
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards sustainable hydrogenation of 5-(hydroxymethyl)furfural: a two-stage continuous process in aqueous media over RANEY® catalysts
Sérgio Lima ,
David Chadwick and
Klaus Hellgardt*
,
David Chadwick and
Klaus Hellgardt*
Department of Chemical Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: k.hellgardt@imperial.ac.uk; Tel: +44 (0)20 759 45577
First published on 19th June 2017
Abstract
The hydrogenation of 5-(hydroxymethyl)furfural (HMF) to 2,5-bis(hydroxymethyl)tetrahydrofuran (DHMTHF) in aqueous media under relatively mild reaction conditions has been investigated over heterogeneous RANEY® Cu and Ni catalysts using a continuous-flow hydrogenation reactor. These RANEY® catalysts were selected following a screening of several catalysts including precious metals supported on carbon for the hydrogenation of HMF. A single-stage versus a two-stage process for the hydrogenation of HMF into DHMTHF, i.e. via 2,5-dihydroxymethylfuran (DHMF) has been evaluated. The best result with an average selectivity of 98% for DHMTHF was obtained using a two-stage process; RANEY® Cu was used as a catalyst for the highly selective hydrogenation of HMF to DHMF (92 mol%) in the first stage and this product was used without further purification for in a second-stage selective hydrogenation of DHMF into DHMTHF using RANEY® Ni as a catalyst. The influence of the HMF concentration in the feeding solution (1–3 wt%), flow rate (0.05–0.25 mL min−1) and total pressure (20–90 bar) were investigated for the first-stage hydrogenation of HMF into DHMF over RANEY® Cu. HMF was found to exert an inhibiting effect on the conversion due to strong adsorption. The RANEY® Ni catalyst used in the second stage gradually deactivated. A procedure for in situ regeneration of the partially deactivated RANEY® Ni catalyst using acetic acid washing was investigated with limited success.
1. Introduction
During the coming decades the world will be challenged to replace a major part of its use of oil based feedstocks for the production of new commodity chemicals and fuels. A non-petrochemical technology platform requires synthesis of next-generation as well as existing functional organic materials from sustainable intermediates. In this context, particular attention is now given to the use of renewable biomass in the chemical industry, where lignocellulosic biomass and non-edible resources have the potential to provide direct access to valuable chemicals.1–3 Ideally, this would employ continuous processes, inexpensive catalysts, and preferentially allow retrofitting of existing industrial processes.5-(Hydroxymethyl)furfural (HMF) can be obtained from carbohydrate biomass, i.e. lignocellulosic biomass, in particular from the dehydration of C-6 carbohydrates (for example D-glucose and D-fructose – constituent lignocellulosic biomass monomers), using homogeneous and heterogeneous acid catalysts.4–6 HMF is considered one of the most promising platform chemicals in the biorefinery, providing access to a wide range of alternative polymer-building blocks.7–9 Among the furanic intermediates that are highly attractive as alternative renewable polymer-building blocks are the HMF-based di-ols 2,5-dihydroxymethylfuran (DHMF) and 2,5-bis(hydroxymethyl)tetrahydrofuran (DHMTHF). DHMF is considered an analogue of m/p-benzenedimethanol, which is currently used for the production of phenolic and aromatic resins, homopolymers, polyesters and polyurethanes, and can be obtained by the selective hydrogenation of HMF, i.e. reductive hydrogenation of the carbonyl group (see Scheme 1(1)).10 DHMTHF is considered an analogue of 1,3/1,4-cyclohexanedimethanol currently used for poly-ester production and can find several applications, for example as a solvent or an intermediate for pharmaceuticals production. It can be obtained by total hydrogenation of HMF shown in Scheme 1. In principle, DHMTHF can be converted to 1,6-hexanediol (1,6HD) by ring opening and hydrogenolysis or hydrodeoxygenation, which opens a route to the industrial production of polyamines such as nylon-6.3,10,11
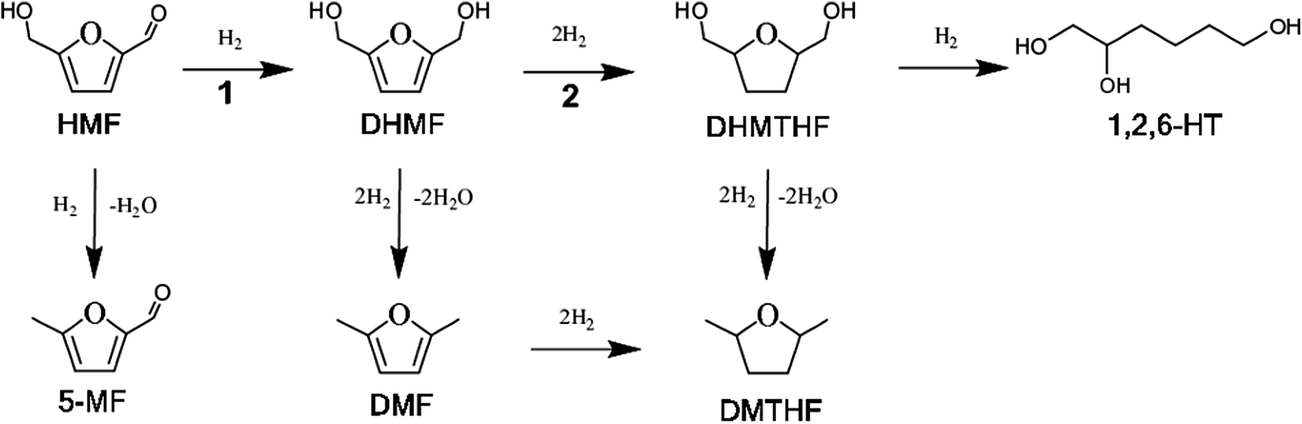 | ||
| Scheme 1 Schematic reductive hydrogenation and hydrogenolysis/hydrodeoxygenation of HMF. HMF: 5-hydroxymethylfurfural, DHMF: 2,5-dihydroxymethylfuran, 5-MF: 5-methylfurfural, DMF: dimethylfurfural, DMTHF: 2,5-dimethyltetrahydrofuran, DHMTHF: 2,5-bis(hydroxymethyl)tetrahydrofuran, 1,2,6-HT: 1,2,6-hexanetriol. | ||
Several studies have been reported concerning the catalytic hydrogenation of HMF to DHMF using metal-based heterogeneous catalysts especially over various metals such as Au, Cu, Ni, Pt, Pd, Ir, Ru – for example bimetallic nickel and iron supported on carbon nanotubes catalyst or copper supported in γ-alumina.10–22 The choice of catalysts and reaction conditions governs the product distribution.10–22 Various organic solvents including 1,4-dioxane, ethanol, 1-propanol or organic solvent–water biphasic mixtures such as n-butanol/water, THF/water or toluene/water have been used in those reported studies.10–22 DHMF selectivities and HMF conversions higher than 96% using 1 wt% Pt on MCM-41 (Pt/MCM-41), gold sub-nano clusters supported on alumina (Au/Al2O3) or ReOx modified SiO2-supported metal catalysts with iridium (Ir–ReOx/SiO2) as catalysts have been reported at temperatures between 30 and 110 °C and pressures of 6.9 to 65 bar after 2–6 hours using water as monophasic solvent system.18–22
A considerable number of studies have been reported for the synthesis of DHMTHF from HMF.11,22,23,23–30 Due to the relatively stable furan ring, typically harsher conditions are required (i.e. higher pressure and temperatures) compared to the reduction of the aldehyde group of HMF to form DHMF.23 Common materials investigated as heterogeneous catalysts include non-noble and noble metals, especially bifunctional catalysts derived from hydrotalcite-like compounds, bimetallic catalysts supported on silica as Ni–Pd/SiO2 and Pd–Ir/SiO2 and RANEY®-type metal catalysts (Co, Cu, Ni) and others using different monophasic organic solvents.11,17,22–26,28
RANEY®-type metals (Cu, Co and Ni) have been investigated as catalyst for total hydrogenation of HMF using 1,4-dioxane as solvent by Kong et al.25 Among the RANEY®-type metals, RANEY® Ni was revealed to be the most selective with a 96% DHMTHF selectivity at full conversion, at 100 °C and 15 bar of H2 after 15 h of reaction.25 RANEY® Ni has also been successfully used to catalyse the hydrogenation of HMF into DHMTHF in other solvents such as methanol or ethanol with yields higher than 95%.11,26 The best result was reported by Connolly et al. using methanol as solvent, achieving 99% selectivity to DHMTHF and over 95% HMF conversion under relatively mild reaction conditions, 60 °C under 4.8 bar of H2 after 4 h of batch operation.26
All the above research has been conducted in batch reactors. To our knowledge only three papers have addressed the synthesis of DHMF or DHMTHF using heterogeneous catalytic continuous hydrogenation of HMF, although indirectly, despite the potential advantages of catalytic continuous-flow hydrogenation for large-scale industrial production compared with batch reactor processes.31–39 The catalysts studied include bifunctional oxides with Brønsted or Lewis acidy such as γ-Al2O3, ZrO2, TiO2, Al2O3/SBA-15, ZrO2/SBA-15, TiO2/SBA-15, H-BEA, Sn-BEA ZrO2, and supported metals such as Fe2O3-supported Pd, and Ni, Pd, and Cu supported on γ-alumina.31–39 Tucker et al. proposed a tandem catalytic approach to continuous production of DHMTHF from fructose, in which fructose is first dehydrated to HMF, then HMF is hydrogenated to DHMTHF.40 A certain amount of DHMTHF is recovered as product, while the remainder is recycled to serve as the co-solvent in the dehydration reaction.40 However, no catalytic results were reported by the authors using a flow hydrogenation reactor.40
In this paper we report the selective catalytic synthesis of DHMTHF from HMF using a continuous hydrogenation reactor system. A single-stage process to DHMTHF and a two-stage process via the selective synthesis of the intermediate DHMF have been investigated. These studies have been performed in aqueous media at the relatively mild conditions of 90 °C and 90 bar H2. The performance of heterogeneous RANEY® Cu and Ni catalysts has been compared with common supported precious metal catalysts. The catalysts were in the form of commercial CatCart® supplied by ThalesNano Inc. RANEY® catalysts have the advantage of being inexpensive compared to precious metal catalysts. We show that the two-stage process using RANEY® Cu in the first stage and RANEY® Ni in the second stage is the most efficient system for selective synthesis of DHMTHF from HMF at the mild conditions used. A pathway for in situ regeneration of RANEY® Ni catalyst is also proposed in this paper.
2. Experimental section
5-(Hydroxymethyl)furfural was purchased from Sigma-Aldrich and used as received without further purification. For the catalytic reactions a fixed-bed bench-top continuous-flow hydrogenation reactor, H-CUBE® model HC 2, developed by ThalesNano Inc. (Budapest, Hungary) was used.35,36 Commercial pre-packed heterogeneous catalyst cartridges (CatCat®) of 4 mm inner diameter and 30 mm length (tubular) filled with ca. 140 mg of catalyst (particle size of 50–400 μm) were used. The general protocol was as follows: the CatCart® was placed vertically in the H-Cube system cartridge holder and rinsed with degassed, deionised water followed by activation by pretreatment with water saturated with hydrogen through the CatCart® by an HPLC-pump (flow rate of 0.25 mL min−1) for 30 min at the selected temperature and pressure, usually 90 bar. The catalytic reaction was performed by feeding a continuous flow of the substrate solution saturated with hydrogen. The performance of the selected CatCart® was evaluated by analysing the liquid samples collected on an hourly basis after the start of the experimental run feeding the substrate solution. Steady state was reached after 1 h of time on stream. The average conversions and product selectivities were calculated, for example over the period of 1–8 h, by averaging the point conversions and selectivities obtained from the liquid samples collected at each hour interval from 1 to 8 h time-on-stream (7 points). The product solutions were analysed by a GC (Hewlett-Packard HP 6890 series) equipped with a 30 m StabilWAX(R)-DA column (30 m length, 0.32 mm internal diameter, and 0.5 μm film thickness) and a flame ionization detector (FID), injection volume 0.5 μL, inlet temperature 250 °C, detector temperature 250 °C, and a split ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. The initial column temperature was 50 °C (5 min) with a temperature rise of 10 °C min−1 and final temperature was 240 °C. Nitrogen was used as the carrier gas. Each peak of the GC chromatogram was properly integrated and the actual concentration of each product was obtained from a pre-calibrated plot of peak area against concentrations. Phenol was used as internal standard. Conversion, selectivities and yields were calculated with the following eqn (eqn (1)–(3)):
:5. The initial column temperature was 50 °C (5 min) with a temperature rise of 10 °C min−1 and final temperature was 240 °C. Nitrogen was used as the carrier gas. Each peak of the GC chromatogram was properly integrated and the actual concentration of each product was obtained from a pre-calibrated plot of peak area against concentrations. Phenol was used as internal standard. Conversion, selectivities and yields were calculated with the following eqn (eqn (1)–(3)):
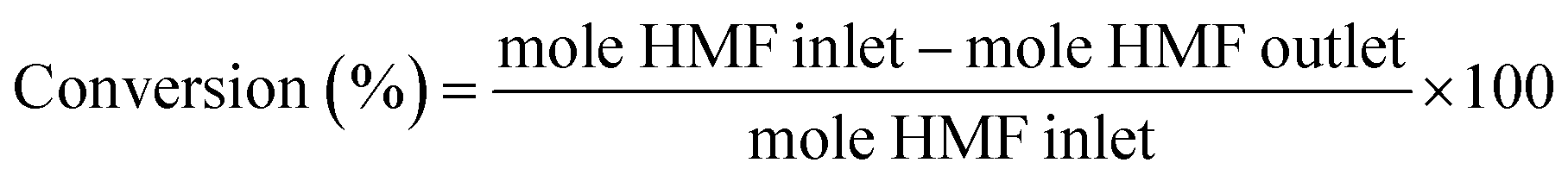 | (1) |
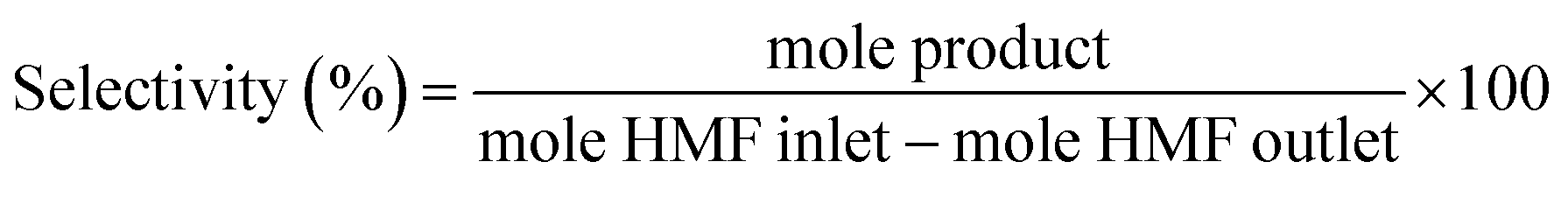 | (2) |
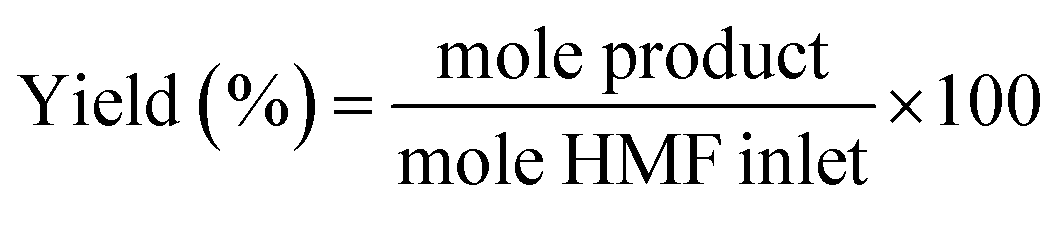 | (3) |
Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analyses were performed using a ICP-OES spectrometer Perkin-Elmer Optima 2000 DV ICP.
3. Results and discussion
The reaction network for the catalytic hydrogenation of HMF is given in Scheme 1. The reactions of interest in the present study are HMF to DHMF (reaction 1) and DHMF to DHMTHF (reaction 2). The hydrogenolysis/hydrodeoxygenation of HMF to 5-methylfurfural (5-MF), and the further conversion of DMF, DHMF and DHMTHF to dimethylfurfural (DMF), 2,5-dimethyltetrahydrofuran (DMTHF) among others are herein referred to as giving rise to by-products.The present catalytic hydrogenation results were obtained in aqueous media using heterogeneous pre-packed catalyst cartridges in a continuous-flow hydrogenation bench-top reactor (H-Cube) as noted above, where molecular hydrogen was generated in situ by the electrolysis of water and continuously mixed into the flowing aqueous solution of feed, for example HMF.
3.1 Single-stage catalytic hydrogenation of HMF
The single-stage hydrogenation of HMF to DHMTHF was investigated using a range of commercially available cartridge catalysts. These included precious metals supported on carbon and SiO2, heterogeneous RANEY® catalysts (Ni, Cu), and Ni/SiO2–Al2O3, 5% Rh/Al2O3 as CatCart® catalysts. The reactions were performed using aqueous solutions of 1 wt% HMF as feeding solution (feed flow rate of 0.05 mL min−1) under mild reaction condition (90 °C, 90 bar H2) and a liquid hourly space velocity (LHSV) of 7.95 h−1, calculated considering the internal cartridge volume = 3.77 × 10−7 m3, equivalent to a mean liquid residence time for this feed flow rate of 4.16 min based on the average dead volume for 30 mm filled CatCart = 0.208 mL (value provided by ThalesNano). The conversion of HMF to DHMF, DHMTHF or by-products using Darco® CatCart® (carbon black) as blank at these conditions was negligible after 8 h on stream.The average conversion and product selectivity for the studied catalysts are given in Table 1. The average conversion of HMF was in range of 70–100% for most of the catalysts, with the exception of Ni/SiO2–Al2O3, 5% Ru/C (Table 1, entry 1 and 4). In general, the catalysts investigated exhibited poor selectivity for the single-stage synthesis of DHMTHF. Average selectivity from 1–8 h on stream was less than 20% for DHMTHF, showing that these catalysts are not effective for a continuous single-stage synthesis of DHMTHF from HMF in aqueous solution under the reaction conditions investigated. The mass balance are not closed for most of catalysts investigated, except for Ni/SiO2–Al2O3 and Rh/Al2O3, which can be consequence of either formation of insoluble by-products and/or because not all by-products formed were detected by the GC-FID analysis. RANEY® Ni catalyst gave a 13% average selectivity to DHMTHF which corresponded to an average productivity of 0.2 mmolDHMTHF.gcatalyst−1 h−1 after 8 h of operation. Nevertheless, RANEY® Cu as catalyst (Table 1, entry 3) showed a stable average selectivity to DHMF of 84% at and an average HMF conversion of 94%. This result suggested that a two-stage continuous process might prove attractive.
| Entry | Catalyst | Conv.b (%) | Bio-products selectivityb,c (%) | Total yieldd (%) | ||||
|---|---|---|---|---|---|---|---|---|
| DMF | 5-MF | DMTHF | DHMF | DHMTHF | ||||
| a Reaction conditions: [HMF]0 = 1 wt% in water, 0.05 mL min−1, 90 °C, 90 bar h.b The average HMF conversion, average selectivities, and average yields were calculated by averaging the moles of HMF or bio-products obtained from the GC-FID analysis of the liquid samples collected at each hour from 1 h to 8 h of time on-stream (7 points). The steady state was reached after 1 h of time on stream.c In brackets is provided the yield.d Total yield was calculated as the sum of the yields for the identified by-products. | ||||||||
| 1 | Ni/SiO2–Al2O3 | 52 | — | 2 (1) | — | 93 (48) | 5 (3) | 52 |
| 2 | RANEY® Ni | 100 | — | 3 (3) | — | 60 (60) | 13 (13) | 76 |
| 3 | RANEY® Cu | 94 | — | 9 (8) | — | 84 (79) | — | 87 |
| 4 | 5% Ru/C | 100 | — | — | 4 (4) | 27 (27) | 18 (18) | 49 |
| 5 | 4.5% Ru 0.5% Pd/C | 96 | — | 4 (4) | 7 (7) | 30 (30) | 6 (6) | 45 |
| 6 | 1% Pt/SiO2 | 69 | — | 5 (3) | — | 78 (54) | — | 57 |
| 7 | 10% Pt/C | 99 | 1 (1) | 10 (10) | 3 (3) | 44 (44) | 1 (1) | 58 |
| 8 | 5% Pd/SiO2 | 100 | — | 2 (2) | 4 (4) | 17 (17) | 16 (16) | 39 |
| 9 | 10% Pd/C | 100 | — | — | 21 (21) | — | 17 (17) | 38 |
| 10 | 5% Rh/Al2O3 | 74 | — | 5 (4) | 7 (5) | 81 (60) | 7 (5) | 74 |
3.2 Two-stages catalytic hydrogenation of HMF
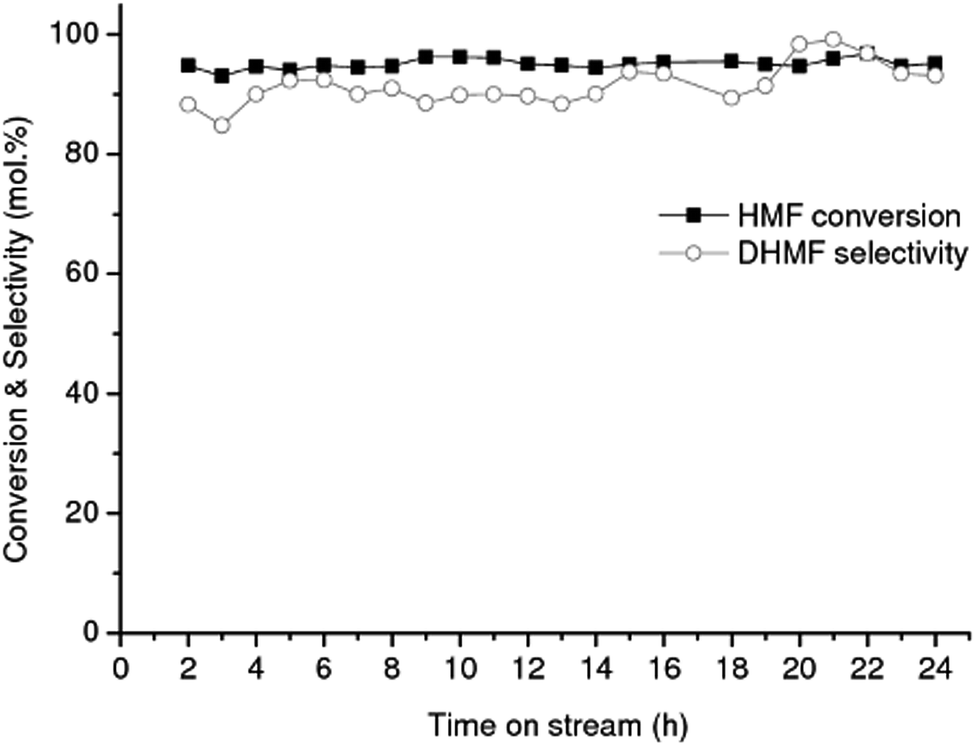 | ||
| Fig. 1 Conversion of HMF into DHMF over RANEY® Cu catalyst. Reaction conditions: flow rate of 0.05 mL min−1, 1 wt% HMF in water, 90 bar, 90 °C. | ||
The effect of total pressure of the system on the HMF conversion to DHMF was investigated in range of 20–100 bar over the RANEY® Cu catalyst at 90 °C, Fig. 2(a). Each point in the plot corresponds to an average value obtained after 8 h of on-stream operation. An excellent linear correlation (R2 = 0.99) for the reaction activity was observed up to 70 bar (flow rate: 0.05 mL min−1, 90 °C, 1 wt% HMF), showing that the hydrogenation of HMF is strongly dependent on the hydrogen concentration up to this pressure (Fig. 2(a)), due to the limiting stoichiometric hydrogen availability up to this pressure (mole fraction of hydrogen per moles of HMF in aqueous phase = 0.74 at 70 bar and 0.96 at 90 bar).41,42 Similar results were reported by Tukacs et al. for the hydrogenation of levulinic acid to γ-valerolactone using Ru/C® and butyl-bis-(m-sulfonated-phenyl)phosphine ligand as co-catalyst in aqueous media.43 The conversions reported in Fig. 2(a) are not limited by thermodynamic equilibrium based on the temperature and pressure dependence of the thermodynamic equilibrium constant for the hydrogenation of HMF to DHMF as previously reported.45,46
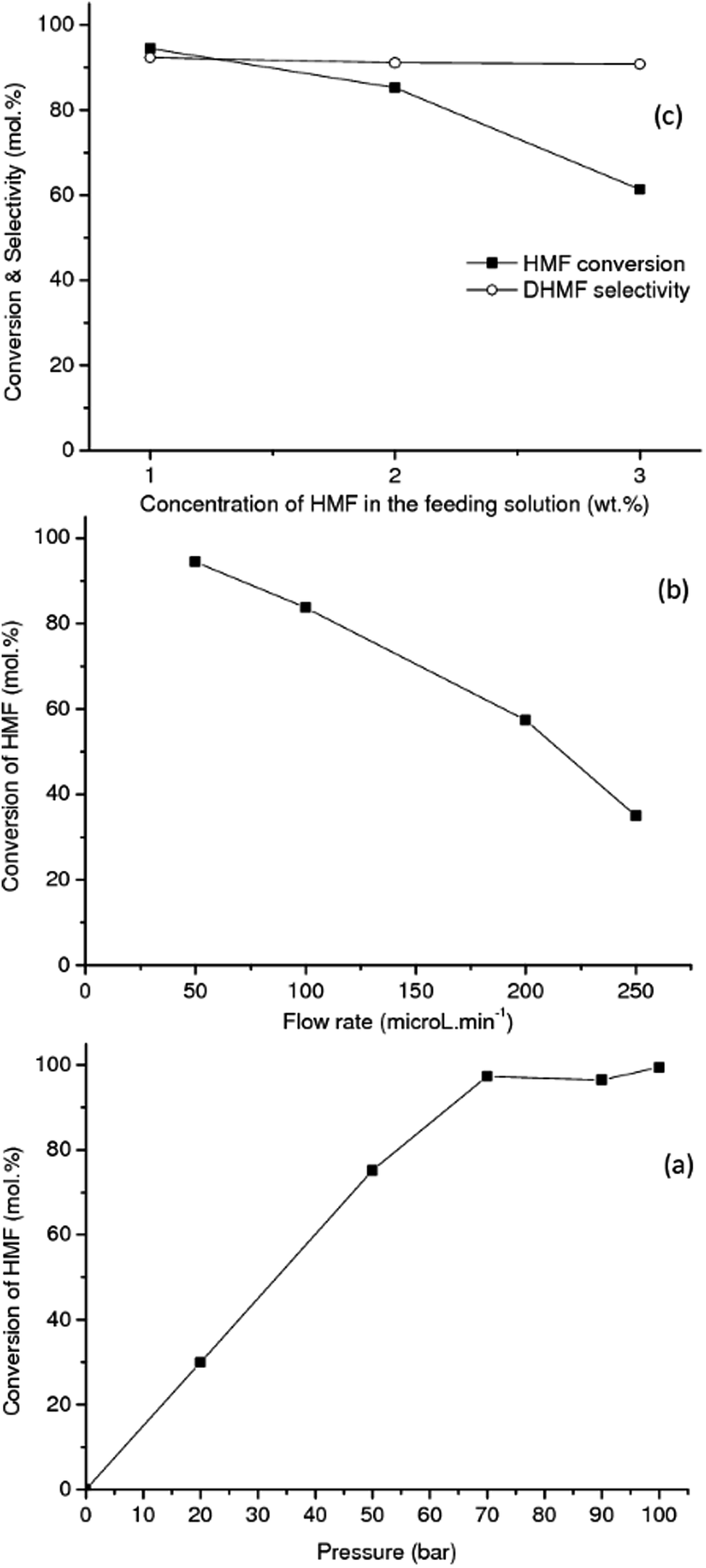 | ||
| Fig. 2 Effect of: (a) total pressure (flow rate: 0.050 mL min−1, 1 wt% HMF), (b) feed flow rate (pressure: 90 bar, 1 wt% HMF in water) and (c) concentration of substrate (flow rate: 0.05 mL min−1, pressure: 90 bar) on the HMF hydrogenation to DHMF in the presence of RANEY® Cu at 90 °C in aqueous media. Each point correspond to an average after 8 h of time on stream. | ||
The influence of the flow rate on the hydrogenation of HMF at a concentration of 1 wt% was investigated in the range of 0.05–0.25 mL min−1 (LHSV: 7.95–39.78 h−1) at 90 °C and 90 bar by using the RANEY® Cu as catalyst (Fig. 2(b)). As the flow rate increases in the range of 0.05–0.25 mL min−1 the conversion of HMF decreases, as expected. An excellent linear correlation (R2 = 0.99) was observed, being consistent with a zero-order kinetic regime in the concentration of HMF. In a similar way, the hydrogenation reaction of furfural to furfuryl alcohol in the liquid-phase, was reported to be negative first-order to zero-order in furfural concentration (0.025–0.1 M), and close to first-order with respect to hydrogen pressure, in the temperature range of 50–90 °C.46 However, closer to first-order dependence was observed with respect to both HMF and hydrogen, reported for the hydrogenation of HMF to DHMF in aqueous media in the concentration range 0.05–0.3 M and temperature range 40–70 °C, over Ru/C.21
The influence of the HMF concentration in the feeding solution was also investigated. Fig. 2(c) shows the results for the hydrogenation of HMF to DHMF with feed concentrations in the range of 1–3 wt% (i.e. from 8.2 to 24.7 mM) at 90 °C and 90 bar total pressure over RANEY® Cu. As the concentration of HMF increases from 1 to 3 wt%, the conversion of HMF decreases, as expected due to strong adsorption of HMF, and matches the observed flow rate dependence. Interestingly, in the range of HMF concentrations investigated, the selectivity to DHMF (average ca. 92%) did not change significantly with the increase of the HMF concentration in the feeding aqueous solutions.
The RANEY® Ni catalyst exhibited exceptional selectivity toward DHMTHF, with an average selectivity of 98% and a high productivity = 19 mmolDHMTHF.gcatalyst−1 h−1 after 15 h of operation, Fig. 3. Measurement of the amount of leaching gave 4.9 ppm of Ni and 0.023 ppm of Al in the product liquid corresponding to the total mass of Ni and Al leached after 15 h of operation of 0.18 wt% and <0.01 wt% respectively, based on the initial specified composition of RANEY® Ni alloy (85 wt% Ni min., 12 wt% Al max.) and 140 mg of catalyst filled in the cartridge. Other two well-known hydrogenating catalysts, 10% Pd/C and 5% Rh/C were also investigated as catalysts for the selective hydrogenation of DHMF to DHMTHF in aqueous phase under the same operation reaction conditions (temperature, pressure and feeding flow rate). An average DHMTHF selectivity less than 5% was observed for 10% Pd/C after 8 h of time on stream and 5% Rh/C catalyst completely deactivated after 2 h (result not shown). When RANEY® Ni was used as catalyst for the single-stage conversion of HMF into DHMTHF (Table 1, entry 2) an average selectivity of only 13% was obtained after 8 h of time on stream.
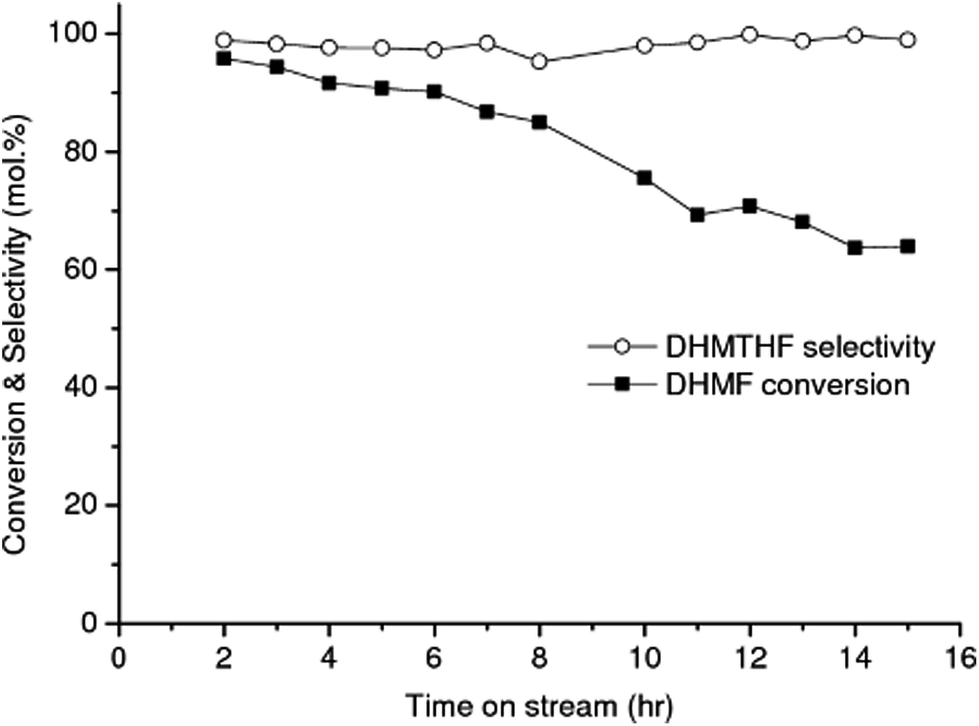 | ||
| Fig. 3 Conversion of DHMF into DHMTHF over RANEY® Ni catalyst. Reaction conditions: flow rate of 0.05 mL min−1, 90 bar, 90 °C. Feed solution: aqueous solution of DHMF (molar ratio DHMF/HMF = 0.92) obtained from the catalytic experiment using RANEY® Cu (first-stage reaction) under the similar operation conditions. | ||
The effect of hydrogen pressure on the DHMF conversion into DHMTHF was also investigated in the range of 20–90 bar using RANEY® Ni at 90 °C, Fig. 4. The hydrogenation is not limited by thermodynamic equilibrium at these conditions.44,45 As can be observed, the highest catalytic performance (conversion and selectivity) for the conversion of DHMF to DHMTHF (Scheme 1(2)) was reached at the same operation pressure as the first-stage reaction, i.e. 90 bar, which is convenient from a process stand point.
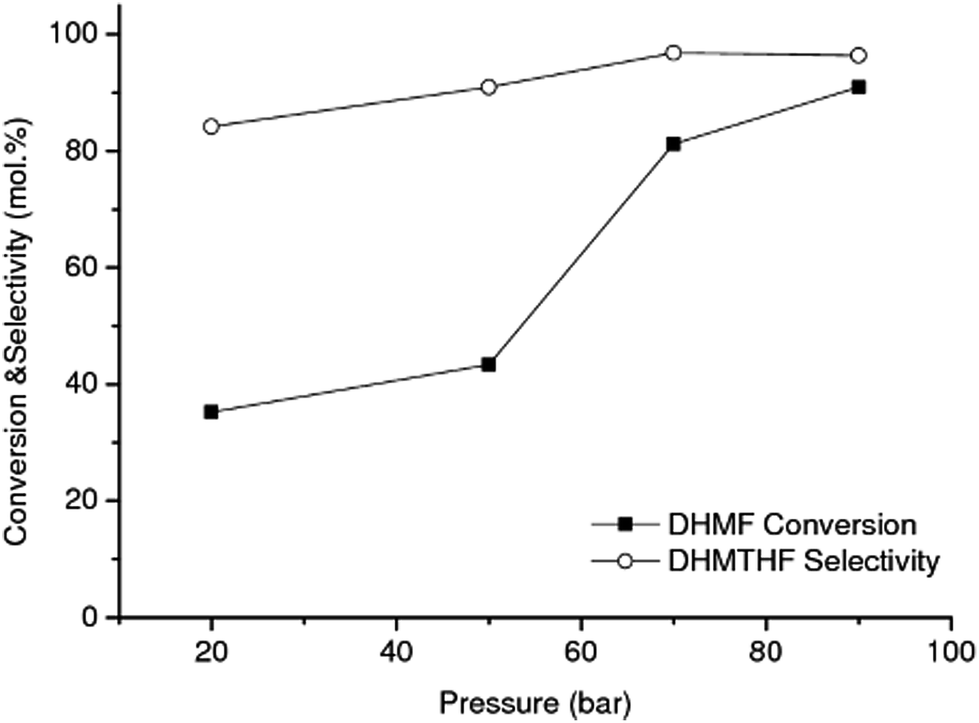 | ||
| Fig. 4 Effect of total pressure on the hydrogenation of DHMF into DHMTHF over RANEY® Ni (reaction conditions: flow rate of 0.050 mL min−1, 1 wt% HMF in water, 90 °C). Feed solution: aqueous solution of DHMF (molar ratio DHMF/HMF = 0.92) from the catalytic reaction using RANEY® Cu as catalyst under similar operation conditions. Each point correspond to an average after 8 h of time on stream. | ||
While the selectivity did not change significantly over the operating time investigated, shown in Fig. 3, the DHMF conversion gradually decreased over time. After 32 h of operation (result not shown), the conversion decreased to ca. 71% of the initial value. Stability of catalyst performance is of extreme importance in industrial processing. From previous studies the main reasons for deactivation of RANEY®-type catalysts are considered to be: (1) loss of active Ni surface by sintering; (2) leaching of Ni and promoter metal into the acidic and chelating reaction mixture. However, the negligible Ni and Al leaching measured after 15 h of operation suggests that the observed deactivation of RANEY® Ni may not be a result of loss of metal and/or promoter species; (3) poisoning of the active Ni surface by organic species principally produced by side reactions.47 Hoffer et al. showed that RANEY®-type Ni catalysts used in hydrogenation of aqueous solutions of D-glucose can be effectively regenerated by a hydrogen treatment at temperatures higher than 120 °C after a very severe washing procedure.47 In order to recover the RANEY® Ni activity we used a procedure based on early literature reported by Hauschild et al. to perform the regeneration in situ, i.e. without unpacking the catalyst from the cartridge.48 The spent RANEY® Ni cartridge was flushed in situ with a degassed, alcoholic solution of acetic acid (acetic acid in methanol/water) saturated with hydrogen (generated in situ by electrolysis of water) at a flow rate of 1 mL min−1 at 40 °C and 50 bar for 2 h, followed by washing the CatCart® with deionised water saturated with hydrogen for another 3 h under the same conditions.
The activity of catalyst treated according to the above procedure was not comparable to the activity of the fresh catalyst, even after repeating the severe washing procedure two times, as shown in Fig. 5. These results show that regeneration of RANEY® Ni was achieved with only limited success. Harsher conditions than those that can be achieved in the H-Cube hydrogenation reactor, for example higher temperature under hydrogen, might be needed to effectively regenerate the Ni surface after being washed.47
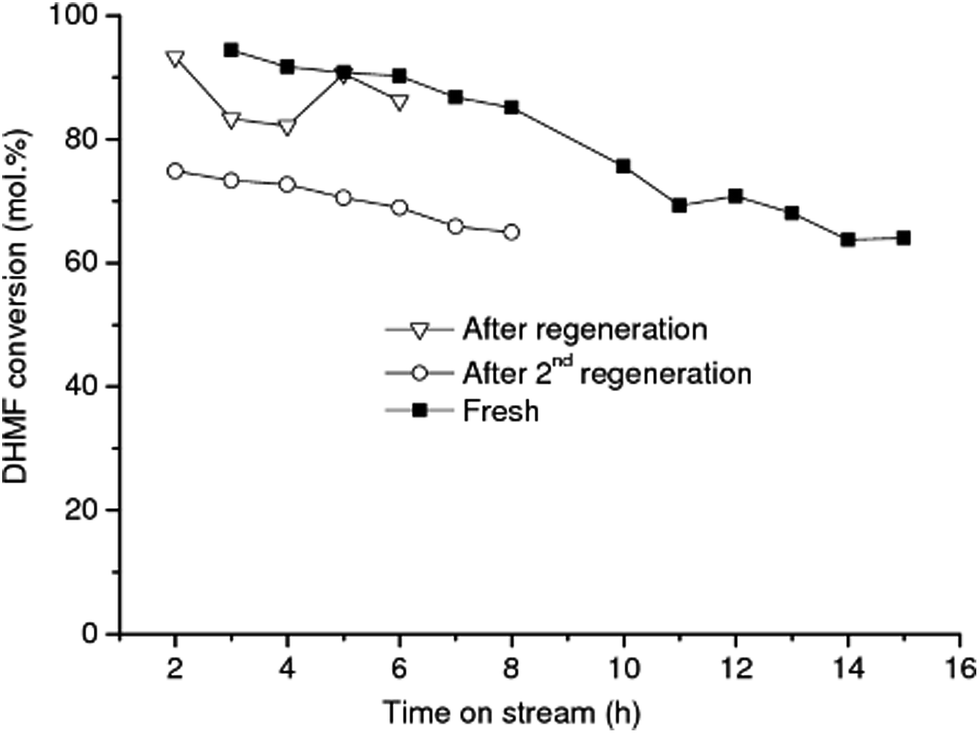 | ||
| Fig. 5 Conversion of DHMF into DHMTHF over RANEY® Ni in aqueous media. Comparison of DHMF conversion after the CatCart® have been regenerated/washed (after regeneration) and after the same CatCart® have been regenerated for second time (after regeneration for the second time). | ||
4. Conclusions
DHMTHF was obtained efficiently from the hydrogenation of HMF in aqueous media using a continuous two-stage catalytic process via DHMF under relatively mild reaction conditions, 90 °C at 90 bar. An average selectivity of 98% was achieved using different non-noble catalysts in each stage: RANEY® Cu to hydrogenate HMF to DHMF and RANEY® Ni to hydrogenate DHMF to DHMTHF under the same reaction conditions (productivity = 19 mmolDHMTHF.gcatalyst−1 h−1 after 15 h of operation). This approach was shown to be preferable to a single-stage process using RANEY® Ni catalyst where only 13% average selectivity to DHMTHF was obtained (average productivity = 0.2 mmolDHMTHF.gcatalyst−1 h−1 after 8 h of operation). The first-stage hydrogenation of HMF to DHMF over the RANEY® Cu catalyst proved to give stable performance, but the RANEY® Ni in the second stage gradually deactivated while maintaining high selectivity to DHMTHF. Negligible Ni and Al leaching occurred over 15 h of operation, suggesting that the observed decrease of activity over time-on-stream might be a result of either poisoning of the active Ni surface by organic species or due to irreversible oxidation of the active Ni surface species. Regeneration of the RANEY® Ni catalyst by washing the spent catalyst with an alcoholic aqueous solution of acetic acid in the presence of hydrogen had only a partial success. The conditions for the regeneration are limited by the H-Cube which would not necessarily apply to an industrial process.The two-stage approach described here can be an important contribution to the development of sustainable catalytic routes to DHMTHF production, where continuous hydrogenation is required for large-scale production of this high value intermediate for polymer manufacture.
Acknowledgements
This work was financially supported by EPSRC(UK) under grant EP/K014749/1.References
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106(9), 4044–4098 CrossRef CAS PubMed.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46(38), 7164–7184 CrossRef CAS PubMed.
- G. Berndes and J. Hansson, Energy Policy, 2007, 35(12), 5965–5979 CrossRef.
- P. K. Rout, A. D. Nannaware, O. Prakash, A. Kalra and R. Rajasekharan, Chem. Eng. Sci., 2016, 142, 318–346 CrossRef CAS.
- A. Chinnappan, C. Baskar and H. Kim, RSC Adv., 2016, 6, 63991–64002 RSC.
- I. Agirrezabal-Telleria, I. Gandarias and P. L. Arias, Catal. Today, 2014, 234, 42–58 CrossRef CAS.
- L. Hu, G. Zhao, W. Hao, X. Tang, Y. Sun, L. Lin and S. Liu, RSC Adv., 2012, 2, 11184–11206 RSC.
- K. Yan and A. Chen, Fuel, 2014, 115, 101–108 CrossRef CAS.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 321, 1933–1938 CrossRef PubMed.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- Y. Zhu, X. Kong, H. Zheng, G. Ding, Y. Zhu and Y.-W. Li, Catal. Sci. Technol., 2015, 5, 4208–4217 CAS.
- G. C. A. Luijkx, N. P. M. Huck, F. van Rantwijk, L. Maat and H. van Bekkum, Heterocycles, 2009, 77, 1037–1044 CrossRef CAS.
- A. J. Kumalaputri, G. Bottari, P. M. Erne, H. J. Heeres and K. Barta, ChemSusChem, 2014, 7, 2266–2275 CrossRef CAS PubMed.
- L. Yu, L. He, J. Chen, J. Zheng, L. Ye, H. Lin and Y. Yuan, ChemCatChem, 2015, 7, 1701–1707 CrossRef CAS.
- Y. Liu, M. A. Mellmer, D. M. Alonso and J. A. Dumesic, ChemSusChem, 2015, 8, 3983–3986 CrossRef CAS PubMed.
- R. Alamillo, M. Tucker, M. Chia, Y. Pagán-Torres and J. A. Dumesic, Green Chem., 2012, 14, 1413–1419 RSC.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2014, 16, 4734–4739 RSC.
- M. Tamura, K. Tokonami, Y. Nakagawa and K. Tomishige, Chem. Commun., 2013, 49, 7034–7036 RSC.
- J. Ohyama, A. Esaki, Y. Yamamoto, S. Arai and A. Satsuma, RSC Adv., 2013, 2, 1033–1036 RSC.
- A. B. Jain and P. D. Vaidya, Int. J. Chem. Kinet., 2016, 48, 318–328 CrossRef CAS.
- A. J. Sanborn and P. D. Bloom, US Pat. number 7393963 B2, 2008.
- K. Weissermel and H.-J. Arpe, Industrielle Organische Chemie, VCH, Weinheim, 4th edn, 1994 Search PubMed.
- X. Kong, R. Zheng, Y. Zhu, G. Ding, Y. Zhu and Y.-W. Li, Green Chem., 2015, 17, 2504–2515 RSC.
- X. Kong, Y. Zhu, H. Zheng, F. Dong, Y. Zhu and Y.-W. Li, RSC Adv., 2014, 4, 60467–60472 RSC.
- T. J. Connolly, J. L. Considine, Z. Ding, B. Fordatz, M. N. Jennings, M. F. MacEwan, K. M. McCoy, D. W. Place, A. Sharma and K. Sutherland, Org. Process Res. Dev., 2010, 14, 459–465 CrossRef CAS.
- J. Chen, R. Liu, Y. Guo, L. Chen and H. Gao, ACS Catal., 2015, 5, 722–733 CrossRef CAS.
- S. Yao, X. Wang, Y. Jiang, F. Wu, X. Chen and X. Mu, ACS Sustainable Chem. Eng., 2014, 2, 173–180 CrossRef CAS.
- Y. Nakagawa and K. Tomishige, Catal. Commun., 2010, 12, 154–156 CrossRef CAS.
- Y. Nakagawa, K. Takada, M. Tamura and K. Tomishige, ACS Catal., 2014, 4, 2718–2726 CrossRef CAS.
- J. Luo, J. Yu, R. J. Gorte, E. Mahmoud, D. G. Vlachos and M. A. Smith, Catal. Sci. Technol., 2014, 4, 3074–3081 CAS.
- D. Scholz, C. Aellig and I. Hermans, ChemSusChem, 2014, 7, 268–275 CrossRef CAS PubMed.
- D. P. Duarte, R. Martínez and L. J. Hoyos, Ind. Eng. Chem. Res., 2016, 55, 54–63 CrossRef CAS.
- B. Xiao, M. Zheng, X. Li, J. Pang, R. Sun, H. Wang, X. Pang, A. Wang, X. Wang and T. Zhang, Green Chem., 2016, 18, 2175–2184 RSC.
- N. G. Anderson, Org. Process Res. Dev., 2012, 16, 852–869 CrossRef CAS.
- R. Jones, L. Gödörházy, N. Varga, D. Szalay, L. Ürge and F. Darvas, J. Comb. Chem., 2006, 8, 110–116 CrossRef CAS PubMed.
- E. A. Artiukha, A. L. Nuzhdin, G. A. Bukhtiyarova, Y. S. Zaytsev, P. E. Plyusnin, Y. V. Shubin and V. I. Bukhtiyarov, Catal. Sci. Technol., 2015, 5, 4741–4745 CAS.
- M. C. Bryan, D. Wernick, C. D. Hein, J. V. Petersen, J. W. Eschelbach and E. M. Doherty, Beilstein J. Org. Chem., 2011, 7, 1141–1149 CrossRef CAS PubMed.
- P. J. Cossar, L. Hizartzidis, M. I. Simone, A. McCluskey and C. P. Gordon, Org. Biomol. Chem., 2015, 13, 7119–7130 CAS.
- M. Tucker, H. R. Alamillo, A. J. Crisci, G. M. Gonzalez, S. L. Scott and J. A. Dumesic, ACS Sustainable Chem. Eng., 2013, 1, 554–560 CrossRef CAS.
- E. L. Stephan, N. S. Hatfield, R. S. Peoples and H. A. H. Pray, USAEC Report BMI-1067, Battelle Memorial Institute, Columbus, Ohio, 1956 Search PubMed.
- H. A. Pray, C. E. Schweickert and B. H. Minnich, Ind. Eng. Chem., 1952, 44(5), 1146–1151 CrossRef CAS.
- J. M. Tukacs, R. V. Jones, F. Darvas, G. Dibó, G. Lezsák and L. T. Mika, RSC Adv., 2013, 3, 16283–16287 RSC.
- R. S. Assary, P. C. Redfern, J. R. Hammond, J. Greeley and L. A. Curtiss, Chem. Phys. Lett., 2010, 497, 123–128 CrossRef CAS.
- S. P. Verevkin, V. N. Emel'yanenko, E. N. Stepurko, R. V. Ralys and D. H. Zaitsau, Ind. Eng. Chem. Res., 2009, 48, 10087–10093 CrossRef CAS.
- H. Rojas, J. J. Martínez and P. Reyes, Dyna, 2010, 163, 151–159 CAS.
- B. W. Hoffer, E. Crezee, F. Devred, P. R. M. Mooijman, W. G. Sloof, P. J. Kooyman, A. D. van Langeveld, F. Kapteijn and J. A. Moulijn, Appl. Catal., A, 2003, 253, 437–452 CrossRef CAS.
- U. Hauschild and H. Nicolaus, US Pat. number 3165478 A, 1965.
| This journal is © The Royal Society of Chemistry 2017 |