have extended the concept of homopolymerization based on in situ borylation/cross coupling. This polymerization route is appealing in the sense that it not only completely eliminates the necessity to synthesize high-purity boron agents, but also can potentially produce high molecular weight polymeric materials through efficient polymerization, which is evidenced by our kinetic investigation. Yet when applied to head-to-tail asymmetrical donor–acceptor macromonomers rather than simple, symmetrical monomers, such homopolymerizations tend to introduce donor–donor, acceptor–acceptor segments into the polymer backbone, apart from the expected alternating donor–acceptor structure. This type of structural complication was shown to occur in the homopolymerization by correlating the experimental UV-vis absorptions with calculated absorptions from TD-DFT. Nevertheless, MALDI-TOF measurements showed the capacity of this homopolymerization in producing polymers with improved structural integrity, compared to the standard Stille copolymerization which most likely suffers from destannylation and/or methyl transfer, affording moderate molecular weights even with prolonged reaction time. This work is to lead to new strategies for the synthesis of high-quality donor–acceptor conjugated polymers, which shall feature improved chemical integrity and high molecular weights.Acknowledgements
This work is part of the research program of the Foundation for Fundamental Research on Matter (FOM), which is part of the Netherlands Organization for Scientific Research (NWO). This is a publication by the FOM Focus Group ‘Next Generation Organic Photovoltaics’, participating in the Dutch Institute for Fundamental Energy Research (DIFFER).References
- Z. Bao, W. K. Chan and L. Yu, J. Am. Chem. Soc., 1995, 117, 12426–12435 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed.
- J. Pei, W.-L. Yu, W. Huang and A. J. Heeger, Macromolecules, 2000, 33, 2462–2741 CrossRef CAS.
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS PubMed.
- V. Farina, V. Krishnamurthy and W. J. Scott, Organic Reactions, John Wiley & Sons, Inc., New York, 1997, vol. 50, p. 46 Search PubMed.
- T. Qin, W. Zajaczkowski, W. Pisula, M. Baumgarten, M. Chen, M. Gao, G. Wilson, C. D. Easton, K. Müllen and S. E. Watkins, J. Am. Chem. Soc., 2014, 136, 6049–6055 CrossRef CAS PubMed.
- H. Zhong, C.-Z. Li, J. Carpenter, H. Ade and A. K.-Y. Jen, J. Am. Chem. Soc., 2015, 137, 7616–7619 CrossRef CAS PubMed.
- K. H. Hendriks, W. Li, G. H. L. Heintges, G. W. P. van Pruissen, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 11128–11133 CrossRef CAS PubMed.
- T. Vangerven, P. Verstappen, J. Drijkoningen, W. Dierckx, S. Himmelberger, A. Salleo, D. Vanderzande, W. Maes and J. V. Manca, Chem. Mater., 2015, 27, 3726–3732 CrossRef CAS.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS PubMed.
- N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295–2300 CrossRef CAS.
- H. Fan, Z. Zhang, Y. Li and X. Zhan, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1462–1470 CrossRef CAS.
- F. Parenti, P. Morvillo, E. Bobeico, R. Diana, M. Lanzi, C. Fontanesi, F. Tassinari, L. Schenetti and A. Mucci, Eur. J. Org. Chem., 2011, 28, 5659–5667 CrossRef.
- A. Giroux, Y. Han and P. Prasit, Tetrahedron Lett., 1997, 38, 3841–3844 CrossRef CAS.
- L. Zhu, J. Duquette and M. Zhang, J. Org. Chem., 2013, 68, 3729–3732 CrossRef PubMed.
- G. Cuny, M. Bois-Choussy and J. Zhu, Angew. Chem., Int. Ed., 2003, 42, 4774–4777 CrossRef CAS PubMed.
- C. F. Nising, U. K. Schmid, M. Nieger and S. Bräse, J. Org. Chem., 2004, 69, 6830–6833 CrossRef CAS PubMed.
- N. Ma, Z. Zhu and Y. Wu, Tetrahedron, 2007, 63, 4625–4629 CrossRef CAS.
- P. B. Dzhevakov, M. A. Topchiy, D. A. Zharkova, O. S. Morozov, A. F. Asachenko and M. S. Nechaev, Adv. Synth. Catal., 2016, 358, 977–983 CrossRef CAS.
- A. Izumi, R. Nomura and T. Masuda, Chem. Lett., 2000, 29, 728–729 CrossRef.
- T. Ishiyama, K. Ishida and N. Miyaura, Tetrahedron, 2001, 57, 9813–9816 CrossRef CAS.
- R. M. Walczak, R. N. Brookins, A. M. Savage, E. M. van der Aa and J. R. Reynolds, Macromolecules, 2009, 42, 1445–1447 CrossRef CAS.
- F. Brouwer, J. Alma, H. Valkenier, T. P. Voortman, J. Hillebrand, R. C. Chiechi and J. C. Hummelen, J. Mater. Chem., 2011, 21, 1582–1592 RSC.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS PubMed.
- C. M. Amb, S. Chen, K. R. Graham, J. Subbiah, C. E. Small, F. So and J. R. Reynolds, J. Am. Chem. Soc., 2001, 133, 10062–10065 CrossRef PubMed.
- J. Hou, H.-Y. Chen, S. Zhang, G. Li and Y. Yang, J. Am. Chem. Soc., 2008, 130, 16144–16145 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- J.-L. Brédas, J. Cornil and A. J. Heeger, Adv. Mater., 1996, 8, 447–452 CrossRef.
- S. D. Hanton, Chem. Rev., 2001, 101, 527–570 CrossRef CAS PubMed.
- M. Jayakannan, J. L. J. van Dongen and R. A. J. Janssen, Macromolecules, 2001, 34, 5386–5393 CrossRef CAS.
- S. M. Weidner and S. Trimpin, Anal. Chem., 2008, 80, 4349–4361 CrossRef CAS PubMed.
- V. Farina, V. Krishnamurthy and W. J. Scott, Organic Reactions, John Wiley & Sons, Inc., New York, 1997, vol. 50, p. 48 Search PubMed.
- V. Farina, V. Krishnamurthy and W. J. Scott, Organic Reactions, John Wiley & Sons, Inc., New York, 1997, vol. 50, p. 47 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectrum, HPLC trace, IR spectra, additional quantum calculation results, AFM images, extra absorbance measurements, and mobility measurements. See DOI: 10.1039/c7ra03539j |
| This journal is © The Royal Society of Chemistry 2017 |