
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Serum amyloid A1 and plasminogen as predictory proteins to monitor the progression of preleukemic diseases towards acute lymphoblastic leukaemia†
Syed Kashif Razaa,
Tahir Shamsi c and
Syed Ghulam Musharraf*ab
c and
Syed Ghulam Musharraf*ab
aDr Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Science, University of Karachi, Karachi – 75270, Pakistan. E-mail: musharraf1977@yahoo.com; musharraf@iccs.edu; Fax: +92-213-34819018; Fax: +92-99261713-4; Tel: +92-21-99261701-2 Tel: +92-21-111222292 (ext. 134)
bH.E.J. Research Institute of Chemistry, International Center for Chemical and Biological Science, University of Karachi, Karachi – 75270, Pakistan
cNational Institute of Blood Diseases, Karachi, Pakistan
First published on 5th June 2017
Abstract
Acute lymphoblastic leukaemia (ALL) is a type of cancer and is the most common form of leukaemia in children. It also occurs in adults around the age of 50 at a low incidence rate. Aplastic anaemia (APA) and myelodysplastic syndrome (MDS), two preleukemic diseases, have been reported to convert into ALL in some case studies. To obtain molecular insight into this progression and identify the proteomic molecules that can associate these preleukemic diseases to ALL, this study is focused on the comparative proteomic profiling of ALL, APA, MDS and the healthy control. A multi-fractionation approach was used for the fractionation of pooled plasma samples of all study groups. Up- and down-fold changes in the concentration of proteins were observed in two-dimensional gel electrophoresis (2D-GE) gels of diseases compared to the healthy group. Among the 34 identified proteins, the eight proteins that were significantly deregulated included serum amyloid A-1 (SAA1), haptoglobin (HPT), C4b-binding protein alpha chain, complement factor 7, apolipoprotein E (ApoE), plasminogen, prothrombin, and complement factor H (CFH), and their links to important cell signalling pathways were found. Validation through enzyme-linked immunosorbent assay (ELISA) showed that SAA1 and plasminogen may be considered as potential molecules that link ALL with APA and MDS.
Introduction
Acute lymphoblastic leukaemia (ALL) is a heterogeneous disease, and numerous genetic mutations lead toward its development. It is the most common cancer in children and is caused by the accumulation of multiple consecutive genetic mutations in the early hematopoietic cells.1 In adults, the incidence peak of ALL is around the age of 50 and although a complete remission is achieved in the children, only 30–40% of the adults can expect a cure from ALL.2 It could be either without signs and symptoms or with life threatening infections, respiratory suffering or haemorrhage.MDS is a group of heterogeneous malignancy of the hematopoietic stem cell, characterized by bone marrow hypercellularity, cytopenias, and increased risk of transformation to acute leukaemia. Usually, one third of the MDS cases transform into acute leukaemia, mainly acute myeloblastic leukaemia (AML). Although conversion to ALL is quite rare, some case studies have been reported in the literature, where MDS has transformed into ALL. This shows that the origin of this disorder is at the same level as a common progenitor cell, which gives rise to both hematopoietic as well as lymphoid origin cells.3–9
The APA is a hematological illness, and the molecular mechanism behind this abnormal immune system and insufficiencies in hematopoietic cells is genetic, namely mutations in repairing genes of the telomeres and the dysregulated pathways of T-cell stimulation.10 Some case studies have reported this type of conversion of APA to ALL.11–19
Currently, molecular genetics, cytogenetics, and morphologic, biochemical and immunologic characteristics of lymphoblasts are needed to make the correct diagnosis and classification,20 and to perform all these tests, a bone marrow puncture is required, which is quite painful and invasive, particularly for children. Therefore, diagnostic procedures need to be improved for timely diagnosis using minimal invasive tests.21 Proteomic analysis provides information about the proteome's dynamicity and rapid changes towards illness and assesses the consequences of diseases using the powerful tool of proteomics.22–24 Our aim in this study is to obtain, for the first time, some proteomic molecules that may determine the relationship of ALL with preleukemic diseases such as APA and MDS using a multidimensional protein fractionation strategy, i.e. fast protein liquid chromatography (FPLC), ZOOM isoelectric focusing (IEF), 2D-GE, and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) analysis.
Experimental
Sample collection and characterization methodology
We selected ALL, APA, MDS and healthy samples for this study. Blood samples were collected from the National Institute of Blood Diseases and Bone Marrow Transplantation (NIBD), Pakistan, after the written informed consent of the participants, in accordance with the ethical standards in the Helsinki declaration. The study was approved by the Ethics Committee of the hospital as per ICH GCP guidelines as well as by the Independent Ethics Committee (IEC) (-022-HB-2017) of the principal investigating institute. All experiments were performed in compliance with the relevant laws and institutional guidelines, and a detailed questionnaire was filled in by every patient and healthy volunteers, which was accepted by the institutional human research ethics committee. Disease samples were classified according to the World Health Organisation (WHO) guidelines. The samples were processed according to the standard protocol of the human proteome organization (HUPO).25 Furthermore, 5 mL whole blood was taken into vacutainers containing K2 ethylene diamine tetra-acetic acid (EDTA) as an anticoagulant. Whole blood was centrifuged at 2200 × g for 10 min at 4 °C to separate the plasma, followed by aliquoting and storage at −80 °C. Equal amount of individual healthy plasma were mixed to obtain the Pakistani pooled plasma of healthy control group, and same strategy was used for the pooling of disease samples.One-dimensional gel electrophoresis
One-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis (1D SDS-PAGE) was performed on an X Cell SureLock system (Invitrogen, USA). Chemicals and reagents were purchased from Invitrogen (USA), containing Precast NuPAGE® bis–tris mini gels 12%, NuPAGE® 2-(N-morpholino-ethanesulfonic acid) MES SDS running buffer 20×, NuPAGE® MOPS SDS running buffer, protein ladder, and colloidal coomassie staining kit. β-Mercaptoethanol, sucrose, and Tris HCl were purchased from Sigma Aldrich (USA).Depletion of abundant proteins
Immunodepletion of the top-7 most abundant proteins was performed using the Multiple Affinity Removal Column (MARS) Hu-7 (4.6 × 50 mm) purchased from Agilent (USA) on an ÄKTA™ Purifier FPLC system, GE Healthcare (Sweden). Protease inhibitors EDTA, leupeptin, pepstatin-A and phenyl methanesulfonyl fluoride (PMSF) were purchased from Sigma Chemicals (USA). A 500 mL Vacuum Filter/Storage Bottle System was purchased from Corning (USA). A total of 300 μL of the plasma was depleted according to the manufacturer's protocol, followed by 1D SDS-PAGE to check the sample recovery.The pooled unbound fraction was enriched through a 5 kDa molecular weight cut-off (MWCO) filter tubes according to the kit protocol. The enrichment efficiency was checked through 1D SDS-PAGE analysis by loading the volume of fractions equivalent to 0.1 μL of the original plasma (before and after concentration).
Proteins in the unbound portion were reduced and alkylated according to a standard protocol with minor modifications.26 Urea, tris, dichlorodiphenyltrichloroethane (DTT), and iodoacetamide (IAM) were purchased from Invitrogen (USA), Boehringer Mannheim (Germany), and SERVA (Germany), respectively. Acetone and trichloroacetic acid (TCA) were purchased from Fisher Scientific (UK) and Scharlau (Spain), respectively. The protein precipitation was performed using the TCA/acetone protocol27 with some minor modifications.
Microscale isoelectric focusing
The ZOOM IEF Fractionator Combo Kit comprising a ZOOM IEF Fractionator was purchased from Invitrogen (USA). The sample pellet was dissolved in the ZOOM buffer along with the addition of 26 μL of the ZOOM carrier ampholytes (pH 3–10) and a small amount of bromophenol blue (BPB). The fractionation of the each pooled unbound fraction was carried out using established protocols.28 The 1D SDS-PAGE analysis of all the five ZOOM fractions of different pH ranges was performed to check the efficiency of IEF and compare the disease samples to the healthy control.2D gel electrophoresis
The proteins from the ZOOM fractions were precipitated according to the above mentioned protocol. The 2D-GE was performed on a Bio-Rad PROTEAN IEF cell (USA). The ReadyPrep 2-D Starter Kit, ReadyStrip IPG Strips, 7 cm, pH 4–7, Ready Gel Precast Gel, mineral oil, and 10× Tris/glycine/SDS buffer were all purchased from Bio-Rad (USA). The entire procedure was performed according to the kit protocol provided by the manufacturer.The gel images were taken through the Gel DOC 800 system Bio-Rad (USA). Analysis was performed in Bio-Rad PDQuest version 8.0.1. Bio-Rad (USA). The images were cropped to the area containing the spots of interest. A master gel was created and the healthy plasma pool was considered as the control. Spot detection, spot matching, and semi-quantitative statistical analysis were performed. The t-test was used to study the differential expression, with the p value < 0.05. Two analysis sets were created, one with a 4× quantity difference and the other with a significance level ≥ 95%; using these analysis sets numerous other Boolean union/intersect analysis sets were created among the gels of the study groups. Some detected spots were manually edited for greater accuracy. The protein spots of interest were extracted using the manual cutting procedure.
Matrix-assisted laser desorption ionization-time-of-flight-mass spectrometry
Analysis was performed using the matrix-assisted laser desorption ionization-time-of-flight-mass spectrometry (MALDI-TOF-MS) (Ultraflex III, Bruker Daltonics Germany). The mass spectrometric profile was obtained using flexAnalysis version 3.0 (Bruker Daltonics). Spot cutting and the in-gel digestion was performed according to the reported protocol.29,30 The digested peptides were analysed using a MALDI-TOF mass spectrometer. Briefly, the samples were mixed in equal proportion with freshly prepared α-cyano-4-hydroxycinnamic acid in acetonitrile (ACN). Instrument calibration was done in a reflector positive mode using a peptide calibrant standard I (Bruker Daltonics). Spectra were obtained over the mass range of 500–3000 m/z with the sum of 2000 laser shots within the same spot (200 shots per position) and an intensity of 20–40%.The protein mass fingerprinting (PMF) of the mass values obtained from MALDI-TOF was performed using the online MASCOT server with Swiss-Prot and NCBInr databases; moreover, carbamidomethylation of cysteine as the fixed and the oxidation of methionine as the variable modification were selected. The maximum number of the missed cleavages was set to 0 and 1, peptide tolerance 100 ppm per 1 Da, and p < 0.05 was used to find the proteins.
Pathway analysis
Differently expressed proteins were further subjected to Gene Ontology (GO)-based analysis to determine their functions, and then the connections among the proteins and their connection with the other proteins were assessed using the STRING: EMBL (European Molecular Biology Laboratory) software.31ELISA analysis
ELISA was performed on individual samples for the five proteins SAA1, CFH, complement C7, plasminogen, and ApoE on Thermo Fischer Scientific™ Multiskan™ FC Microplate Photometer (USA). The ELISA kits were purchased from Crystalchem (USA) and Assaypro (USA). Plasma samples of 20 ALL, 15 AA, 15 MDS, and 15 healthy individuals were diluted according to the kit protocol. The entire procedure was performed according to the manufacturer's given methodology, followed by statistical analysis using GraphPad Prism 7.0.Results
Auxological data
A total of 65 plasma samples of ALL, 41 AA, and 17 MDS were included in this study, while 50 healthy male and 50 female healthy volunteers were selected for the control group. The classification of healthy samples on the basis of age and number of samples are shown in Table 1. All the healthy individuals were free of any hematological diseases at the time of sampling with normal vital signs and with no family history of ALL, AA, and MDS.| Healthy | Number of samples | |||
|---|---|---|---|---|
| Male | Female | |||
| Age group | 20–30 years | Group I | 20 | 20 |
| 30–40 years | Group II | 10 | 10 | |
| 40–50 years | Group III | 10 | 10 | |
| 50–above years | Group IV | 10 | 10 | |
| Total number of samples | 100 | |||
Immunodepletion of top-7 abundant proteins
The pool plasma samples were depleted of the top-7 most abundant proteins using a MARS column. The resultant FPLC spectrum showed a clear separation of the unbound and bound proteins, in full agreement with the figure provided in the manufacturer's protocol (Fig. S1†). The depletion efficiency was shown to be effective through the one-dimensional gel electrophoresis (1D-GE) analysis (Fig. S2†). Then, the unbound portions of each group were enriched through 5 kDa MWCO tubes, and the efficiency showed acceptable protein enrichment (Fig. S3†).Microscale isoelectric focusing
Fractioned proteins were further separated using the ZOOM IEF over a pH range of 3.0–10 into several fractions with different pH ranges; pH: 3.0–4.6, pH: 4.6–5.4, pH: 5.4–6.2, pH: 6.2–7.0, and pH: 7.0–10.0. The 1D-GE results showed that the two fractions of pH: 5.4–6.2 and pH: 6.2–7.0 from all the study groups had a large number of the protein bands (Fig. S4†). The comparison of these two fractions is shown in Fig. 1. These two fractions were pooled to make a single pooled fraction over the pH range from 5.4 to 7.0 from each group, followed by protein precipitation. | ||
| Fig. 1 (A) Comparison of fraction-3 (pH: 5.4–6.2) after ZOOM IEF. (1) Protein ladder, (2) blank, (3) ALL, (4) APA, (5) MDS and (6) healthy. (B) Comparison of fraction-4 (pH: 6.2–7.0) after ZOOM IEF. (1) Protein ladder, (2) blank, (3) ALL, (4) APA, (5) MDS and (6) healthy. The amount of fractions loaded into the gel was equivalent to 3 μL of the original plasma. | ||
2D gel electrophoresis and mass spectrometry
The resultant (2D-GE) maps of all the study groups are shown in Fig. 2. The master gel was used for the comparison of ALL gels with the preleukemic and the healthy gel images (Fig. S5†). The ALL pool was compared with the pool of the two preleukemic diseases and with the healthy pool, separately. Boolean interception sets were created for each comparison separately and their Boolean union was created, which had 56 spots, for a combined comparison. The bar graphs of these 56 spots are shown in Fig. S6.† Among the 56 spots, we chose only those spots that had a pattern of up- or down-regulation in the leukemic and preleukemic diseases compared to the healthy sample.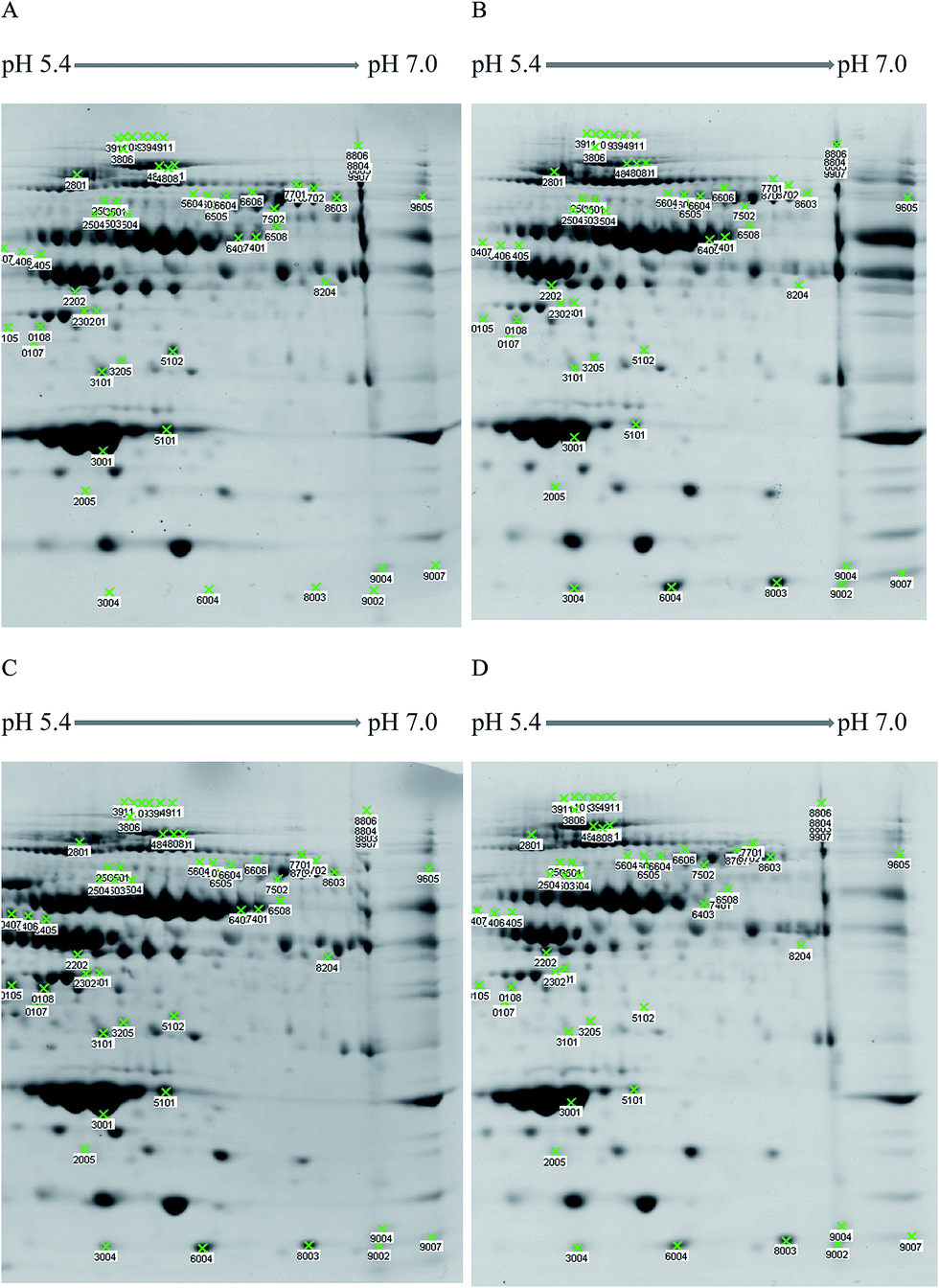 | ||
| Fig. 2 2D-Gel analysis of the pools. (A) Healthy pool, (B) acute lymphoblastic leukaemia, (C) aplastic anemia and (D) myelodysplastic syndrome. Green crosses are spots that are 95% significant and 4× up- or down-regulated. | ||
Altogether, a total of 182 gel spots were analysed using MALDI-TOF MS, which led to the identification of 34 distinct proteins and/or their respective isoforms and subunits (Fig. 3). The list of identified proteins is shown in Table 2. We identified eight proteins that are significant and consistently deregulated: ApoE, complement factor 7, C4b-binding protein alpha chain, plasminogen, prothrombin, SAA1, CFH, and HPT. The former five proteins were found to be down-regulated, while the latter three were up-regulated, as shown in the column graphs (Fig. S6†).
 | ||
| Fig. 3 Identified variants and subunits of proteins in 2-D gel image through MALDI-MS and MASCOT database searching [(1, 39 and 46) hemopexin, (2) alpha-1β-glycoprotein, (3 and 20) kininogen-1, (4) Vit-D-binding protein, (5) transthyretin, (6 and 9) apolipoprotein A-I, (7, 8 and 52) ceruloplasmin, (10, 51 and 55) complement factor H, (11) human serum amyloid-P component, (12, 13, 14 and 32) apolipoprotein E, (15) apolipoprotein A-IV, (16, 35 and 44) haptoglobin, (17 and 53) alpha 2-macroglobulin, (18) fibronectin, (21) complement factor 7, (22) CD5 antigen-like, (23 and 40) prothrombin, (24) complement C1r-subcomponent, (25) complement factor B, (26) complement C4-A, (27) alpha-1 microglobulin, (28, 29 and 48) fibrinogen gamma chain, (30, 31 and 47) fibrinogen beta chain, (33, 49 and 50) plasminogen, (34, 41 and 43) serum amyloid A-I, (36) retinol-binding protein, (37 and 39) C4b-binding protein alpha chain, (38) gelsolin, (39) human serum albumin, (42) serum amyloid A-IV, (43) hemoglobin β-component, (45) complement C4 and (54) fibrinogen alpha chain]. | ||
| SSP IDs | Protein name | Accession no. | Database | Sequence coverage (%) | Mr | pI | Matched peptides no. | Expect value | Score |
|---|---|---|---|---|---|---|---|---|---|
| 3803, 3907–3911, 4801–4804, 4911, 4912, 5701, 5702, 5801, 5802, 6801–6803, 7701, 7801–7803 | Alpha-2-macroglobulin | gi|224053 | NCBInr | 29% | 162![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 072 072 |
5.92 | 38/146 | 3.1 × 10−7 | 120 |
| 3804, 3805, 3901, 4807–4809, 4902, 5901 | Complement factor H | CFAH_HUMAN | SwissProt | 35% | 143680 |
6.21 | 45/136 | 6.4 × 10−11 | 145 |
| 0701, 0702, 0706, 1601, 1602, 1701, 1801, 1802, 2801–2803 | Ceruloplasmin | CERU_HUMAN | SwissProt | 26% | 122983 |
5.44 | 19/107 | 1.4 × 10−5 | 92 |
| 5604, 6601, 6603, 6604, 7601, 7602, 8601, 8602 | Complement factor B | CFAB_HUMAN | SwissProt | 30% | 86847 |
6.67 | 20/90 | 3.3 × 10−6 | 98 |
| 2501, 3501, 3503, 4501–4503 | Complement C1r subcomponent | C1R_HUMAN | SwissProt | 21% | 81606 |
5.82 | 16/64 | 6.7 × 10−4 | 75 |
| 2503, 2504, 3504 | Prothrombin | THRB_HUMAN | SwissProt | 34% | 71475 |
5.64 | 18/88 | 6.4 × 10−8 | 115 |
| 5504, 6404, 6507, 6508, 7402, 6402, 6403, 7401 | C4b-binding protein alpha chain | C4BPA_HUMAN | SwissProt | 39% | 69042 |
7.15 | 19/131 | 2.5 × 10−7 | 109 |
| 0401, 0408, 1401, 2401 | Alpha-1β-glycoprotein | A1BG_HUMAN | SwissProt | 35% | 54790 |
5.56 | 18/125 | 9.5 × 10−5 | 83 |
| 0301, 0302, 1201, 2201, 2203, 3301 | Vitamin D-binding protein | VTDB_HUMAN | SwissProt | 41% | 54526 |
5.40 | 15/111 | 2.5 × 10−4 | 79 |
| 6402, 6403, 7401, 2402, 3401, 4401, 5401, 5402, 6401 | Hemopexin | HEMO_HUMAN | SwissProt | 41% | 52385 |
6.55 | 19/127 | 2 × 10−9 | 130 |
| 2202, 2204, 3201, 3402, 4201, 5201, 5301, 6204, 6208, 6405, 9205 | Fibrinogen gamma chain | gi|223170 | NCBInr | 31% | 46823 |
5.54 | 10/66 | 7.5 × 10−3 | 76 |
| 0405, 0406 | Kininogen 1 | gi|37748641 | NCBInr | 44% | 48954 |
6.29 | 17/115 | 2 × 10−6 | 112 |
| 0105, 0107, 0108, 2005, 2101, 2103, 3103, 3104, 4002, 4107, 5103, 6001, 8002 | Haptoglobin | HPT_HUMAN | SwissProt | 36% | 45861 |
6.13 | 15/102 | 8 × 10−8 | 114 |
| 3806 | Fibronectin | FINC_HUMAN | SwissProt | 18% | 266052 |
5.46 | 43/179 | 0.0079 | 64 |
| 2301, 2302, 3302 | CD5 antigen-like | CD5L_HUMAN | SwissProt | 55% | 39603 |
5.28 | 17/80 | 1.3 × 10−7 | 112 |
| 0101, 0102, 1001, 2001, 3001, 4101, 5101 | Chain A, apolipoprotein A-I | gi|90108664 | NCBInr | 62% | 28061 |
5.27 | 15/77 | 9.9 × 10−9 | 135 |
| 2106, 3108, 3109, 4106, 5104 | Serum amyloid P-component | SAMP_HUMAN | SwissProt | 29% | 25485 |
6.10 | 9/76 | 2 × 10−3 | 70 |
| 0002, 3002, 5001 | Chain A, transthyretin | gi|14719497 | NCBInr | 66% | 12671 |
5.26 | 6/81 | 4.8 × 10−3 | 78 |
| 0201–0203, 1202 | Apolipoprotein A-IV | APOA4_HUMAN | SwissProt | 38% | 45371 |
5.28 | 15/72 | 1.1 × 10−5 | 92 |
| 2105, 3101, 3205, 4102, 4105, 5102, 6102 | Apolipoprotein E | APOE_HUMAN | SwissProt | 50% | 36246 |
5.65 | 16/92 | 1.3 × 10−7 | 112 |
| 0002, 3002, 5001 | Transthyretin | TTHY_HUMAN | SwissProt | 48% | 15991 |
5.52 | 4/95 | 0.04 | 57 |
| 7301, 8201, 8202, 8204, 8301, 8401, 9302, 9303 | Fibrinogen beta chain | FIBB_HUMAN | SwissProt | 60% | 56577 |
8.54 | 28/108 | 8 × 10−15 | 184 |
| 6605–6607, 7603, 8701, 8702 | Complement component C7 | CO7_HUMAN | SwissProt | 23% | 96550 |
9.06 | 17/91 | 0.0034 | 68 |
| 9402 | Fibrinogen alpha chain precursor | gi|11761629 | NCBInr | 36% | 70227 |
8.23 | 23/147 | 0.0016 | 83 |
| 9002 | Hemoglobin subunit beta | HBB_HUMAN | SwissProt | 49% | 16102 |
6.75 | 6/78 | 0.72 | 44 |
| 9004 | Serum amyloid A-4 | SAA4_HUMAN | SwissProt | 40% | 14851 |
9.17 | 6/60 | 0.016 | 61 |
| 8803, 8804, 8806, 9601, 9605, 9907 | Plasminogen | PLMN_HUMAN | SwissProt | 39% | 93247 |
7.04 | 27/112 | 1 × 10−14 | 183 |
| 6501, 6505, 7501, 7505 | Gelsolin | GELS_HUMAN | SwissProt | 17% | 86043 |
5.90 | 13/105 | 0.46 | 46 |
| 3004, 6004, 8003, 9002, 9007 | Serum amyloid A-1 | SAA1_HUMAN | SwissProt | 46% | 13581 |
6.28 | 5/70 | 0.0079 | 64 |
| 0105, 0108, 2101, 3103, 4107 | Retinol-binding protein | RET4_HUMAN | SwissProt | 51% | 23337 |
5.76 | 9/63 | 0.11 | 53 |
| 2501, 3501, 3503, 4501–4503 | Complement C1s subcomponent | C1S_HUMAN | SwissProt | 25% | 78174 |
4.86 | 18/113 | 0.29 | 48 |
| 6402, 6403, 7401 | Serum albumin | ALBU_HUMAN | SwissProt | 39% | 71317 |
5.92 | 21/129 | 7.7 × 10−6 | 94 |
ELISA validation
We selected SAA1, ApoE, plasminogen, complement factor 7 and CFH proteins for further validation through ELISA.Serum amyloid A1
One-way analysis of variance (ANOVA) test showed significant differences among the means of these four groups with a p value < 0.0001. The mean value was 1.188 ± 0.2694 mg dL−1, 1.238 ± 0.3705 mg dL−1, 1.175 ± 0.3594 mg dL−1 for ALL, APA, and MDS, respectively which are high compared to the healthy group i.e., 0.6953 ± 0.1962 mg dL−1 (Fig. 4). Tukey's multiple comparisons test showed no significant difference among the ALL and preleukemic diseases but showed a significant difference when these three groups were compared to the healthy group (Fig. 4). This protein followed the up-regulated pattern in ALL, APA and MDS compared to the healthy group, which is quite similar to the 2D-GE results. | ||
| Fig. 4 Left side: Scatter plot with standard deviation achieved through ELISA results of individual samples of healthy, APA, MDS and ALL subjects. (A1) SAA1 protein and (B1) plasminogen. Right side: Confidence intervals (Cis) for graphing ordinary one-way ANOVA. (A2) SAA1 and (B2) plasminogen. | ||
Apolipoprotein E
The validation results of individual samples through ELISA did not follow the same pattern as expected from the 2D-GE results (down-regulated in all three diseases compared to healthy group), but showed up-regulation in the ALL group compared to the healthy group, while the results of APA and MDS were in accordance with the 2D-GE results. One-way ANOVA test showed a significant difference among the means of the study groups with a p value of 0.0012. The mean values of the ALL, APA, MDS, and healthy samples were 904 ± 516.8 μg mL−1, 551 ± 358.4 μg mL−1, 443.5 ± 5 μg mL−1, and 504.4 ± 214.2 μg mL−1, respectively. Similarly, Tukey's multiple comparison test showed a significant difference among APA and ALL, MDS and ALL, and healthy and ALL pairs, but no significant difference between MDS and APA, healthy and APA, and healthy and MDS pairs.Complement factor C7
The results were found to be up-regulated in both the leukemic and preleukemic conditions, opposite to the 2D-GE results (down-regulated in all three diseases compared to the healthy group). One-way ANOVA test showed no significant difference, with a p value of 0.1559, and the mean values were 105.4 ± 27.54 μg mL−1, 100.3 ± 23.03 μg mL−1, 102 ± 37.26 μg mL−1, and 84.62 ± 17.92 μg mL−1 for ALL, APA, MDS, and healthy samples, respectively. Tukey's multiple comparisons test also showed no significant difference.Complement factor H
These results are not similar to the 2D-GE results (up-regulated). A one-way ANOVA test showed no significant difference among the study groups with a p value of 0.1399. The mean values were 4180 ± 374.5 μg mL−1, 4265 ± 164.6 μg mL−1, 4324 ± 256.3 μg mL−1, and 4381 ± 109.2 μg mL−1 for the ALL, APA, MDS, and healthy samples, respectively. Tukey's multiple comparisons test also showed no significant difference.Plasminogen
This protein showed a similar behaviour of down-regulation in both leukemic and preleukemic conditions, quite similar to the 2D-GE results (Fig. 4). One-way ANOVA test and Tukey's multiple comparisons test showed no significant difference (Fig. S6†). The mean values of plasminogen were 201 ± 81.45 μg mL−1, 271.8 ± 117.6 μg mL−1, 251.7 ± 89.35 μg mL−1, and 269.8 ± 58.74 μg mL−1 of the ALL, APA, MDS, and healthy samples, respectively. All these statistics indicate that the plasminogen protein followed a pattern of down-regulation in ALL and MDS compared to the healthy groups (similar to 2D-GE results), but up-regulation in APA.Discussion
We have investigated differential protein patterns between leukemic, preleukemic conditions, and the healthy group in order to examine certain proteins or the group of proteins that might act as a useful indicator of the link between ALL and two preleukemic diseases, APA and MDS, when there is a history of progression of these preleukemic diseases towards ALL.We first depleted the samples from the top-7 most abundant proteins in plasma including albumin, immunoglobulin G (IgG), immunoglobulin A (IgA), fibrinogen, transferrin, anti-trypsin and HPT, followed by 1D-GE. We noticed that the depletion was not 100% because we identified albumin, three chains of fibrinogen (αβγ), and haptoglobin in the unbound fraction (Fig. 3). This issue has also been reported in other studies.32 From the observed 2D-GE images, it was found that several proteins consisted of more than one spot, and their change in pI or the mass shifts between the theoretical and the experimentally calculated values might be because of the post translational modifications (PTMs), particularly glycosylation, affecting the electrophoretic mobility of the proteins, as has already been reported (Fig. 3).33
According to Gene Ontology, the functions of the total identified proteins could be categorized as 21.24% protein binding, 4.42% antioxidant activity, 2.65% phosphatidylcholine-sterol O-acyltransferase activator, 4.42% alcohol binding, 5.31% peptidase regular activity, 8.85% receptor binding, 2.65% cholesterol transport, 4.42% heparin binding, 7.96% enzyme regulator, 4.42% endopeptidase inhibitor, 3.54% steroid binding, 5.31% enzyme inhibitor, 3.54% lipid transport, 1.77% haemoglobin binding, 6.19% lipid binding, 2.65% cholesterol binding, 1.77% complement component C1q binding, 2.65% copper ion binding, 2.65% chaperone binding, 1.77% phosphatidylcholine binding, and 1.77% vitamin transport. To gain an understanding of the biological functions and the interactive links that are most significantly associated with the differentially expressed proteins in the dataset, the STRING software program was used with an interaction score of medium confidence 0.400, using stringent criteria, and only experimental protein–protein interactions and the pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases were taken into account. The blue lines show that known interactions are from the KEGG pathways, whereas purple lines designate experimental evidence (Fig. S7†). The pathways involved in these differentially regulated proteins are mentioned in Table S1.†
In a STRING pathway analysis, links were found to the phosphatidylinositol 3′-kinase (PI3K)-Akt, the p53, and Rap1 signalling pathways. The cumulative effects of the three pathways are cell cycle arrest, apoptosis, protein synthesis, metabolism, cell proliferation, cell survival, and gene activation. Links were also found to mitotic cell cycle division. Recently, various mechanisms associated with abnormal proliferation or block differentiation by disruption of cell cycle and production of inhibitory oncogenic fusion proteins, which make normal differentiation regulatory proteins inactive, have already been reported in cancers.34 We infer from these findings that our differentially expressed proteins may be deregulated under the disturbance of these signalling pathways or during the cell cycle arrest in disease conditions and may predict the underlying picture. Pathway analysis also created links to the genetic mutation behind ALL (Fig. S8†). According to these findings, our differential expressed proteins have some type of very important link to leukemic and preleukemic diseases.
HPT and fibrinogen (α, β, and γ chain) were not quantified through ELISA because the depletion column has immunoaffinity towards them. The proteins that showed satisfactory validation results are SAA1 and plasminogen. The SAA1 protein is found in plasma in response to malignancy and can be used as an indicator biomarker for late-stage diseases.35–37 Its links with a number of malignancies have already been reported via involvement with tumour pathogenesis and gene polymorphism of SAA1.37,38 Some say that SAA1 affects the tumour microenvironment and adds to metastasis.39 In our study, SAA1 was found to be up-regulated in ALL, MDS, and APA compared to healthy samples. Results show significant concordance with 2D-GE. These promising results may help in progression studies of APA and MDS to ALL (Fig. 4). Plasminogen deficiency leads to the progression of pseudo membranes, with consequent end-organ damage of the affected tissue.40 Components of the plasminogen–plasmin system are found in most malignancies and their expression conveys a prognostic worth.41 Plasminogen levels were found down-regulated in ALL and MDS in comparison with healthy group (in accordance with 2D-GE results), but results did not follow the same trend for ALL and APA.
ApoE, CFH, and complement factor C7 showed no significant results in accordance with the 2D-GE results, and the reason behind their difference could be the presence of several subunits of a single protein. In 2D-GE not all subunits of a protein were found to be deregulated, and possibly a specific unit was deregulated in the disease condition while ELISA technique is able to quantify a single protein which nullifies the effect of that subunit. The second reason could be that in the disease condition, some of the subunits of that specific subunit of protein might have changed because of PTMs and changed the position in the 2D-GE maps, resulting in a low intensity of those spots because of the changed pI and molecular mass. Therefore, in the light of these points, we recommend that further protein profiling of this type should be done on a large number of samples for a better statistical correlation.
Conclusion
Plasma proteomic analysis is an intricate task because of factors that range from the large number of protein analytes to the marked variation in the physical properties of the proteins in the human proteome. This study shows that employing a multi-fractionation strategy like immunodepletion, ZOOM IEF, 2D-GE and MALDI-MS is a powerful approach for detecting low abundance proteins in plasma. This strategy has been successfully applied for the protein profiling of ALL, AA, MDS and the healthy control group. Differentially expressed proteins in our study have shown links to some signalling pathways, particularly p53, which are important in carcinogenesis, and the results of this study show that SAA1 and plasminogen proteins can be used as characteristic molecules, which may be used as additional predictory molecules when there is a case report of progression of preleukemic diseases towards the ALL. We further recommend validation on a large number of individual samples, particularly on those patients who have a history of preleukemic disease before ALL.Funding
The study was financially supported by Higher Education Commission (no. 4493), Pakistan. No funding from NIH.Conflict of interest
Authors of the study declare no conflict of interest.Acknowledgements
Our special thanks to all healthy participants who voluntarily gave their blood samples for this research project.References
- M. Greaves, Infection, immune responses and the aetiology of childhood leukaemia, Nat. Rev. Cancer, 2006, 6(3), 193–203 CrossRef CAS PubMed.
- A. Zamo and D. Cecconi, Proteomic analysis of lymphoid and hematopoietic neoplasms: There's more than biomarker discovery, J. Proteomics, 2010, 73(3), 508–520 CrossRef CAS PubMed.
- C. S. P. Lima, C. A. d. Souza, I. A. Cardinalli and I. Lorand-Metze, Lymphoblastic transformation of myelodysplastic syndrome, Sao Paulo Med. J., 1997, 115, 1508–1512 CrossRef CAS PubMed.
- N. Sato, T. Nakazato, M. Kizaki, Y. Ikeda and S. Okamoto, Transformation of myelodysplastic syndrome to acute lymphoblastic leukemia: A case report and review of the literature, Int. J. Hematol., 2004, 79(2), 147–151 CrossRef PubMed.
- A. A. Algarni, M. Akhtari and K. Fu, Myelodysplastic syndrome with myelofibrosis transformed to a precursor b-cell acute lymphoblastic leukemia: a case report with review of the literature, Case Rep. Hematol, 2012, 2012, 207537 Search PubMed.
- S. Serefhanoglu, H. Goker, Y. Buyukasik, N. Sayinalp and O. I. Ozcebe, Transformation of adult myelodysplastic syndrome-refractory anemia to acute t-cell lymphoblastic leukemia, J. Natl. Med. Assoc., 2009, 101(4), 370–372 CrossRef PubMed.
- V. Gupta and B. Bhatia, Transformation of myelodysplastic syndrome to acute lymphoblastic leukemia in a child, Indian J. Hematol. Blood Transfus., 2010, 26(3), 111–113 CrossRef PubMed.
- N. Sato, T. Nakazato, M. Kizaki, Y. Ikeda and S. Okamoto, Transformation of myelodysplastic syndrome to acute lymphoblastic leukemia: A case report and review of the literature, Int. J. Hematol., 2004, 79(2), 147–151 CrossRef PubMed.
- P. A. Kouides and J. M. Bennett, Transformation of chronic myelomonocytic leukemia to acute lymphoblastic leukemia: case report and review of the literature of lymphoblastic transformation of myelodysplastic syndrome, Am. J. Hematol., 1995, 49(2), 157–162 CrossRef CAS PubMed.
- N. S. Young, R. T. Calado and P. Scheinberg, Current concepts in the pathophysiology and treatment of aplastic anemia, Blood, 2006, 108(8), 2509–2519 CrossRef CAS PubMed.
- Y. H. Matloub, R. D. Brunning, D. C. Arthur and N. K. C. Ramsay, Severe aplastic anemia preceding acute lymphoblastic leukemia, Cancer, 1993, 71(1), 264–268 CrossRef CAS PubMed.
- S. W. Horsley, S. Colman, M. McKinley, C. M. Bateman, M. Jenney and T. Chaplin, et al., Genetic lesions in a preleukemic aplasia phase in a child with acute lymphoblastic leukemia, Genes, Chromosomes Cancer, 2008, 47(4), 333–340 CrossRef CAS PubMed.
- K. Kelly and P. Murphy, Aplastic anaemia preceding acute lymphoblastic leukaemia in an adult with isolated deletion of chromosome 9q, Leuk. Res., 2008, 32(12), 1936–1938 CrossRef CAS PubMed.
- F. Suzan, C. Terré, I. Garcia, J. N. Bastie, E. Baumelou and E. Gluckman, et al., Three cases of typical aplastic anaemia associated with a philadelphia chromosome, Br. J. Haematol., 2001, 112(2), 385–387 CrossRef CAS PubMed.
- L. Villarreal-Martínez, J. C. Jaime-Pérez, M. Rodríguez-Martínez, O. González-Llano and D. Gómez-Almaguer, Acute lymphoblastic leukemia of childhood presenting as aplastic anemia: report of two cases, Revista Brasileira de Hematologia e Hemoterapia, 2012, 34, 165–167 CrossRef PubMed.
- R. Liang, T. K. Chan and D. Todd, Childhood acute lymphoblastic leukaemia and aplastic anaemia, Leuk. Lymphoma, 1994, 13(5–6), 411–415 CAS.
- H. Hasle, S. Heim, H. Schroeder, K. Schmiegelow, E. Ostergaard and G. Kerndrup, Transient pancytopenia preceding acute lymphoblastic leukemia (pre-all), Leukemia, 1995, 9(4), 605–608 CAS.
- Y. Nakamori, M. Takahashi, Y. Moriyama, A. Httori, A. Shibata and T. Watanabe, et al., The aplastic presentation of adult acute lymphoblastic leukemia, Br. J. Haematol., 1986, 62(4), 782–783 CrossRef CAS PubMed.
- E. Orlandi, E. P. Alessandrino, D. Caldera and C. Bernasconi, Adult leukemia developing after aplastic anemia: report of 8 cases, Acta Haematol., 1988, 79(3), 174–177 CrossRef CAS PubMed.
- V. Conter, C. Rizzari, A. Sala, R. Chiesa and M. Citterio, Acute lymphoblastic leukemia, Orphanet Encyclopedia, 2004 Search PubMed.
- Y. Bai, H. Zhang, X. Sun, C. Sun and L. Ren, Biomarker identification and pathway analysis by serum metabolomics of childhood acute lymphoblastic leukemia, Clin. Chim. Acta, 2014, 436, 207–216 CrossRef CAS PubMed.
- I. M. Cristea, S. J. Gaskell and A. D. Whetton, Proteomics techniques and their application to hematology, Blood, 2004, 103(10), 3624–3634 CrossRef CAS PubMed.
- A. A. Zada, M. H. Geletu, J. A. Pulikkan, C. Muller-Tidow, V. A. Reddy and M. Christopeit, et al., Proteomic analysis of acute promyelocytic leukemia: Pml-raralpha leads to decreased phosphorylation of op18 at serine 63, Proteomics, 2006, 6(21), 5705–5719 CrossRef CAS PubMed.
- L. Shi, J. Zhang, P. Wu, K. Feng, J. Li and Z. Xie, et al., Discovery and identification of potential biomarkers of pediatric acute lymphoblastic leukemia, Proteome Sci., 2009, 7(1), 7 CrossRef PubMed.
- A. J. Rai, C. A. Gelfand, B. C. Haywood, D. J. Warunek, J. Yi and M. D. Schuchard, et al., Hupo plasma proteome project specimen collection and handling: Towards the standardization of parameters for plasma proteome samples, Proteomics, 2005, 5(13), 3262–3277 CrossRef CAS PubMed.
- H.-Y. Tang, L. A. Beer and D. W. Speicher, In-depth analysis of a plasma or serum proteome using a 4d protein profiling method, Methods Mol. Biol., 2011, 728, 47–67 CAS.
- E. Fic, S. Kedracka-Krok, U. Jankowska, A. Pirog and M. Dziedzicka-Wasylewska, Comparison of protein precipitation methods for various rat brain structures prior to proteomic analysis, Electrophoresis, 2010, 31(21), 3573–3579 CrossRef CAS PubMed.
- N. S. Vasudev, R. E. Ferguson, D. A. Cairns, A. J. Stanley, P. J. Selby and R. E. Banks, Serum biomarker discovery in renal cancer using 2-de and prefractionation by immunodepletion and isoelectric focusing; increasing coverage or more of the same?, Proteomics, 2008, 8(23–24), 5074–5085 CrossRef CAS PubMed.
- B. de Roos, S. J. Duthie, A. C. Polley, F. Mulholland, F. G. Bouwman and C. Heim, et al., Proteomic methodological recommendations for studies involving human plasma, platelets, and peripheral blood mononuclear cells, J. Proteome Res., 2008, 7(6), 2280–2290 CrossRef PubMed.
- F. Bouwman, D. Suylen, J. Renes and E. Mariman, Evaluation and improving the success rate of protein identification by peptide mass fingerprinting using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry, Rapid Commun. Mass Spectrom., 2005, 19(17), 2465–2468 CrossRef CAS PubMed.
- L. J. Jensen, M. Kuhn, M. Stark, S. Chaffron, C. Creevey and J. Muller, et al., String 8–a global view on proteins and their functional interactions in 630 organisms, Nucleic Acids Res., 2009, 37, D412–D416 CrossRef CAS PubMed.
- M. Zhou, D. A. Lucas, K. C. Chan, H. J. Issaq, E. F. Petricoin III and L. A. Liotta, et al., An investigation into the human serum “interactome”, Electrophoresis, 2004, 25(9), 1289–1298 CrossRef CAS PubMed.
- S. Barrabes, A. Sarrats, E. Fort, R. De Llorens, P. M. Rudd and R. Peracaula, Effect of sialic acid content on glycoprotein pi analyzed by two-dimensional electrophoresis, Electrophoresis, 2010, 31(17), 2903–2912 CrossRef CAS PubMed.
- J. K. Kanaujiya, S. Lochab, P. Pal, M. Christopeit, S. M. Singh and S. Sanyal, et al., Proteomic approaches in myeloid leukemia, Electrophoresis, 2011, 32(3–4), 357–367 CrossRef CAS PubMed.
- C. M. Uhlar and A. S. Whitehead, Serum amyloid A, the major vertebrate acute-phase reactant, Eur. J. Biochem., 1999, 265(2), 501–523 CrossRef CAS PubMed.
- C. Gabay and I. Kushner, Acute-phase proteins and other systemic responses to inflammation, N. Engl. J. Med., 1999, 340(6), 448–454 CrossRef CAS PubMed.
- E. Malle, S. Sodin-Semrl and A. Kovacevic, Serum amyloid A: an acute-phase protein involved in tumour pathogenesis, Cell. Mol. Life Sci., 2009, 66(1), 9–26 CrossRef CAS PubMed.
- H. L. Lung, O. Y. Man, M. C. Yeung, J. M. Ko, A. K. Cheung and E. W. Law, et al., SAA1 polymorphisms are associated with variation in antiangiogenic and tumor-suppressive activities in nasopharyngeal carcinoma, Oncogene, 2015, 34(7), 878–889 CrossRef CAS PubMed.
- M. T. Hansen, B. Forst, N. Cremers, L. Quagliata, N. Ambartsumian and B. Grum-Schwensen, et al., A link between inflammation and metastasis: Serum amyloid A1 and A3 induce metastasis, and are targets of metastasis-inducing S100A4, Oncogene, 2015, 34(4), 424–435 CrossRef CAS PubMed.
- R. Mehta and A. D. Shapiro, Plasminogen deficiency, Haemophilia, 2008, 14(6), 1261–1268 CrossRef CAS PubMed.
- H. C. Kwaan and B. McMahon, The role of plasminogen–plasmin system in cancer, in Coagulation in cancer, ed. C. H. Kwaan and D. Green, 2009, Springer, US, Boston, MA, pp. 43–66 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03445h |
| This journal is © The Royal Society of Chemistry 2017 |