
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Renewable sources from plants as the starting material for designing new terpene chiral ionic liquids used for the chromatographic separation of acidic enantiomers†
Joanna Feder-Kubis *a,
Jolanta Fliegerb,
Małgorzata Tatarczak-Michalewskab,
Anita Płazińskac,
Anna Madejskab and
Marta Swatko-Ossord
*a,
Jolanta Fliegerb,
Małgorzata Tatarczak-Michalewskab,
Anita Płazińskac,
Anna Madejskab and
Marta Swatko-Ossord
aFaculty of Chemistry, Wrocław University of Science and Technology, 50-370 Wrocław, Poland. E-mail: joanna.feder-kubis@pwr.edu.pl
bDepartment of Analytical Chemistry, Medical University of Lublin, 20-093 Lublin, Poland
cDepartment of Biopharmacy, Medical University of Lublin, 093 Lublin, Poland
dDepartment of Biochemistry and Biotechnology, Medical University of Lublin, 20-093 Lublin, Poland
First published on 23rd June 2017
Abstract
Synthesis of cheap and natural resources is an important topic in green chemistry. For that reason, new chiral ionic liquids (CILs) containing a bicyclic terpene moiety were designed and prepared from renewable plant resources. The synthesis route was comprehensively described, especially a specific type of Menschutkin reaction, which gives high energy efficiency during the process. The physicochemical characteristics of the obtained chiral salts, including the spectral properties, melting point, crystal shape, specific rotation and solubility in various solvents were examined. The study presents the effect of new chiral ionic liquids as mobile phase additives on the chiral recognition of acidic enantiomers on a teicoplanin-based chiral stationary phase (CSP). Based on the van't Hoff relationships, the thermodynamic functions were determined. The plots revealed that the chiral recognition was enthalpy driven. The chiral salts obtained exhibited a synergistic effect with the teicoplanin-based stationary phase in the chromatographic system, enhancing the resolution of acidic enantiomers. Structural task-specific properties of the new terpene-based chiral ionic liquids were confirmed by molecular modeling and docking simulations.
Introduction
Ionic liquids (ILs) are defined as salts with melting points below 100 °C and have attracted researchers' attention in various fields of the chemical industry for the last few decades. They have partially replaced the organic solvents and are growing in popularity due to their ecological nature and the possibility of designing them with specific properties. They have also brought many benefits to analytical chemistry. Concerning applications in separation techniques, ILs have been used for either modification of stationary phases in gas-liquid chromatography (GLC)1,2 and capillary electrophoresis3 or as mobile phase additives in liquid chromatography4–7 and electrophoresis as background electrolyte modifiers.8,9 ILs are used as solvents in different extraction techniques. Whereas water-insoluble ILs are useful in liquid–liquid microextractions10,11 and as modifiers of adsorbents for solid phase extraction (SPE),12 water-soluble ILs can be used as extracting solvents, thus creating aqueous biphasic systems (ABS).13–15 The overviews of the various applications of ILs in separation techniques are described extensively in review articles.16–18One of the most interesting developments of ILs appears to be searching for new task-specific ILs by expanding their particular properties. For instance there exists a possibility of creating chiral ionic liquids (CILs) by introducing chiral centers either in the cations or anions. The application of chiral ILs for separation of enantiomers has been described previously mainly in electrophoretic techniques.19–23 It should be emphasized, however, that the much better results of enantiomers' resolution could be achieved using a few chiral selectors simultaneously, making use of the synergistic effects of such combined systems. For that purpose a few ILs or system containing chiral ILs and β-cyclodextrins or antibiotics (vancomycin) have been applied to improve enantiorecognition.22,24,25
Only a few papers have described the potential of CILs as chiral selectors in liquid chromatography.26–31 In the study by Yuan et al.,26 the (R)-N,N,N-trimethyl-2-aminobutanol bis(trifluoromethylsulfonyl)imide ionic liquid was applied in high performance liquid chromatography (HPLC), as a chiral mobile phase modifier using C18 column. Zhou et al.,31 for the first time, applied an IL-based chiral selector as a stationary phase. For that purpose, new ILs functionalized β-CDs were bonded to silica gel. Recently, the research on the immobilized chiral ionic liquids on a HPLC column has been described by Qian et al.32 In this work, tropine-type CILs with a proline anion were immobilized on silica gel. The chiral ionic liquid-modified silica gel saturated by Cu(II) was used in the resolution of racemic amino acids.
Currently particular efforts are made to design a ‘fully green’ ionic liquid. For this reason recently, the issues of biodegradability and toxicity of ILs have recently become of primary interest.33,34 The synthesis of ionic liquids using renewable compounds as the starting materials, e.g. terpenes,35 sugar,36 amino alcohols,37 hydroxy acids,38 or amino acids39 seems to be extremely desirable.
Here we report the synthesis, characterization and application of chiral ionic liquids containing a bicyclic monoterpene moiety. As starting materials, renewable sources from plants were selected: (1S)-endo-(−)-borneol and (1R)-endo-(+)-fenchol. CILs with bicyclic terpene moieties were used as chiral selectors for the first time. The described synergistic system basing on a glycopeptide stationary phase with CILs as mobile phase additives has never been reported before in LC. The specific cooperative effect was used for the enantiomeric resolution of acids. The effect of different parameters on resolution was explored in detail, along with the associated thermodynamics. Moreover, in this work the molecular mechanism of stereoselective binding of enantiomers to a teicoplanin structure in the presence of CILs is discussed for the first time.
Experimental
Materials and techniques
The materials used were sourced as follows: imidazole (≥99%), 1-methylimidazole (99%), (1S)-endo-(−)-borneol (97%), (1R)-endo-(+)-fenchol (96%), paraformaldehyde (powder, 95%), sodium (cubes, contains mineral oil, 99.9% trace metals basis), 1-bromopentane (98%), hydrochloric acid (35–38%), sulfuric acid (≥96%) were purchased from Sigma-Aldrich. Racemic standards of mandelic acid, phenyllactic acid, 4-hydroxy-3-methoxymandelic acid (vanilmandelic) acid were also obtained from Sigma Aldrich (St. Louis, MO, USA). HPLC grade methanol (MeOH) was purchased from Merck (Darmstadt, Germany). HPLC water was obtained from Barnstead deionising system (Dubuque, IA, USA). All mobile phases were filtered with Nylon 66 membrane filters (0.45 μm) by Whatman (Maidstone, England) by the use of a filtration apparatus. Stock solutions of individual racemic acids at 1.0 mg L−1 were prepared in methanol and stored in glass vials in refrigerator.For NMR analysis, deuterated chloroform (CDCl3) purchased from Merck was used. All the solvents were dried before use. All reagents were dried and purified before use by the usual procedures; reactions were performed under anhydrous conditions.
Melting points were determined by using an electrothermal digital-melting-point apparatus model JA 9100 (temperature resolution ± 0.1 °C; accuracy ± 1%; choice of ramp rate of 1.0 °C min−1). The type and the shape of the crystals were analysed via an optical microscope AxioImager M1m (Zeiss) in reflection mode. Specific rotations at 578 nm were measured using an Optical Activity Ltd. Model AA-5 automatic polarimeter (resolution ± 0.01°, reproducibility ± 0.01°, accuracy ± 0.01°, temperature probe measurement accuracy ± 0.1 °C, the equipment provide four results for each measurement). The structure and purity of all of the synthesized salts were confirmed by spectral analysis. Elemental analyses were carried out for all of the synthesized substances using VARIO EL-III. The 1H NMR and 13C NMR spectra were recorded on a Bruker DRX instrument with tetramethylsilane as standard (at 600 and 75 MHz, respectively). HRMS analyses were performed on LCT Premier XE Waters apparatus, on mode ESI+ (TOF MS ES+). Elemental analyses were carried out for all of the synthesised substances using a VARIO EL-III.
General methods for synthesis
Imidazole was freshly recrystallized from benzene (mp 90–91 °C). 1-Methylimidazole was purchased, while 1-pentylimidazole was obtained following the published method.40 These 1-alkylimidazoles were purified by vacuum distillation before use in a quaternization reaction.Synthesis of the quaternary agents
The amount of each of chloromethyl terpenyl ethers in the final product was determined by the alkalimetric method (alkalimetric titration of the HCl obtained as a result of ether hydrolysis), according to the procedure described earlier by our group for chloromethyl (1R,2S,5R)-(−)-menthyl ether,41 as follows: 1 g crude product (ether) was added to 10 mL acetone at −20 °C. The possible free HCl absorbed in the sample was quickly neutralized with 0.02 M KOH in MeOH. Hot water (5 mL) was then added. HCl as a product of hydrolysis of ether was neutralized with 0.2 M KOH in MeOH. The crude product contained 95.0% and 97.0% of chloromethyl (1S)-endo-(−)-bornyl ether (1a) and chloromethyl (1R)-endo-(+)-fenchyl (2a) respectively. The chloromethyl terpenyl ethers obtained were purified by vacuum distillation to give clear liquids and the process was repeated each time before used them as quaternary agents in the Menschutkin reaction.
(1S)-endo-(−)-Bornyl chloromethyl ether (1a). Vacuum distillation conditions: bp 85–86 °C at 1 mmHg (yield: 93.5%, 0.318 mol, 64.467 g). Elemental analysis calc. (%) for C11H19ClO (202.75): C 65.16, H 9.46, found: C 65.22, H 9.53.
Chloromethyl (1R)-endo-(+)-fenchyl ether (2a). Vacuum distillation conditions: bp 76–77 °C at 1 mmHg (yield: 96.5%, 0.328 mol, 66.535 g). Elemental analysis calc. (%) for C11H19ClO (202.75): C 65.16, H 9.46, found: C 65.14, H 9.56.
Synthesis of the chiral ionic liquids – Menschutkin quaternization
1-[(1S)-endo-(−)-Borneoxymethyl]-3-methylimidazolium chloride [C1-Im-CH2O-Bor][Cl] (1b). Yield: 99.4%, 0.0298 mol, 8.495 g. 1H NMR: (600 MHz, CDCl3): δ = 0.76 (s, 3H, bor.), 0.79–0.84 (m, 6H, bor.), 0.86–0.92 (m, 1H, bor.), 1.15–1.24 (m, 2H, bor.), 1.635–1.65 (m, 1H, bor.), 1.67–1.72 (m, 1H, bor.), 1.86–1.90 (m, 1H, bor.), 2.21–2.25 (m, 1H, bor.), 3.83 (d, J = 9.0 Hz, 1H, bor. CH–O), 4.14 (s, 3H, N–CH3), 5.69 and 5.79 (d, J = 10.2 Hz, 2H, AB system, O–CH2–N), 7.46 (s, 1H, imi.), 7.64 (s, 1H, imi.), 10.81 (s, 1H, imi.). 13C NMR (151 MHz, CDCl3): δ = 13.55 (bor.), 18.95 (bor.), 19.6 (bor.), 26.4 (bor.), 28.0 (bor.), 35.9 (bor.), 36.8 (N–CH3), 44.9 (bor.), 47.7 (bor.), 49.3 (bor.), 78.7 (bor. CH–O), 86.0 (O–CH2–N), 120.7 (imi.), 123.9 (imi.), 138.2 (imi.). HRMS (ESI+): m/z (%) calcd for C15H25N2O: 249.1967, found: 249.1960. Elemental analysis calc. (%) for C15H25ClN2O (284.87): C 63.24, H 8.86, N 9.84, found: C 63.35, H 8.99, N 9.70.
1-[(1S)-endo-(−)-Borneoxymethyl]-3-pentylimidazolium chloride [C5-Im-CH2O-Bor][Cl] (1c). Yield: 97.6%, 0.0293 mol, 9.984 g. 1H NMR: (600 MHz, CDCl3): δ = 0.755 (s, 3H, bor.), 0.82–0.90 (m, 10H, 7H: bor., 3H: N–CH2CH2CH2CH2
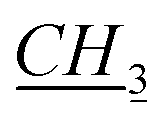 ), 1.15–1.25 (m, 2H, bor.), 1.305–1.39 (m, 4H, N–CH2CH2
), 1.15–1.25 (m, 2H, bor.), 1.305–1.39 (m, 4H, N–CH2CH2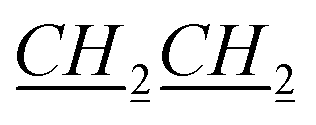 CH3), 1.64–1.65 (m, 1H, bor.), 1.69–1.71 (m, 1H, bor.), 1.865–1.965 (m, 3H, 1H: bor., 2H: N–CH2
CH3), 1.64–1.65 (m, 1H, bor.), 1.69–1.71 (m, 1H, bor.), 1.865–1.965 (m, 3H, 1H: bor., 2H: N–CH2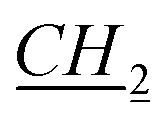 CH2CH2CH3), 2.21–2.23 (m, 1H, bor.), 3.86–3.88 (m, 1H, bor. CH–O), 4.36–4.38 (m, 2H, N–
CH2CH2CH3), 2.21–2.23 (m, 1H, bor.), 3.86–3.88 (m, 1H, bor. CH–O), 4.36–4.38 (m, 2H, N–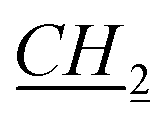 CH2CH2CH2CH3), 5.74 and 5.85 (d, J = 10.2 Hz, 2H, AB system, O–CH2–N), 7.47 (s, 1H, imi.), 7.475 (s, 1H, imi.), 11.01 (s, 1H, imi.). 13C NMR (151 MHz, CDCl3): δ = 13.5 (bor.), 13.8 (N–CH2CH2CH2CH2
CH2CH2CH2CH3), 5.74 and 5.85 (d, J = 10.2 Hz, 2H, AB system, O–CH2–N), 7.47 (s, 1H, imi.), 7.475 (s, 1H, imi.), 11.01 (s, 1H, imi.). 13C NMR (151 MHz, CDCl3): δ = 13.5 (bor.), 13.8 (N–CH2CH2CH2CH2![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H3), 18.95 (bor.), 19.6 (bor.), 22.0 (N–CH2CH2CH2H2CH3), 26.4 (bor.), 28.0 (bor.), 28.2 (N–CH2CH2H2CH2CH3), 29.9 (N–CH2H2CH2CH2CH3), 35.9 (bor.), 44.9 (bor.), 47.7 (bor.), 49.3 (bor.), 50.15 (N–H2CH2CH2CH2CH3), 78.7 (bor. CH–O), 86.0 (O–CH2–N), 121.0 (imi.), 122.6 (imi.), 138.5 (imi.). HRMS (ESI+): m/z (%) calcd for C19H33N2O: 305.2593, found: 305.2605. Elemental analysis calc. (%) for C19H33ClN2O (340.99): C 66.92, H 9.77, N 8.22, found: C 66.83, H 9.91, N 8.30.
H3), 18.95 (bor.), 19.6 (bor.), 22.0 (N–CH2CH2CH2H2CH3), 26.4 (bor.), 28.0 (bor.), 28.2 (N–CH2CH2H2CH2CH3), 29.9 (N–CH2H2CH2CH2CH3), 35.9 (bor.), 44.9 (bor.), 47.7 (bor.), 49.3 (bor.), 50.15 (N–H2CH2CH2CH2CH3), 78.7 (bor. CH–O), 86.0 (O–CH2–N), 121.0 (imi.), 122.6 (imi.), 138.5 (imi.). HRMS (ESI+): m/z (%) calcd for C19H33N2O: 305.2593, found: 305.2605. Elemental analysis calc. (%) for C19H33ClN2O (340.99): C 66.92, H 9.77, N 8.22, found: C 66.83, H 9.91, N 8.30.
1-[(1R)-endo-(+)-Fenchoxymethyl]-3-methylimidazolium chloride [C1-Im-CH2O-Fen][Cl] (2b). Yield: 99.6%, 0.0299 mol, 8.512 g. 1H NMR: (600 MHz, CDCl3): δ = 0.83 (s, 3H, fen.), 0.92–1.01 (m, 7H, fen.), 1.06–1.08 (m, 1H, fen.), 1.34–1.40 (m, 1H, fen.), 1.44–1.46 (m, 1H, fen.), 1.61–1.64 (m, 3H, fen.), 3.19 (s, 1H, fen. CH–O), 4.14 (s, 3H, N–CH3), 5.68 and 5.72 (d, J = 10.8 Hz, J = 10.3 Hz, 2H, AB system, O–CH2–N), 7.455 (s, 1H, imi.), 7.72 (s, 1H, imi.), 10.82 (s, 1H, imi.). 13C NMR (151 MHz, CDCl3): δ = 19.4 (fen.), 21.0 (fen.), 25.75 (fen.), 25.8 (fen.), 31.3 (fen.), 36.7 (N–CH3), 39.4 (fen.), 41.0 (fen.), 48.4 (fen.), 48.9 (fen.), 79.3 (fen. CH–O), 93.7 (O–CH2–N), 120.9 (imi.), 124.05 (imi.), 138.2 (imi.). HRMS (ESI+): m/z (%) calcd for C15H25N2O: 249.1967, found: 249, 1972. Elemental analysis calc. (%) for C15H25ClN2O (284.87): C 63.24, H 8.86, N 9.84, found: C 63.17, H 8.94, N 9.90.
1-[(1R)-endo-(+)-Fenchoxymethyl]-3-pentylimidazolium chloride [C5-Im-CH2O-Fen][Cl] (2c). Yield: 98.8%, 0.0296 mol, 10.107 g. 1H NMR: (600 MHz, CDCl3): δ = 0.84 (s, 3H, fen.), 0.88–0.90 (m, 3H, N–CH2CH2CH2CH2
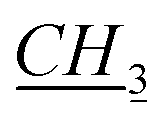 ), 0.935–1.02 (m, 7H, fen.), 1.02–1.09 (m, 1H, fen.), 1.31–1.42 (m, 5H, 1H: fen., 4H: N–CH2CH2
), 0.935–1.02 (m, 7H, fen.), 1.02–1.09 (m, 1H, fen.), 1.31–1.42 (m, 5H, 1H: fen., 4H: N–CH2CH2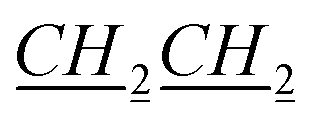 CH3), 1.47–1.49 (m, 1H, fen.), 1.625–1.66 (m, 3H, fen.), 1.91–1.92 (m, 2H, N–CH2
CH3), 1.47–1.49 (m, 1H, fen.), 1.625–1.66 (m, 3H, fen.), 1.91–1.92 (m, 2H, N–CH2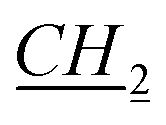 CH2CH2CH3), 3.24 (s, 1H, fen. CH–O), 4.37–4.40 (m, 2H, N–
CH2CH2CH3), 3.24 (s, 1H, fen. CH–O), 4.37–4.40 (m, 2H, N–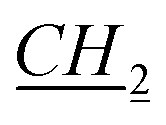 CH2CH2CH2CH3), 5.74 and 5.79 (d, J = 10.2 Hz, 2H, AB system, O–CH2–N), 7.46 (t, J = 1.8 Hz, 1H, imi.), 7.47 (t, J = 1.8 Hz, 1H, imi.), 10.89 (s, 1H, imi.). 13C NMR (151 MHz, CDCl3): δ = 13.8 (N–CH2CH2CH2CH2H3), 19.4 (fen.), 21.0 (fen.), 22.0 (N–CH2CH2CH2H2CH3), 25.75 (fen.), 25.8 (fen.), 28.25 (N–CH2CH2H2CH2CH3), 29.9 (N–CH2H2CH2CH2CH3), 31.2 (fen.), 39.4 (fen.), 41.0 (fen.), 48.4 (fen.), 49.0 (fen.), 50.2 (N–H2CH2CH2CH2CH3), 79.5 (fen. CH–O), 93.9 (O–CH2–N), 121.0 (imi.), 122.2 (imi.), 138.5 (imi.). HRMS (ESI+): m/z (%) calcd for C19H33N2O: 305.2593, found: 305.2601. Elemental analysis calc. (%) for C19H33ClN2O (340.99): C 66.92, H 9.77, N 8.22, found: C 67.02, H 9.82, N 8.15.
CH2CH2CH2CH3), 5.74 and 5.79 (d, J = 10.2 Hz, 2H, AB system, O–CH2–N), 7.46 (t, J = 1.8 Hz, 1H, imi.), 7.47 (t, J = 1.8 Hz, 1H, imi.), 10.89 (s, 1H, imi.). 13C NMR (151 MHz, CDCl3): δ = 13.8 (N–CH2CH2CH2CH2H3), 19.4 (fen.), 21.0 (fen.), 22.0 (N–CH2CH2CH2H2CH3), 25.75 (fen.), 25.8 (fen.), 28.25 (N–CH2CH2H2CH2CH3), 29.9 (N–CH2H2CH2CH2CH3), 31.2 (fen.), 39.4 (fen.), 41.0 (fen.), 48.4 (fen.), 49.0 (fen.), 50.2 (N–H2CH2CH2CH2CH3), 79.5 (fen. CH–O), 93.9 (O–CH2–N), 121.0 (imi.), 122.2 (imi.), 138.5 (imi.). HRMS (ESI+): m/z (%) calcd for C19H33N2O: 305.2593, found: 305.2601. Elemental analysis calc. (%) for C19H33ClN2O (340.99): C 66.92, H 9.77, N 8.22, found: C 67.02, H 9.82, N 8.15.
Solubility of chiral ionic liquids
The solubility of obtained CILs was determined according to Vogel's Textbook of Practical Organic Chemistry.42 The measurements were conducted for all of the synthesized chlorides at 20 °C and at 50 °C under ambient pressure for popular representative solvents. The term complete solubility refers to those salts which follow the rule: 0.1 g of each salts is soluble in 1 mL of solvent, while the phrase limited solubility means that 0.1 g of each of the investigated chlorides is dissolved in 3 mL of solvent. The term insoluble specifies those CILs which are not soluble (0.1 g of each) in 3 mL of solvent.Chromatographic instrument and conditions
Experiments were performed using a LaChrom HPLC Merck Hitachi (E.Merck, Darmstadt, Germany) model equipped with diode array detector (DAD). The column (150 mm × 4.6 mm I.D.) was packed with 5 μm Astec Chirobiotic T (Sigma-Aldrich, St. Louis, MO, USA). The void volume was 1.2 mL established by injection of blank mobile phase. Retention data were recorded at a mobile phase flow-rate of 1 mL min−1 with online degassing using L-7612 solvent degasser. The column temperature was controlled by a JetStream II Plus thermostat. In all experiments, the column temperature was kept at 20 °C, except when the effect of temperature was evaluated. In the latter case, the temperature varied from 5 to 35 °C. The signals on HPLC system with a multiple wavelength UV detector were recorded at 220 nm. Typical injection volumes were 5 μL. The elution was carried out in the isocratic mode by mobile phase consisted of 10% methanol in water and appropriate concentration of additives ranging from 5 to 20 mM.Thermodynamics
The thermodynamics was evaluated through the determination of chromatographic retention parameter k at different column temperatures using the following equation:
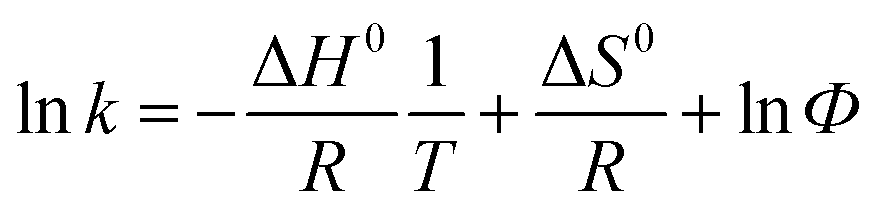 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k vs. 1/T, −ΔH0/R represents the slope and ΔS0/R + lnΦ represents intercept of the above relationship. Multiplying the slope and the intercept values by R, standard enthalpy change and entropy variations (ΔS0*) biased by R lnΦ could be calculated respectively. The term ΔS0* can be used to represent the entropy change directly. This bias is eliminated for Δ(ΔS) value considering the fact that the bias is constant for the particular column.
k vs. 1/T, −ΔH0/R represents the slope and ΔS0/R + lnΦ represents intercept of the above relationship. Multiplying the slope and the intercept values by R, standard enthalpy change and entropy variations (ΔS0*) biased by R lnΦ could be calculated respectively. The term ΔS0* can be used to represent the entropy change directly. This bias is eliminated for Δ(ΔS) value considering the fact that the bias is constant for the particular column.
Docking methodology
The structure of teicoplanin was modeled on the basis of the available crystal structure (PDB: 5AWV).43 The crystal structure of teicoplanin was additionally functionalized by adding the three carbohydrate moieties (O1-linked N-acetyl-β-D-glucosamine, O1-linked N-decane-β-D-glucosamine and O1-linked α-D-mannose), which corresponds to teicoplanin immobilized on the stationary phase [Astec Chirobiotic T column (Sigma-Aldrich, St. Louis, MO, USA)]. The structural model of teicoplanin was built by using Avogadro 1.1.1 (ref. 44) and optimized within the UFF force field.45 In the next step, the geometry of teicoplanin was reoptimized at the PM6 (ref. 46) level of theory using Gaussian09.47 The carboxyl and amine moieties of teicoplanin were set as charged, according to the condition under physiological pH. The enantiomers, as well as the chiral ionic liquids, were prepared using Avogadro and optimized using the UFF force field. Docking simulations were performed in the Molegro Virtual Docker software (MVD v. 2010.4.0.0), according to the methodology described in our previous paper.48 The flexible, optimized ligands of chiral ionic liquids and two enantiomers of mandelic acid, vanilmandelic and phenyllactic acid were docked into the sphere that included all four binding cavities of teicoplanin, Fig. 3. The estimation of the ligand–protein interactions was described by the MVD implemented scoring function (MolDock Score).49 The predicted positions of the ligands docked into teicoplanin were characterized by a simultaneous lowering of the scoring function value, MolDock Score [kJ mol−1]; this corresponds to the high values of the ligand binding energy. The enantiomers of mandelic acid, vanilmandelic and phenyllactic acid were docked into: (i) the teicoplanin molecule and (ii) the chiral ionic liquid–teicoplanin complex. Regarding step (ii), the chiral ionic liquid was initially docked to teicoplanin and the complexes obtained (characterized by the lowest values MolDock Score) were optimized in Avogadro (UFF force field).Results and discussion
The synthesis of the chiral ionic liquids containing the natural bicyclic terpene moiety
Several chiral imidazolium salts with a terpene moiety and chloride anion were prepared by Menschutkin reaction (Scheme 1). As starting materials, renewable sources from plants were selected: (i) (1S)-endo-(−)-borneol (1) and (ii) (1R)-endo-(+)-fenchol (2) being bicyclic compounds.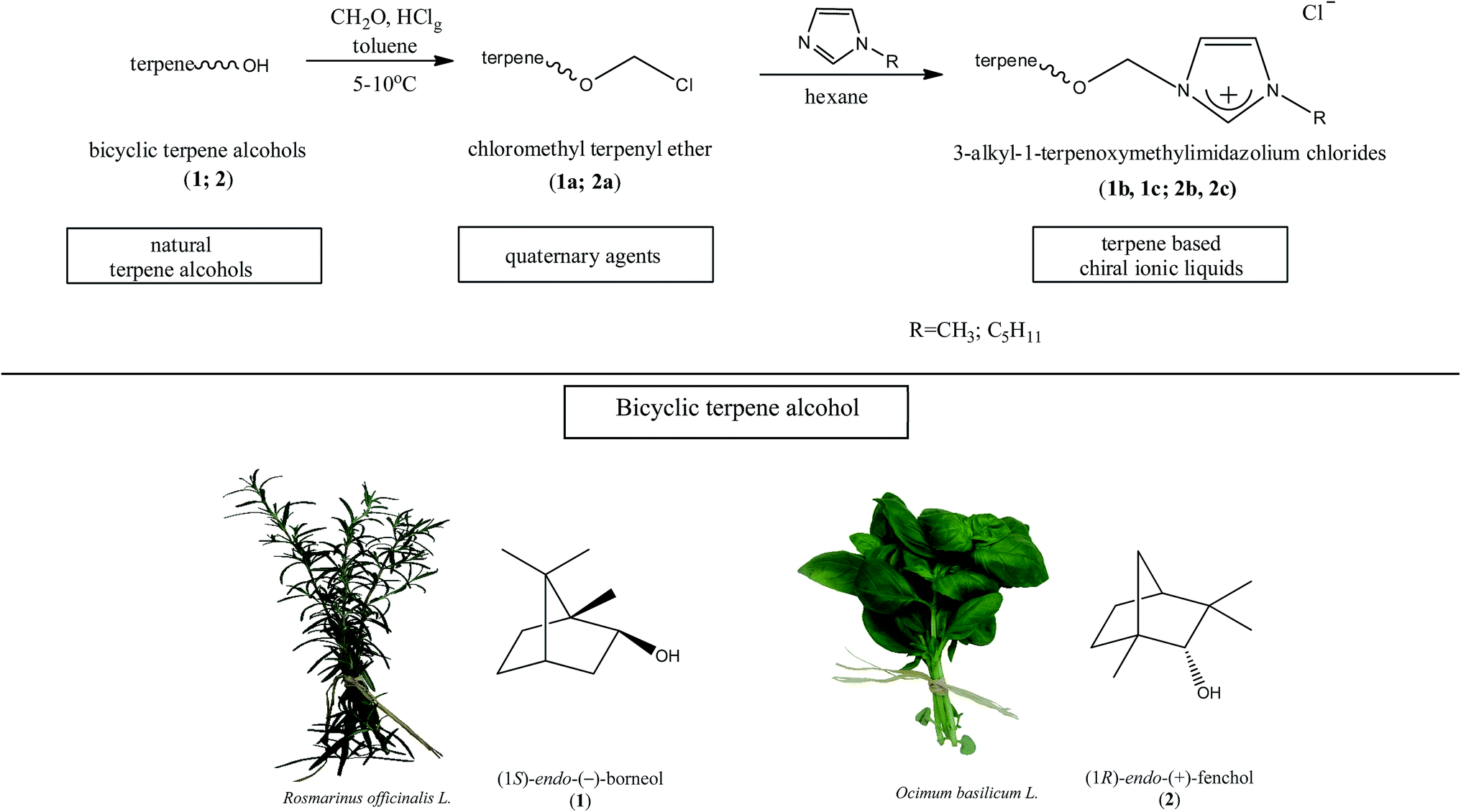 | ||
| Scheme 1 The synthesis of the chiral ionic liquids with bicyclic monoterpene moiety. | ||
Chloromethyl terpenyl ethers were prepared first, namely (1S)-endo-(−)-bornyl chloromethyl ether (1a) and chloromethyl (1R)-endo-(+)-fenchyl ether (2a). These quaternary agents were obtained by chloromethylation of the appropriate terpene alcohol. Such ethers are excellent reagents for quaternization, but they readily hydrolyze to HCl, CH2O, and proper terpene alcohol. Therefore the process of performing chloromethyl terpenyl ethers should be conducted under strictly anhydrous conditions. The obtained ethers were purified by vacuum distillation to give final products with satisfactory yields, greater than from 93.5% in each case (Table 1).
| Compound no | Structure and name of chiral quaternary agents | Type of the terpene | Distillation conditions (°C) | Quaternary agent content before and after distillationa,b (%) | Yieldc,d (%) |
|---|---|---|---|---|---|
| a Isolated amount of quaternary agent in the product by alkalimetric titration before and after vacuum distillation.b Accuracy ± 0.5%.c Isolated yield after vacuum distillation.d Accuracy ± 0.5%. | |||||
| 1a | 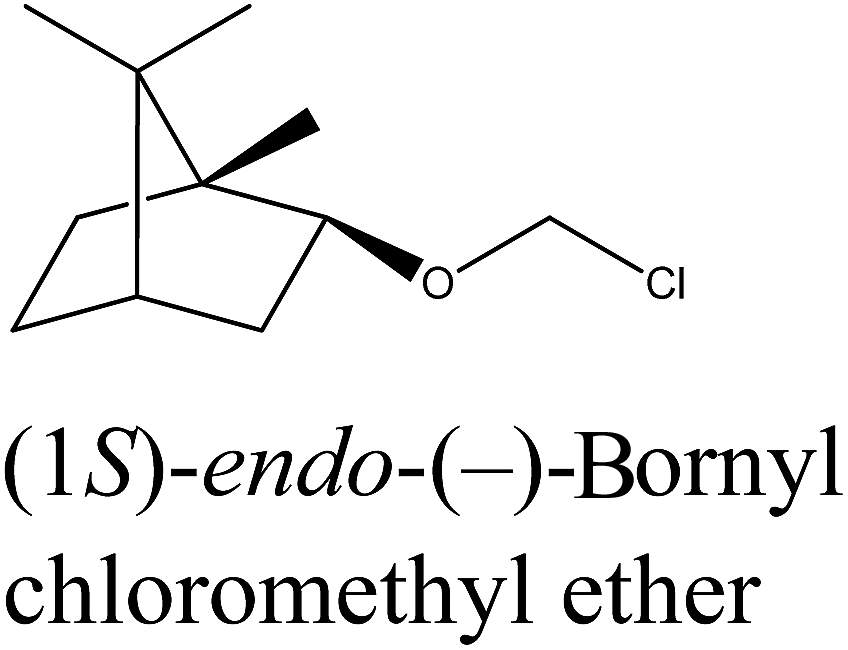 |
Bicyclic monoterpene | 85–86@1 mmHg | 95.0/99.5 | 93.5 |
| 2a | 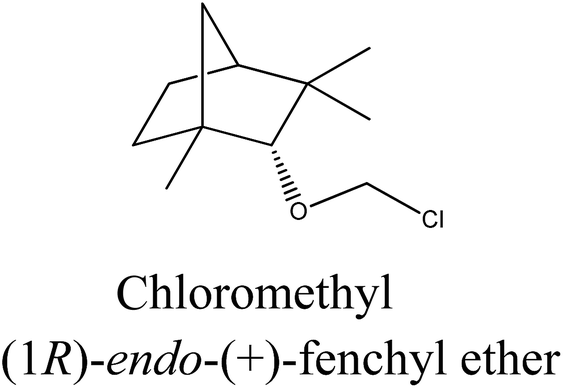 |
Bicyclic monoterpene | 76–77@1 mmHg | 97.0/99.5 | 96.5 |
The chiral imidazolium chlorides containing terpene moiety were prepared by the Menschutkin reaction. Quaternization was achieved using distilled 1-alkylimidazole and freshly distilled appropriate chloromethyl terpenyl ether under strictly anhydrous conditions. The reaction was carried out in anhydrous hexane at room temperature, and the product precipitated from such solution. Quaternization takes place immediately and proceeds readily at room temperature according to an SN1 mechanism.41 This reaction has a high product yield of 97.6 to 99.6%. Due to the fact that used chloromethyl terpenyl ethers (1a and 2a) are very reactive and quaternization takes place immediately, particular attention should be paid to increasing temperature during the process. For this reason quaternization should be carried out using a reflux condenser.
Table 2 contains a list of 3-alkyl-1-[(1S)-endo-(−)-borneoxymethyl]imidazolium chlorides ([C1-Im-CH2O-Bor][Cl] and [C5-Im-CH2O-Bor][Cl]) and 3-alkyl-1-[(1R)-endo-(+)-fenchoxymethyl]imidazolium chlorides ([C1-Im-CH2O-Fen][Cl] and [C5-Im-CH2O-Fen][Cl]) which have not been described in the literature before. All of the synthesized 3-alkyl-1-terpenoxymethylimidazolium chlorides are hydrophilic and are stable both in air and in aqueous solutions. The purities of the discussed crystallized chlorides were determined by a direct two-phase titration technique (EN ISO 2871-2: 2010 norm) and range from 99.1 to 99.7%. The rest consists of water. All details are given in the Table 3. The melting point, crystallization condition, and specific rotation of each imidazolium salt are also presented in Table 3.
| CILs | Structure | R | Name of chiral ionic liquids based on terpene | Abbreviation [Cn-Im-CH2O-Terpene][Cl] | Yielda,b (%) |
|---|---|---|---|---|---|
| a Isolated yield after purification and drying.b Accuracy ± 0.1%. | |||||
| (1S)-endo-(−)-Borneol based chiral ionic liquids | |||||
| 3-Alkyl-1-[(1S)-endo-(−)-borneoxymethyl]imidazolium chlorides | |||||
| 1b | 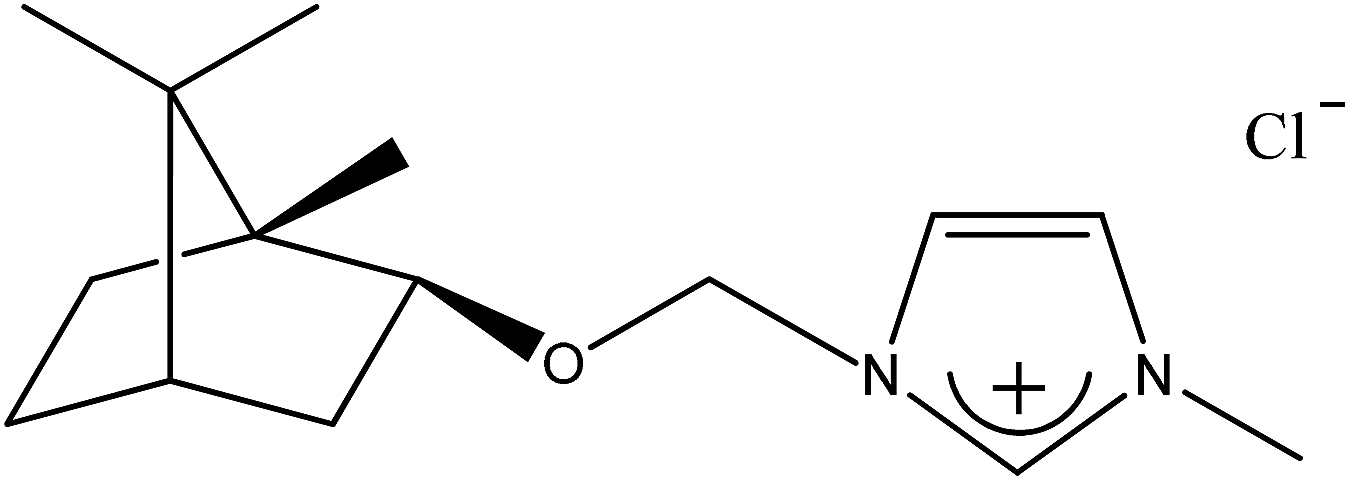 |
CH3 | 1-[(1S)-endo-(−)-Borneoxymethyl]-3-methylimidazolium chloride | [C1-Im-CH2O-Bor][Cl] | 99.4 |
| 1c | 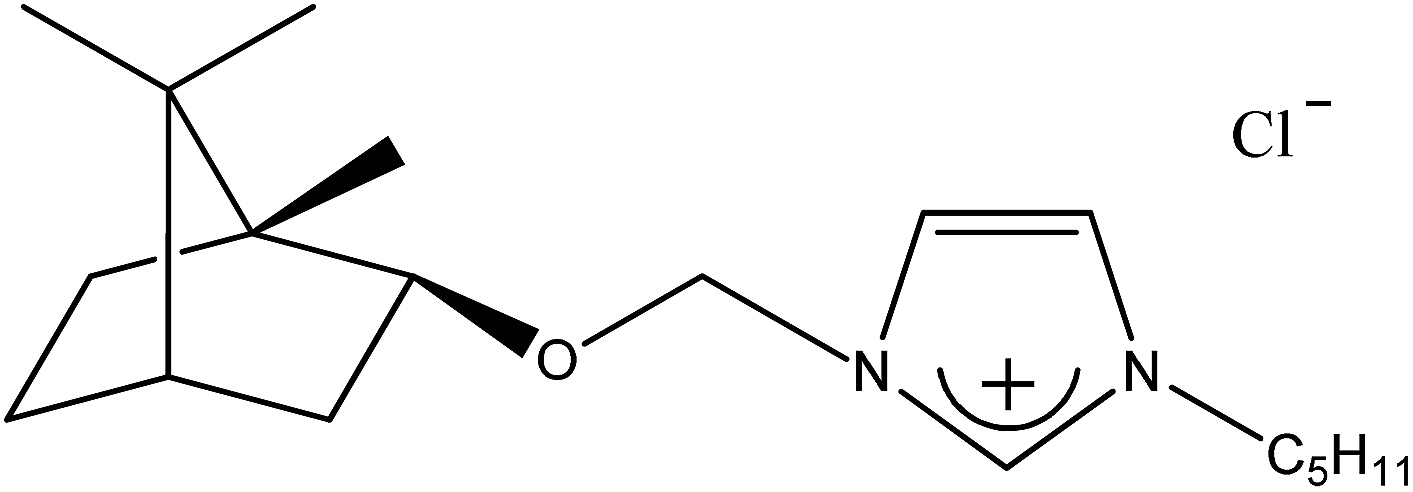 |
C5H11 | 1-[(1S)-endo-(−)-Borneoxymethyl]-3-pentylimidazolium chloride | [C5-Im-CH2O-Bor][Cl] | 97.6 |
|
|||||
| (1R)-endo-(+)-Fenchol based chiral ionic liquids | |||||
| 3-Alkyl-1-[(1R)-endo-(+)-fenchoxymethyl]imidazolium chlorides | |||||
| 2b | 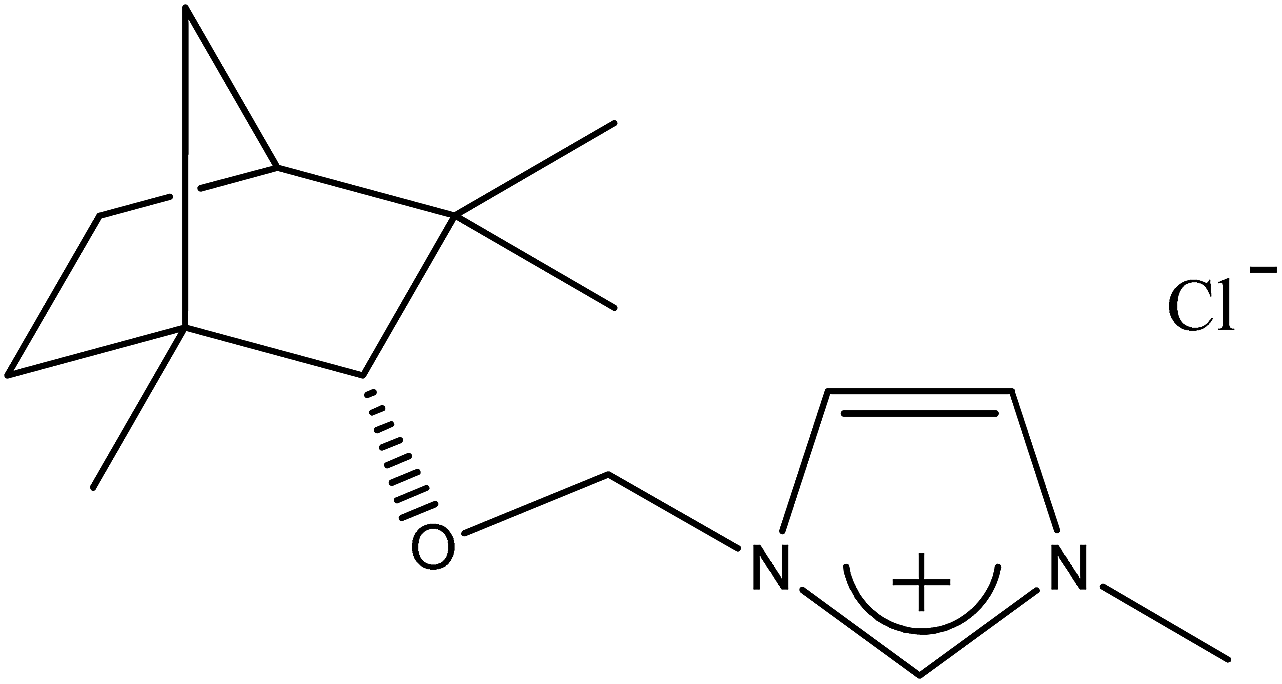 |
CH3 | 1-[(1R)-endo-(+)-Fenchoxymethyl]-3-methylimidazolium chloride | [C1-Im-CH2O-Fen][Cl] | 99.6 |
| 2c | 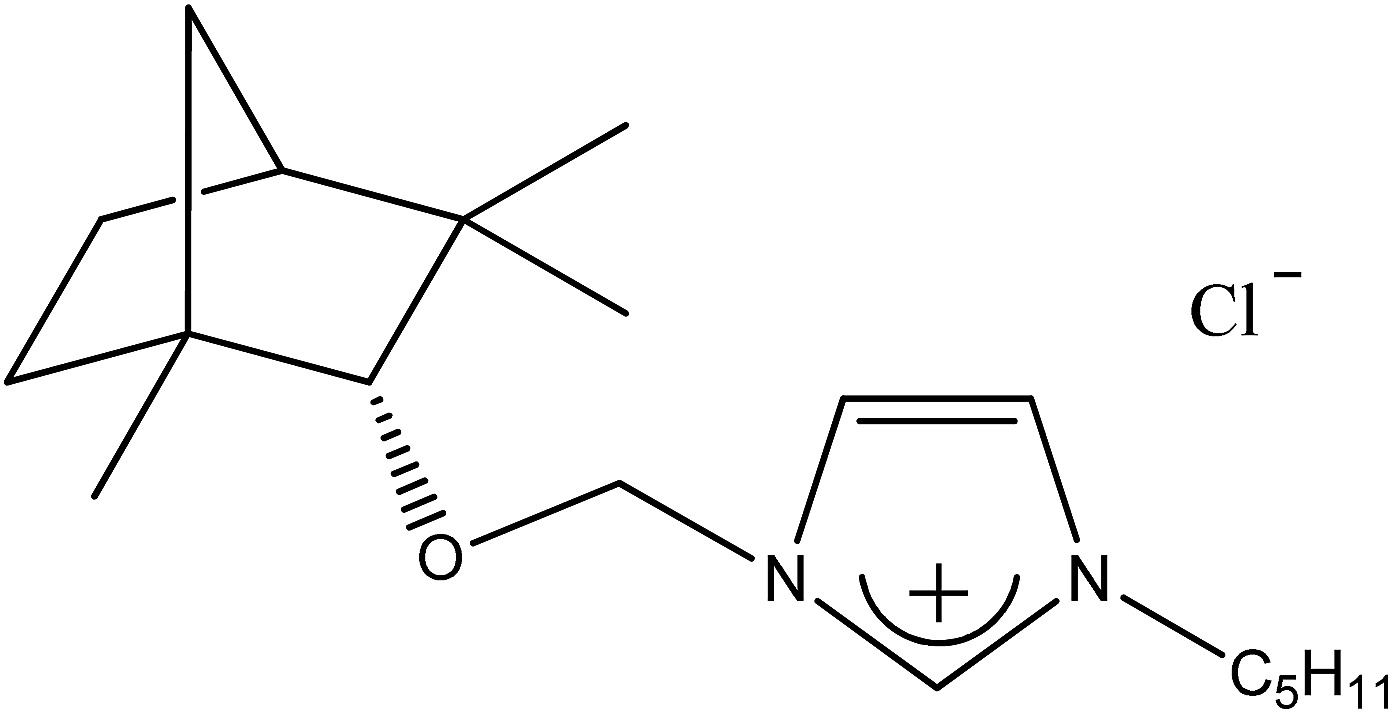 |
C5H11 | 1-[(1R)-endo-(+)-Fenchoxymethyl]-3-pentylimidazolium chloride | [C5-Im-CH2O-Fen][Cl] | 98.8 |
| CILs | Abbreviation [Cn-Im-CH2O-Terpene][Cl] | Melting pointa (°C) | Crystallization solvents | Crystal shape | Surfactant contentb (%) | Specific rotationc,d,e [α]25D |
|---|---|---|---|---|---|---|
| a Accuracy ± 0.1 °C.b Accuracy ± 0.1%.c c in methylene chloride.d Standard uncertainty for specific rotation u is u(α) = ±0.5°.e Standard uncertainty for concentration u is u(c) = ±0.002 g/100 mL. | ||||||
| (1S)-endo-(−)-Borneol based chiral ionic liquids | ||||||
| 3-Alkyl-1-[(1S)-endo-(−)-borneoxymethyl]imidazolium chlorides | ||||||
| 1b | [C1-Im-CH2O-Bor][Cl] | 103.7–104.5 | Ethyl acetate/acetone | Squared, regular, plates | 99.7 | −47.72 (c 1.035) |
| 1c | [C5-Im-CH2O-Bor][Cl] | 81.9–82.5 | Ethyl acetate/acetone | Long, regular needles | 99.2 | −36.80 (c 0.996) |
|
||||||
| (1R)-endo-(+)-Fenchol based chiral ionic liquids | ||||||
| 3-Alkyl-1-[(1R)-endo-(+)-fenchoxymethyl]imidazolium chlorides | ||||||
| 2b | [C1-Im-CH2O-Fen][Cl] | 137.9–138.2 | Ethyl acetate/acetone/ethanol | Regular needles | 99.6 | +23.125 (c 0.995) |
| 2c | [C5-Im-CH2O-Fen][Cl] | 60.7–61.4 | Ethyl acetate/acetone | Squared, regular, plates | 99.1 | +26.51 (c 1.007) |
All of the synthesized chlorides are solid. The melting point of chlorides with methyl substituent exceed 100 °C, but it should be noted that the melting point of [C1-Im-CH2O-Bor][Cl] is only slightly higher then 100 °C and is of: 103.7–104.5 °C. On the other hand the chlorides with pentyl substituent melt below 100 °C and these salts should be considered as ionic liquids, according to the definition.50 The general trend that can be observed for discussed CILs is that the chlorides with longer alkyl chain have lower melting temperatures than their analogs with a shorter alkyl chain.
The solubility test has been performed using popular polar and nonpolar solvents according to the method described by Vogel et al.42 The results are presented in Table 4. All of obtained chlorides are soluble in water, methanol, propanol, acetonitrile, chloroform, dimethylformamide and dimethyl sulfoxide at 20 °C. The solubility of discussed salts in acetone, ethyl acetate, tetrahydrofuran and toluene at 20 °C is limited or completely not observed. The raising of the temperature of the test to 50 °C and/or changing the substituent from methyl to pentyl of each salt significantly affect the increase of the solubility in conferred solvents. The synthesized salts are not soluble in diethyl ether and hexane even at elevated temperatures.
| CILs | Solvent | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ab | Bb | Cb | Db | Dc | Eb | Fb | Gb | Hb | Ib | Jb | Kb | Kc | Lb | Lc | Mb | Mc | |
| a A – water, B – methanol, C – propanol, D – acetone, E – acetonitrile, F – diethyl ether, G – hexane, H – chloroform, I – dimethylformamide (DMF), J – dimethyl sulfoxide (DMSO), K – ethyl acetate, L – tetrahydrofuran (THF), M – toluene, ‘+’ – complete soluble, ‘±’ – limited solubility, ‘−’ – insoluble.b At 20 °C.c At 50 °C. | |||||||||||||||||
| 1b | + | + | + | ± | + | + | − | − | + | + | + | − | − | − | − | − | ± |
| 1c | + | + | + | + | + | + | − | − | + | + | + | − | ± | ± | + | − | + |
| 2b | + | + | + | − | + | + | − | − | + | + | + | − | − | − | − | − | − |
| 2c | + | + | + | + | + | + | − | − | + | + | + | − | + | ± | + | ± | + |
The structure and purity of all of the synthesized salts was confirmed by spectral analysis (for spectra see ESI†). The purity was assessed by elemental analysis and HRMS analysis. All of the discussed quaternary chlorides were characterized by 1H and 13C NMR.
There are two characteristic doublets observed in 1H NMR spectra of all of the discussed salts: one doublet around 5.60–5.75 ppm and second doublet around 5.70–5.90 ppm, depending on considered chloride spectra. These signals described CH2 groups, which link the terpene moiety and imidazolium derivative, appear in the spectra in the form of two doublets as an AB spin system. This is a typical situation of the presence of diastereotopic protons, which are generally seen in the CH2 group of chiral molecules.
Optimization of HPLC measurements
Teicoplanin is widely used as the chiral stationary phase (CSP). Similarly to other macrocyclic glycopeptides, it can work either in normal phase (NP), reversed phase (RP) or polar ionic (PIM) modes. Depending on the mobile phase composition, different interactions such as hydrophobic interactions, electrostatic forces, steric hindrance or hydrogen bonding could be dominant in enantiorecognition. Studies performed by Thompson et al.51 using a crown ether column, Flieger et al.7 on a teicoplanin column and Sanganyado et al.52 on a vancomycin based column showed that enantioresolution could be positively affected by addition of the liophilic ions to the mobile phase. As it was emphasized by the above researchers, the influence of ions on enantioselectivity followed the Hofmeister series.In this experiment, the cooperative effect of liophilic ions, achiral ionic liquids (ACILs), four new (1S)-endo-(−)-borneol and (1R)-endo-(+)-fenchol-type CILs with a teicoplanin based CSP was evaluated. Acidic enantiomers of mandelic acid, vanilmandelic acid, phenyllactic acid were chosen as model analytes. The role of the mobile phase additives was examined by altering their kind and concentration. Fig. 1 presents the effect of all the investigated mobile phase modifiers on the difference between the retention times of both enantiomers for each analyte. It can be observed that the influence of the modifiers on the Δtr value varied according to their type.
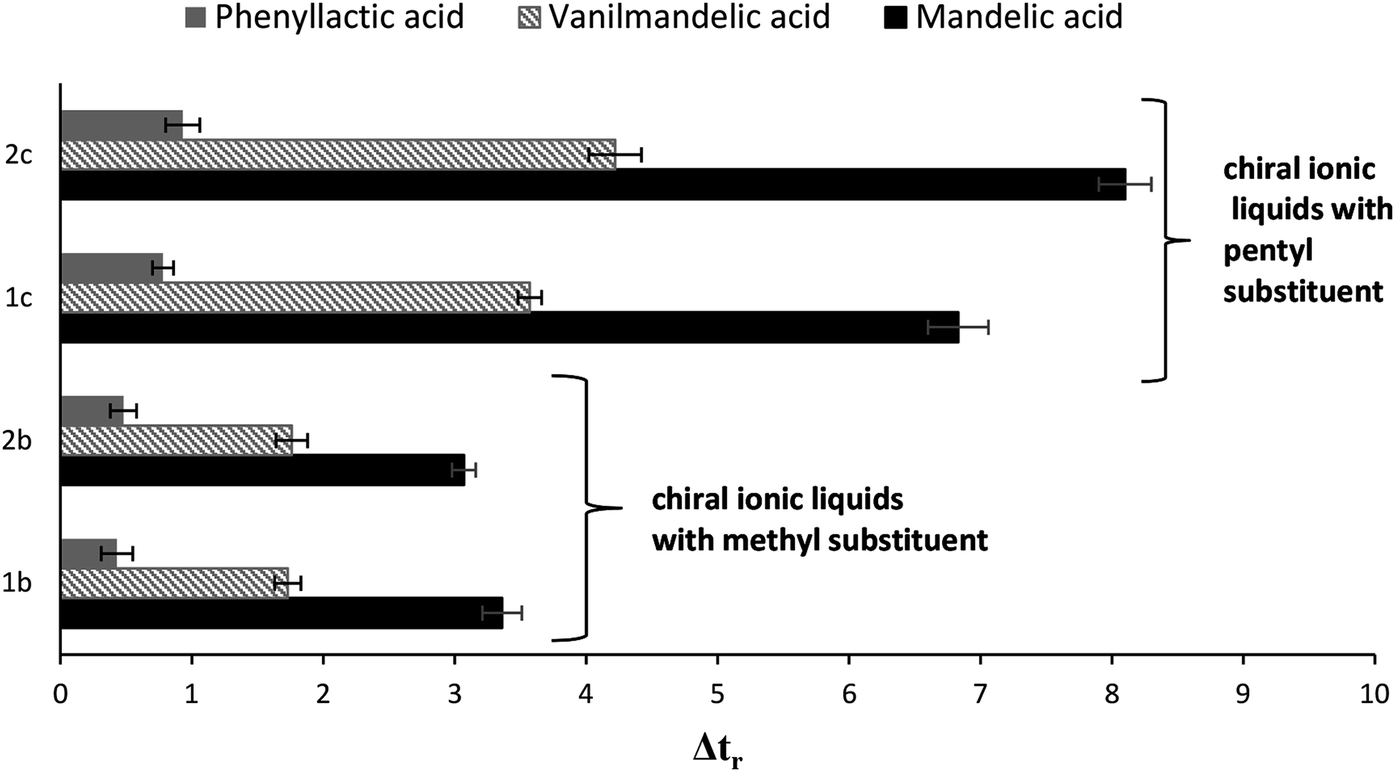 | ||
| Fig. 1 Effect of type and amount of the mobile phase modifiers on retention time difference between enantiomers of investigated acids (Δtr). Experimental conditions: the column: Astec Chirobiotic T, the mobile phase 10% MeOH/water, detection wavelength: 220 nm, flow rate: 1 mL min−1, temperature 20 °C. | ||
The effect of chaotropic ionic additives in the mobile phase on the retention and enantioresolution on a teicoplanin column is known and has been described previously.7,51,52
A similar trend was previously described for achiral ionic liquid (ACILs) additives.53 Comparing the effects of ACILs, the less hydrophobic cations revealed a smaller influence on enantioresolution than more hydrophobic ones. It should be emphasized that the effect of ACILs was much stronger than one of chaotropic salts.
Comparison of data obtained for systems modified with new additives (Fig. 1) indicates that addition of the chiral ionic liquids (CILs) to the mobile phase is responsible for the highest improving of enantioselectivity. This phenomenon could be explained by the overall synergistic effect of both chiral components of the system with either teicoplanin or CIL. Considering CILs, it should be stressed that the most important parameter influencing enantioseparation appears to be the length of the hydrocarbon chain at the N3 position of imidazolium ring. CILs possessing pentyl substituent at the N3 position exhibited a more beneficial effect on enantioselectivity than other derivatives with a methyl substituent, independently of the kind of terpene component. In other words, presence of (1S)-endo-(−)-borneol, (1R)-endo-(+)-fenchol gives similar beneficial effects and without doubt, it contributes to the improvement of enantioseparation. However, the strategic importance is presented by the substituent at the N3 position.
Chromatogram shown in Fig. 2 that both the retention and separation of enantiomers increased with the increasing concentration of CILs in the mobile phase. Whereas, in order to obtain optimal enantioselectivity, 60 mM of chaotropic salts should be added to the mobile phase, whereas in the case of CILs, a concentration of 20 mM was sufficient.
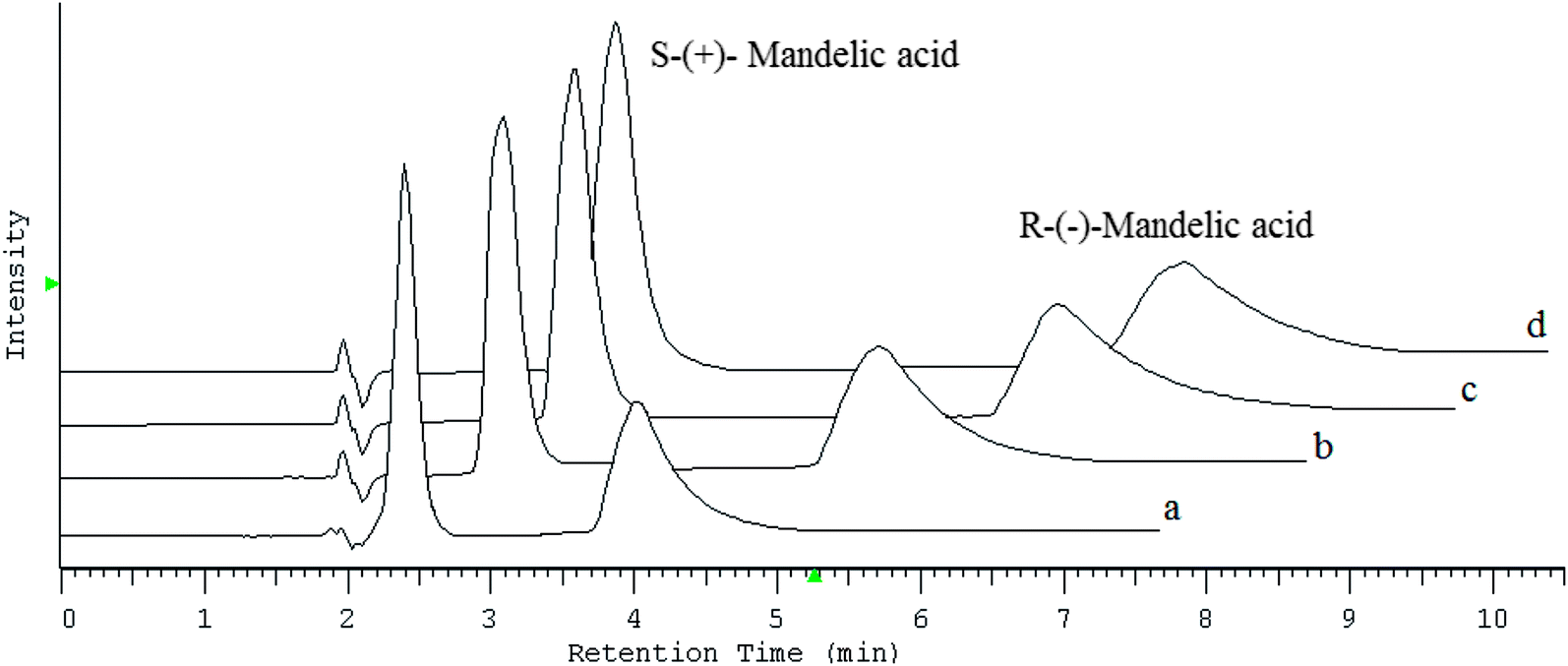 | ||
| Fig. 2 Chromatogram of mandelic acid enantiomers resolution analyzing by the use of synergistic system containing teicoplanin and different concentration of 1b CIL (a) 5 mM of CIL in 10% MeOH; (b) 10 mM of CIL in 10% MeOH; (c) 15 mM of CIL in 10% MeOH; (d) 20 mM of CIL in 10% MeOH. | ||
Thermodynamics
The effect of temperature was evaluated over the range of 5–35 °C. The enantioselectivity for investigated analytes using different CILs at the same concentration 20 mM in the mobile phase increased when the temperature was decreased. In this study, we varied the mobile phase modifiers and measured the effect of temperature on the analytes' retention factor. Linear van't Hoff curves representing lnk against 1/T were obtained for the analytes. The determination coefficient R2 values of linear plots ranged from 0.9672 to 0.9998 indicating uniform and constant retention mechanism of all analytes within the studied temperature range (Table 5). The thermodynamic functions were calculated for the binding interactions of enantiomers transferring from the mobile phase to teicoplanin bonded stationary phase in the presence of CILs acting as additional chiral selectors.
| CILs | Investigated compounds | ΔS0* (kJ mol−1 K−1) | Δ(ΔS0) (kJ mol−1 K−1) | ΔH0 (kJ mol−1) | Δ(ΔH0) (kJ mol−1) | ΔG0 (kJ mol−1) | Δ(ΔG0) (kJ mol−1) | r2 | |
|---|---|---|---|---|---|---|---|---|---|
| 1b | Mandelic acid | S(+) | −0.0282(±0.0009) | 0.0164 | −10.1639(±0.5914) | 6.8939 | −1.9023(±0.0322) | 2.0924 | 0.9911 |
| R(−) | −0.0446(±0.0027) | −17.0578(±0.8626) | −3.9947(±0.0648) | 0.9917 | |||||
| Vanilmandelic acid | S(+) | −0.0456(±0.0031) | 0.0088 | −15.8190(±0.6552) | 3.8677 | −2.4622(±0.0553) | 1.2881 | 0.9981 | |
| R(−) | −0.0544(±0.0053) | −19.6867(±0.9991) | −3.7503(±0.0608) | 0.9988 | |||||
| Phenyllactic acid | S(+) | −0.0296(±0.0015) | 0.0038 | −10.9903(±0.4276) | 1.2812 | −2.2973(±0.0440) | 0.1750 | 0.9862 | |
| R(−) | −0.0334(±0.0022) | −12.2715(±0.6115) | −2.4723(±0.0596) | 0.9949 | |||||
| 1c | Mandelic acid | S(+) | −0.0219(±0.0014) | 0.0188 | −9.5220(±0.4883) | 7.9125 | −3.0950(±0.0670) | 2.3938 | 0.9706 |
| R(−) | −0.0407(±0.0011) | −17.4345(±0.3792) | −5.4888(±0.0728) | 0.9965 | |||||
| Vanilmandelic acid | S(+) | −0.0435(±0.0061) | 0.0077 | −16.3728(±0.7188) | 3.6132 | −3.6211(±0.0773) | 1.3573 | 0.9964 | |
| R(−) | −0.0512(±0.0019) | −19.9860(±0.5343) | −4.9784(±0.0242) | 0.9989 | |||||
| Phenyllactic acid | S(+) | −0.0251(±0.0016) | 0.0053 | −10.8664(±0.7112) | 1.8050 | −3.5027(±0.0691) | 0.2674 | 0.9845 | |
| R(−) | −0.0304(±0.0020) | −12.6714(±0.7386) | −3.7701(±0.0526) | 0.9851 | |||||
| 2b | Mandelic acid | S(+) | −0.0310(±0.0018) | 0.0198 | −10.6519(±0.2946) | 8.0496 | −1.5705(±0.0307) | 2.2492 | 0.9972 |
| R(−) | −0.0508(±0.0027) | −18.7015(±0.7562) | −3.8197(±0.0628) | 0.9998 | |||||
| Vanilmandelic acid | S(+) | −0.0496(±0.0021) | 0.0111 | −16.6654(±0.5526) | 4.5968 | −2.1409(±0.0492) | 1.3401 | 0.9965 | |
| R(−) | −0.0607(±0.0035) | −21.2622(±1.0006) | −3.4810(±0.0715) | 0.9970 | |||||
| Phenyllactic acid | S(+) | −0.0326(±0.0011) | 0.0056 | −11.6188(±0.5559) | 1.9189 | −2.0655(±0.0229) | 0.2628 | 0.9927 | |
| R(−) | −0.0382(±0.0022) | −13.5377(±0.5913) | −2.3283(±0.0490) | 0.9936 | |||||
| 2c | Mandelic acid | S(+) | −0.0172(±0.0009) | 0.0187 | −8.0962(±0.3026) | 7.8858 | −3.0447(±0.0611) | 2.4096 | 0.9672 |
| R(−) | −0.0359(±0.0016) | −15.9820(±0.2989) | −5.4543(±0.0630) | 0.9983 | |||||
| Vanilmandelic acid | S(+) | −0.0392(±0.0029) | 0.0085 | −15.1348(±0.8004) | 3.8560 | −3.6351(±0.0801) | 1.3606 | 0.9943 | |
| R(−) | −0.0477(±0.0033) | −18.9908(±0.8591) | −4.9957(±0.0785) | 0.9981 | |||||
| Phenyllactic acid | S(+) | −0.0200(±0.0013) | 0.0037 | −9.3915(±0.4215) | 1.3735 | −3.5326(±0.0694) | 0.2923 | 0.9838 | |
| R(−) | −0.0237(±0.0015) | −10.7650(±0.5100) | −3.8249(±0.0726) | 0.9836 | |||||
The standard changes in enthalpy (ΔH0), entropy (ΔS0*) and Gibbs free energy (at 20 °C) (ΔG0) were calculated from the slopes and intercepts of the van't Hoff relationship.
The ΔH0 values calculated for all enantiomers were negative, indicating that the transfer of the enantiomers is enthalpically favored. The ΔH0 values were in the range from −10.16 to −21.26 kJ mol−1 for “b” series of CILs and from −8.10 to −19.99 kJ mol−1 for series “c”.
Generally, the ΔH0 values, for the R configuration were smaller than that for the S enantiomers. This means that the interaction between the R enantiomer was more favorable on the teicoplanin CSP in the presence of CILs. These results were reflected in the order of elution of the enantiomers, as the S enantiomer was always eluted first. Similar trend was observed by other researchers applying glicopeptide CSPs with only one chiral selector.54 Regarding the ΔS0 values, negative values were also obtained. The ΔS0 values for the S enantiomers were more positive than for the R ones. Considering the presence of CIL as second chiral selector, the ΔS0 values were always more negative for the system containing series “b” of CILs with a methyl functional group at the imidazolium ring regardless of the kind of terpene component in their structure. Change of CILs from the “c” in a series of “b” was associated with a decrease in entropy change. This change is related to the number of solvent molecules solvating the CSP, CIL and the analyte in the mobile phase and releasing during adsorption, or a reduced ability to move or rotate of the adsorbed analytes.
It should be emphasized that a change in the type of CILs in synergistic chromatographic system entailed a much larger difference in the value of ΔS0 than that observed for the ΔH0.
The total free energy change, ΔG0, was more negative for synergistic system containing CILs of a series “c”. The smaller ΔG0 was visible in stronger retention of the analytes and better enantioresolution.
The Δ(ΔH0), Δ(ΔS0) values can be calculated as the following differences ΔH0R − ΔH0S and ΔS0R − ΔS0S. The Δ(ΔH0) and Δ(ΔS0) values decreased with changing a series “b” into a series “c” of fenchol based CILs. Similar trend was observed for menthol based CILs.53 With the changing in Δ(ΔH0) and Δ(ΔS0), the difference in free energy, Δ(ΔG0), increased always with changing a series “b” into “c” of CILs regardless of the terpene component in their structure. Because the values (ΔΔG0) contribute to enantioseparation in accordance with the following equation: ΔΔG0 = −RTlnα, the best resolution of enantiomers for system containing a series “c” supports obtained results.
Furthermore, ΔΔG0 (at 20 °C) values derived from van't Hoff plots are in excellent agreement with the enantioselectivity factor α measured in chromatographic experiments:
| ΔΔG0 = 1.4078 (±0.0874)α − 1.2444 (±0.1654), R2 = 0.9629, se = 0.31, F = 259.62, n = 10 |
The docking simulations
The teicoplanin molecule contains four binding cavities (A–D), Fig. 3. All the ligands docked to teicoplanin in similar manner and they bind only to cavities B, C and D, Fig. 4.
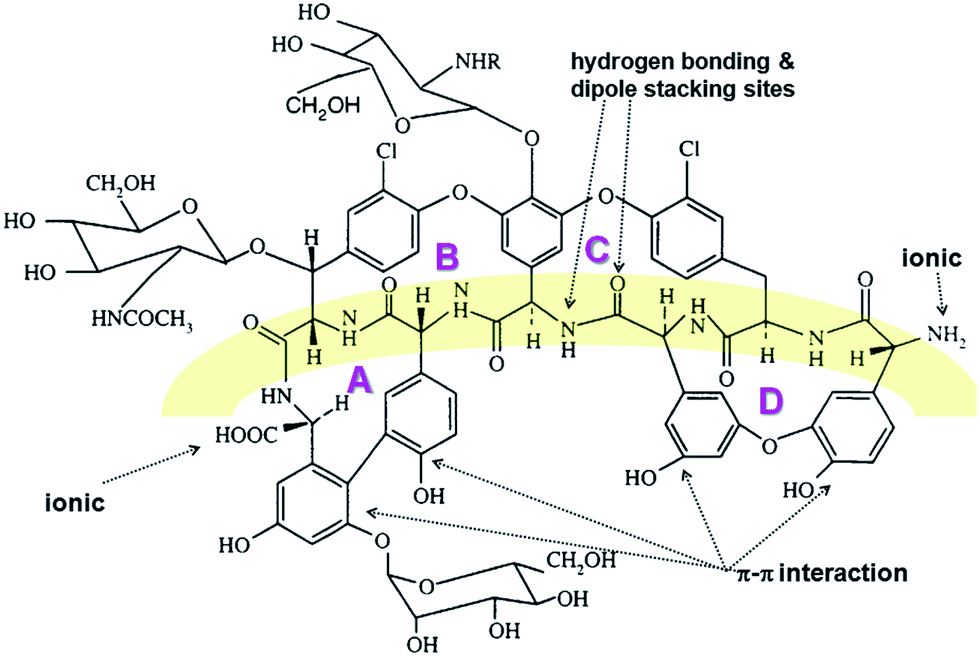 | ||
| Fig. 3 The molecular structure of teicoplanin. The distinct four binding cavities are indicated (A, B, C and D). Additionally, the groups responsible for the most relevant interactions with IL are shown. R denotes CH3-decanoic acid. | ||
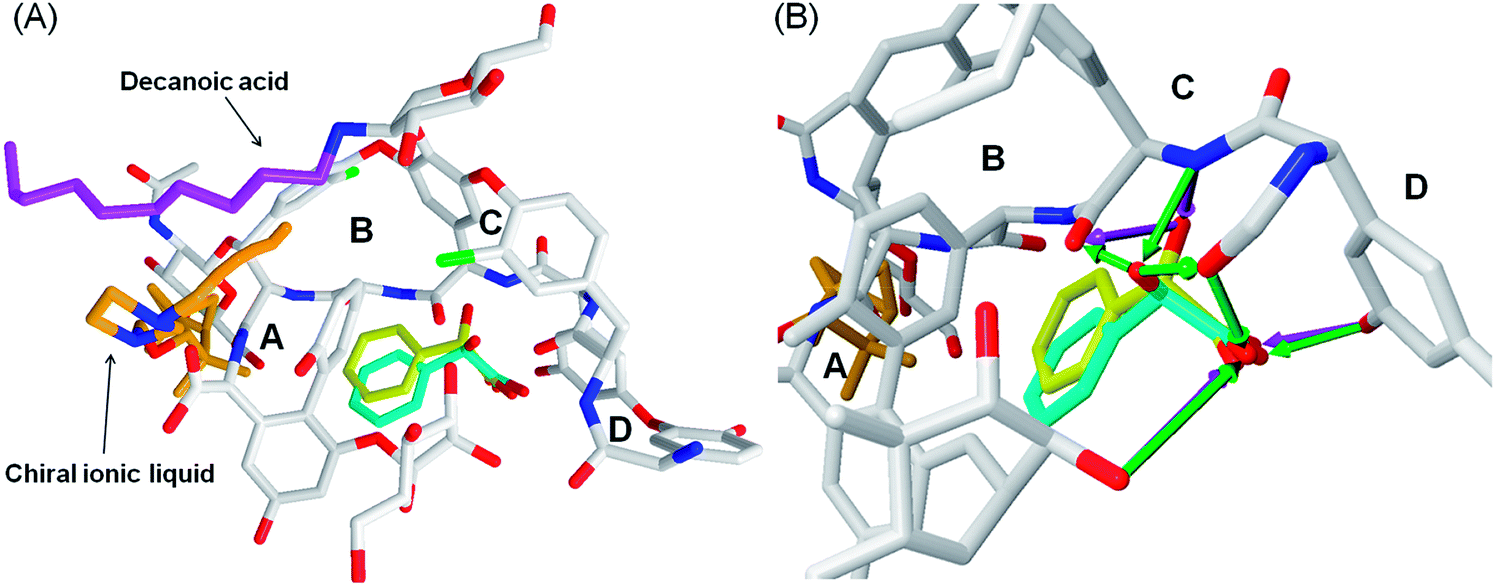 | ||
| Fig. 4 The chiral ionic liquid 2c-teicoplanin-R,S-mandelic acid complex. The 2c compound binds to cavities B, C and D. The hydrogen bonds were denoted as the green (teicoplanin-R-mandelic acid) and magenta (teicoplanin-S-mandelic acid) arrows. R-Mandelic acid is colored in blue, S-mandelic acid in yellow, teicoplanin in gray (except of most relevant oxygen and nitrogen atoms) and 2c compound in orange. The decanoic acid chain is shown in purple. | ||
The positions of the R- and S-mandelic acid molecules in the teicoplanin molecule are very similar to each other, however R-enantiomer can create more hydrogen bonds than the S-enantiomer. In the case of R-mandelic acid molecules the hydroxyl group, located at the asymmetric carbon atom, can create three hydrogen bonds (HBs): (i) one as an acceptor with the nitrogen of secondary amine; (ii) two HBs as a donor with oxygen of the diethyl ether moieties of teicoplanin, Fig. 4. Moreover, the stereoconfiguration of the molecule can determine the position of the carboxyl group of the given enantiomer and interactions created by this group with teicoplanin. The carboxyl oxygen atoms of R-mandelic acid can create three HBs (with hydroxyphenyl, 2-OH glucose moieties and oxygen of the diethyl ether moiety). In the case of S-mandelic acid, the carboxyl group is exploited as an two HBs acceptor, Fig. 4. The comparison of MolDock Score values characterizing the complexes including R- and S-enantiomers suggests that the complexes formed with R-enantiomer are more stable; in each of the considered cases, the complexes including R-enantiomer exhibit the lower MolDock Score value in comparison to complexes with S-enantiomer, Table 6. In the case of both R- and S-vanilmandelic acid, the addition of the hydroxyl and methoxyl moieties to the phenyl ring results in the creation of additional HBs with teicoplanin. These compounds can bind simultaneously to the A, B, C and D binding cavities (Fig. S1†). Moreover, for R- and S-vanilmandelic acid, more favorable values of MolDock Score were observed in contrast to mandelic acid. The respective differences are close to ∼2 kJ mol−1, independently of the enantiomer.
| Complex | MolDock Score (kJ mol−1) | Δ = complex with R-enantiomer − complex with S-enantiomer (kJ mol−1) |
|---|---|---|
| Teicoplanin-R-mandelic acid | −64.3 | −7.6 |
| Teicoplanin-S-mandelic acid | −56.7 | |
| Teicoplanin-R-vanilmandelic acid | −61.4 | −2.2 |
| Teicoplanin-S-vanilmandelic acid | −59.2 | |
| Teicoplanin-R-phenyllactic acid | −58.6 | −3.5 |
| Teicoplanin-S-phenyllactic acid | −55.1 | |
| 1b-teicoplanin-R-mandelic acid | −60.8 | −8.4 |
| 1b-teicoplanin-S-mandelic acid | −52.4 | |
| 1c-teicoplanin-R-mandelic acid | −63.1 | −10.2 |
| 1c-teicoplanin-S-mandelic acid | −52.9 | |
| 1b-teicoplanin-R-vanilmandelic acid | −59.5 | −3.9 |
| 1b-teicoplanin-S-vanilmandelic acid | −55.6 | |
| 1c-teicoplanin-R-vanilmandelic acid | −61.6 | −4.2 |
| 1c-teicoplanin-S-vanilmandelic acid | −57.4 | |
| 1b-teicoplanin-R-phenyllactic acid | −59.1 | −3.8 |
| 1b-teicoplanin-S-phenyllactic acid | −55.3 | |
| 1c-teicoplanin-R-phenyllactic acid | −63.1 | −4.1 |
| 1c-teicoplanin-S-phenyllactic acid | −59.0 | |
| 2b-teicoplanin-R-mandelic acid | −61.1 | −8.9 |
| 2b-teicoplanin-S-mandelic acid | −52.2 | |
| 2c-teicoplanin-R-mandelic acid | −64.2 | −11.9 |
| 2c-teicoplanin-S-mandelic acid | −52.3 | |
| 2b-teicoplanin-R-vanilmandelic acid | −63.4 | −4.6 |
| 2b-teicoplanin-S-vanilmandelic acid | −58.8 | |
| 2c-teicoplanin-R-vanilmandelic acid | −66.2 | −4.9 |
| 2c-teicoplanin-S-vanilmandelic acid | −60.3 | |
| 2b-teicoplanin-R-phenyllactic acid | −59.7 | −3.6 |
| 2b-teicoplanin-S-phenyllactic acid | −56.1 | |
| 2c-teicoplanin-R-phenyllactic acid | −60.2 | −3.8 |
| 2c-teicoplanin-S-phenyllactic acid | 56.4 |
Moreover, theoretical calculations showed that the separation-enhancement abilities of ILs can be significantly altered upon changing the length of the alkyl chains included in their molecular structure. The length of this chain affects differences between binding energies of R- and S-mandelic acid to the chiral IL-teicoplanin complexes, expressed as MolDock Score values, Table 6. Both the experimental and theoretical studies indicate that the alkyl chain length in the ionic liquid molecules is positively correlated with the stability of the mandelic acid–IL–teicoplanin complex, Table 6. Docking simulations revealed the molecular details of interactions between chiral IL and teicoplanin. The imidazole ring of chiral ILs creates the ionic bridge with the carboxyl moiety of teicoplanin, Fig. 5. Moreover, the alkyl group of the chiral IL can interact (hydrophobic interactions) with the decanoic aliphatic chain of teicoplanin. This interaction stabilizes the conformation of the teicoplanin decanoic chain and expose the binding cavities of teicoplanin to interaction with potential ligands. Moreover, we observed the steric adjustment between the IL terpene substituent and beta-D-glucosamine ring of teicoplanin (in 4C1 chair conformation), Fig. 5. The exocyclic moieties of both these rings exhibit the equatorial arrangement, which facilitates the contact between them. The change of the chiral IL stereoconfiguration is associated with the equatorial–axial inversion and will support the reorientation of IL molecule in the vicinity of the ring, as well as loosing the favorable interactions with teicoplanin. The scheme of the molecular interactions within the mandelic acid–IL–teicoplanin complex is common for all studied chiral ILs, (Fig. S2†).
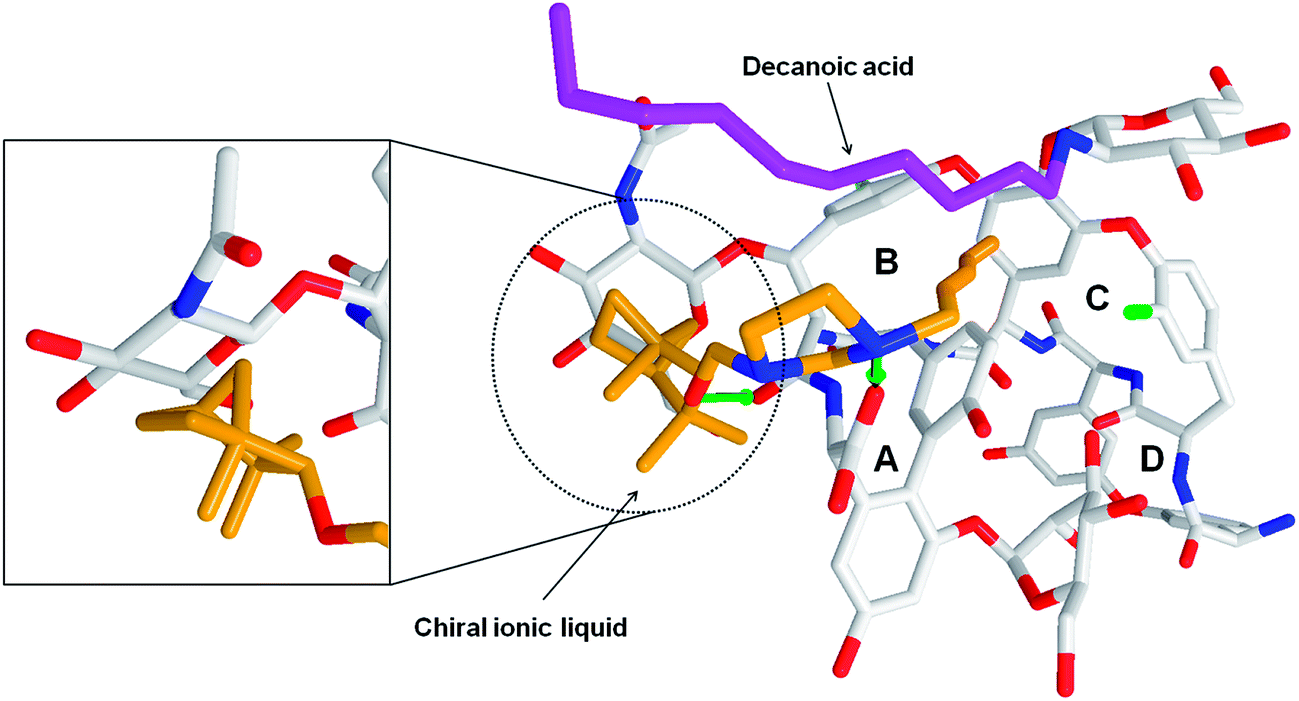 | ||
| Fig. 5 The teicoplanin-2c complex. The imidazolum moiety of 2c forms the ionic bridge with the carboxyl group of teicoplanin. Teicoplanin is presented in gray (except of most relevant oxygen and nitrogen atoms). The 2c compound was colored in orange. The decanoic acid chain was given in purple. The left panel shows the steric adjustment between the terpene conformation of IL and beta-D-glucosamine moiety of teicoplanin. | ||
Conclusion
In summary, a novel group of terpene-based CILs derived from renewable sources from plants were prepared and characterized. As starting materials bicyclic terpene alcohols: (1S)-endo-(−)-borneol and (1R)-endo-(+)-fenchol were selected. The quaternization (a specific type of Menschutkin reaction) does not required energy input. The physicochemical characteristics of obtained CILs, including spectral properties, melting point, type of the crystal shape, specific rotation and solubility in various solvents were comprehensively studied.To our knowledge, this is the first study on the synergistic effect of chiral ionic liquid additives and a teicoplanin-based CSP in LC. The chiral recognition mechanism in such a synergistic system containing two chiral selectors has not been previously investigated.
We experimentally and theoretically proved that the addition of the chiral ionic liquids with a natural terpene substituent and elongation of the alkyl group of the chiral ILs affects the increasing differences in the binding energy of R-enantiomer in comparison to S-enantiomer binds to teicoplanin, thus determining better separations of the studied enantiomers.
The results of docking simulations clearly show the differences in the binding modes of acidic enantiomers interacting with teicoplanin. We indicated that: (i) both of the mandelic acid enantiomers bind into the C and D binding cavities of teicoplanin; (ii) R-mandelic acid exhibits more favorable interactions with teicoplanin in comparison to S-mandelic acid which is correlated with the number of intermolecular hydrogen bonds. We revealed that the presence of chiral ionic liquid affects the mandelic acid enantiomers binding into teicoplanin. Namely, it increases the stability of the teicoplanin–mandelic acid complex which is due to the greater amount of hydrogen bonding that appears in such complex. Secondly, the difference between stabilities of the complexes including R- and S-enantiomers of mandelic acid is enhanced in the presence of CIL. Finally, the elongation of the alkyl chain present in the IL molecules additionally promotes both the binding strength and the enantiospecific effects that occur in mandelic acid binding to teicoplanin. We also observed that the stereoconfiguration of IL may influence its interaction with teicoplanin.
Acknowledgements
This research was financed by the National Science Centre (Poland) grant no. 2013/09/D/ST5/03904.References
- Q. Q. Baltazar, S. K. Leininger and J. L. Anderson, J. Chromatogr. A, 2008, 1182, 119–127 CrossRef CAS PubMed.
- J. L. Anderson and D. W. Armstrong, Anal. Chem., 2003, 75, 4851–4858 CrossRef CAS PubMed.
- W. Qin, H. Wei and S. F. Y. Li, J. Chromatogr. A, 2003, 985, 447–454 CrossRef CAS PubMed.
- X. Xiao, L. Zhao, X. Liu and S. Jiang, Anal. Chim. Acta, 2004, 519, 207–211 CrossRef CAS.
- J. Flieger and A. Czajkowska-Żelazko, J. Sep. Sci., 2011, 34, 733–739 CrossRef CAS PubMed.
- Y. Wang, M. Tian, W. Bi and K. H. Row, Int. J. Mol. Sci., 2009, 10, 2591–2610 CrossRef CAS PubMed.
- J. Flieger, Anal. Lett., 2009, 42, 1632–1649 CrossRef CAS.
- V. Maier, J. Horáková, J. Petr, D. Drahoňovský and J. Ševcík, J. Chromatogr. A, 2006, 1103, 337–343 CrossRef CAS PubMed.
- P. Laamanen, S. Busi, M. Lahtinen and R. Matilainen, J. Chromatogr. A, 2005, 1095, 164–171 CrossRef CAS PubMed.
- J. G. Huddleston, H. D. Willauer, R. P. Swatloski, A. E. Visser and R. D. Rogers, Chem. Commun., 1998, 1765–1766 RSC.
- J. F. Liu, G. B. Jiang, Y. G. Chi, Y. Q. Cai, Q. X. Zhou and J. T. Hu, Anal. Chem., 2003, 75, 5870–5876 CrossRef CAS PubMed.
- N. Fontanals, F. Borrull and R. M. Marce, TrAC, Trends Anal. Chem., 2012, 41, 15–26 CrossRef CAS.
- J. Flieger, M. Tatarczak-Michalewska, A. Groszek and El. Blicharska, Anal. Lett., 2016, 49, 1997–2005 CrossRef CAS.
- J. Flieger and A. Czajkowska-Żelazko, Food Chem., 2015, 166, 150–157 CrossRef CAS PubMed.
- K. E. Gutowski, G. A. Broker, H. D. Willauer, J. G. Huddleston, R. P. Swatloski, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2003, 125, 6632–6633 CrossRef CAS PubMed.
- D. Han and K. H. Row, Molecules, 2010, 15, 2405–2426 CrossRef CAS PubMed.
- J. Flieger, El. B. Grushka and A. Czajkowska-Zelazko, Austin J. Anal. Pharm. Chem., 2014, 1, 1009 Search PubMed.
- A. Berthod, M. J. Ruiz-Ángel and S. Carda-Broch, J. Chromatogr. A, 2008, 1184, 6–18 CrossRef CAS PubMed.
- C. D. Tran and I. Mejac, J. Chromatogr. A, 2008, 1204, 204–209 CrossRef CAS PubMed.
- S. Rizvi and S. A. Shamsi, Anal. Chem., 2006, 78, 7061–7069 CrossRef CAS PubMed.
- Y. Francois, A. Varenne, E. Juillerat, D. Villemin and P. Gareil, J. Chromatogr. A, 2007, 1155, 134–141 CrossRef CAS PubMed.
- B. Wang, J. He, V. Bianchi and S. A. Shamsi, Electrophoresis, 2009, 30, 2812–2819 CrossRef CAS PubMed.
- J. Zhang, Y. Du, Q. Zhang and Y. Lei, Talanta, 2014, 119, 193–201 CrossRef CAS PubMed.
- Y. Francois, A. Varenne, E. Juillerat, D. Villemin and P. Gareil, J. Chromatogr. A, 2007, 1155, 134–141 CrossRef CAS PubMed.
- Y. Zhang, S. Du, Z. Feng, Y. Du and Z. Yan, Anal. Bioanal. Chem., 2016, 408, 2543–2555 CrossRef CAS PubMed.
- L. M. Yuan, Y. Han, Y. Zhou, X. Meng, Z. Li, M. Zi and Y. Chang, Anal. Lett., 2006, 39, 1439–1449 CrossRef CAS.
- P. Kodali and A. M. Stalcup, J. Liq. Chromatogr. Relat. Technol., 2014, 37, 893–906 CrossRef CAS.
- Q. Liu, K. Wu, F. Tang, L. Yao, F. Yang, Z. Nie and S. Yao, Chem.–Eur. J., 2009, 15, 9889–9896 CrossRef CAS PubMed.
- Y. X. Yang, J. Li and X. Y. Jiang, J. Cent. South Univ., 2013, 20, 1173–1177 CrossRef CAS.
- H. Qing, X. Jiang and J. Yu, Chirality, 2014, 26, 160–165 CrossRef CAS PubMed.
- Z. Zhou, X. Li, X. Chen and X. Hao, Anal. Chim. Acta, 2010, 678, 208–214 CrossRef CAS PubMed.
- G. Qian, H. Song and S. Yao, J. Chromatogr. A, 2016, 1429, 127–133 CrossRef CAS PubMed.
- J. Pernak, M. Niemczak, Ł. Chrzanowski, Ł. Ławniczak, P. Fochtman, K. Marcinkowska and T. Praczyk, Chem.–Eur. J., 2016, 22, 12012–12021 CrossRef CAS PubMed.
- J. Neumann, S. Steudte, C.-W. Cho, J. Tröming and S. Stolte, Green Chem., 2014, 16, 2174–2184 RSC.
- J. Feder-Kubis, B. Szefczyk and M. Kubicki, J. Org. Chem., 2015, 80, 237–246 CrossRef CAS PubMed.
- K. Erfurt, I. Wandzik, K. Walczak, K. Matuszek and A. Chrobok, Green Chem., 2014, 16, 3508–3514 RSC.
- J. M. M. Araújo, C. Florindo, A. B. Pereiro, N. S. M. Vieira, A. A. Matias, C. M. M. Duarte, L. P. N. Rebelo and I. M. Marrucho, RSC Adv., 2014, 4, 28126–28132 RSC.
- Z. Wang, Q. Wang, Y. Zhang and W. Bao, Tetrahedron Lett., 2005, 46, 4657–4660 CrossRef CAS.
- J. C. Plaquevent, J. Levillain, F. Guillen, C. Malhiac and A. C. Gaumont, Chem. Rev., 2008, 108, 5035–5060 CrossRef CAS PubMed.
- J. Pernak, J. Feder-Kubis, A. Cieniecka-Rosłonkiewicz, C. Fischmeister, S. Griffin and R. D. Rogers, New J. Chem., 2007, 31, 879–892 RSC.
- J. Pernak and J. Feder-Kubis, Chem.–Eur. J., 2005, 11, 4441–4449 CrossRef CAS PubMed.
- A. I. Vogel, A. R. Tatchell, B. S. Furnis, A. J. Hannaford and P. W. G. Smith, Vogel's Textbook of Practical Organic Chemistry, Longman, 5th edn, 1989 Search PubMed.
- Y. C. Liu, Y. S. Li, S. Y Lyu, L. J. Hsu, Y. H. Chen, Y. T. Huang, H. C. Chan, C. J. Huang, G. H. Chen, C. C. Chou, M. D. Tsai and T. L. Li, Nat. Chem. Biol., 2011, 7, 304–309 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17–33 CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. Plazinska, M. Kolinski, I. W. Wainer and K. Jozwiak, J. Mol. Model., 2013, 19, 4919–4930 CrossRef CAS PubMed.
- A. Roy, A. Kucukural and Y. Zhang, Nat. Protoc., 2011, 5, 725–738 CrossRef PubMed.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS PubMed.
- R. A. Thompson, Z. Ge, N. Grinberg, D. Ellison and P. Tway, Anal. Chem., 1995, 67, 1580–1587 CrossRef CAS.
- E. Sanganyado, Z. Lu and J. Gan, J. Chromatogr. A, 2014, 1368, 82–88 CrossRef CAS PubMed.
- J. Flieger, J. Feder-Kubis, M. Tatarczak-Michalewska, A. Płazińska, A. Madejska and M. Swatko-Ossor, J. Sep. Sci. DOI:10.1002/jssc.201700197 , in press.
- A. Péter, G. Török, D. W. Armstrong, G. Toth and D. Tourwe, J. Chromatogr. A, 1998, 828, 177–190 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03310a |
| This journal is © The Royal Society of Chemistry 2017 |