
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Superhemophobic titania nanotube array surfaces for blood contacting medical devices
Kevin Bartleta,
Sanli Movafaghia,
Arun Kota ab and
Ketul C. Popat*ab
ab and
Ketul C. Popat*ab
aDepartment of Mechanical Engineering, Colorado State University, Campus Delivery 1374, Fort Collins, CO 80523, USA. E-mail: ketul.popat@colostate.edu
bSchool of Biomedical Engineering, Colorado State University, Campus Delivery 1376, Fort Collins, CO 80523, USA
First published on 17th July 2017
Abstract
Hemocompatibility of blood-contacting medical devices is necessary to prevent device failure. As soon as a material encounters blood, proteins and platelets will adsorb and attach to its surface. This leads to thrombosis and clot formation on the surfaces, restricting blood flow and in some cases leading to inflammation and device failure. To avoid these complications, patients receiving blood-contact devices are prescribed blood thinning medications, which must be taken for the rest of the patient's life. Some devices can be pre-clotted to improve hemocompatibility, but the benefits will not last the device's entire life. Enhancing hemocompatibility has been a focus of recent research. Proposed methods have included diamond-like carbon surfaces, heparin-based surface coatings, modified polymer surfaces, and titania nanotube arrays. These methods have all shown some enhancement of hemocompatibility initially, but no approach has proven durable over long periods of time. Superhemophobic surfaces are a new approach to improving hemocompatibility, but the interactions of blood components with these surfaces have not been studied in depth. In this study, we have developed superhemophobic surfaces by modifying the surface topography and surface chemistry of titanium. The surface topography was modified by creating titania nanotube arrays through a well-documented anodization technique. Superhemophobicity was induced by modifying the titania nanotube arrays with two different silanes using chemical vapor deposition. The investigations of blood interactions with superhemophobic surfaces showed reduced protein adsorption and platelet adhesion/activation, indicating this a potential approach for enhancing material hemocompatibility.
1. Introduction
Materials that come into contact with blood have several issues which can cause medical devices to fail. For example, thrombosis is a major challenge for vascular stents as it obstructs the flow of blood.1,2 Leaflets of artificial heart valves are also prone to thrombosis, preventing them from opening or closing properly.3–5 Also, the forces exerted by the leaflets on the blood and its components tend to damage the platelets, further promoting thrombosis.6,7 Catheters may be obstructed by blood clots formed on the material surface and are one of the leading sources for bloodstream infections.8 Due to complications such as these, hemocompatibility is a challenge for blood-contacting medical devices. There is not a single material that is truly compatible with blood and its components.9 When blood contacts a material, proteins from the plasma in the blood, particularly fibrinogen, will begin to adsorb on the surface within a few minutes of contact.10 The fibrinogen will convert into fibrin, which along with other blood serum proteins will promote the attachment of platelets on the material surface. After adhering, the platelets will begin to activate, forming a platelet–immune complex.10,11 The platelet–immune complexes signal leukocytes to attach on the material surface as part of the immune response, leading to further clot formation.12,13 The clot can remain on the surface and grow restricting the blood flow, or it can detach from the material surface into the bloodstream, travelling throughout the body and potentially causing major complications for the patient.10,12To deal with these adverse effects, patients receiving these medical devices are often prescribed blood thinning medications. These medications must be continued for the rest of the patient's life and increase the risks of heart failure and internal bleeding, and thus are not an ideal solution. One other method employed in clinical situations is to pre-clot the medical device surface by exposing it to the patient's blood prior to implantation.14 This method, however, can only be used for porous implants such as vascular grafts, so is not applicable for valves and catheters.14 Further, over time the pre-clotted material surface will wear off, exposing the device to the same issues discussed previously. Thus, there is an unmet need to prevent these complications by designing hemocompatible materials for blood contacting medical devices.
Recent studies have examined several strategies to improving material hemocompatibility.11,15–17 Heparin has been investigated as a coating for blood contacting devices, particularly for stents and catheters.18–21 It is a common blood thinning medication and has been shown to improve the hemocompatibility of stents by reducing the activation of thrombin, an enzyme that begins clot formation.19,22 However, because of its dense negative charge, it is also thought to initiate the intrinsic pathway of blood coagulation.23 Polymers modified with surface coatings, such as polyethylene terephthalate coated with polydopamine, have been investigated in attempts to enhance hemocompatibility.16,24 The addition of different coatings has been shown to reduce blood cell adhesion and plasma protein adsorption.17,24,25 Additionally, carbon films have been studied because of their biological inertness and inherent hemocompatibility.26 All these strategies have shown some increase in hemocompatibility, but are not effective over long periods of time.20 Thus, there is a need to develop material surfaces that interact with blood and its components appropriately and remain hemocompatible over long time periods.
In this study, we have developed superhemophobic surfaces as a potential approach for enhancing the hemocompatibility of titanium based blood contacting medical devices. Titanium has been shown to be biocompatible but blood clots readily form on titanium devices, leading to their failure.27 Superhemophobic surfaces were fabricated by first modifying the topography and then the chemistry of titanium surfaces. The surface topography was modified by fabricating titania nanotube arrays on titanium surface. Previous studies have shown enhanced hemocompatibility of these titania nanotubes arrays.10,28 These titania nanotube arrays were further modified with alkyl and fluorinated silanes using a chemical vapor deposition technique to fabricate superhemophobic surfaces. Platelet adhesion studies on these surfaces have shown promising results, however, the way in which blood and its individual components interacts with these surfaces is not known.29 In this study, superhemophobic surfaces were characterized using scanning electron microscopy (SEM), contact angle goniometry, X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD). Protein adsorption, material surface cytotoxicity, platelet/leukocyte adhesion, platelet activation and hemolysis was investigated on superhemophobic surfaces. The results presented here indicate improved hemocompatibility of superhemophobic surfaces when compared to that of control surfaces.
2. Materials and methods
2.1 Fabrication of superhemophobic titania nanotube arrays
Titania nanotube arrays were fabricated from titanium sheets (0.1 cm thick) cut into 2.5 cm × 2.5 cm squares. The titanium substrates were first cleaned in acetone using a sonicator for 7 min. They were then rinsed with M90 detergent and cleaned with isopropyl alcohol in a sonicator for 3 min. The titania nanotube arrays were fabricated using the anodization process described elsewhere.10,28,30–32 The electrolyte used for anodization was composed of 95% v/v diethylene glycol (DEG, Alfa) and 2% v/v hydrofluoric acid (HF, Alfa) by volume in de-inoized (DI) water. The titanium sheet was used as the anode and platinum foil was used as the cathode. The anodization was done for 24 h at 60 V. After anodization, the titania nanotube arrays were rinsed three times with DI water, dried with nitrogen gas, and annealed for 3 h at 530 °C with a ramp rate of 15 °C in ambient oxygen. The titania nanotube arrays (and unmodified titanium) were further cut into 0.5 cm × 0.5 cm which were used for all subsequent studies.Superhemophobic titania nanotube arrays were fabricated by modifying the surface using chemical vapor deposition. Prior to surface modification, titania nanotube arrays were etched in plasma atmosphere at 200 V in 10 cm3 min−1 of oxygen gas for 10 min. The titania nanotube arrays were placed on a hot plate next to a glass slide with 100–120 μl of either octadecyltrichlorosilane (referred to as S1, Gelest) or (heptadecafluoro-1,1,2,2-tetrahydrodecyl)trichlorosilane (referred to as S2, Gelest). The titania nanotube arrays and glass slide were covered with a glass bowl, and heated for 1 h at 120 °C. The superhemophobic titania nanotube arrays were then rinsed with DI water, dried and stored in desiccator until further use.
The following nomenclature will be used in the rest of the manuscript for all the substrates: unmodified titanium (referred to as Ti), unmodified titania nanotube arrays (referred to as NT), and titania nanotube arrays coated with the two silanes (referred to as NT-S1 and NT-S2). Prior to all the biological experiments, the substrates were sterilized. They were incubated in ethanol for 30 min, followed by rinsing with DI water.
2.2 Characterization of titania nanotube arrays
The surface morphology was characterized using a field emission scanning electron microscope (SEM, JEOL JSM-6500). All surfaces were coated with a 10 nm layer of gold prior to imaging and imaged at 15 kV.10,32–34 The average nanotube diameter was measured using ImageJ.The hemophobicity was characterized by measuring the contact angle of whole blood on different surfaces. An approximately 10 μl droplet of blood was formed on the tip of a syringe and lowered until it contacted and detached onto the surface. An image of the droplet was taken using a goniometer (Ramé-Hart Model 250) connected to a camera.33 Images were captured within 5 s of contact between the droplet and the surface. Images for advancing and receding contact angles were also taken by slowly adding and removing water from the droplet, and roll off angles were acquired by placing a droplet on the surface and tilting the goniometer. The images were analyzed using the goniometer software to measure the contact angles.
The surface chemistry was characterized using X-ray photoelectron spectroscopy (XPS). Scans were taken for all four substrates. Survey spectra were collected from 0 to 1100 eV with a pass energy of 187.85 eV. High resolution spectra were collected for titanium and oxygen using a pass energy of 10 eV. Surface elemental composition was calculated using peak fit analysis in the Multipack software.35
The presence of anatase and rutile crystal phases on different substrates was detected through GAXRD (Bruker D8).30 XRD scans were collected at θ = 1.5° and 2θ ranges were chosen based on significant peak intensities. Detector scans were run at a step size of 0.01 with a time per step of 1 s. Peaks were filtered and correlated to crystal structures using DIFFRACT.EVA software.
2.3 Protein adsorption on different surfaces
Protein adsorption on sterilized substrates was characterized using the process described elsewhere.11 Sterilized substrates were incubated in 48-well plates with 100 μg ml−1 of a protein solution on a horizontal shaker plate (100 rpm) at 37 °C and 5% CO2 for 2 h. The two proteins investigated were human serum albumin (Pierce Biotechnology) and fibrinogen (Pierce Biotechnology). After 2 h of incubation, the protein solution was aspirated followed by 3 rinses with PBS to remove any non-adherent proteins. The adsorbed proteins on the surface was characterized using X-ray photoelectron spectroscopy (XPS). High resolution spectra were collected for carbon and nitrogen using a pass energy of 10 eV. Peak fit analysis for high resolution carbon (C 1s) peak was done using the Multipack software.35 Further, the protein-adsorbed substrates were air dried and coated with a 10 nm layer of gold and imaged at 15 kV.2.4 Isolation of human platelet rich plasma (PRP)
Whole blood from healthy individual volunteers, acquired through venipuncture, was drawn into standard 6 ml vacuum tubes coated with the anti-coagulant ethylenediaminetetraacetic acid (EDTA). The protocol for blood isolation from healthy individuals was approved by Colorado State University Institutional Review Board. All procedures were performed in compliance with the National Institutes of Health's “Guiding Principles for Ethical Research”. Informed consents were obtained from human participants of this study. The first tube was discarded to account for the skin plug and locally activated platelets resulting from the needle insertion, following the protocol described elsewhere.10,11,33 Whole blood was isolated by centrifuging the tubes at 150 g for 15 min to separate the PRP from the red blood cells. The PRP was then collected and pooled in a separate tube for further use. All the studies discussed below were repeated at least three time with blood drawn from a minimum of three different healthy individuals, however, for each experiment the PRP was only pooled from the same donor. This is because there is donor-to-donor variability in the number of platelets and it is not possible to compare the absolute values from different donors.2.5 Cytotoxicity of different surfaces
The cytotoxicity of the different substrates was investigated using a Cayman LDH assay (Cayman Chemical). Sterilized samples were incubated with 500 μl of human PRP at 37 °C and 5% CO2 for 2 h in a 48-well plate so that all the samples are completely immersed in PRP. After incubation, 100 μl of the PRP was placed in a new clear polystyrene U-bottom 96-well plate (Greiner Bio-One). 100 μl of the LDH reaction solution, mixed according to the manufacturer's instructions, was added to each well.33 The 96-well plate was placed in an incubator at 37 °C and 5% CO2 for 30 min. The absorbance of the PRP-assay solution was then read at 490 nm using a plate reader (BMG Labtech).2.6 Cell adhesion on different surfaces
Cell adhesion was characterized using fluorescence microscopy. The adhered cells were stained using calcein-AM stain (Invitrogen). Sterilized substrates were incubated in a 48-well plate with 500 μl of PRP at 37 °C and 5% CO2 on a horizontal shaker plate at 100 rpm for 2 h. After incubation, the PRP was aspirated from the substrates. The substrates were rinsed 3 times with sterile PBS to remove any unattached platelets. This was followed by incubating the substrates with a 2 μM calcein-AM solution at 37 °C and 5% CO2 (in PBS) for 30 min. The substrates were rinsed once more with PBS and imaged using a fluorescence microscope (Zeiss).28,32,36 ImageJ was used to calculate the cell coverage on the substrates.The calcein-AM stain will stain both platelets and leukocytes green, so additional studies were performed to distinguish between the two cell types. Cell adhesion was characterized by staining the cell cytoskeleton protein actin using rhodamine-phalloidin (Invitrogen). Cell nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole, Invitrogen). The DAPI will stain cell nuclei blue in leukocytes, whereas the rhodamine-phalloidin will stain the actin red in both platelets and leukocytes, thus distinguishing the two cell types. Substrates were incubated in PRP using the same conditions as described earlier. After incubation, the PRP was aspirated off and the substrates were rinsed twice in sterile PBS. The substrates were moved to a new 48-well plate and fixed in a solution of 3.7% formaldehyde in PBS at room temperature for 15 min. The substrates were rinsed twice in sterile PBS, sitting for 5 min in PBS each time. Next the substrates were moved to clean wells and submerged in a permeative of 1% Triton X in PBS for 3 min.10,28,32 The substrates were rinsed twice more in sterile PBS and moved to a new 48-well plate. 200 μl of rhodamine phalloidin (actin) solution in PBS (at a concentration of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200) was added to each well.33 The substrates were incubated in this solution for 25 min. Next, 21 μl of DAPI stain was added to each well and the substrates were incubated for 5 more mins. The substrates were then rinsed twice in sterile PBS and imaged using a fluorescence microscope. ImageJ was used to calculate the actin cell coverage and number of nuclei on the substrates.
:200) was added to each well.33 The substrates were incubated in this solution for 25 min. Next, 21 μl of DAPI stain was added to each well and the substrates were incubated for 5 more mins. The substrates were then rinsed twice in sterile PBS and imaged using a fluorescence microscope. ImageJ was used to calculate the actin cell coverage and number of nuclei on the substrates.
2.7 Platelet activation on different surfaces
Sterilized substrates were incubated with 500 μl of human PRP at 37 °C for 2 h on a shaker plate. The substrates were rinsed in sterile PBS to remove any unattached platelets. The substrates were then incubated in a primary fixative – a solution of 3% glutaraldehyde (Sigma), 0.1 M sodium cacodylate (Polysciences), and 0.1 M sucrose (Sigma) in DI water for 45 min. Then the substrates were placed in the secondary fixative – 0.1 M sodium cacodylate and 0.1 M sucrose in DI water – for 10 min. Next the substrates were dehydrated in consecutive solutions of ethanol – 35, 50, 70, 95, and 100% – for 10 min each. The last dehydration step was to soak the solution in hexamethyldisilazane (HMDS) (Sigma) for 10 min.10,28,32,34The substrates were coated with 10 nm of gold and imaged using SEM at 2 kV. The platelets were characterized into un-activated, short-dendritic, and long-dendritic morphologies.11 Un-activated platelets were defined to be spherical with compact central bodies. Short-dendritic platelets were defined to be partially activated with small dendrites extending from the bodies. Long-dendritic platelets were defined to be fully activated with substantial dendrites extending from the bodies.
2.8 Hemolysis on different surfaces
Hemolysis investigated using the process described elsewhere.11 Whole human blood was drawn and a 5 μl drop was placed on each sterilized substrate in a 24-well plate. The substrates were left alone for 15, 30, and 60 min. After the designated time, 500 μl of DI water was added to the wells. The substrates were agitated gently on a horizontal shaker plate for 5 min to dissolve any un-clotted blood, releasing the free hemoglobin. The absorbance of the solution was then measured using a plate reader at 540 nm.2.9 Statistical analysis
Surface characterization was reconfirmed on 2 different samples of each substrate. SEM images and contact angle measurements were taken for 5 samples of each substrate (n = 5). Protein adsorption was reconfirmed on 3 different samples of each substrate. The LDH assay, calcein-AM stains, and rhodamine-phalloidin, and DAPI stains were repeated twice with five samples of each substrate (n = 10). SEM images for platelet activation were taken for at least six samples of each substrate (n = 6). At least five images were taken for each sample and were quantified to evaluate level of platelet activation (n = 30). The hemolysis studies were repeated three times with five samples of each surface (n = 15). The quantitative results were analyzed using either one-way or two-way anova tests as appropriate. Results were considered statistically significant with a p-value < 0.05.The studies were repeated with blood drawn from a minimum of three different healthy individuals. The data that is presented (i.e., the arithmetic mean and standard deviation) is only from one donor (from a minimum of three repetitive samples of each surface). This is because there is donor-to-donor variability in the number of platelets and it is not possible to compare the absolute values from different donors. However, similar trends were observed for blood used from each donor for all the resulted presented here, which implies the reproducibility of the trends observed in our experiments.
3. Results and discussion
Hemocompatibility is a challenge for all medical devices since to date there is not a material that is truly compatible with blood. Contact between a material with blood may lead to complications including clots forming on the material surface. These clots can detach, causing heart failure and strokes among other complications. Additionally, the clots may not detach, instead continuing to grow on the material surface causing inflammation and eventually leading to device failure. Current strategies for reducing clot formation have significant drawbacks. In this study, we propose superhemophobic surfaces for enhancing hemocompatibility by modifying the material's surface topography and chemistry.3.1 Characterization of titania nanotube arrays
SEM was used to characterize surface morphology of titania nanotube arrays. The results indicate that vertically oriented and uniformly distributed titania nanotube arrays with an average diameter of 155.9 nm and length of 3.5 μm (Fig. 1(a)). The nanotube arrays were further modified with S1 and S2 to make them superhemophobic. In this study, the goal was to investigate the influence of the molecular structure (specifically, alkyl (S1) vs. fluoroalkyl (S2)) on platelet rich plasma. Fluoroalkyl groups are known to possess lower surface energy,37 but are considered less “slippery” compared to alkyl groups.38 Thus, in order to better understand the role of surface energy and “slipperiness” of the surface on hemophobicity, titania nanotube arrays were modified with an alkyl silane (S1)) and a fluoroalkyl silane (S2). The results indicate no visible changes in the morphology of the nanotube arrays after modification with S1 and S2, however, the average diameter increased to 167.2 nm for NT-S1 (p ≤ 0.05) and 169.8 nm for NT-S2 (p ≤ 0.05) (Fig. 1(b)). Using the inner and outer diameters of titania nanotube arrays along with a length of ∼3.5 μm, the number of nanotubes per unit area were estimated to be ∼8 nanotubes per μm2. Further, the roughness factor r (ratio of the actual surface area to the projected surface area) for NT, NT-S1 and NT-S2 was estimated. The results indicate that there is not a significant difference in the surface area of titania nanotube arrays upon modification with S1 and S2.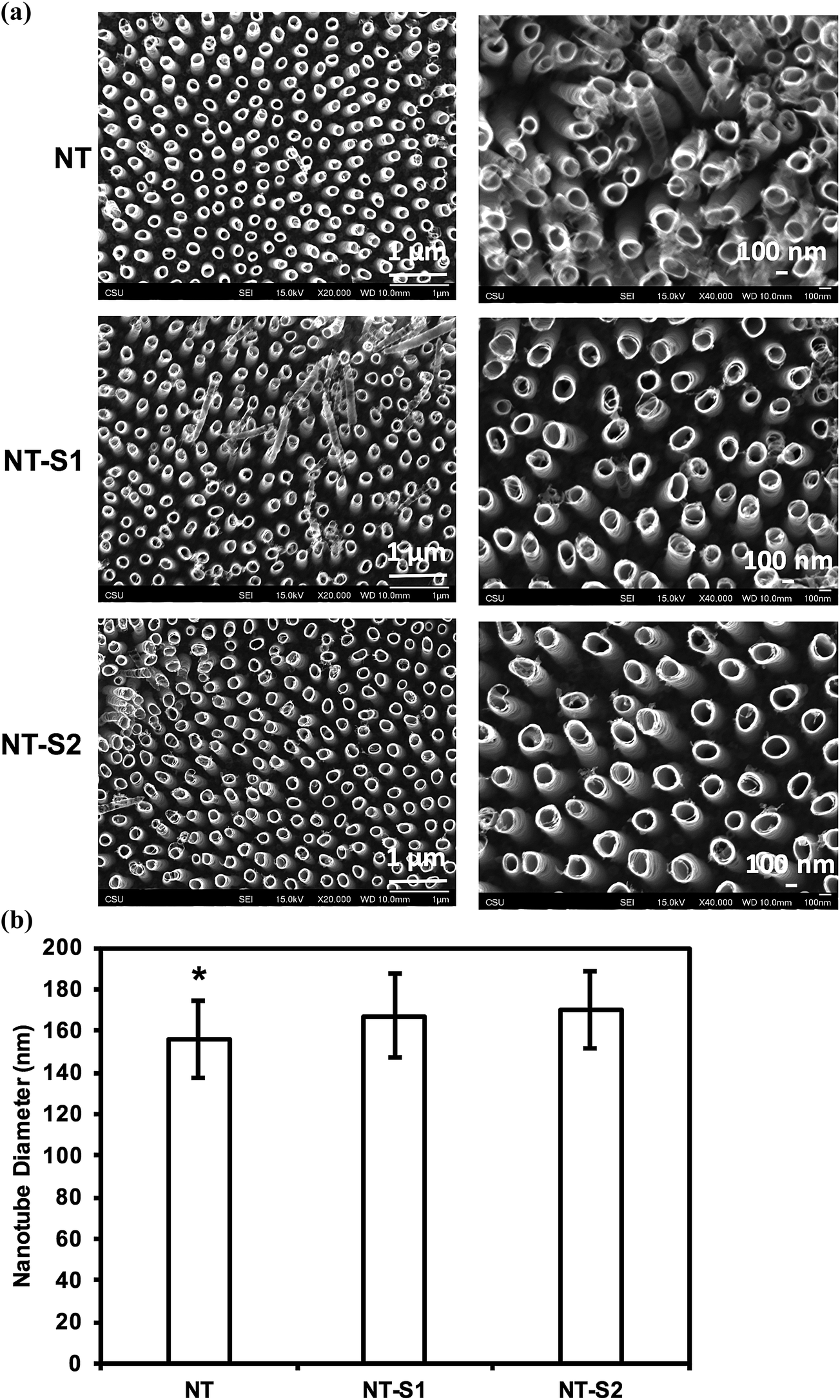 | ||
| Fig. 1 (a) SEM images of titania nanotube arrays fabricated by anodization and modified using chemical vapor deposition. (b) Average diameter of titania nanotube arrays before and after silane modification. The average diameters for NT were smaller than NT-S1 or NT-S2 diameters (p ≤ 0.05). | ||
Contact angle goniometry was used to characterize the surface hemophobicity. The results indicate a static contact angle of 61° for Ti, 28° for NT, 154° for NT-S1, and 157° for NT-S2 (Fig. 2). The contact angle measurements for NT-S1 and NT-S2 were significantly higher than Ti and NT (p < 0.05) (Fig. 2). The roll off angles were measured as 16° for NT-S1 and 9° for NT-S2, while the blood did not roll off the Ti or NT surfaces. A contact angle greater than 150° and roll off angle less than 10° indicates that a surface is superhemophobic. This means that the NT-S2 is the most superhemophobic, followed by NT-S1. A droplet on a textured surface will enter either the Wenzel state or the Cassie–Baxter state. The Wenzel state is where the droplet spreads throughout the surface features, completely wetting the surface.29 This state is expected for Ti and NT. The Cassie–Baxter state is when air pockets remain among the surfaces features, leading to the droplet being suspended above the surface. The Cassie–Baxter state is characterized by high contact angles due to the reduced contact between the droplet and surface due to the presence of the air pockets. This state is expected for NT-S1 and NT-S2. We hypothesize that by minimizing the contact between blood and a surface we can reduce the formation of clots.
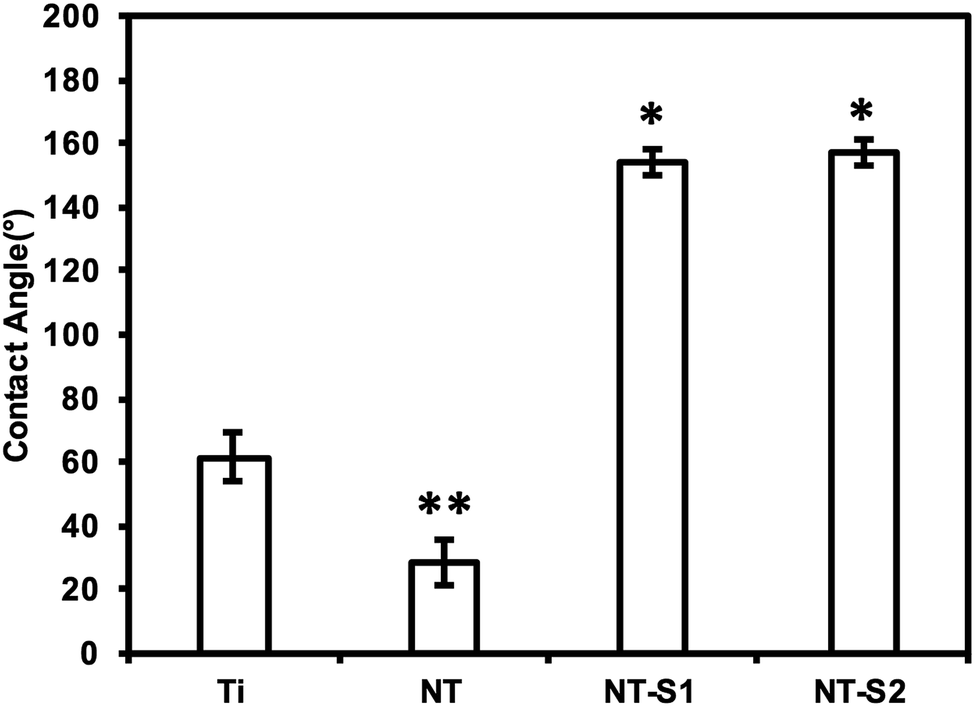 | ||
| Fig. 2 Average whole blood contact angles for different surfaces. The contact angles for NT-S1 and NT-S2 are significantly higher than that for Ti and NT (p ≤ 0.05). | ||
XPS was used to characterize the surface chemistry of different substrates. XPS will show the relative compositions of different elements on the surface of a material. The results indicate O 1s, Ti 2p3/2, and C 1s peaks present on all four surfaces (Fig. 3). The C 1s peak was present on Ti because contamination in the XPS chamber and some trace amounts of carbon present on the Ti surface. The C 1s peak was reduced on NT because of the electrochemical etching and oxidation process. Further, the C 1s peak increased for both NT-S1 and NT-S2 since the silanes contain significant amounts of carbon. Similarly, the Ti 2p3/2 peak is present on Ti, and the peak increases on NT because of the anodization process exposing a higher amount of titanium. After silanization, the Ti 2p3/2 peak decreased for both NT-S1 and NT-S2. Additionally, Si 1s peaks are present on both NT-S1 and NT-S2 since both the silanes contain silicon, along with a F 1s peak on NT-S2 since S2 contains fluorine (Fig. 3). The Ti 2p peak decreases after silanization because the silanes are deposited on the surfaces, reducing the relative composition of titanium on the surface.
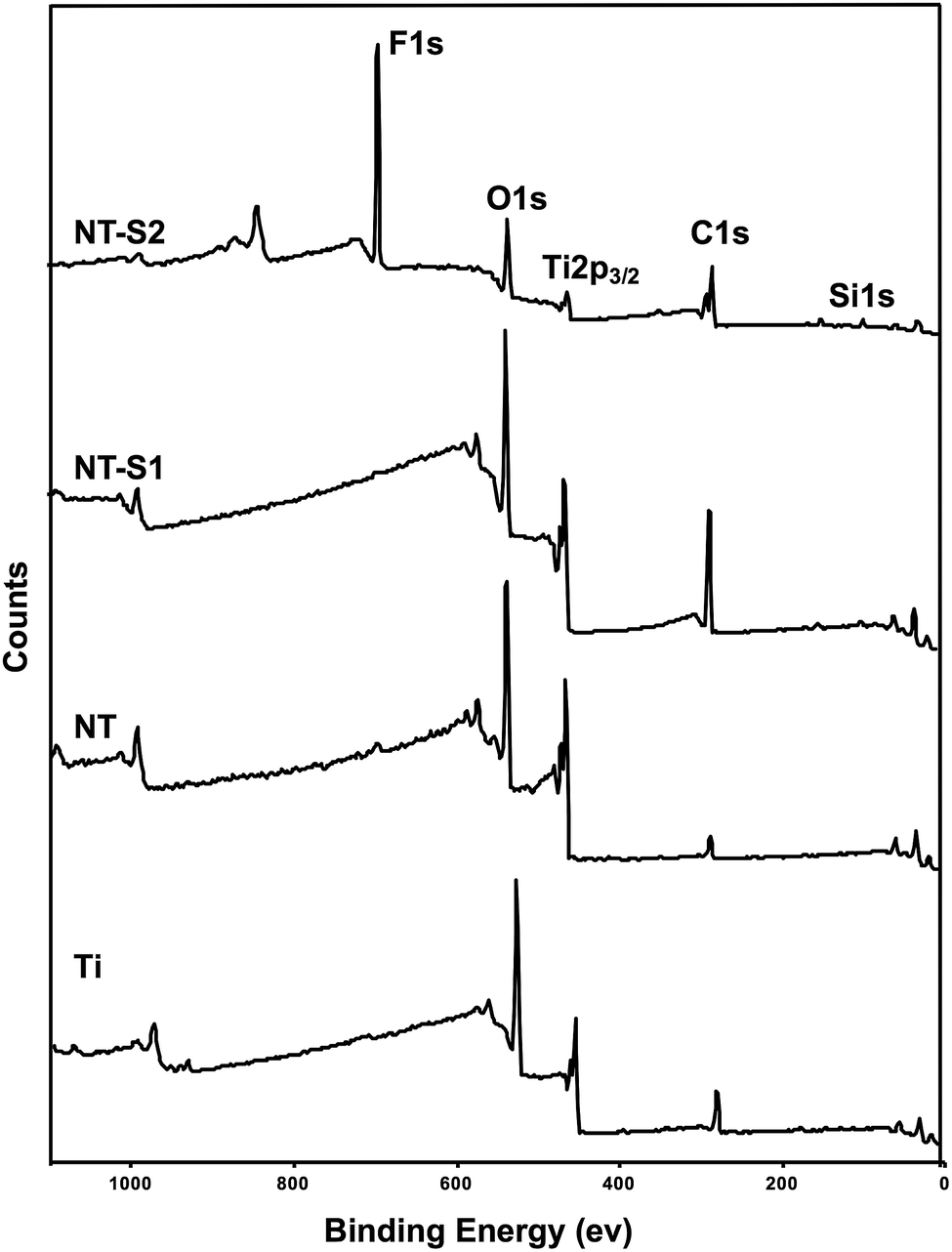 | ||
| Fig. 3 XPS survey scans for different surfaces. | ||
GAXRD was used to characterize the crystal structures on different surfaces. NT, NT-S1, and NT-S2 all have anatase and rutile crystal phases that are not present on Ti (Fig. 4). These crystal phases are formed during the annealing process for the surfaces. The rutile phases are the most stable phase, but previous work has shown that a higher presence of rutile phases will cause the nanotube arrays to fall apart.30 The anatase crystal structures have been shown to be metastable compare to rutile phase, and to be biocompatible, which is important for this application.39,40 However, the crystal structure does not differ between the NT, NT-S1, and NT-S2 surfaces. The results indicate that the silanization does not affect the crystal structure of the titania nanotube arrays.
 | ||
| Fig. 4 XRD scans for different surfaces. | ||
3.2 Protein adsorption on different surfaces
Protein adsorption on different surfaces was investigated by incubating the surfaces in solutions of human serum albumin and human fibrinogen and using XPS to determine how much protein was adsorbed onto the surface (Fig. 5). Albumin is a passivating protein which, when present on a surface, reduces the tendency of platelets to adhere on a surface. The high-resolution C 1s scans indicated three peaks: C–C, C–N, and N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O for all the substrates. A C–F peak is present for only the NT-S2 due to the presence of fluorine in S2. The N–CO peak is the amide peak, and is the characteristic peak for proteins on the surface.11 A precise way to characterize proteins adsorbed on the surface is to determine the contribution of N–CO (amide) peak (the N–CO peaks is at a shift 1.8 eV from the C–C peak) in the overall C 1s peak (Table 1). The results indicate that the Ti had the highest albumin adsorption, followed by NT, NT-S1 and NT-S2. (Table 1 (N–CO peak contribution), Fig. 5(a)). Previous studies have shown that titania nanotubes exhibit reduced protein adsorption compared to unmodified titanium.11 The high-resolution N 1s peak, which is characteristic to proteins as it is not inherently present on any surface, followed similar trend as that of N–CO peak (Fig. 5(a)). The SEM images of the surfaces exposed to albumin show that there was not significant visible adsorption on the NT-S1 and NT-S2 surfaces, while the albumin, a globular protein (shown by arrows in the images) was crystalized and adsorbed onto the Ti and NT (Fig. 5(a)). Fibrinogen is the main protein involved in thrombosis. When fibrinogen adsorbs onto a surface, it allows platelets to adhere, recruit leukocytes, and activate, which are the initial steps in clot formation. Fibrinogen is long and narrow protein, contrasting with albumin's globular structure, so it adsorbed more on all surfaces as compared to albumin. However, the fibrinogen adsorption followed similar trend as that of albumin adsorption with higher adsorption on Ti, followed by NT, NT-S1 and NT-S2 (Table 1 (N–CO peak contribution), Fig. 5(b)). The SEM images of surfaces exposed to fibrinogen that there was not significant visible adsorption on the NT-S1 and NT-S2 surfaces, while the fibrinogen, a planar protein (shown by arrows in the images), was crystalized and adsorbed onto the Ti and NT (Fig. 5(b)).
O for all the substrates. A C–F peak is present for only the NT-S2 due to the presence of fluorine in S2. The N–CO peak is the amide peak, and is the characteristic peak for proteins on the surface.11 A precise way to characterize proteins adsorbed on the surface is to determine the contribution of N–CO (amide) peak (the N–CO peaks is at a shift 1.8 eV from the C–C peak) in the overall C 1s peak (Table 1). The results indicate that the Ti had the highest albumin adsorption, followed by NT, NT-S1 and NT-S2. (Table 1 (N–CO peak contribution), Fig. 5(a)). Previous studies have shown that titania nanotubes exhibit reduced protein adsorption compared to unmodified titanium.11 The high-resolution N 1s peak, which is characteristic to proteins as it is not inherently present on any surface, followed similar trend as that of N–CO peak (Fig. 5(a)). The SEM images of the surfaces exposed to albumin show that there was not significant visible adsorption on the NT-S1 and NT-S2 surfaces, while the albumin, a globular protein (shown by arrows in the images) was crystalized and adsorbed onto the Ti and NT (Fig. 5(a)). Fibrinogen is the main protein involved in thrombosis. When fibrinogen adsorbs onto a surface, it allows platelets to adhere, recruit leukocytes, and activate, which are the initial steps in clot formation. Fibrinogen is long and narrow protein, contrasting with albumin's globular structure, so it adsorbed more on all surfaces as compared to albumin. However, the fibrinogen adsorption followed similar trend as that of albumin adsorption with higher adsorption on Ti, followed by NT, NT-S1 and NT-S2 (Table 1 (N–CO peak contribution), Fig. 5(b)). The SEM images of surfaces exposed to fibrinogen that there was not significant visible adsorption on the NT-S1 and NT-S2 surfaces, while the fibrinogen, a planar protein (shown by arrows in the images), was crystalized and adsorbed onto the Ti and NT (Fig. 5(b)).
 | ||
| Fig. 5 (a) High resolution C 1s and N 1s scans and SEM images for albumin adsorption on different surfaces (b) high resolution C 1s and N 1s scans and SEM images for fibrinogen adsorption on different surfaces. | ||
O and C–F peaks in the overall C 1s peak are given as percentages
| C–C | C–N | N–CO |
C–F | ||
|---|---|---|---|---|---|
| Albumin | Ti | 55.34 | 23.15 | 21.51 | 0 |
| NT | 59.3 | 22.52 | 18.18 | 0 | |
| NT-S1 | 69.14 | 17.07 | 13.79 | 0 | |
| NT-S2 | 43.47 | 16.05 | 11.00 | 29.48 | |
| Fibrinogen | Ti | 46.31 | 27.17 | 26.52 | 0 |
| NT | 50.69 | 26.55 | 22.75 | 0 | |
| NT-S1 | 55.08 | 24.86 | 20.6 | 0 | |
| NT-S2 | 40.67 | 27.19 | 17.45 | 14.69 | |
3.3 Cytotoxicity of different surfaces
An LDH (lactose dehydrogenase) assay was used to investigate the cytotoxicity of different surfaces. LDH is a chemical generated by cells when they are exposed to toxic environments. When the cells die the LDH is released, and thus the amount of LDH can be measured to determine how toxic a given substrate is. The assay catalyzes a reaction which uses the LDH present in a solution to create a formazan salt that absorbs at 490 nm. Because the amount of absorbance will be proportional to the amount of LDH present, the absorbance reading will indicate the cytotoxicity of the substrate. The results indicate that the PRP exposed to NT-S1 and NT-S2 showed higher amounts of LDH present compared to Ti and NT (p < 0.05) (Fig. 6). None of the substrates, however, caused enough LDH to be released to be considered cytotoxic (note: the high absorbance values for LDH were due to the use of path correction feature in the manufacturer provided software for the plate reader).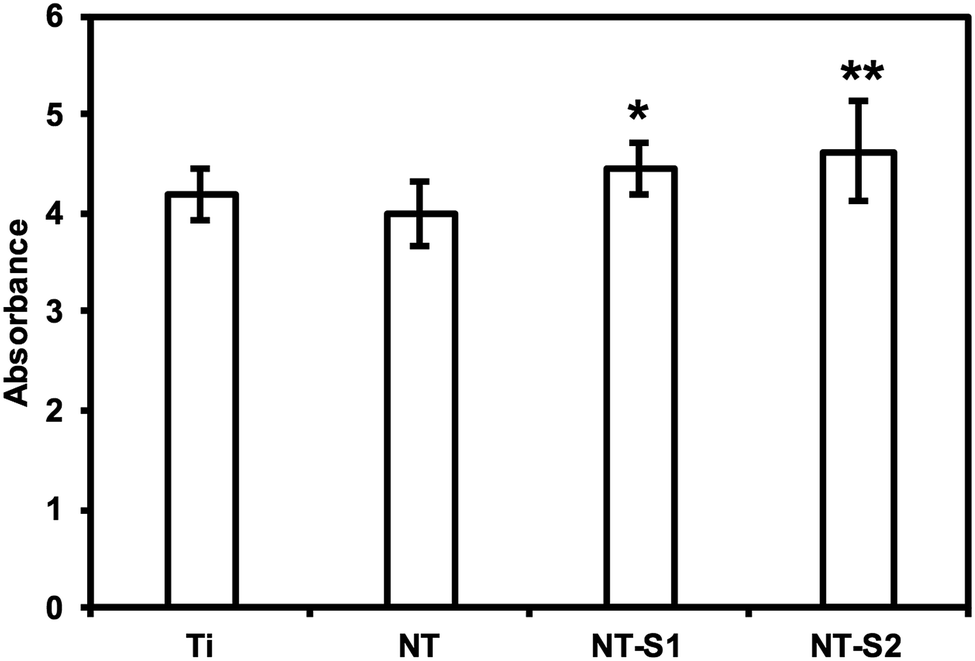 | ||
| Fig. 6 Cytotoxicity of different surfaces measured using LDH assay. NT-S1 and NT-S2 have significantly higher cytotoxicity than Ti and NT (p ≤ 0.05). | ||
3.4 Cell adhesion on different surfaces
Platelet and leukocyte adhesion was investigated by fluorescence microscopy. Platelet adhesion has previously been shown to be reduced on titania nanotube arrays compared to unmodified titanium.10 It was expected that the modification of titania nanotube arrays with silanes would further reduce the adhesion of platelets. The results indicate that NT-S1 and NT-S2 had lower cell adhesion compared to Ti and NT (Fig. 7(a)). The percent area of adhered platelets was calculated from the fluorescence microscopy images (Fig. 7(b)). Ti had the highest cell coverage, followed by NT. The NT-S1 and NT-S2 surfaces had lower cell coverage compared to Ti and NT (p ≤ 0.05) (Fig. 7(b). This result was expected because the superhydrophobicity of the NT-S1 and NT-S2 that minimizes the contact between the PRP and surface. Additionally, the reduced fibrinogen adsorption on NT-S1 and NT-S2 also will affect platelet adhesion. | ||
| Fig. 7 (a) Fluorescence microscopy images of calcein-AM stained cells on different surfaces. (b) The percentage surface area covered by cells on different surfaces. NT-S1 and NT-S2 have significantly lower cell adhesion than Ti or NT (p ≤ 0.05). | ||
PRP contains two types of cells – platelets and leukocytes. However, the calcein stain does not distinguish between the two types of cell since it stains the cytoplasm of cells. The adhesion of leukocytes and platelets was determined by staining the cells with rhodamine-phalloidin and DAPI, which stain cell cytoskeleton (actin) and cell nuclei respectively. Since platelets do not have nucleus, the DAPI stain indicates leukocytes, whereas the rhodamine-phalloidin will stain both platelets and leukocytes. Similar to the calcein stained images, the results of rhodamine-phalloidin stained images indicate that NT-S1 and NT-S2 had lower cell adhesion compared to Ti and NT (Fig. 8(a) and (b)). However, the DAPI stained images indicate that Ti had fewer leukocytes compared to NT, NT-S1, and NT-S2 (p ≤ 0.05) (Fig. 8(a) and (c)). The higher leukocyte adhesion on titania nanotube arrays was likely due to the presence of nanotopography on the surface, as previous studies have shown that surface roughness promotes the adhesion of leukocytes.33
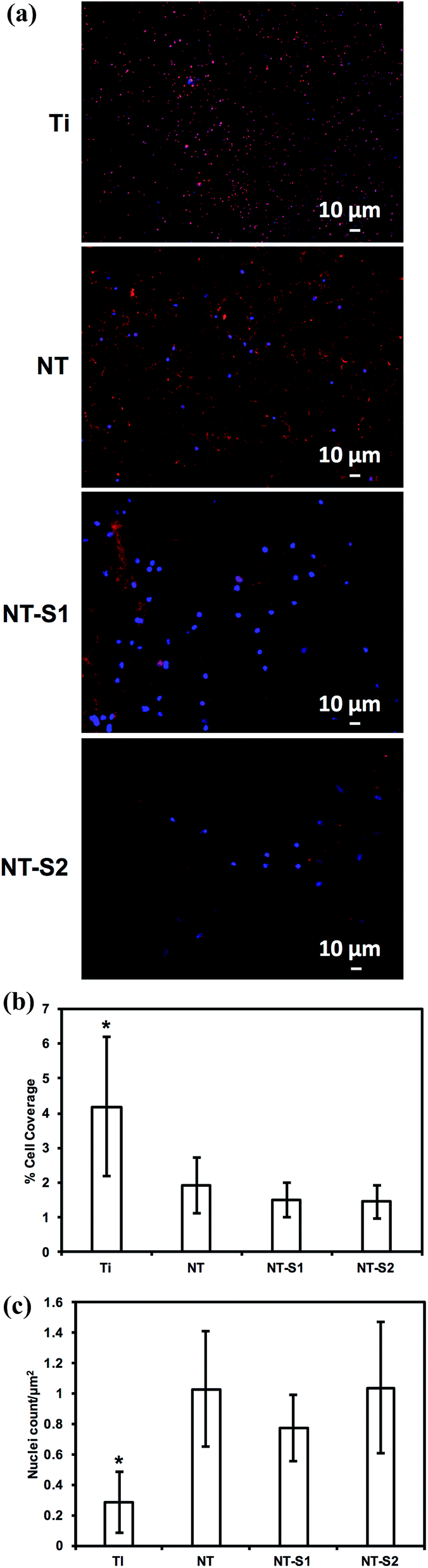 | ||
| Fig. 8 (a) Fluorescence microscopy images of rhodamine-phalloidin stained cells on different surfaces. (b) The percentage surface area covered by cells on different surfaces. (c) Nuclei count per μm2 on different surfaces. Ti have significantly lower leukocyte adhesion than NT, NT-S1 and NT-S2 (p ≤ 0.05). | ||
3.5 Platelet activation on different surfaces
SEM was used to investigate platelet activation on different surfaces. The degree of platelet activation was characterized through changes in the platelet shapes, extension of dendrites, and aggregation. A larger number of activated and aggregated platelets indicates a greater propensity for thrombosis. The SEM images were also used to count the number of un-activated, partially activated, and fully activated platelets. Platelet aggregation was seen on both the Ti and NT surfaces (Fig. 9(a)). The results indicate that Ti and NT had fewer un-activated platelets than partially or fully activated platelets (p ≤ 0.05) (Fig. 9(b)). Minimal platelet aggregation and adhesion was seen on NT-S1 and NT-S2 (Fig. 9(a)). Of the platelets that were attached, there were no significant differences between the number of activated and un-activated platelets NT-S1 and NT-S2 (Fig. 9(b)). | ||
| Fig. 9 (a) SEM images of cells on different surfaces. (b) Platelet count on different surfaces. For Ti and NT, the number of un-activated platelets was significantly lower than partially activated and fully activated platelets (p ≤ 0.05). NT-S1 and NT-S2 had lower counts of all morphologies of platelets (p ≤ 0.05). | ||
3.6 Hemolysis on different surfaces
The hemolysis of whole human blood on the different substrates was investigated by allowing drops of blood to clot on different surfaces for 15, 30, and 60 minutes and then measuring the absorbance of hemoglobin in the un-clotted blood. After the specific time interval, DI water was added to the well plate to lyse the red blood cells in un-clotted blood and release their hemoglobin. The absorbance of the solution is directly proportional to the amount of hemoglobin dissolved in the DI water, so a higher absorbance value means there is more hemoglobin dissolved in the water, corresponding to less blood clotted on the surface. After 15 min of blood clotting, Ti and NT had lower amounts of free hemoglobin than NT-S1 and NT-S2 indicating more clotting on the surface (p ≤ 0.05) (Fig. 10). The trends were similar after 30 and 60 min of hemolysis on different surfaces. However, after 60 min there was significantly lower amounts of free hemoglobin on Ti and NT, indicating significant clotting on these surfaces (p ≤ 0.05), whereas the amount of free hemoglobin for NT-S1 and NT-S2 was the same, indicating minimal clotting on these surfaces. This was expected because the superhemophobic surfaces have minimal contact with the blood due to the air pockets trapped between the blood and surface (note: the high absorbance values for hemoglobin shown in Fig. 10 were due to the use of path correction feature in the manufacturer provided software for the plate reader).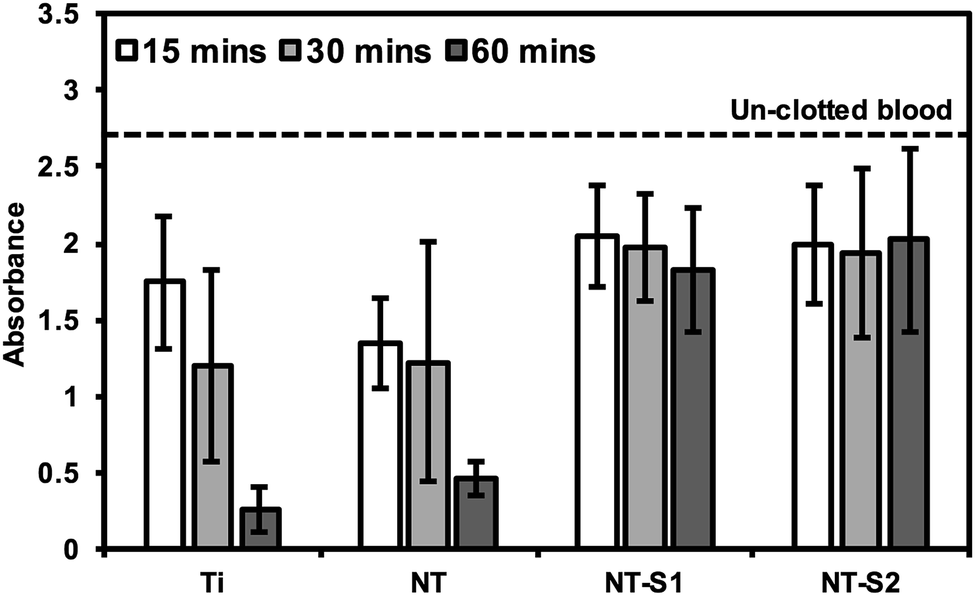 | ||
| Fig. 10 Whole blood clotting on different surfaces for up to 60 min. The dotted line represents the absorbance of free hemoglobin in un-clotted blood. | ||
4. Conclusion
Hemocompatibility remains a challenge for any blood-contacting medical device. Thrombosis is a major complication for stents, heart valves, and similar devices as it will obstruct the flow of blood. Reducing the incidence of thrombosis and clot formation on device surfaces is important to reduce device failure. Though there is no truly hemocompatible material, titania nanotube arrays have been shown to enhance hemocompatibility. Previous work with superhemophobic titania nanotube arrays has shown promising reductions in PRP cell adhesion, but the interactions of individual blood components have not been reported. In this work, we have fabricated superhemophobic titania nanotube arrays using anodization of titanium followed by modifying the surface chemistry by chemical vapor deposition of two silanes. The hemocompatibility of these surfaces was evaluated by measuring blood protein adsorption, cytotoxicity, platelet/leukocyte adhesion, platelet activation, and clotting. The results indicate that albumin and fibrinogen adsorption was reduced on superhemophobic surfaces as compared unmodified surfaces and that none of the surfaces were cytotoxic. Further, the superhemophobic surfaces significantly reduced PRP cell adhesion and platelet activation. These results indicate that superhemophobic surfaces are a potential approach for enhancing hemocompatibility of blood-contacting devices. The interactions of blood with superhemophobic surfaces should be investigated further, including for longer time periods than the 1–3 hour periods investigated in this study.Acknowledgements
The authors would like to acknowledge Prof. Ana Paula Rosifini Alves Claro from UNESP-Guaratingueta, Brazil and Prof. Paulo Soares from PUCPR, Brazil with their assistance with XRD analysis.References
- W. Schmidt, P. Behrens, C. Brandt-Wunderlich, S. Siewert, N. Grabow and K. P. Schmitz, Cardiovasc. Revasc. Med., 2016, 17, 375–383 CrossRef PubMed.
- H. Liu, C. Pan, S. Zhou, J. Li, N. Huang and L. Dong, Mater. Sci. Eng., C, 2016, 69, 1175–1182 CrossRef CAS PubMed.
- N. C. Hansson, E. L. Grove, H. R. Andersen, J. Leipsic, O. N. Mathiassen, J. M. Jensen, K. T. Jensen, P. Blanke, T. Leetmaa, M. Tang, L. R. Krusell, K. E. Klaaborg, E. H. Christiansen, K. Terp, C. J. Terkelsen, S. H. Poulsen, J. Webb, H. E. Bøtker and B. L. Nørgaard, J. Am. Coll. Cardiol., 2016, 68, 2059–2069 CrossRef PubMed.
- R. R. Makkar, G. Fontana, H. Jilaihawi, T. Chakravarty, K. F. Kofoed, O. de Backer, F. M. Asch, C. E. Ruiz, N. T. Olsen, A. Trento, J. Friedman, D. Berman, W. Cheng, M. Kashif, V. Jelnin, C. A. Kliger, H. Guo, A. D. Pichard, N. J. Weissman, S. Kapadia, E. Manasse, D. L. Bhatt, M. B. Leon and L. Søndergaard, N. Engl. J. Med., 2015, 373, 2015–2024 CrossRef CAS PubMed.
- E. De Marchena, J. Mesa, S. Pomenti, C. Marin Y Kall, X. Marincic, K. Yahagi, E. Ladich, R. Kutz, Y. Aga, M. Ragosta, A. Chawla, M. E. Ring and R. Virmani, JACC Cardiovasc. Interv., 2015, 8, 728–739 CrossRef PubMed.
- L. Ge, L. P. Dasi, F. Sotiropoulos and A. P. Yoganathan, Ann. Biomed. Eng., 2008, 36, 276–297 CrossRef PubMed.
- H. a. Simon, H.-L. Leo, J. Carberry and A. P. Yoganathan, Ann. Biomed. Eng., 2004, 32, 1607–1617 CrossRef PubMed.
- J.-F. Timsit, Y. Dubois, C. Minet, A. Bonadona, M. Lugosi, C. Ara-Somohano, R. Hamidfar-Roy and C. Schwebel, Ann. Intensive Care, 2011, 1, 34 CrossRef PubMed.
- C. Werner, M. F. Maitz and C. Sperling, J. Mater. Chem., 2007, 17, 3376–3384 RSC.
- B. S. Smith, P. Capellato, S. Kelley, M. Gonzalez-Juarrero and K. C. Popat, Biomater. Sci., 2013, 1, 322–332 RSC.
- V. Leszczak, B. S. Smith and K. C. Popat, J. Biomater. Sci., Polym. Ed., 2013, 37–41 Search PubMed.
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86–100 CrossRef CAS PubMed.
- S. Franz, S. Rammelt, D. Scharnweber and J. C. Simon, Biomaterials, 2011, 32, 6692–6709 CrossRef CAS PubMed.
- M. Hisagi, T. Nishimura, M. Ono, S. Gojo, K. Nawata and S. Kyo, J. Artif. Organs, 2010, 13, 174–177 CrossRef CAS PubMed.
- N. A. Riedel, B. S. Smith, J. D. Williams and K. C. Popat, Mater. Sci. Eng., C, 2012, 32, 1196–1203 CrossRef CAS.
- E. A. Bender, M. D. Adorne, L. M. Colomé, D. S. P. Abdalla, S. S. Guterres and A. R. Pohlmann, Int. J. Pharm., 2012, 426, 271–279 CrossRef CAS PubMed.
- N. R. Kuznetsova, C. Sevrin, D. Lespineux, N. V. Bovin, E. L. Vodovozova, T. Mészáros, J. Szebeni and C. Grandfils, J. Controlled Release, 2012, 160, 394–400 CrossRef CAS PubMed.
- W. C. Lin, T. Y. Liu and M. C. Yang, Biomaterials, 2004, 25, 1947–1957 CrossRef CAS PubMed.
- C.-C. Chou, S.-W. Hsin, H.-C. Lin, C.-H. Yeh, R. Wu and W.-J. Cherng, Surf. Coat. Technol., 2016, 303, 277–282 CrossRef CAS.
- C. C. Chou, H. J. Zeng and C. H. Yeh, Thin Solid Films, 2013, 549, 117–122 CrossRef CAS.
- N. Weber, H. P. Wendel and G. Ziemer, Biomaterials, 2002, 23, 429–439 CrossRef CAS PubMed.
- Z. Zheng, W. Wang, X. Huang, Q. Lv, W. Fan, W. Yu, L. Li and Z. Zhang, Artif. Organs, 2016, 40, E219–E229 CrossRef CAS PubMed.
- R. Blezer, B. Fouache, G. M. Willems and T. Lindhout, J. Biomed. Mater. Res., 1997, 37, 108–113 CrossRef CAS PubMed.
- X. Jin, J. Yuan and J. Shen, Colloids Surf., B, 2016, 145, 275–284 CrossRef CAS PubMed.
- P. Hamerli, T. Weigel, T. Groth and D. Paul, Biomaterials, 2003, 24, 3989–3999 CrossRef CAS PubMed.
- R. K. Roy, H. W. Choi, J. W. Yi, M. W. Moon, K. R. Lee, D. K. Han, J. H. Shin, A. Kamijo and T. Hasebe, Acta Biomater., 2009, 5, 249–256 CrossRef CAS PubMed.
- V. B. Damodaran, D. Bhatnagar, V. Leszczak and K. C. Popat, RSC Adv., 2015, 5, 37149–37171 RSC.
- B. S. Smith, S. Yoriya, T. Johnson and K. C. Popat, Acta Biomater., 2011, 7, 2686–2696 CrossRef CAS PubMed.
- S. Movafaghi, V. Leszczak, W. Wang, J. A. Sorkin, L. P. Dasi, K. C. Popat and A. K. Kota, Adv. Healthcare Mater., 2016, 1600717 Search PubMed.
- J. A. Sorkin, S. Hughes, P. Soares and K. C. Popat, Mater. Sci. Eng., C, 2015, 49, 735–745 CrossRef CAS PubMed.
- P. Capellato, A. L. Escada, K. C. Popat and A. P. Claro, J. Biomed. Mater. Res., Part A, 2013, 2147–2156 Search PubMed.
- P. Capellato, B. S. Smith, K. C. Popat and A. P. R. A. Claro, Mater. Sci. Eng., C, 2012, 32, 2060–2067 CrossRef CAS.
- V. Leszczak and K. C. Popat, ACS Appl. Mater. Interfaces, 2014, 6, 15913–15924 CAS.
- N. A. Riedel, J. D. Williams and K. C. Popat, J. Mater. Sci., 2011, 46, 6087–6095 CrossRef CAS.
- B. S. Smith, S. Yoriya, L. Grissom, C. A. Grimes and K. C. Popat, J. Biomed. Mater. Res., Part A, 2010, 95(2), 350–360 CrossRef PubMed.
- N. A. Riedel, S. L. Bechara, K. C. Popat and J. D. Williams, Mater. Lett., 2012, 81, 158–161 CrossRef CAS.
- S. R. Coulson, I. S. Woodward, J. P. S. Badyal, S. A. Brewer and C. Willis, Chem. Mater., 2000, 12, 2031–2038 CrossRef CAS.
- O. M. Roscioni, L. Muccioli, A. Mityashin, J. Cornil and C. Zannoni, J. Phys. Chem. C, 2016, 120, 14652–14662 CAS.
- D. Zhang, G. Li, H. Wang, K. M. Chan and J. C. Yu, Cryst. Growth Des., 2010, 10, 1130–1137 CAS.
- M. M. Hasan, A. S. M. A. Haseeb, R. Saidur, H. H. Masjuki, World Acad. Sci. Eng. Technol., 2008, vol. 16, pp. 221–225 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |