
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe protective effects of Shikonin on lipopolysaccharide/D-galactosamine-induced acute liver injury via inhibiting MAPK and NF-κB and activating Nrf2/HO-1 signaling pathways
Ye Tian†
,
Zheng Li†,
Bingyu Shen,
Lin Wu,
Lu Han,
Qiaoling Zhang and
Haihua Feng *
*
Key Laboratory of Zoonosis, Ministry of Education, College of Veterinary Medicine, Jilin University, Changchun, Jilin 130062, PR China. E-mail: fhh70@163.com; Fax: +86 431 87836160; Tel: +86 431 87836161
First published on 11th July 2017
Abstract
Shikonin (SHK) has various biological and pharmacological activities, including anticancer, antibacterial and anti-inflammation activities. However, the protective effects and mechanism of SHK on lipopolysaccharide (LPS) and D-galactosamine (D-GalN) induced acute liver injury remain unclear. In this study, LPS/D-GalN caused acute liver injury by intraperitoneal injection. SHK was administrated for 1 h. Then, LPS/D-GalN was given to C57BL/6 mice for 3 h. Our results showed that SHK treatment distinctly decreased serum TNF-α, IL-1β, IL-6 and IFN-γ inflammatory cytokine production, reduced serum ALT, AST, hepatic MPO and ROS production levels, and tissue histology harmful effects, inhibited JNK1/2, ERK1/2, p38 and NF-κB (p65) phosphorylation, and suppressed IκBα phosphorylation and degradation. Furthermore, our research showed that SHK could dramatically increase SOD and GSH production, as well as reduce ROS production, through up-regulating the protein expression of HO-1, Nqo1, Gclc and Gclm, which was related to the induction of Nrf2 nuclear translocation. These results showed that SHK exerted anti-inflammatory activity, which was associated to the inhibition of inflammatory production via down-regulation of the MAPK and NF-κB signaling pathways, and anti-oxidative effects were connected with GSH and SOD activation through up-regulation of the Nrf2/HO-1 signaling pathways.
Introduction
Acute liver injury is regarded as severe and life-threatening and can lead to awful hemorrhagic necrosis and hepatocellular death.1 It is also a devastating syndrome that is characterized by a high rate of mortality.2 Although the treatment strategies of acute liver injury have been comprehensively studied, there is still no appropriate therapy for it.3,4 Lipopolysaccharides (LPS) are the main membrane component of Gram-negative bacteria, and have been reported to play critical roles in the activity of endotoxins.5 The capacity of D-GalN could enlarge liver injury induced by LPS in a moment.6 LPS and D-GalN-induced acute liver injury in mice is similar to the pathological process of human liver damage and has been widely considered as a representative experimental model.7 LPS and D-GalN could induce various inflammatory responses which result in release of inflammatory mediators such as TNF-α, IL-1β, IL-6, and IFN-γ.8 Massive evidence have revealed that inflammatory mediators are activated by nuclear factor-kappa B (NF-κB) and three mitogen-activated protein kinase (MAPK) pathways, including the c-Jun NH2-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 pathways overexpression.9,10 These inflammatory cytokines also can lead to the apoptosis of hepatocytes and liver failure.11 The features of hepatitis are mainly associated with the release of inflammatory cytokines, the elevation of aspartate aminotransferase (AST) and alanine aminotransferase (ALT), and hepatocyte necrosis. Although many treatment manners are currently used in the clinic, the therapeutic effect is not ideal. Therefore, safe and effective treatments need to be explored.Nuclear factor erythroid-2-related factor 2 (Nrf2) is a redox sensitive transcription factor, and it plays a critical role in regulating cellular defenses against endogenous or exogenous stresses by enhancing the expression of various anagotoxic and antioxidant genes, such as heme oxygenase 1 (HO-1).12 Due to the discovery of cytoprotective properties, HO-1 has gained comprehensive attention. HO-1 and its enzymatic metabolites can protect bodies against oxidative damage.13,14 In recent studies, HO-1 has been reported to be a novel enzyme with potent antioxidant, anti-inflammatory, and anti-proliferative effects.14–16
Previous studies have confirmed that Nrf2 could attenuate the LPS-induced liver damage.17 Thus, Nrf2 may be regarded as a prospective target for treating liver diseases.18 Recent reports suggest that LPS can enhance the development of reactive oxygen species (ROS) and extensively be applied to inducing pharmacological research models of ALI as inflammatory mediators.19
ROS is connected with the metabolism of cells and various oxidative stress diseases.20 High concentrations of ROS may create cells damage and are related to a series of adverse result for cell, such as inflammation, apoptosis, necrosis, and cancer diseases.21 Interestingly, low concentration of ROS can produce protective enzymes and endogenous antioxidants that regulate the balance of intracellular homeostasis in mammalian cells.22 Previous studies reported that Nrf2 is reliable for modulating the antioxidant response element (ARE)-driven expression of antioxidant enzymes, which results in the induction of many cytoprotective proteins, such as HO-1, NAD(P)H:quinone oxidoreductase 1 (Nqo1), and glutamate-cysteine ligase, catalytic (Gclc) and glutamate-cysteine ligase, modifier (Gclm).23,24
Shikonin (SHK), a natural crystalline powder extracted from the roots of Lithospermum erythrorhizon, has many pharmacological effects, including anticancer, anti-inflammatory, and antibacterial functions.25 Recently, SHK has been reported to play an important role in regulating the processes of inflammation and exerting strong anti-inflammatory effects. Lee and colleagues found that SHK could effectively suppress allergic airway inflammation in a model of asthma and inhibit bone marrow-derived dendritic cell (BM-DC) maturation in vitro.26 SHK exerted anti-oxidative effects via Unique Modulation of the Nrf2/ARE Pathway by Induced HL-60 Cell Differentiation, as described by Bo Zhang and colleagues.27 Xiong et al. also found that SHK could decrease the release of pro-inflammatory cytokines in cerulein-induced acute pancreatitis in mice model.28 However, the mechanism of action of SHK in a model of LPS/D-GalN-induced acute liver injury remains unclear.
Materials and methods
Materials
Shikonin (purity ≥ 98%) was purchased from the Chengdu Pufei De Biotech co., Ltd (Chengdu, China). Dimethylsulfoxide (DMSO), LPS (Escherichia coli lipopolysaccharide, 055:B5) were purchased from Sigma-Aldrich (St. Louis, MO, USA). We purchased D-galactosamine hydrochloride from Aladdin Industrial Corporation (Shanghai, China). Aspartate aminotransferase (AST), alanine aminotransferase (ALT) superoxide dismutase (SOD), reduced glutathione (GSH), MPO (myeloperoxidase) detection kits were provided by the Jiancheng Bioengineering Institute of Nanjing (Nanjing, Jiangsu, China). Mouse TNF-α, IL-1β, INF-γ and IL-6 enzyme-linked immunosorbent assay (ELISA) kits were provided by BioLegend (CA, USA). Mouse reactive oxygen species (ROS) ELISA kit was provided by HoraBio Biological Technology co., LTD (Shanghai, China). Antibodies against Gclc, Gclm, Nqo1, p-(ERK1/2), ERK1/2, p-(JNK1/2), JNK1/2, p-p38, p38, IκB, p-IκB, and p-P65 were purchased from Cell signal technology (Boston, MA, USA). Anti-HO-1 and anti-Nrf2 monoclonal antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Anti-Lamin B, anti-β-actin monoclonal antibodies were purchased from Proteintech Group Inc. (Boston, MA, USA), and HRP-conjugated goat anti-rabbit and goat anti-mouse antibodies were provided by GE Healthcare (Buckinghamshire, UK). All other chemicals were of reagent grade.Animals and experimental design
C57BL/6 mice (male, 6–8 weeks old, weighing approximately 18 to 22 g each) were purchased from the Liaoning Changsheng Biotechnology (Liaoning, China). These mice were given adequate food and water ad libitum and housed in clean cages for a week. The laboratory temperature was 24 ± 1 °C and relative humidity was 40–80%. All studies were performed in accordance with the National Institutes of Health guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. This study was reviewed and approved by the Animal Welfare and Research Ethics Committee at Jilin University.Acute liver injury was induced by intraperitoneal (IP) injection of LPS (30 μg kg−1) together with D-GalN (600 mg kg−1) dissolved in phosphate-buffered saline, which can increase the sensitivity of hepatocytes. Blood was collected from retro-orbital venous plexus at 3 h after the injection of LPS/D-GalN. Then mice were dissected and liver tissues were removed immediately for subsequent protein extraction and histological detection. Normal phosphate-buffered saline was used in control group mice. SHK (12.5 and 25 mg kg−1) were dissolved in dimethylsulfoxide (DMSO) and injected intraperitoneally to each treated mouse 20 μl at 1 h before the injection of LPS/D-GalN. The concentration of DMSO was 5%. As a negative control, SHK (12.5 mg kg−1) only was injected containing the same concentration of DMSO into the matched group mice.
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST)
Blood sample was collected stationary for one night at 4 °C. Then the serum was isolated after centrifugation at 3000 rpm for 10 min at 4 °C. The ALT and AST levels in serum were determined by test kits purchased from Jiancheng Bioengineering Institute of Nanjing according to the instructions.ELISA assays
According to previous study, the blood was collected for measuring TNF-α, IL-1β, IFN-γ, and IL-6 at 3 h after LPS/D-GalN injection. Then the serum was parted from blood by centrifugation at 3000 rpm at 4 °C for 10 min. These cytokines were measured by using mouse enzyme-linked immunosorbent assay (ELISA) kits (Biolegend) on the base of the manufacturer's instructions.Liver histology analyses
Liver tissues were fixed in 10% neutral buffered formalin, dehydrated and embedded with paraffin, and then sliced into 4 μm-thickness sections. Liver tissue sections were deparaffinized, rehydrated and stained with hematoxylin and eosin (H&E) to evaluate the liver tissue pathology.Biochemical analyses
All of the mice were sacrificed at 3 h after LPS/D-GalN treatment, and mice liver tissues were homogenized and dissolved in extraction buffer to analyze the GSH, ROS, MPO, and SOD levels according to the manufacturer's instructions. MPO has ability of reducing hydrogen peroxide, by which we can determine the activity of MPO and the number of neutrophil granulocytes. After the hydrogen donor offered hydrogen, a yellow compound was created. Determine the amount at 460 nm by colorimetry, so MPO activity levels were known. All results were normalized by the total protein concentration in each sample. The liver homogenate SOD levels were quantified using an SOD activity kit, which is a sensitive kit using WST-1 that produces a water-soluble formazan dyeupon reduction with superoxide anion. The rate of the reduction with a superoxide anion is linearly related to the xanthine oxidase (XO) activity and is inhibited by SOD. Therefore, the inhibition activity of SOD can be determined by a colorimetric method. These results were also normalized by the total protein concentration in each sample. GSH is a kind of low-molecular-weight free radicals scavenge, which could clear O2−, H2O2, and LOOH. GSH could react with dithiobisnitrobenzoicacid. Then a yellow compound was produced. GSH content was determined at 405 nm by the quantitative colorimetric. ROS come from aerobic cells, and it can produce O2−, H2O2, H2O–, and –OH. ROS cause cell apoptosis and death by oxidative stress. We obtained ROS level by ELISA method.Western blot analysis
Liver tissue samples were grinded by tissue grinder, the tissue homogenate lysed in Radio Immunoprecipitation Assay (RIPA) buffer containing 1 mM phenylmethanesulfonyl fluoride (PMSF). Nuclear and cytoplasmic proteins of the liver tissues were extracted from livers by using Nuclear and cytoplasmic protein Extraction kit (beyotime, Beijing, China). Protein concentrations were measured using the bicinchoninic acid protein assay kit (Thermo Scientific, Rockford, IL, USA). Then 30 μg of proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane following separation on 12% SDS-polyacryl-amide-gel. The membrane was blocked with 5% nonfat milk shaken continuously on the table for 2 h at room temperature. Then the membrane was incubated overnight at 4 °C with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 dilution of primary antibodies. Then the membrane was incubated in a 1:5000 dilution of secondary antibodies conjugated to horseradish peroxidase at room temperature for 1 h and visualized by chemiluminescence (ECL) western blotting detection system and band intensities were quantified using ImageJ gel analysis software.
:1000 dilution of primary antibodies. Then the membrane was incubated in a 1:5000 dilution of secondary antibodies conjugated to horseradish peroxidase at room temperature for 1 h and visualized by chemiluminescence (ECL) western blotting detection system and band intensities were quantified using ImageJ gel analysis software.
Statistical analysis
All data were expressed as mean ± SD. The statistical analysis was performed by the two-tailed Student's t-test and one-way ANOVA using the GraphPad Prism 6.0 software. Statistical significance was accepted at P < 0.05 or P < 0.01.Results
Effects of SHK on the mortality of LPS/D-GalN treated mice
Acute liver injury had a high death rate in mice; therefore, we assessed SHK on LPS (30 μg kg−1)/D-GalN (600 mg kg−1) induced mortality. The survival rate of the LPS/D-GalN plus SHK-treated mice were monitored for 24 hours. As is shown in Fig. 1B, mice treated only by SHK (12.5 and 25 mg kg−1) all survived as the control group. Mice injected only with LPS/D-GalN suffered successive death in 8 hours and the mortality rate was 100% (10/10). However, we found the mortality rate of pretreatment with SHK (12.5 and 25 mg kg−1) on LPS/D-GalN-induced liver damage significantly decreased. Respectively the survival rates of 30% (3/10) and 80% (8/10) were measured. But we found that SHK (50 mg kg−1) dramatically improved the mortality rate to 50% (5/10) without LPS/D-GalN involvement. These results suggested that SHK (12.5 and 25 mg kg−1) effectively protected against LPS/D-GalN-induced high mortality. But SHK (50 mg kg−1) was toxic, and SHK (12.5 and 25 mg kg−1) were not toxic for mice. So we selected SHK (12.5 and 25 mg kg−1) for future experiments.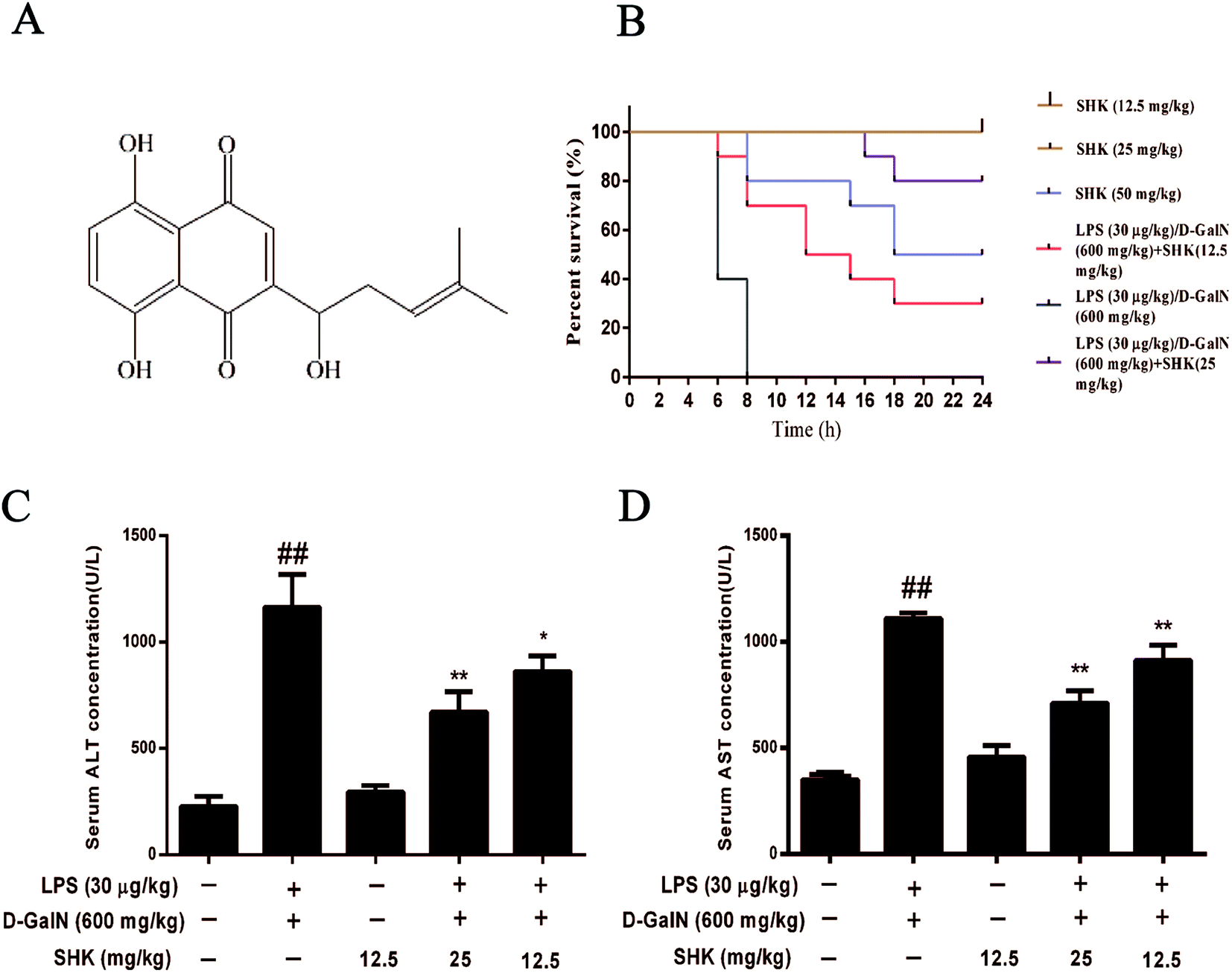 | ||
| Fig. 1 Effects of SHK on the mortality of LPS/D-GalN as well as serum ALT and AST levels in mice. Mice were administrated different concentrations of SHK in existence or shortage of LPS (30 μg kg−1) and D-GalN (600 mg kg−1). (A) The chemical structure of SHK. (B) Mice were administered SHK (12.5 and 25 mg kg−1) for 1 h before LPS/D-GalN stimulation and determined the mortality rate. In order to exploring drug toxicity, pretreatment with SHK only (12.5, 25, and 50 mg kg−1) were given to mice. Survival rate of mice received the results, followed by accepting a lethal dose of LPS (30 μg kg−1) and D-GalN (600 mg kg−1). The data are expressed as the percentage of surviving mice at each time point (n = 10 in each group). (C and D) After the SHK administered 1 h, LPS (30 μg kg−1) and D-GalN (600 mg kg−1) were injected for 3 h (n = 5/group). Then, the serum levels of ALT and AST were measured, as shown. ##p < 0.01 vs. the control group; *p < 0.05 and **p < 0.01 vs. the LPS/D-GalN group. | ||
Effects of SHK on LPS/GalN-induced serum ALT and AST levels in mice
In order to investigate the effects of SHK on LPS/D-GalN-induced liver damage, we evaluated the effects of SHK on LPS/D-GalN-induced hepatotoxicity. Thus the serum was collected for detecting ALT and AST levels. As is shown in Fig. 1C and D, SHK (12.5 mg kg−1) did not affect the levels of serum ALT and AST compared to the control group. However, LPS/D-GalN dramatically increased the serum AST and ALT levels, whereas SHK (12.5 and 25 mg kg−1) reduced these increases in a dose-dependent manner. The decreasing enzyme of evidence indicates that SHK could reduce LPS/D-GalN-induced hepatotoxicity.Effects of SHK treatment on LPS/D-GalN-induced MPO, ROS, SOD and GSH production in mice
Next, we investigated whether SHK protected against hepatotoxicity by mitigating oxidative stress. Our results showed that SHK (12.5 and 25 mg kg−1) treatment reduced the ROS and MPO levels (Fig. 2B and C) and markedly enhanced the GSH and SOD levels (Fig. 2A and D), which play critical roles in protecting against LPS/D-GalN-induced inflammatory and oxidative stress, suggesting that SHK could inhibit the capacity of oxidative stress, such as ROS, SOD, and GSH. Furthermore, SHK also decreased inflammatory effects by MPO levels.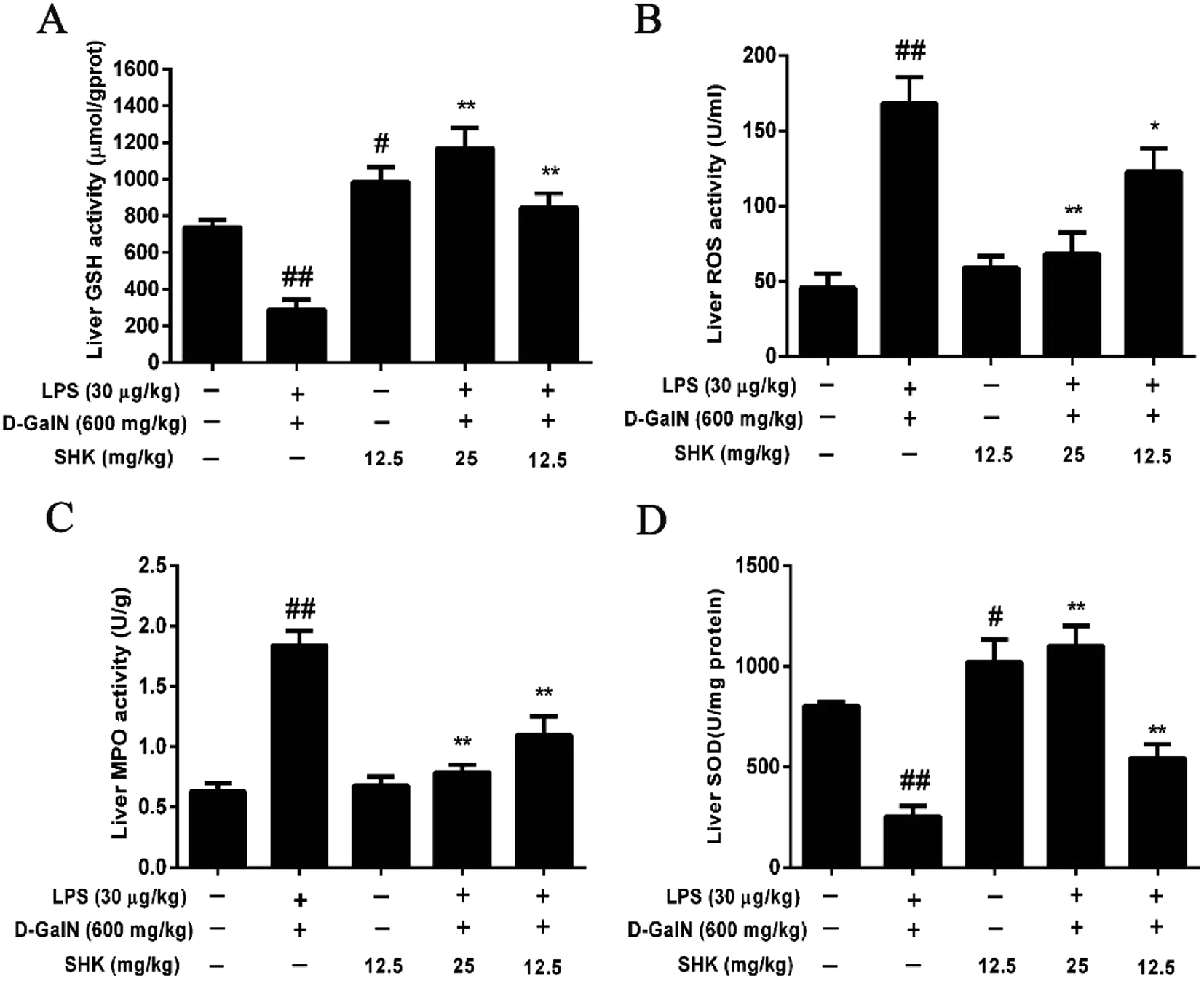 | ||
| Fig. 2 Effects of SHK treatment on LPS/D-GalN-induced MPO, ROS, SOD and GSH production in mice. Mice were injected with SHK (12.5 and 25 mg kg−1) in existence or shortage of LPS (30 μg kg−1) and D-GalN (600 mg kg−1). Mice were administered SHK i.p. for 1 h. Then, the mice were injected with LPS/D-GalN for 3 h. (A) SHK improved GSH production of liver. GSH levels were assayed by using the Griess reaction. (B) SHK inhibited the release of LPS/D-GalN-induced ROS in liver. The protein expression of ROS was determined using a ROS ELISA kit. (C) SHK significantly inhibited LPS/D-GalN-induced MPO production in liver. (D) SHK improved the release of SOD in liver. MPO and SOD levels were assayed by using the Griess reaction. All data were presented as means ± SD of three independent experiments. ##p < 0.01 and #p < 0.05 vs. the control group; *p < 0.05 and **p < 0.01 vs. the LPS/D-GalN group. | ||
Effects of SHK in decreasing inflammatory responses and hepatotoxicity on LPS/D-GalN-induced acute liver injury
The study of liver injury is closely connected with the release of pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, and IFN-γ. The levels of serum TNF-α, IL-1β, IL-6, and IFN-γ were measured by ELISA. As is shown in Fig. 3A, SHK (12.5 mg kg−1) did not affect the levels of serum TNF-α, IL-1β, IL-6, and IFN-γ compared to the control group. However, LPS/D-GalN-treated group significantly increase pro-inflammatory cytokines than that of the control group. Before 1 h LPS/D-GalN simulation, SHK (12.5 and 25 mg kg−1) was injected to mice. SHK pretreatment dramatically inhibited the release of these pro-inflammatory cytokines compare to the LPS/D-GalN group in a dose-dependent manner. Furthermore, we checked the extent of liver injury to confirm the protective effects of SHK. As is shown in Fig. 3b, the mice treated with LPS (30 μg kg−1) and D-GalN (600 mg kg−1) showed severe intrahepatic hemorrhage and necrosis, and SHK (12.5 and 25 mg kg−1) treatment ameliorated these changes (Fig. 3c and d). In addition, SHK (12.5 mg kg−1) only treatment has no more change than that of normal liver (Fig. 3a and e). These results suggested that SHK effectively protected against LPS/D-GalN-induced hepatotoxicity and inflammatory responses.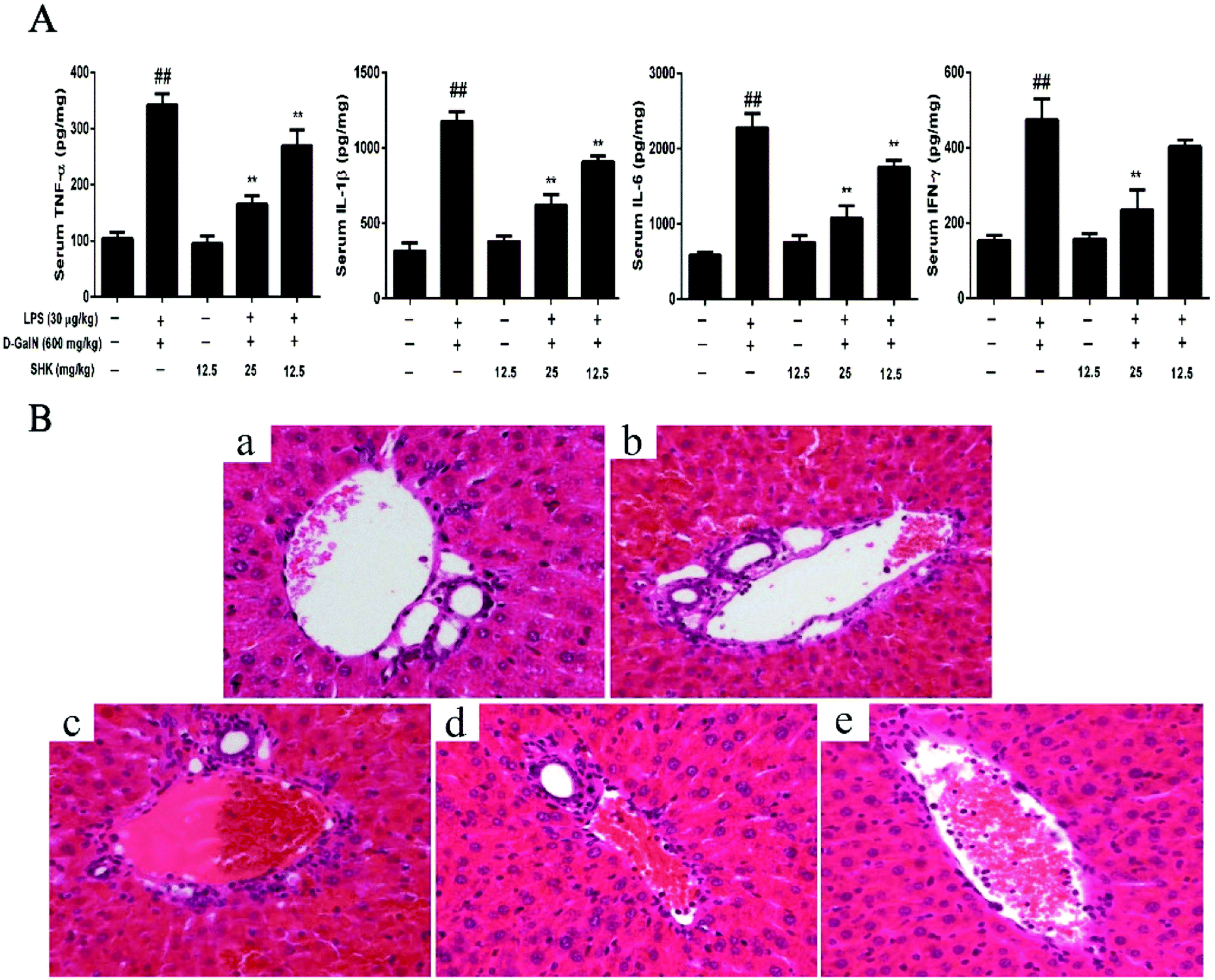 | ||
| Fig. 3 Effects of SHK on LPS/D-GalN-induced inflammatory responses and liver injuries. Mice were injected with SHK (12.5 and 25 mg kg−1) in existence or shortage of LPS (30 μg kg−1) and D-GalN (600 mg kg−1). Mice were administered SHK i.p. for 1 h. Then, the mice were injected with LPS/D-GalN for 3 h. (A) Effects of SHK on serum TNF-α, IL-1β, IL-6, and IFN-γ levels. The results were assayed by using ELISA kit. All data were presented as means ± SD of three independent experiments. ##p < 0.01 vs. the control group; *p < 0.05 and **p < 0.01 vs. the LPS/D-GalN group. (B) Representative histological images of H & E staining of liver tissues obtained in different groups. All of the data shown represent the average from three independent experiments. Representative histological changes of liver obtained from mice of different groups. (a) Control group, (b) LPS/D-GalN group, (c) LPS/D-GalN + SHK (12.5 mg kg−1) group, (d) LPS/D-GalN + SHK (25 mg kg−1) group, and (e) SHK (12.5 mg kg−1) group (HE ×200). | ||
Effects of SHK treatment on LPS/D-GalN-induced MAPK activation in mice
MAPK pathways play a vital role in the inflammatory course and are activated by the release of pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, and IFN-γ. Thus, we checked whether SHK was associated with the MAPK pathways on LPS/D-GalN-induced acute liver injury. As is shown in Fig. 4, treatment of LPS/D-GalN obviously activated AMPK phosphorylation. Moreover, SHK alleviated the phosphorylation of AMPK induced by LPS/D-GalN. It is well known that MAPK family members include JNK1/2, p38, and ERK1/2. Furthermore, our western blot results demonstrated that SHK (12.5 and 25 mg kg−1) ameliorated LPS/D-GalN-induced phosphorylation of JNK1/2, ERK1/2 and p38. However, pretreatment with SHK (12.5 mg kg−1) did not stimulate MAPK family phosphorylated.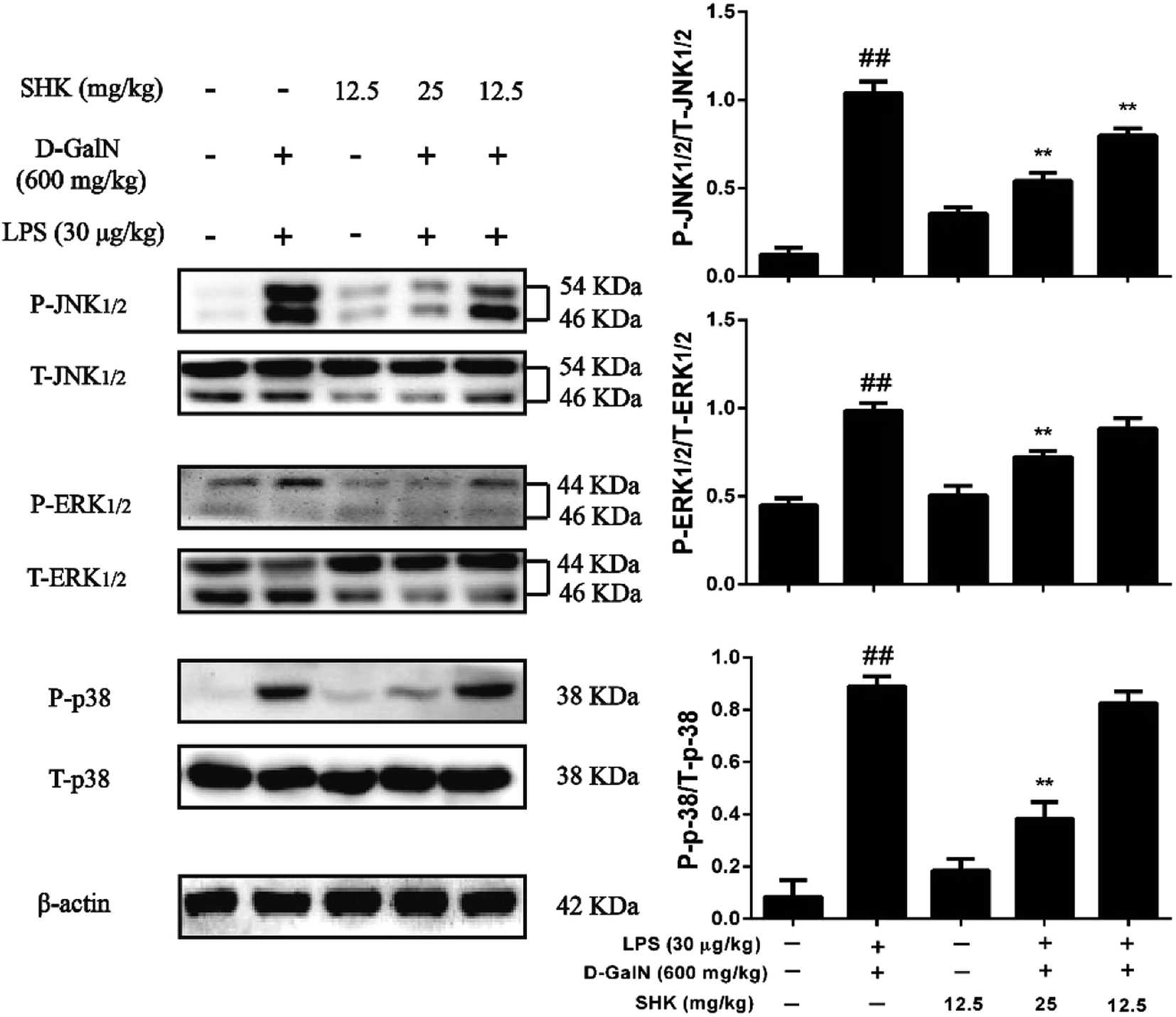 | ||
| Fig. 4 Effects of SHK treatment on LPS/D-GalN-induced MAPK activation in mice. Mice were injected with SHK (12.5 and 25 mg kg−1) in existence or shortage of LPS (30 μg kg−1) and D-GalN (600 mg kg−1). At 1 h after the administering of SHK, the mice were injected with LPS/D-GalN. After 3 h, liver tissue homogenates were analyzed by western blot. Quantifications of the relative expression of P-JNK1/2/T-JNK1/2, P-ERK1/2/T-ERK1/2 and P-p38/T-p38. ##p < 0.01 vs. the control group; **p < 0.01 vs. the LPS/D-GalN group. | ||
Effects of SHK treatment on LPS/D-GalN-induced NF-κB activation in mice
Because the NF-κB signaling pathway is one of typical inflammatory pathways, we investigate the effects of SHK treatment on LPS/D-GalN-induced NF-κB activation. As is shown in Fig. 5, LPS/D-GalN aggravated phosphorylation of NF-κB and IκB compared to the control group. Compared to the LPS/D-GalN-stimulation group, 12.5 and 25 mg kg−1 of SHK pretreatment decreased IκBα phosphorylation and degradation and inhibited the phosphorylation of NF-κB (p65) in a dose-dependent manner.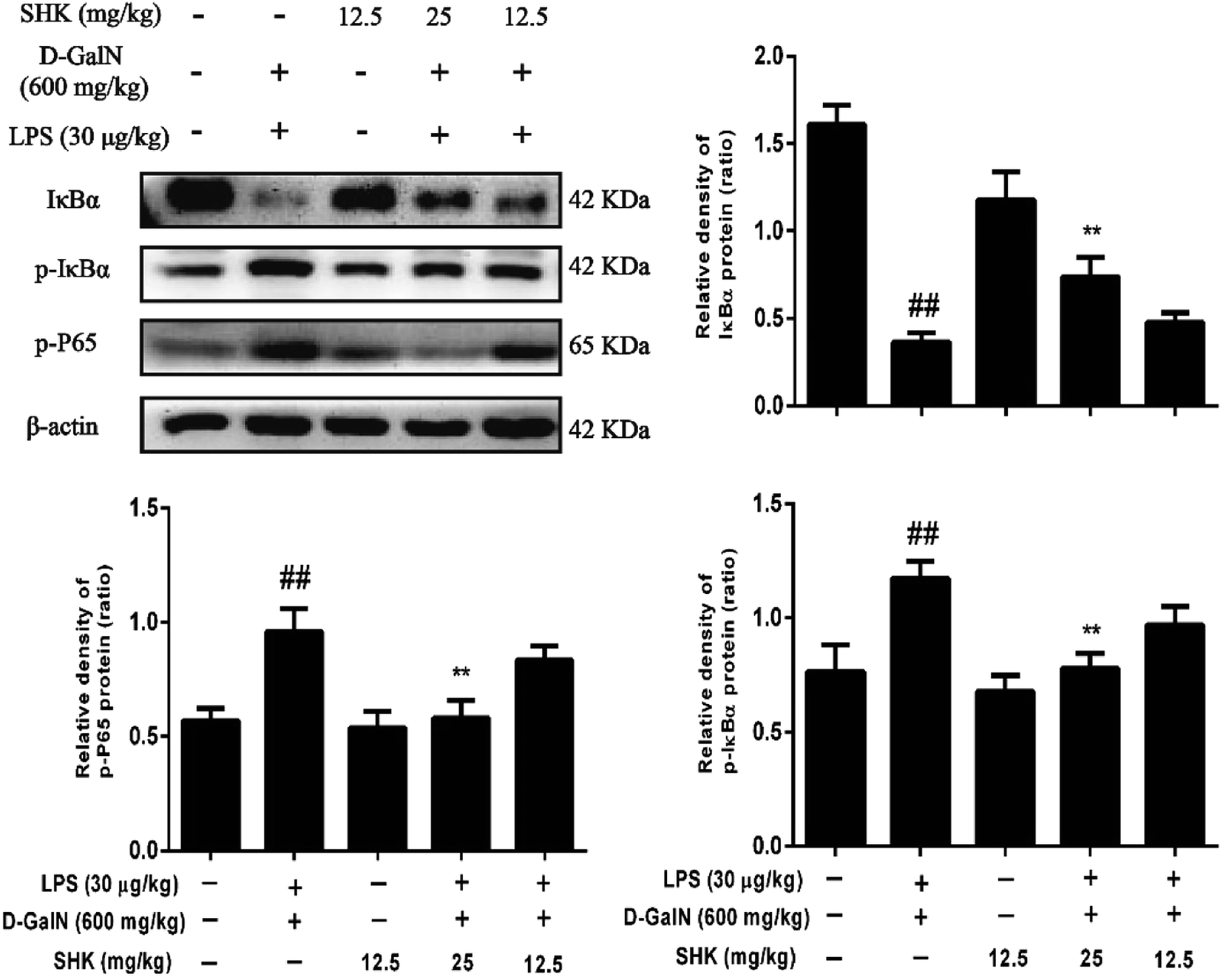 | ||
| Fig. 5 Effects of SHK treatment on LPS/D-GalN induced NF-κB activation in mice. The dose of SHK (12.5 and 25 mg kg−1) was administered to the mice at 1 h before LPS (30 μg kg−1) and D-GalN (600 mg kg−1) injected. After 3 h, the liver tissue homogenates were analyzed by western blot. Quantifications of IκBα, P-IκBα, and P-NF-κB (p65) expression were normalized than that of β-actin. Typical data were obtained in three independent experiments, and one of the three representative experiments is shown. ##p < 0.01 vs. the control group; *p < 0.05 and **p < 0.01 vs. the LPS/D-GalN group. | ||
Effect of SHK on LPS/D-GalN-induced Nrf2 protein expression in mice
Nrf2 regulates the expression of antioxidant proteins that protect against oxidative damage triggered by injury and inflammation. Consequently, we investigated whether SHK could induce Nrf2 protein change in nucleus and cytoplasm. As is shown in Fig. 6, we found that only treatment of SHK (12.5 mg kg−1) group could elevate rate of Nrf2 protein nuclear translocation compared to the control group. In addition, the release of the nuclear proteins Nrf2 was also activated by SHK (12.5 and 25 mg kg−1) in LPS/D-GalN-induced liver damage. Whereas, the LPS/D-GalN simulation group had a little decline compared to the control group and there were no obvious changes. Furthermore, SHK (12.5 and 25 mg kg−1) administration increased the expression and nuclear translocation of Nrf2.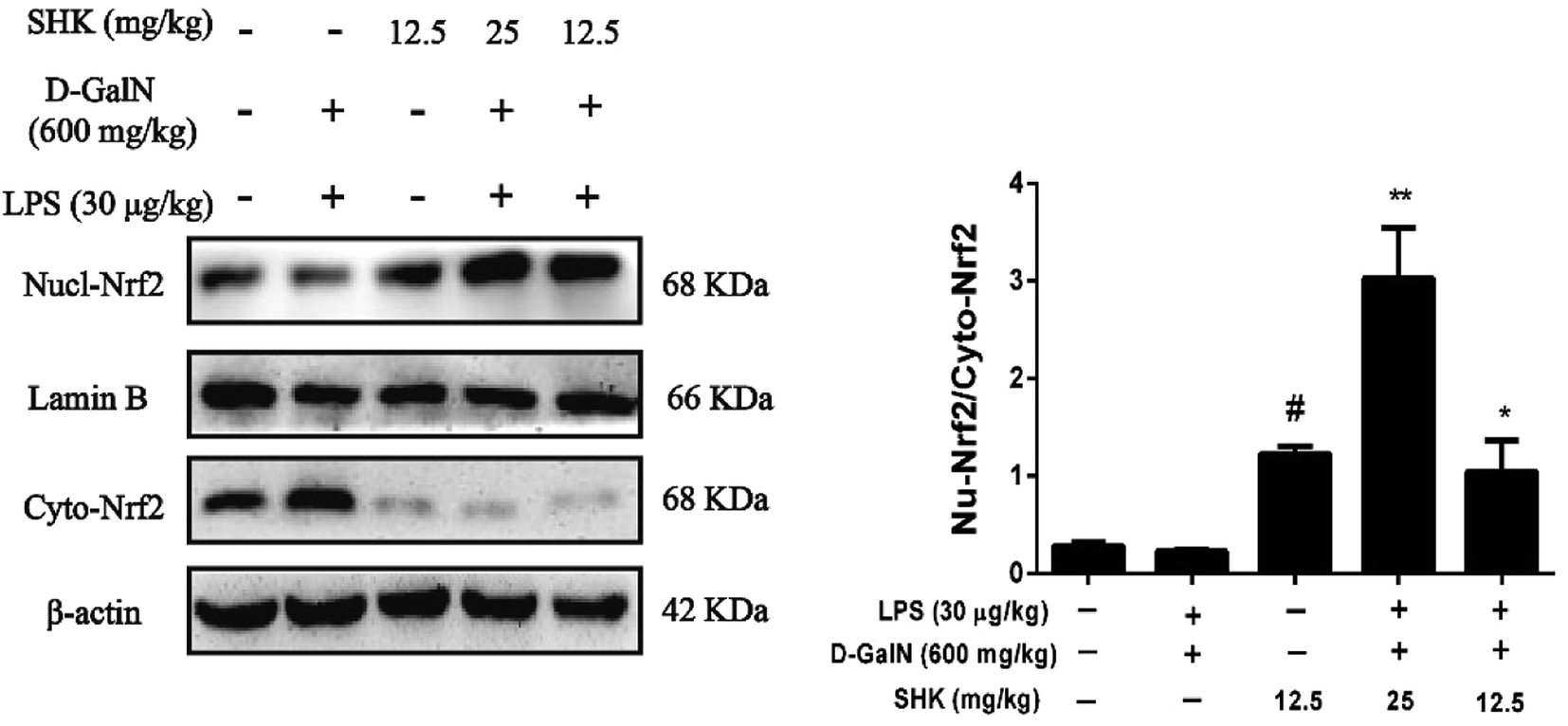 | ||
| Fig. 6 Effect of SHK on LPS/D-GalN-induced Nrf2 protein expression in mice. The dose of SHK (12.5 and 25 mg kg−1) was administered to the mice at 1 h before LPS (30 μg kg−1) and D-GalN (600 mg kg−1) injected. After 3 h, the nuclear and cytoplasmic levels of Nrf2 were checked by western blot. Quantifications of the relative expression of nucl-Nrf2/cyto-Nrf2. All of the data were presented as means ± SD of three independent experiments. ##p < 0.01 vs. the control group; *p < 0.05 and **p < 0.01 vs. the LPS/D-GalN group. | ||
Effects of SHK treatment on HO-1, Nqo1, Gclc, and Gclm protein expression in mice
When Nrf2 developed nuclear translocation, important upstream gene of antioxidant enzymes for ARE-driven also increase, including HO-1, Nqo1, Gclc, and Gclm. Previous experiment found that SHK induced Nrf2 activation. So, we examined HO-1, Nqo1, Gclc and Gclm protein expression. HO-1 is essential for inhibiting inflammatory responses. We examined whether the SHK elevated HO-1 expression and enhanced the resistance of LPS/D-GalN-induced inflammatory damage. As is shown in Fig. 7, we found LPS/D-GalN could inhibit HO-1 expression in mice. In addition, Nqo1, Gclc, and Gclm were also blocked due to LPS/D-GalN induced oxidative and inflammatory damage. We next verified whether SHK improved HO-1, Nqo1, Gclc, and Gclm expression. Our western blot results indicated that LPS/D-GalN treatment reduced the protein expression of HO-1, Nqo1, Gclc, and Gclm, but pretreatment with SHK repaired these changes.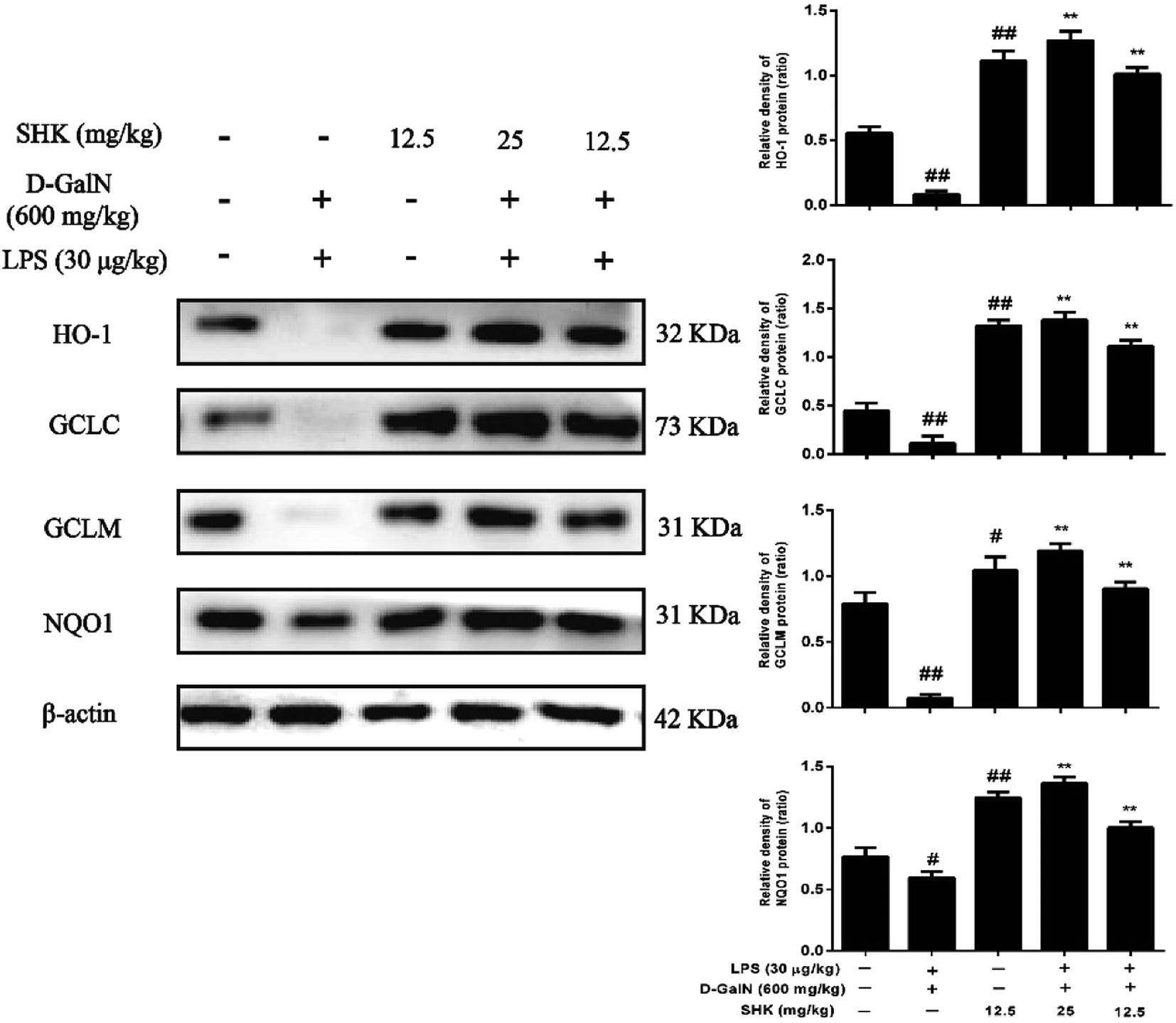 | ||
| Fig. 7 Effects of SHK treatment on HO-1, Nqo1, Gclc, and Gclm protein expression in mice. The dose of SHK (12.5 and 25 mg kg−1) was administered to the mice at 1 h before LPS (30 μg kg−1) and D-GalN (600 mg kg−1) injected for 3 h. the liver tissue homogenates were analyzed by western blotting. The expressed of HO-1, Nqo1, Gclc, and Gclm protein were analyze. β-Actin was used as an internal control. All of data were presented as means ± SD of three independent experiments. #p < 0.05 and ##p < 0.01 vs. the control group; **p < 0.01 vs. the control group. | ||
Discussion
In recent years, LPS/D-GalN-induced acute liver injury has been described as xenobiotic induced by hepatotoxicity and often used for screening anti-hepatotoxic or hepatoprotective agent. The LPS/D-GalN induced acute liver injury is similar to that of viral hepatitis.29,30 Nevertheless, liver injuries induced by other substances, such as CCL4, concanavalin A, and acetaminophen, are all different from viral hepatitis. There is urgent necessity for the search of safe and effective hepatoprotective agent of viral hepatitis. LPS/D-GalN also can induce high level of oxidative stress and alter liver biomarker enzymes.31 LPS/D-GalN improves the release of inflammatory cytokines and which leads to endotoxemia; thus it causes fulminant hepatitis.32 D-GalN plays a critical role in impairing ionic pumps, which enhances damage sensibility in the calcium pumps and results in cell damage.30 These evidences certify LPS/D-GalN-induced acute liver injury is closely associated with inflammatory and oxidative stress. These may be one point of penetration for preventing liver damage.Over release of reactive oxygen species (ROS) can cause oxidative stress, and inflict lipids, proteins, and DNA damage that destroy essential cellular macromolecules. These damage results in several human diseases, including cancer, inflammation, rheumatoid arthritis, atherosclerosis, and neurodegenerative diseases.33 Previous reports certified that LPS/D-GalN exposure could lead to oxidative stress by increasing ROS formation. ROS is associated with liver damage and chronic disease development.34,35 Therefore, accelerating the clearance of ROS and oxidative stress inhibition may play critical roles in protecting against various diseases. GSH, a powerful antioxidant, cofactor, and coenzyme, has an important effect in being closely involving in the clearance of ROS.37 Our experiment revealed that SHK could improve GSH production and inhibit the release of ROS. Furthermore, the overproduction of Gcl enhances total GSH contents, including Gclc and Gclm, and protects against H2O2-induced cell death in human granulose tumour cells.36 Meanwhile, Nqo1 is a prototypical Nrf2 target gene that catalyzes the reduction and detoxification of highly reactive quinones that can cause redox cycling and oxidative stress.38 Previous research has reported that induction of Nqo1 is mediated solely through the Keap1/Nrf2/ARE.39 For example, previous reports showed that syringin treated fulminant hepatic failure induced by D-galactosamine and lipopolysaccharide through increasing the release of GSH levels.40 Hence, the purpose of this study was aimed at investigating the antioxidant effect of SHK through reducing oxidant-induced liver damage in mice. Our experiment revealed that SHK enhanced Nrf2 expression by activating Nqo1, Gclc, and Gclm proteins expression.
As a nuclear translation factor, the activation of Nrf2 will result in the induction of many cytoprotective proteins, including HO-1, Nqo1 Gclc, and Gclm. However, based on these observations, we next verified whether SHK could change Nrf2 and HO-1 protein expression on LPS/D-GalN-induced liver injury. HO-1 is a Nrf2 target gene that has been shown to protect from a variety of diseases, including sepsis, hypertension, atherosclerosis, acute lung injury, kidney injury, and pain.41 HO-1 is an enzyme that catalyzes the disassociation of heme into the antioxidant biliverdin and resists cell oxidative damage. Our western blot results indicated that different concentrations and exposure periods of SHK treatment obviously augmented HO-1 induction in mice liver. Furthermore, LPS/D-GalN stimulation dramatically inhibited HO-1 protein expression. For purpose of investigating the transcriptional activation of Nrf2, we detected Nrf2 induction in nucleus and cytoplasm. In this research, we found that SHK treatment increased Nrf2 protein expression in liver. Moreover, SHK treatment markedly promoted the nuclear translocation of Nrf2 and was directly proportional to the decrease in the cytoplasm of Nrf2. There were a lot of evidences that SHK attenuated oxidative damage on LPS/D-GalN-induced acute liver damage through activating Nrf2 expression.
Previous study has reported oxidative stress and inflammation are closely linked.33 Strong oxidative damage can cause subsequent inflammatory occurrence. So we investigated whether LPS/D-GalN could break out inflammatory mediators in mice, including TNF-α, IL-1β, IL-6, and IFN-γ. We found that LPS/D-GalN significantly improved the release of inflammatory cytokines, but pretreatment with SHK alleviated this adverse outcome. NF-κB, an essential regulatory transcription factor protein, is primarily activated by TNF-α, IL-1β, and IL-6.42 Activation of NF-κB is accompanied by IκB phosphorylation and subsequent degradation by kinase (IKK). On dissociation from the inhibitor IκBα, NF-κB can enter into the nucleus and promote the expression of pro-inflammatory mediators, such as TNF-α, IL-1β, and IL-6.43 Thus, in order to exploring inflammatory effect, the ratio of phospho-IκBα and phosphop65 was detected by western blot. These results showed that SHK markedly inhibited the phosphorylation and subsequent dissociation of IκBα and suppressed NF-κB (p65) phosphorylation. Furthermore, mass reports have confirmed that MAPKs, three kinds of signaling molecules that include p38, ERK1/2 and JNK1/2, play a critical role in pro-inflammatory process.44 JNK1/2, ERK1/2 and p38 phosphorylation are chief factors for the adjustment of inflammatory cytokines expression.45 Various cytokines could stimulate MAPK activation, such as TNF-α. In addition, as a signal pathway connector, phosphorylation of MAPK could induce IκBα degradation and NF-κB activation by phosphorylation intrusion nucleus, and subsequently induced inflammation. Moreover, previous studies have reported that Nrf2-dependent HO-1 has an impact on lipopolysaccharide (LPS)-mediated inflammatory responses in RAW264.7 or mouse peritoneal macrophage-derived foam cell macrophages. Nrf2/HO-1 plays a major role in anti-inflammatory function.46 Our results suggested that SHK significantly depressed MAPK and NF-κB on LPS/D-GalN-induced acute liver injury. On the side, SHK maybe mediated by activating Nrf2 and lead to the inhibition of NF-κB signaling.
Conclusion
In conclusion, our findings revealed that Shikonin (SHK) treatment effectively prevented LPS/D-GalN-induced death in mice. SHK induced the expression of GCLC, GCLM, HO-1 and NQO1, which was largely dependent on the upregulation of the Nrf2 signaling pathway. SHK also inhibited MAPK and NF-κB phosphorylation, thereby protected mice against LPS/D-GalN-induced oxidative stress and inflammatory damage. These events were not only connected with reduced ROS production and MPO formation but also increased the SOD and GSH levels. In the future, combination therapies with anti-oxidative and anti-inflammatory effects, may be a good strategy that effectively abolish LPS/D-GalN hepatotoxicity. Hence, SHK might contribute to the treatment of virus hepatitis.Conflict of interest
The authors declare no conflicts of interest.Abbreviations
| SHK | Shikonin |
| SD | Standard deviations |
| PVDF | Polyvinylidene fluoride |
| LPS | Lipopolysaccharide |
| D-GalN | D-Galactosamine |
| ALT | Alanine transaminase |
| AST | Aspartate aminotransferase |
| MAPK | Mitogen-activated protein kinase |
| JNK | c-Jun N-terminal kinase |
| ERK | Extracellular signal-regulated kinase |
| Nrf2 | Nuclear factor (erythroid-derived 2)-like 2 |
| TNF-α | Tumour necrosis factor α |
| IL-1β | Interleukin 1β |
| IL-6 | Interleukin 6 |
| IFN-γ | Interferon γ |
| HO-1 | Heme oxygenase 1 |
| Gclc | Glutamate cysteine ligase, catalytic |
| Gclm | Glutamate cysteine ligase, modifier |
| Nqo1 | NAD(P)H dehydrogenase quinone 1 |
| NF-κB | Nuclear factor κB |
| IκBα | Inhibitor of κBα |
| GSH | Reduced glutathione |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| MPO | Myeloperoxidase |
Acknowledgements
This work was supported by a grant from National Natural Science Foundation of China (No. 31572347, No. 31372478).Notes and references
- S. J. Kim, J. K. Kim, D. U. Lee, J. H. Kwak and S. M. Lee, Genipin protects lipopolysaccharide-induced apoptotic liver damage in D-galactosamine-sensitized mice, Eur. J. Pharmacol., 2010, 635, 188–193 CrossRef CAS PubMed.
- G. Splendiani, V. Mazzarella, S. Cipriani, S. Pollicita, F. Rodio and C. U. Casciani, Dialytic treatment of rhabdomyolysis-induced acute renal failure: our experience, Renal Failure, 2001, 23, 183–191 CrossRef CAS PubMed.
- T. Fukuda, A. Mogami, H. Tanaka, T. Yoshikawa, M. Hisadome and H. Komatsu, Y-40138, a multiple cytokine production modulator, protects against D-galactosamine and lipopolysaccharide-induced hepatitis, Life Sci., 2006, 79, 822–827 CrossRef CAS PubMed.
- B. Z. Yan, L. Y. Chen, L. Kang, X. R. Wang, M. R. Bi, W. Wang and B. S. Yang, Hepatoprotective effects of cathepsin B inhibitor on acute hepatic failure induced by lipopolysaccharide/D-galactosamine in mice, Hepatobiliary Pancreatic Dis. Int., 2013, 12, 80–86 CrossRef CAS.
- D. Heumann and T. Roger, Initial responses to endotoxins and Gram-negative bacteria, Clin. Chim. Acta, 2002, 323, 59–72 CrossRef CAS.
- W. X. Ding and X. M. Yin, Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury, J. Cell. Mol. Med., 2004, 8, 445–454 CrossRef CAS PubMed.
- Z. Li, H. Z. Li, G. Xia, F. L. Luo, B. Wang, H. Ning, C. D. Wang, Z. Zhuo and J. Y. Wan, Protective effects of Asiaticoside on acute liver injury induced by lipopolysaccharide/D-galactosamine in mice, Phytomedicine, 2010, 17, 811–819 CrossRef PubMed.
- T. Hochepied, F. G. Berger, H. Baumann and C. Libert, Alpha(1)-acid glycoprotein: an acute phase protein with inflammatory and immunomodulating properties, Cytokine Growth Factor Rev., 2003, 14, 25–34 CrossRef CAS PubMed.
- R. B. Devlin, Regulation of cytokine production in human alveolar macrophages and airway epithelial cells in response to ambient air pollution particles: further mechanistic studies, Toxicol. Appl. Pharmacol., 2005, 207, 269–275 CrossRef PubMed.
- J. B. Kim, A. R. Han and E. Y. Park, Inhibition of LPS-induced iNOS, COX-2 and cytokines expression by poncirin through the NF-kappaB inactivation in RAW 264.7 macrophage cells, Biol. Pharm. Bull., 2007, 30, 2345 CAS.
- G. E. Arteel, Oxidants and antioxidants in alcohol-induced liver disease, Gastroenterology, 2003, 124, 778–790 CrossRef CAS PubMed.
- Q. Ma, Role of nrf2 in oxidative stress and toxicity, Annu. Rev. Pharmacol., 2013, 53, 401 CrossRef CAS PubMed.
- Y. P. Hwang, J. H. Choi, J. M. Choi, Y. C. Chung and H. G. Jeong, Protective mechanisms of anthocyanins from purple sweet potato against tert-butyl hydroperoxide-induced hepatotoxicity, Food Chem. Toxicol., 2011, 49, 2081–2089 CrossRef CAS PubMed.
- T. Takahashi, K. Morita, R. Akagi and S. Sassa, Heme oxygenase-1: a novel therapeutic target in oxidative tissue injuries, Curr. Med. Chem., 2004, 11, 1545–1561 CrossRef CAS PubMed.
- D. A. Tulis, W. Durante, K. J. Peyton, A. J. Evans and A. I. Schafer, Heme oxygenase-1 attenuates vascular remodeling following balloon injury in rat carotid arteries, Atherosclerosis, 2001, 155, 113–122 CrossRef CAS PubMed.
- M. D. Maines, Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications, FASEB J., 1988, 2, 2557–2568 CAS.
- M. Nakasone, K. Nakaso, Y. Horikoshi, T. Hanaki, Y. Kitagawa, T. Takahashi, Y. Inagaki and T. Matsura, Preconditioning by Low Dose LPS Prevents Subsequent LPS-Induced Severe Liver Injury via Nrf2 Activation in Mice, Yonago Acta Med., 2016, 59, 223–231 Search PubMed.
- A. M. Bataille and J. E. Manautou, Nrf2: A Potential Target for New Therapeutics in Liver Disease, Clin. Pharmacol. Ther., 2012, 92, 340–348 CrossRef CAS PubMed.
- A. Matsuzawa, K. Saegusa, T. Noguchi, C. Sadamitsu, H. Nishitoh, S. Nagai, S. Koyasu, K. Matsumoto, K. Takeda and H. Ichijo, ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity, Nat. Immunol., 2005, 6, 587 CrossRef CAS PubMed.
- J. Y. Han, S. S. Cho, H. Y. Ji, K. M. Kim, H. J. Chang, E. P. Da, J. S. Bang, Y. S. Jung and S. H. Ki, The chalcone compound isosalipurposide (ISPP) exerts a cytoprotective effect against oxidative injury via Nrf2 activation, Toxicol. Appl. Pharmacol., 2015, 287, 77 CrossRef CAS PubMed.
- A. M. El, J. Angulo and L. Rodríguez-Mañas, Oxidative stress and vascular inflammation in aging, Free Radical Biol. Med., 2013, 65, 380–401 CrossRef PubMed.
- X. L. Chen and C. Kunsch, Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: a new therapeutic approach for the treatment of inflammatory diseases, Curr. Pharm. Des., 2004, 10, 879–891 CrossRef CAS PubMed.
- K. Itoh, J. Mimura and M. Yamamoto, Discovery of the negative regulator of Nrf2, Keap1: a historical overview, Antioxid. Redox Signaling, 2010, 13, 1665–1678 CrossRef CAS PubMed.
- H. Tanaka, T. Nishi and Y. Wada, The Nrf2-Antioxidant Response Element Signaling Pathway and its Activation by Oxidative Stress, J. Biol. Chem., 2009, 284, 13291 CrossRef PubMed.
- L. Lu, A. Qin, H. Huang, P. Zhou, C. Zhang, N. Liu, S. Li, G. Wen, C. Zhang and W. Dong, Shikonin extracted from medicinal Chinese herbs exerts anti-inflammatory effect via proteasome inhibition, Eur. J. Pharmacol., 2011, 658, 242–247 CrossRef CAS PubMed.
- C.-N. W. Chen-Chen Lee, Y.-T. Lai, J.-J. Kang, J.-W. Liao, B.-L. Chiang, H.-C. Chen and Y.-W. Cheng, Shikonin inhibits maturation of bone marrow-derived dendritic cells and suppresses allergic airway inflammation in a murine model of asthma, Br. J. Pharmacol., 2010, 161, 1496–1511 CrossRef PubMed.
- I. Andújar, M. C. Recio, T. Bacelli, R. M. Giner and J. L. Ríos, Shikonin reduces oedema induced by phorbol ester by interfering with IkappaBalpha degradation thus inhibiting translocation of NF-kappaB to the nucleus, Br. J. Pharmacol., 2010, 160, 376–388 CrossRef PubMed.
- J. Xiong, J. Ni, G. Hu, J. Shen, Y. Zhao, L. Yang, J. Shen, G. Yin, C. Chen and G. Yu, Shikonin ameliorates cerulein-induced acute pancreatitis in mice, J. Ethnopharmacol., 2013, 145, 573 CrossRef CAS PubMed.
- S. S. Chaung, C. C. Lin, J. Lin, K. H. Yu, Y. F. Hsu and M. H. Yen, The hepatoprotective effects of Limonium sinense against carbon tetrachloride and β-D-galactosamine intoxication in rats, Phytother. Res., 2003, 17, 784–791 CrossRef PubMed.
- R. Nakagiri, E. Hashizume, S. Kayahashi, Y. Sakai and T. Kamiya, Suppression by Hydrangeae Dulcis Folium of D-Galactosamine-induced Liver Injury in Vitro and in Vivo, Biosci., Biotechnol., Biochem., 2003, 67, 2641–2643 CrossRef CAS PubMed.
- X. Tang, L. Gao, J. Gao, Y. Fan, L. Xu, X. Zhao and Q. Xu, Mechanisms of Hepatoprotection of Terminalia catappa L. Extract on D-Galactosamine-Induced Liver Damage, Am. J. Chin. Med., 2004, 32, 509–519 CrossRef PubMed.
- Y. Endo, M. Shibazaki, K. Yamaguchi, K. Kai, S. Sugawara, H. Takada, H. Kikuchi and K. Kumagai, Enhancement by galactosamine of lipopolysaccharide (LPS)-induced tumour necrosis factor production and lethality: its suppression by LPS pretreatment, Br. J. Pharmacol., 1999, 128, 5–12 CrossRef CAS PubMed.
- S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Oxidative stress, inflammation, and cancer: How are they linked?, Free Radical Biol. Med., 2010, 49, 1603–1616 CrossRef CAS PubMed.
- C. W. Pan, Z. Z. Pan, J. J. Hu, W. L. Chen, G. Y. Zhou, W. Lin, L. X. Jin and C. L. Xu, Mangiferin alleviates lipopolysaccharide and D-galactosamine-induced acute liver injury by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation, Eur. J. Pharmacol., 2016, 770, 85–91 CrossRef CAS PubMed.
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, Oxidative DNA damage: mechanisms, mutation, and disease, FASEB J., 2003, 17, 1195–1214 CrossRef CAS PubMed.
- M. M. Corteswanstreet, E. Giedzinski, C. L. Limoli and U. Luderer, Overexpression of glutamate-cysteine ligase protects human COV434 granulosa tumour cells against oxidative and gamma-radiation-induced cell death, Mutagenesis, 2009, 24, 211 CrossRef CAS PubMed.
- D. M. Townsend, K. D. Tew and H. Tapiero, The importance of glutathione in human disease, Biomed. Pharmacother., 2003, 57, 145–155 CrossRef CAS.
- R. Venugopal and A. K. Jaiswal, Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1gene, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14960 CrossRef CAS.
- A. T. Dinkova-Kostova, W. D. Holtzclaw, R. N. Cole, K. Itoh, N. Wakabayashi, Y. Katoh, M. Yamamoto and P. Talalay, Direct Evidence that Sulfhydryl Groups of Keap1 are the Sensors Regulating Induction of Phase 2 Enzymes that Protect against Carcinogens and Oxidants, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11908 CrossRef CAS PubMed.
- X. Gong, L. Zhang, R. Jiang, C. D. Wang, X. R. Yin and J. Y. Wan, Hepatoprotective effects of syringin on fulminant hepatic failure induced by D-galactosamine and lipopolysaccharide in mice, J. Appl. Toxicol. JAT, 2014, 34, 265 CrossRef CAS PubMed.
- T. Jarmi and A. Agarwal, Heme oxygenase and renal disease, Curr. Hypertens. Rep., 2009, 11, 56–62 CrossRef CAS PubMed.
- T. Lawrence, D. A. Willoughby and D. W. Gilroy, Anti-inflammatory lipid mediators and insights into the resolution of inflammation, Nat. Rev. Immunol., 2002, 2, 787 CrossRef CAS PubMed.
- T. D. Gilmore, Introduction to NF-kappaB: players, pathways, perspectives, Oncogene, 2006, 25, 6680 CrossRef CAS PubMed.
- E. Cario, I. M. Rosenberg, S. L. Brandwein, P. L. Beck, H. C. Reinecker and D. K. Podolsky, Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing toll-like receptors, J. Immunol., 2000, 164, 966–972 CrossRef CAS.
- M. Guha and N. Mackman, LPS induction of gene expression in human monocytes, Cell. Signalling, 2001, 13, 85–94 CrossRef CAS PubMed.
- A. M. Kuhn, N. Tzieply, M. V. Schmidt, A. von Knethen, D. Namgaladze, M. Yamamoto and B. Brune, Antioxidant signaling via Nrf2 counteracts lipopolysaccharide-mediated inflammatory responses in foam cell macrophages, Free Radical Biol. Med., 2011, 50, 1382–1391 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |