
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Large-scale synthesis of Li3V2(PO4)3@C composites by a modified carbothermal reduction method as cathode material for lithium-ion batteries
Li Zhang†
a,
Lei Hu†b,
Linfeng Fei *c,
Jianquan Qid,
Yongming Hu*a,
Yu Wange and
Haoshuang Gua
*c,
Jianquan Qid,
Yongming Hu*a,
Yu Wange and
Haoshuang Gua
aHubei Collaborative Innovation Center for Advanced Organic Chemical Materials, Hubei Key Lab of Ferro- & Piezoelectric Materials and Devices, Faculty of Physics & Electronic Science, Hubei University, Wuhan 430062, PR China. E-mail: huym@hubu.edu.cn
bSchool of Energy and Power Engineering, Huazhong University of Science and Technology, Wuhan 430074, PR China
cDepartment of Applied Physics, The Hong Kong Polytechnic University, Hong Kong SAR, China. E-mail: feilinfeng@gmail.com
dDepartment of Materials Sciences and Engineering, Northeastern University at Qinhuangdao Branch, Qinhuangdao, Hebei Province 066004, PR China
eSchool of Materials Science and Engineering, Nanchang University, Nanchang 330031, PR China
First published on 11th May 2017
Abstract
Carbon coated Li3V2(PO4)3 composites were prepared by a modified carbothermal reduction method. The method has the advantages of being simple and scalable, while the whole synthesis is devoid of any reducing/protecting gases, therefore, it can be directly carried out in muffle furnace. The obtained Li3V2(PO4)3@C composites have particles sizes from 500 nm to 3 μm, and with homogeneous carbon coating layer thickness of about 7 nm. The battery based on this Li3V2(PO4)3@C composites cathode exhibits a specific discharge capacity of 130.9 mA h g−1 at 0.1 C, and exhibits an initial specific discharge capacity of 102.8 mA h g−1 even at 10 C and almost keeps 96.6% of initial capacity retention after 500 cycles. The performance enhancement can be directly ascribed to the robust structure merits of the Li3V2(PO4)3@C composites synthesized by the modified carbothermal reduction method. Furthermore, the as-developed method has the potential to be expanded to other lithium transition-metal phosphates (such as lithium iron phosphate), which conventionally need to be sintered in reductive or protective atmospheres.
Introduction
Since 1997, rechargeable lithium-ion batteries (LIBs) have been considered as one of the most attractive power sources for a wide range of applications, such as cellular phones, notebook computers, electric vehicles (EV) and hybrid electric vehicles (HEV), etc., due to their high specific capacities, long cycle lives, high power densities and environmental friendliness.1 At present, it is crucial to seek the state-of-the-art cathode materials, since their great importance in determining the whole performances of batteries.2 Amongst the prospective cathode materials for large-scale next-generation LIBs applications, lithium transition-metal phosphates have aroused widespread concerns in recent decade, e.g. olivine LiFePO4, because of its thermodynamically stable structure,3 high specific capacity (the theoretical capacity is 170 mA h g−1) and abundance in nature. However, its low redox potential (only 3.4 V vs. Li/Li+), as well as low gravimetric energy density, has become the obstacles for the practical application of LiFePO4 cathode, although the great progress has been made. Therefore, tremendous efforts have been devoted to developing other lithium transition-metal phosphates with high operating voltages such as LiMnPO4(∼4.1 V vs. Li/Li+),4 LiCoPO4(∼4.8 V vs. Li/Li+),5 LiNiPO4(∼5.2 V vs. Li/Li+),6 and Li3V2(PO4)3(∼4.8 V vs. Li/Li+).7 Among them, monoclinic phase Li3V2(PO4)3 with the highest theoretical specific capacity (197 mA h g−1 for complete extraction/insertion of 3 Li+ in redox reactions at the voltage window of 3.0–4.8 V) has been the most widely studied system in recent years due to its high-power density, high operating voltage (4.8 V)8,9 and natural abundance,10,11 these advantages make it become highly prospective12–16 for high-energy LIBs.However, the applications of Li3V2(PO4)3 cathode are still far, because of its low electronic conductivity, which will significantly limit electrochemical properties of the electrode. Tremendous efforts have been devoted to overcoming the deficiency, such as reduction of particle size and coating conductive carbon.17,18 The pathways for Li+ to transport across the particles can be enlarged and the distance can be shorted, moreover, the electronic contacts between electrode and electrolyte can be increased through decreasing the size.19 Nevertheless, it has negative effects on the morphology and structure of Li3V2(PO4)3 particles, and the crystallinity of Li3V2(PO4)3,20 so it has not achieved a satisfactory level to be fully utilized. Another feasible technique for enhancing electronic conductivity of Li3V2(PO4)3 cathode is coating conductive carbon. It has been extensively researched for its economical and practical features (such as high electronic conductivity, excellent chemistry/electrochemistry stability, low cost, etc.).21 It can modify the surface chemistry and form more efficient electron pathways,4,22,23 so the active materials can be largely utilized at high current rates,24 and it can also alleviate the growing up and aggregation of Li3V2(PO4)3 particles during the high temperature calcination. Additionally, carbon can act as a reducing agent to reduce V5+ to V3+ during the reaction process.25 Meanwhile, various methods are used to synthesize Li3V2(PO4)3@C composites, such as sol–gel method,26 hydrothermal synthesis,27 freeze-drying method28 and rheological phase reaction synthesis,29 etc. they all have great potential for manufacturing, but there are also some drawbacks of restricting their mass productions, for instance, sol–gel and hydrothermal method often demand low reaction temperatures, which will result in low crystallinity of materials, and the rheological phase reaction synthesis demand complicated operations and high cost. It is necessary to further optimize and modify these methods.
On the other hand, Li3V2(PO4)3@C composites can be commonly synthesized by conventional high temperature solid state reaction.30,31 Carbothermal reduction method,32 one of the conventional high temperature solid state reactions, has become one of the best choices for industrial production since the advantages of high yield and simple process. Nevertheless, large consumption of energy and enormous waste of protective gases or reductive gases (including H2, it will bring the risk of explosion) are the drawbacks of this synthetic method. In this work, a modified carbothermal reduction method is employed to prepare Li3V2(PO4)3@C composites, they can be large-scale synthesized without using reduction gas, and reducing the risk of hydrogen explosion in muffle furnace. Meanwhile, it has the merits of ease of using, energy conservation and low consumption. The chief merit among them is that it does not sacrifice its stable structure and excellent electrochemical performances in mass production. In addition, it has the potential to expand to other cathode of lithium transition-metal phosphates, which are also need to be sintered in reduction or protective atmosphere, such as lithium iron phosphate.
Experimental details
Synthesis
Li3V2(PO4)3@C composites were synthesized via a two-step carbothermal reduction approach including the pre-sintering and the subsequent heat treatment. Firstly, according to the molar ratio of Li3V2(PO4)3 (NLi![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NV:NP = 3:2:3), analytic grade CH3COOLi·2H2O (25.3 g), NH4VO3 (49.0 g), NH4H2PO4·2H2O (33.1 g) and sucrose (C12H22O11, 43.2 g) were added into ethanol (100 ml). Secondly, the precursors were obtained after the mixtures were ball-milled for 12 h and dried for 12 h. Thirdly, the precursor powders were put into a smaller skittle pot, and wrap it with an aluminum foil before being tossed upside down into a larger one, and pour a certain amount of reduction powders which are the mixtures of carbon powders, iron powders and silica sands with the molar ratio of 1.5:1:2 into the extra space. Finally, skittle pot was put into muffle furnace to sinter. The precursors were pre-sintered at 450 °C for 8 h to carbonize sucrose and followed by subsequently sintering at 950 °C for 12 h to crystallize. After cooling down, Li3V2(PO4)3@C composites were obtained.
:NV:NP = 3:2:3), analytic grade CH3COOLi·2H2O (25.3 g), NH4VO3 (49.0 g), NH4H2PO4·2H2O (33.1 g) and sucrose (C12H22O11, 43.2 g) were added into ethanol (100 ml). Secondly, the precursors were obtained after the mixtures were ball-milled for 12 h and dried for 12 h. Thirdly, the precursor powders were put into a smaller skittle pot, and wrap it with an aluminum foil before being tossed upside down into a larger one, and pour a certain amount of reduction powders which are the mixtures of carbon powders, iron powders and silica sands with the molar ratio of 1.5:1:2 into the extra space. Finally, skittle pot was put into muffle furnace to sinter. The precursors were pre-sintered at 450 °C for 8 h to carbonize sucrose and followed by subsequently sintering at 950 °C for 12 h to crystallize. After cooling down, Li3V2(PO4)3@C composites were obtained.
Materials characterization
The surface chemistries of Li3V2(PO4)3 composites were characterized by the following techniques: the amount of residual carbon was determined by TG/DTA (DIAMOND TG/DTA) in flowing air with the temperature ranging from 30 to 800 °C at a heating rate of 10 °C min−1. The crystalline phase of the synthesized materials was characterized by the X-ray diffractometer (XRD, BRUKERD8-A25) with 2θ between 10° and 60°, at the scan rate of 0.02°·sec−1, the voltage and current were 40 kV and 40 mA, respectively. Raman scattering spectroscopy was recorded on a SPM-RAMAN (Ntegra upright, NT-MDT). The morphologies of the samples were observed by using scanning electron microscope (SEM, JSM-7100F) and the morphologies of surface carbon layers and rates of elements were obtained by transmission electron microscopy (HRTEM, Tecnai G2 20 U-Twin) coupled with an Energy Dispersive Spectrometer (EDS).Electrochemical measurements
The electrochemical characterizations were carried out in two electrode coin cells (CR2025). The cells were assembled in a dry Ar-filled glove box according to the order of cathode electrode, electrolyte, separator, and lithium plate and nickel foam. Each component of cells would be introduced as followed: the cathode electrode contained 80 wt% of active material, 10 wt% of acetylene black and 10 wt% of PVDF (polyvinylidene fluoride), to provide sufficient Li+. The cathode slurry was prepared by thoroughly mixing a N-methyl-2-pyrrolidine solution with the active material, acetylene black, PVDF. Subsequently, the obtained slurry was deposited on aluminum foil (thickness of 20 μm) and rolling pressed before the entire assembly was dried in a vacuum oven at 120 °C overnight. The average loading of the electrode was 3.8–5.1 mg cm−2.21 The electrolyte contributing to the diffusion of Li+ was made up of 1 M LiPF6 dissolved in a solvent mixture of ethylene carbonate (EC), dimethyl carbonate (DMC) and ethyl methyl carbonate (EMC) with the volume ratio of 1:1:1. A porous polypropylene film (Celgard 2400) was used to protect inner circuit by only permitting the pass of Li+. Pure metallic lithium foil was served as the anode electrode, which was acted as counter electron and reference electrode. Nickel foam played the role of supporting stabilization of cells. The following electrochemical measurements were conducted. Electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) were analyzed on an electrochemical workstation (ZAHNER CIMPS-2). The impedance data were recorded with an AC voltage of 2 mV as the vibration voltage in the frequency from 100 mHz to 1 MHz. The CV measurements were performed in the potential window of 3.0–4.3 V with a scan rate of 0.2 mV s−1. Galvanostatic cycling and rate capacity performances were gauged on a Land automatic battery tester (LAND CT2001A) in the potential window of 3.0–4.3 V.
Results and discussion
Synthesis of the Li3V2(PO4)3@C composites
The Li3V2(PO4)3@C composites are synthesized via the modified carbothermal reduction method, combining with the ball-milling procedure and the high temperature sintering reaction, the detailed fabrication process is schematically shown in Fig. 1. The reduction powders are used to build the reducing atmosphere in the whole process, instead of using reduction gas, that can reduce the risk of explosion. The carbon powders and iron powders are used to isolate from oxygen into the smaller skittle pot, so they have the effect of preventing precursors from being oxidized, the silica sands are used to prevent iron powders and carbon powders from being sintered into blocks at high temperature which can weak the ability of isolating oxygen, the aluminum foil has the functionality to further isolate oxygen, it can ensure that reduction powders will not mix with precursor powders simultaneously. What's more, they can be utilized repeatedly. The next prominent merit is hundreds of grams Li3V2(PO4)3@C composites can be synthesized in laboratory at a time, it is hundreds of times of production synthesized via conventional carbothermal reduction method. In general, all of these make it easier to operate and have greater output than conventional carbothermal reduction method, moreover, it can be easily scaled-up in manufacturing.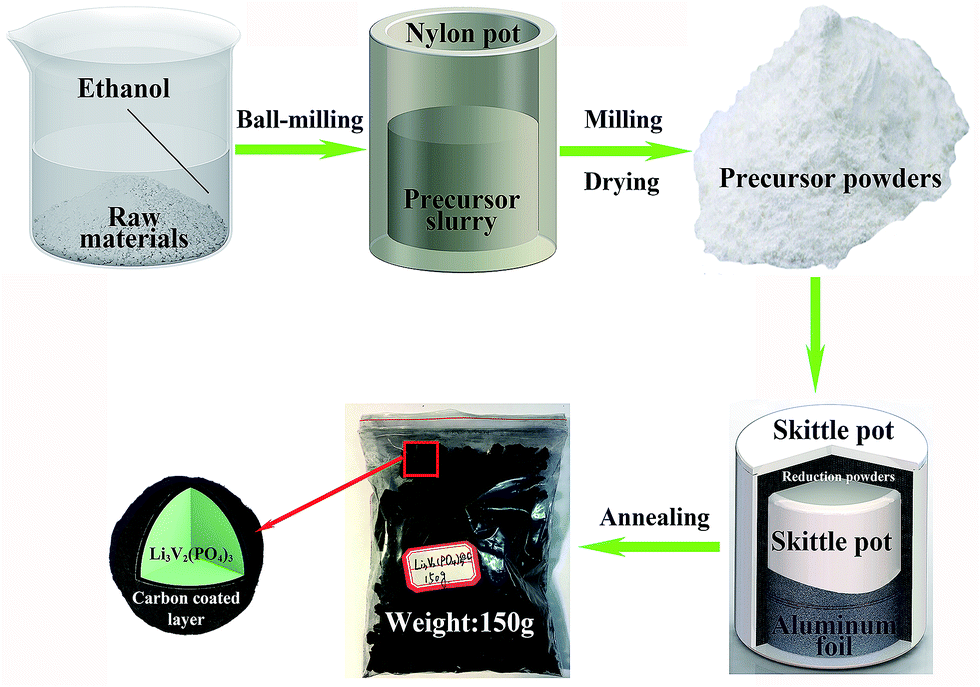 | ||
| Fig. 1 Schematically formation process of carbon coated Li3V2(PO4)3 composites. | ||
Physical characterizations of Li3V2(PO4)3@C composites
Fig. 2a shows the TG curves of Li3V2(PO4)3@C composites measured from RT to 800 °C under an air flow. The weight loss is mainly due to the combustion of carbon and oxidation of vanadium elements in the composites (from V3+ to V5+). The weight of the carbon content is estimated to be about 4.18%.33 | ||
| Fig. 2 (a) TG/DTA curves of the composites recorded from room temperature to 800 °C at a heating rate of 10 °C min−1 under an air flow of 80 mL min−1. (b) X-ray diffraction pattern of carbon coated Li3V2(PO4)3 materials. (c) Raman scattering spectrum of carbon coated Li3V2(PO4)3 materials. | ||
Fig. 2b shows the XRD pattern of Li3V2(PO4)3@C composites. The sharp diffraction peaks of the sample indicate that the composites are well crystallized, which are in good agreement with that of monoclinic Li3V2(PO4)3 (JCPDS card no. 01-072-7074, space group of P21/n). The Miller indexes are added in the XRD pattern. There are no impure diffraction peaks, which suggests that the residual carbon in materials are in amorphous state. The refined lattice parameters of Li3V2(PO4)3@C composites are a = 8.6049(5), b = 8.6056(3), c = 11.9920(8) Å and β = 90.813(8)° with an Rwp = 4.73% and the cell parameters are consistent with those in previous reports.12
Fig. 2c exhibits the Raman scattering spectrum of the carbon coated Li3V2(PO4)3 particles with wavenumbers from 1000 to 1800 cm−1. It can be observed that the characteristic carbon signatures at 1351 and 1600 cm−1 corresponding to a disorder-induced phonon band (D-band) and the graphite band (G-band) respectively. The D-band and the G-band are associated with sp3-type carbon and sp2-type carbon, respectively. The ID/IG ratio provides useful information about the crystallinity of the carbon coated over Li3V2(PO4)3 particles. As previously reported,34 the improvement of the electronic conductivity of the material is corresponding to the decline of ID/IG, which means a drop in the ratio of sp3/sp2. The ratio of ID/IG is 0.96, reflects the good electronic conductivity of the obtained samples.
Fig. 3 shows the SEM and TEM images with different magnifications of Li3V2(PO4)3@C composites. The sizes of these small particles are about 500 nm to 3 μm, and they are uniformly distributed as the Fig. 3a shown. Fig. 3b shows the high-magnification SEM image of the composites, it reflects that the carbon is homogeneously coated on the surface of the particles, which are helpful to enlarge the pathways and shorten the distance for Li+ to transport across the Li3V2(PO4)3 particles. It can be clearly seen from Fig. 3c that the Li3V2(PO4)3 particles are indeed coated with a rough amorphous carbon layer. To further understand the microstructure of the composites and check the carbon coating on the particles, the magnified HRTEM images of the Li3V2(PO4)3@C composites are presented, Fig. 3d and e show the high resolution TEM images of the composites, reveal that the average thickness of the carbon layer is 7 nm, the presence of carbon layers can well control the shape of composite particles and modify the surface chemistry without agglomeration.35 The lattice fringes are clearly observed in the inset figure within Fig. 3d, it clearly reveals that regular lattice fringes of the (020) planes corresponding to the Li3V2(PO4)3 d-spacing value of 0.432 nm through the selected area electron diffraction scan. Fig. 3f shows EDS profile of Li3V2(PO4)3@C composites. The EDS spectrum confirms the existence of C, V, O and P elements in the Li3V2(PO4)3@C composites, and further verifies a rough molar ratio of 2:3 for vanadium and phosphorus elements in accordance with the chemical formula. No obvious peaks corresponded to other impurity elements can be observed (the element Li cannot be identified by EDS detector).
 | ||
| Fig. 3 SEM and TEM images of Li3V2(PO4)3@C composites with different magnifications. (a and b) SEM images; (c–e) TEM and HRTEM images; (f) The EDS profile acquired from Li3V2(PO4)3@C composites. | ||
Electrochemical performances of Li3V2(PO4)3@C composites cathode
To understand the property of Li3V2(PO4)3@C composites synthesized by the modified carbothermal reduction method more deeply, tests are conducted in more details.Fig. 4a displays the galvanostatic charge–discharge potential profiles of the sample between 3.0 and 4.3 V. At the constant current densities of 0.1 C, 1 C, 2 C, 5 C and 10 C (1C = 133 mA g−1), the Li3V2(PO4)3@C composites cathode exhibits the initial specific discharge capacities of 130.9, 127.7, 118.6, 109.8 and 102.8 mA h g−1 (based on the removal of two Li+), corresponding to 98.4%, 96%, 89.2%, 82.6% and 77.3% of the theoretical specific capacity (133 mA h g−1), respectively, which are higher than the previous reported.36 When the sample is cycled at a current density of 10 C, the specific discharge capacity of the sample is about 98.5 mA h g−1 even after 500 cycles as Fig. 4a shown, and it occupies almost 96.6% of initial capacity. It proves that Li3V2(PO4)3@C composites synthesized by the modified carbothermal reduction method can obtain good capacities retention, even at high C-rates.
 | ||
| Fig. 4 (a) Nyquist plots of Li3V2(PO4)3@C composites cathode measured in an AC voltage as a vibration of 2 mV in the frequency range from 100 mHz to 1 MHz and equivalent circuit of the Li3V2(PO4)3@C composites cathode; (b) CV curves of Li3V2(PO4)3@C composites cathode at 0.2 mV s−1 in the potential range of 3.0–4.3 V; (c) cycle performance of the Li3V2(PO4)3@C composites cathode in the potential range of 3.0–4.3 V; (d) initial discharge potential curves of the Li3V2(PO4)3@C composites cathode at different rates (0.1C, 1C, 2C, 5C, 10C) in the potential range of 3.0–4.3 V; (e) initial charge–discharge potential curves of the Li3V2(PO4)3@C composites cathode at 0.1C in the potential range of 3.0–4.3 V; (f) rate performance at various C-rates from 0.1C to 10C each for 20 cycles for the Li3V2(PO4)3@C composites cathode. | ||
The results of Fig. 4b show that Li3V2(PO4)3@C composites cathode deliver the specific discharge capacities of 130.9 mA h g−1 (0.1 C), 127.7 mA h g−1 (1 C), 118.6 mA h g−1 (2 C), 109.8 mA h g−1 (5 C) and 102.8 mA h g−1 (10 C). Obviously, the discharge voltage platforms keep better when the discharge current density is lower. On the contrary, it becomes less conspicuous. It implies that charging/discharging in high current density will limit amount of the Li+ being participated in insertion/de-insertion action, which will reduce the capacity and reversibility of the battery. It explains well why the capacities in smaller current densities are higher than in larger current densities.
Subsequently, the initial charge–discharge potential profiles of the Li3V2(PO4)3@C composites cathode at a current rate of 0.1 C in potential window of 3.0–4.3 V are presented in Fig. 4c, the three clear charge plateaus are 3.62, 3.71 and 4.11 V respectively, and three clear discharge plateaus are 3.56, 3.67 and 4.02 V respectively. In addition, the initial Coulomb efficiency is 99%, which embodies an excellent reversibility of the battery.
Fig. 4d describes the rate capability test of Li3V2(PO4)3@C composites cathode at C-rates ranging from 0.1C to 10C, and back to 0.1C. It exhibits a specific discharge capacity of 130.2 mA h g−1 at 0.1 C and still remains 99.8 mA h g−1 at 10 C, which corresponds to 76.7% of initial specific discharge capacity. When back to 0.1 C, the specific discharge capacity is 128.1 mA h g−1, and the Li3V2(PO4)3@C composites cathode retains 98.4% of initial specific discharge capacity. It is explained that the structure of Li3V2(PO4)3@C composites are stable and the formation of dead Li+ are temporary, which generated at high rate performance, so the capacities and reversibility of the battery will decline at the larger current densities, and will return when back to 0.1 C.
Fig. 4e displays the CV curve of the sample at the scan rate of 0.2 mV s−1 in the potential window of 3.0–4.3 V. The CV curve is composed of three apparent redox couples, due to the insertion/de-insertion of Li+ with only two of them being removed, which means that all of the three couples of current peaks are assigned to the V3+/V4+ redox couples, and it corresponds to the intercalation and extraction of Li+ as the stoichiometric ranges: x = 0.0–0.5, 0.5–1.0 and 1.0–2.0 in Li3−xV2(PO4)3@C, respectively. The three oxidation peaks of Li3V2(PO4)3@C composites cathode are located at about 3.62, 3.70 and 4.12 V respectively, and the three reduction peaks are located at 3.57, 3.66 and 4.02 V respectively, which are in agreement with the three couples of charge/discharge plateaus in Fig. 4c. The extraction and intercalation potentials are similar to previous report.37 The well-defined sharper oxidation/reduction current peaks of the sample are ascribed to the two-phase reaction mechanism, and the broad peak potentials corresponding to the solid-solution behaviors and small potential intervals show low electrochemical polarization reflect excellent reversibility of Li3V2(PO4)3@C composites cathode in the charge/discharge processes.38
EIS measurements are performed on the Li3V2(PO4)3@C composites cathode. Firstly, the coins are galvanostatic charged/discharged for at least 5 cycles to ensure that the SEI films on the surface of the active particles are fully formed, and the coins are then measured with an AC vibration voltage of 2 mV in the frequency range from 100 mHz to 1 MHz. Nyquist plots of the Li3V2(PO4)3@C composites cathode and an equivalent circuit are shown in Fig. 4f. The EIS data are fitted by using Zview2 software. As seen in the figure, an intercept at high frequency on the Z′ axis represents Re of battery, which contains the resistance of electrolyte, separator and electrode. A depressed semicircle in the high-middle frequency region is attributed to the charge transfer resistance, which is associated with the Li+ diffusion in the Li3V2(PO4)3@C particles, and the straight line in the low frequency region stands for the diffusions controlled by Warburg. Rct and W0 are used to denote them in the equivalent circuit. A constant phase element CPE is placed to represent the double layer capacitance and passivation film capacitance,39,40 and the capacitance resistance is so small that it is negligible. Rct of the Li3V2(PO4)3@C composites cathode is 55 ohm, smaller than previous report,41 which is in favor of rapid electrochemical reaction, reflects the excellent electrochemical reaction of the composites cathode. It is explained that the Li3V2 (PO4)3@C composites synthesized by the modified carbothermal reduction method indeed has better structure and good electronic conductivity, which contributes to the diffusion of these Li+ in the Li3V2(PO4)3@C particles.
Among repeated observations, Li3V2(PO4)3 cathode materials with conductive coating in this literature as Fig. 1 shown, possess both outstanding cycling stability and rate capability. The quality of the Li3V2(PO4)3@C composites synthesized by the modified carbothermal reduction method are evidently remarkable. The exceptional performance can be ascribed to the following structural factors. Firstly, the high quality of crystallization, good dispersion and small size of the Li3V2(PO4)3 phase, so it allows the full access of its maximum capacity. Secondly, the carbon coating layer functions as a shell which accommodates the Li3V2(PO4)3 particles just like eggs packed in the shell. With such affection of coated layer, the stress generated due to volume change of Li3V2(PO4)3 caused by the Li+ de-insertion/insertion during the charging/discharging processes will become much localized, thus the phase change and/or decomposition of the particles will be thoroughly eliminated, and the stability of the composites structure will be highly improved upon repeated charging/discharging, the morphology and size of these particles are also greatly reserved even after the deep cycling. Thirdly, the conductive and continuous carbon matrix of the surface carbon coating can enhance the efficient electronic connection between Li3V2(PO4)3 particles. In general, the superior electrochemical performances of Li3V2(PO4)3 composites cathode in this literature is benefit from the core–shell structure of Li3V2(PO4)3 composites synthesized by the modified carbothermal reduction method.
Conclusions
In summary, Li3V2(PO4)3@C composites can be successfully synthesized by the modified carbothermal reduction method. This approach is facile and cost-saving since the Li3V2(PO4)3@C composites are directly sintered without any protective gases or reductive gases, and can produce a large amount at a time, the production is hundreds of times of conventional carbothermal reduction method. The batteries based on this Li3V2(PO4)3@C composites cathode exhibit remarkable reversibility and excellent rate capability, which can be directly ascribed to the robust structure merits of the Li3V2(PO4)3@C composites, it suggests that structure and electrochemical performance is not affected by mass production. In addition, ease of use, energy conservation and low consumption of this approach will greatly improve the efficiency of the industrial production, effectively solve the problems of the traditional method. What's more, this method can be easily extended to other high-performance cathode materials, and can be one of the most promising candidates for future industrial production.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 61274073, 11474088 and 51562026), the Key Project of Education Department in Hubei Province (Grant No. D20151005).Notes and references
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552–6591 CrossRef CAS PubMed.
- L. H. Liao, J. Xie, S. C. Zhang, G. S. Cao and X. B. Zhao, RSC Adv., 2015, 5, 99632–99639 RSC.
- N. N. Bramnik, K. Nikolowski, C. Baehtz, K. G. Bramnik and H. Ehrenberg, Chem. Mater., 2007, 19, 908–915 CrossRef CAS.
- J. Wolfenstine and J. Allen, J. Power Sources, 2005, 142, 389–390 CrossRef CAS.
- Q. Liu, L. B. Ren, C. J. Cong, F. Ding, F. X. Guo, D. W. Song, J. Guo, X. X. Shi and L. Q. Zhang, Electrochim. Acta, 2016, 187, 264–276 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- Y. Q. Qiao, J. P. Tu, X. L. Wang, D. Zhang, J. Y. Xiang, Y. J. Mai and C. D. Gu, J. Power Sources, 2011, 196, 7715–7720 CrossRef CAS.
- J. Kim, J. K. Yoo, Y. S. Jung and K. Kang, Adv. Energy Mater., 2013, 3, 1004–1007 CrossRef CAS.
- J. Su, X. L. Wu, J. S. Lee, J. Kim and Y. G. Guo, J. Mater. Chem. A, 2013, 1, 2508–2514 CAS.
- H. Huang, S. C. Yin, T. Kerr, N. Taylor and L. F. Nazar, Adv. Mater., 2002, 14, 1525–1528 CrossRef CAS.
- M. X. Jing, J. Q. Li, Z. C. Pi, H. A. Zhai, L. L. Chen, S. S. Yao, J. Xiang, X. Q. Shen, X. M. Xi and K. S. Xiao, Electrochim. Acta, 2016, 212, 898–904 CrossRef CAS.
- P. Li, L. Y. Shao, P. F. Wang, X. Zheng, H. X. Yu, S. S. Qian, M. Shui, N. B. Long and J. Shu, Electrochim. Acta, 2015, 180, 120–128 CrossRef CAS.
- C. C. Yang, S. H. Kung, S. J. Lin and W. C. Chien, J. Power Sources, 2014, 251, 296–304 CrossRef CAS.
- L. Chen, B. Yan, H. Y. Wang, X. F. Jiang and G. Yang, J. Power Sources, 2015, 287, 316–322 CrossRef CAS.
- L. F. Shen, H. S. Li, E. Uchaker, X. G. Zhang and G. Z. Cao, Nano Lett., 2012, 12, 5673–5678 CrossRef CAS PubMed.
- H. Q. Li and H. S. Zhou, Chem. Commun., 2012, 48, 1201–1217 RSC.
- N. Raver, Y. Chouinard, J. F. Maganan, S. Besner, M. Gauthier and M. Armand, J. Power Sources, 2001, 97–98, 503–707 Search PubMed.
- Y. G. Wang, P. He and H. S. Zhou, Energy Environ. Sci., 2011, 4, 805–817 CAS.
- L. F. Fei, L. Sun, W. Lu, M. Guo, H. T. Huang, J. P. Wang, H. L. W. Chan, S. S. Fan and Y. Wang, Nanoscale, 2014, 6, 12426–12433 RSC.
- H. D. Liu, G. Yang, X. F. Zhang, P. Gao, L. Wang, J. H. Fang, J. Pinto and X. F. Jiang, J. Mater. Chem., 2012, 22, 11039–11047 RSC.
- H. Wu, G. H. Yu, L. J. Pan, N. Liu, M. T. McDowell, Z. N. Bao and Y. cui, Nat. Commun., 2013, 4, 1943 Search PubMed.
- W. F. Mao, J. Yan, H. Xie, Z. Y. Tang and Q. Xu, J. Power Sources, 2013, 237, 167–171 CrossRef CAS.
- X. H. Rui, C. Li and C. H. Chen, Electrochim. Acta, 2009, 54, 3374–3380 CrossRef CAS.
- L. Chen, B. Yan, H. Y. Wang, X. F. Jiang and G. Yang, J. Power Sources, 2015, 287, 316–322 CrossRef CAS.
- F. Teng, Z. H. Hu, X. H. Ma, L. C. Zhang, C. X. Ding, Y. Yu and C. H. Chen, Electrochim. Acta, 2013, 91, 43–49 CrossRef CAS.
- Y. Q. Qiao, X. L. Wang, Y. J. Mai, X. H. Xia, J. Zhang, C. D. Gu and J. P. Tu, J. Alloys Compd., 2012, 536, 132–137 CrossRef CAS.
- Y. Wu, X. M. Zhao, Z. H. Song, L. P. Lin, C. Q. Du and Z. Y. Tang, J. Power Sources, 2015, 274, 782–790 CrossRef CAS.
- P. Fu, Y. M. Zhao, Y. Z. Dong, X. N. An and G. P. Shen, Electrochim. Acta, 2006, 52, 1003–1008 CrossRef CAS.
- L. J. Wang, X. C. Zhou and Y. L. Guo, J. Power Sources, 2010, 195, 2844–2850 CrossRef CAS.
- Y. Q. Qiao, J. P. Tu, X. L. Wang and C. D. Gu, J. Power Sources, 2012, 199, 287–292 CrossRef CAS.
- J. J. Chen and M. S. Whittingham, Electrochem. Commun., 2006, 8, 855–858 CrossRef CAS.
- T. Muraliganth, A. V. Murugan and A. Manthiram, Chem. Commun., 2009, 19, 7360–7362 RSC.
- W. He, X. D. Zhang, X. Y. Du, Y. Zhang, Y. Z. Yue, J. X. Shen and M. Li, Electrochim. Acta, 2013, 112, 295–303 CrossRef CAS.
- X. Y. Du, W. He, X. D. Zhang, Y. Z. Yue, H. Liu, X. G. Zhang, D. D. Min, X. X. Ge and Y. Du, J. Mater. Chem., 2012, 22, 5960–5969 RSC.
- Q. Kuang, Y. M. Zhao, X. N. An, J. M. Liu, Y. Z. Dong and L. Chen, Electrochim. Acta, 2010, 55, 1575–1581 CrossRef CAS.
- Y. Z. Li, X. Liu and J. Yan, Electrochim. Acta, 2007, 53, 474–479 CrossRef CAS.
- X. H. Rui, N. Yesibolati, S. R. Li, C. C. Yuan and C. H. Chen, Solid State Ionics, 2011, 187, 58–63 CrossRef CAS.
- C. X. Chang, J. F. Xiang, X. X. Shi, X. Y. Han, L. J. Yuan and J. T. Sun, Electrochim. Acta, 2008, 53, 2232–2237 CrossRef CAS.
- Q. L. Wei, Q. Y. An, D. D. Chen, L. Q. Mai, S. Y. Chen, Y. L. Zhao, K. M. Hercule, L. Xu, A. Minhas-Khan and Q. J. Zhang, Nano Lett., 2014, 14, 1042–1048 CrossRef CAS PubMed.
Footnote |
| † L. Zhang and L. Hu contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |