
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced activity of CuO/K2CO3/MgAl2O4 catalyst for lean NOx storage and reduction at high temperatures†
Yaoyao Liua,
Lihong Guoab,
Dongyue Zhaoa,
Xingang Li *a,
Zhongnan Gaoa,
Tong Ding*a,
Ye Tiana and
Zheng Jiangc
*a,
Zhongnan Gaoa,
Tong Ding*a,
Ye Tiana and
Zheng Jiangc
aCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin Key Laboratory of Applied Catalysis Science and Engineering, School of Chemical Engineering & Technology, Tianjin University, Tianjin 300072, P. R. China. E-mail: xingang_li@tju.edu.cn
bSchool of Chemistry and Chemical Engineering, Henan University of Technology, Zhengzhou 450001, P. R. China
cShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai, 201800, P. R. China
First published on 23rd May 2017
Abstract
Herein, we designed a new NOx storage and reduction CuO/K2CO3/MgAl2O4 catalyst operating within the high temperature region of 350–550 °C. Compared with the Al2O3 supported catalyst with the same Cu and K loading, it exhibits superior NOx storage and reduction performance. The NOx reduction percentage (NRP) of the CuO/K2CO3/MgAl2O4 catalyst remains above 90% over a wide temperature range (400–550 °C), and reaches the highest NRP of 99.9% at 450 °C with the N2 selectivity of 99.7%. Uncovered CuO particles with better reducibility exist on the CuO/K2CO3/MgAl2O4 catalyst, with the high NOx oxidation and reduction ability above 400 °C. Potassium carbonates on the CuO/K2CO3/MgAl2O4 catalyst mainly exist in three forms, including free ionic carbonate, bridging bidentate carbonate and chelating bidentate carbonate. Under lean-burn conditions, most of carbonates on the CuO/K2CO3/MgAl2O4 catalyst can store NOx to form nitrates, but only parts of them participate in NOx storage on the CuO/K2CO3/Al2O3 catalyst. The MgAl2O4 support offers additional sites for NOx adsorption, while the formed nitrate on it shows low thermal stability. So, NOx is mainly stored on K2CO3 at high temperatures, because MgAl2O4 can enhance the thermal stability of the supported K2CO3 on it. Our results show that the thermal stability of K2CO3 directly determines the thermal stability of the formed nitrates. Accordingly, the CuO/K2CO3/MgAl2O4 catalyst shows the high NSR activity because of the efficient redox ability of CuO and high thermal stability of K2CO3 at high operating temperatures.
1. Introduction
Modern lean-burn engines usually operate at a high air/fuel ratio, which results in a high fuel utilization efficiency.1 Unfortunately, the emitted NOx (NO and NO2) is hardly removed over the conventional three way catalysts (TWC) because of the presence of excess oxygen. With the increasing NOx emission limit, it is urgent to develop effective catalytic after-treatment systems.2,3 To solve this problem, methodologies for NOx storage and reduction (NSR) and selective catalytic reduction (SCR) of NOx are being developed.4,5 Presently, the NSR technology is a preferred choice especially for light duty lean-burn engines.6,7The process of NSR, also known as lean NOx trap (LNT), is operated in alternative lean-burn/fuel-rich atmospheres, which mainly includes: NOx is captured by alkali/alkali earth components (e.g. barium and potassium) within a long lean-burn period (1–2 min); the trapped NOx species is released and then reduced to harmless N2 by precious group metals (e.g. Pt and Rh) within a subsequent short fuel-rich period (3–20 s).4,8 High NOx removal efficiency has been achieved over the Pt/BaO/Al2O3 catalyst developed by the Toyota company within a narrow temperature window of 300–400 °C.9–15 However, some newly developed lean-burn gasoline engine technologies, such as gasoline direct injection (GDI), are required to operate at higher temperatures than normal. Under this condition, the activity of the traditional Pt/BaO/Al2O3 NSR catalyst significantly drops.16,17 When the operating temperature exceeds 400 °C, the solid-phase reaction between BaO and Al2O3 to form BaAl2O4 will lower the surface area and reduce the NOx storage sites.18 Pt sintering also hampers the reduction of NOx from cycle to cycle.19,20 Furthermore, the low thermal stability of barium nitrates declines its application at high operating temperatures, which is believed to be the essential factor to limit the NOx storage capacity.18,21 Thus, it is urgent to develop a NSR catalyst with good high-temperature performance.
It was reported that mixing an alkaline earth metal oxide with Al2O3 could enhance the basicity of the support and also improve the thermal stability of the trapped nitrates.16 MgAl2O4, as one of these mixed oxides, has been employed as support of high-temperature NSR catalysts. Through supported on MgAl2O4, the stability of nitrate species is strongly improved whether on potassium sites or barium sites,16,18 and Pt sintering can also be effectively inhibited.17 Additionally, the MgAl2O4 support can provide extra storage sites for NOx.22 Hence, we chose MgAl2O4 as the support of NSR catalysts operated at high temperatures.
Additionally, to lower the price of NSR catalysts, it is valuable to substitute Pt with non-noble metal components. In the previous study, the perovskite-type oxides were used in replacement of platinum-based catalyst, such as BaFeO3−x, LaCoO3 and La1−xSrxCoO3.23–28 The transition metal elements of manganese and copper were also studied as substitutes for platinum.29,30 It was reported that Cu/K2Ti2O5 could be applied over a wide temperature range (200–600 °C) with the mutual transformation of different structures.31
As for the storage material, it is generally admitted that potassium-based catalysts have higher ability to storage NOx at high temperatures (above 400 °C) than barium-based ones due to its stronger basicity and better mobility.32 Potassium is more suitable as the storage element for the high-temperature NSR catalyst.17
Based on the above analysis, it is interesting to design a new NSR catalyst with CuO as the NOx oxidation and reduction center, potassium as the NOx storage element and MgAl2O4 as the support material. To the best of our knowledge, the reasonably designed CuO/K2CO3/MgAl2O4 catalyst herein has not been reported before.
In this study, we prepared the CuO/K2CO3/MgAl2O4 catalyst by successive impregnation method. The Al2O3-supported catalyst with the same Cu and K loading was also prepared for comparison. The NOx storage/reduction performance within 350–550 °C of the two catalysts was comparatively investigated. Then, we characterized the catalysts by the X-ray diffraction (XRD), X-ray absorption near-edge structures (XANES), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), Fourier-transforming infrared spectra (FT-IR), H2 temperature-programmed reduction (H2-TPR), temperature-programmed desorption of CO2 (CO2-TPD) and temperature-programmed desorption of NOx (NOx-TPD). Through the results of the above characterizations, we investigated the states of K- and Cu-species in CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts. We also discussed the thermal stability of nitrate on the catalysts and revealed how the CuO/K2CO3/MgAl2O4 catalyst improved the NSR performance at high temperatures.
2. Experimental section
2.1. Support and catalyst preparation
For comparison, the CuO/K2CO3/Al2O3 catalyst was also prepared by incipient wetness impregnation with the same process. The weight loadings of Cu and K were both 10%. The K2CO3/MgAl2O4, K2CO3/Al2O3, CuO/MgAl2O4 and CuO/Al2O3 samples were prepared with the same method. The weight loadings of Cu or K was also 10%.
2.2. Catalyst characterization
The measurement of the specific surface area (SBET) was carried out at −196 °C on a Quantachrome QuadraSorb SI instrument. Before measurements, the samples were degassed in vacuum at 300 °C for 3 h to remove the adsorbed species.XRD analysis was conducted on an X'pert Pro rotatory diffractometer (PANAlytical Company, Cu Kα radiation λ = 0.15418 nm) operating at 40 mA and 40 kV. The diffraction data were collected in the 10 to 90° range at a step size of 0.02°. The crystallite sizes of CuO were calculated by using Scherrer equation:

XPS measurements were carried out by using a PHI-1600 ESCA spectrometer with Mg Kα (1253.6 eV) as radiation source. The base pressure in sample chamber was 5 × 10−8 Pa. The binding energy (BE) peak of C 1s at 284.6 eV was employed to be standard to calibrate the recorded spectra.
The tests of XANES were performed on the 14 W1 beamline of shanghai Synchrotron Radiation Facility. Tests were operated at 250 mA and 3.5 GeV. A Si (1 1 1) double-crystal monochromator was employed to monochromatize X-ray. A copper foil was used for energy calibration.
The morphologies of the catalysts were observed by SEM (S-4800, Hitachi). Before the SEM test, the samples were coated on a thin Pt layer to improve the electrical conductivity.
H2-TPR experiments were carried out on the TP-5079 TPDRO apparatus (Xian quan). The reduction gas is 8 vol% H2/N2 with a flow rate of 30 mL min−1. The weight of sample used for test is 30 mg. The sample was heated from room temperature (RT) to 900 °C, and the heating rate is 10 °C min−1.
CO2-TPD experiments derived from carbonate decomposition were carried out on a Thermo-Finnigan TPDRO 1100. The samples were heated in highly pure helium gas (20 mL min−1) from RT to 900 °C. The heating rate was 10 °C min−1.
NOx-TPD test was conducted in a quartz-tubular continuous flow reactor (i.d. = 4 mm). The samples were heated in pure N2 (400 mL min−1) from 50 °C to 750 °C and the temperature ramp is 5 °C min−1. Before the measurements, the samples were saturated with NOx in the lean gas (400 ppm of NO, 5% O2, balanced by N2) at 450 °C.
The Fourier-transforming infrared spectroscopy (FT-IR) experiment was performed on a Thermo Nicolet Nexus spectrometer. The fresh sample and KBr were mixed with a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, and pressed into a pellet. The spectra based on 32 scans were collected in 400–4000 cm−1 with resolution of 4 cm−1. The spectra were recorded in air at RT.
:100, and pressed into a pellet. The spectra based on 32 scans were collected in 400–4000 cm−1 with resolution of 4 cm−1. The spectra were recorded in air at RT.
2.3. Activity tests
The isothermal NOx storage and reduction experiments of catalysts were carried out in a quartz-tubular continuous flow reactor (i.d. = 4 mm) using 240 mg of the fresh catalysts (40–60 mesh) from 350 to 550 °C with an increment of 50 °C. It was measured by 20 lean/rich (L/R) cycles (L/R = 50/10 s; lean gas 400 ppm of NO, 5% O2, balanced by N2; rich gas 1000 ppm C3H6, balanced by N2). The NOx concentrations were monitored online by a chemiluminescence NO–NO2–NOx analyzer (Model 42i-HL, Thermo Scientific). The total gas flow rate was 400 mL min−1, corresponding to a weight hourly space velocity of 100000 mL g−1 h−1. Meanwhile, the concentration of the byproduct N2O was monitored online by a N2O modular gas analyzer (S710, SICK MAIHAK).
The isothermal NOx storage experiments of the catalysts were carried out in the lean atmosphere (400 ppm of NO, 5% O2, balanced by N2) in the same reactor as above mentioned. The NOx concentrations were also monitored by the same analyzer. The total gas flow rate was 400 mL min−1, corresponding to a weight hourly space velocity of 100000 mL g−1 h−1.
The NOx reduction percentage (NRP) was calculated according to the steady lean/rich cycle as the following formula:

The NSC was taken after the NOx storage process prolonged 60 min and calculated as the following formula:
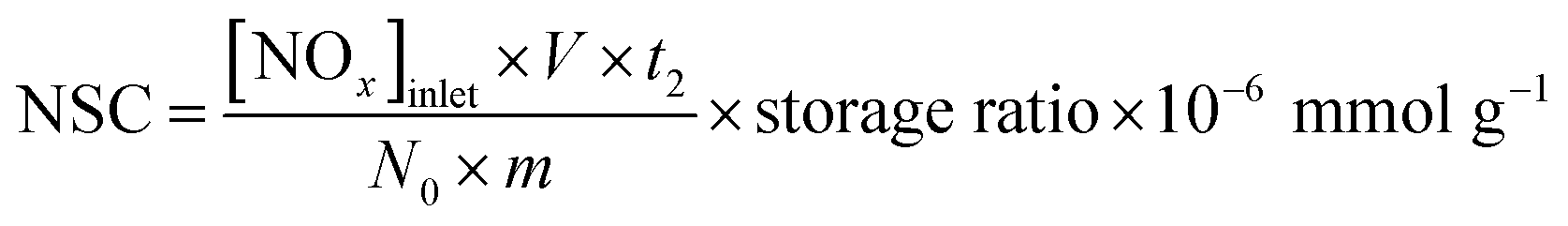
The storage ratio was calculated as the following formula:
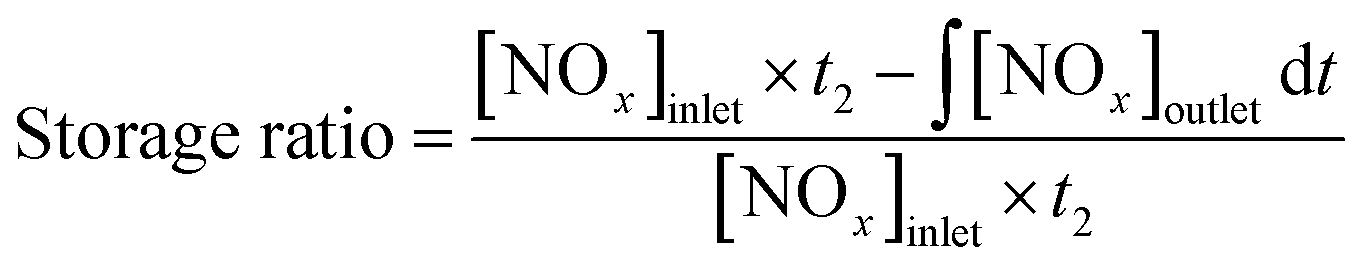
3. Results and discussion
3.1. NOx storage capacity measurement
NOx storage tests were carried out in the temperature range of 350–550 °C. The evolution of outlet NOx is depicted in Fig. 1. The NSC was calculated and summarized in Table 1. Fig. 1a shows the NSC profiles of the CuO/K2CO3/MgAl2O4 catalyst at the different temperatures. The NOx storage capacity of the CuO/K2CO3/MgAl2O4 catalyst varies with the increased reaction temperature. At 350 °C, the NOx trapping kinetics is too slow, inducing the low NSC. Fig. 1a shows that after introducing the feeding gas above 400 °C, a rapid NOx storage behavior occurs and the NOx signal diminishes sharply. Subsequently, the “lean trap” period is observed, namely the stage during which NOx is completely captured on the catalysts and no NOx is released. Then, the NOx concentration gradually recovers to the inlet level until saturation. Probably, CuO is activated above 400 °C for NO oxidation to generate NO2, which is beneficial to NOx storage under lean-burn conditions.34,35 Meanwhile, the stored NOx gets thermally unstable. With the rising of the reaction temperature, the desorption rate of the released NOx from the catalysts is accelerated, as well as the equilibrium of the NOx storage process. The largest NSC uptake of the CuO/K2CO3/MgAl2O4 catalyst is obtained at 450 °C, which is the optimum temperature for the NOx storage reaction. The “lean trap” period lasts for more than 20 min and the NSC value is 1.56 mmol g−1 at 450 °C, which is comparable to or even better than the literature data.17,36 When the reaction temperature continues to increase, the NSC of the CuO/K2CO3/MgAl2O4 catalyst decreases slightly because a small amount of the trapped NOx decomposes at high temperatures. Fig. 1b shows the NOx concentration profiles on the CuO/K2CO3/Al2O3 catalyst at the different temperatures. Similarly, the CuO/K2CO3/Al2O3 catalyst also presents a volcano-type tendency of NSC values in the temperature range of 350–550 °C. Maximum storage capacity is achieved at 400 °C on CuO/K2CO3/Al2O3 and the NSC value is 0.88 mmol g−1. | ||
| Fig. 1 Isothermal NOx storage curves of the (a) CuO/K2CO3/MgAl2O4 and (b) CuO/K2CO3/Al2O3 catalysts at the different temperatures. | ||
| Catalysts | T (°C) | NRP (%) | NSC (mmol g−1) | NO to NO2 conversion (%) |
|---|---|---|---|---|
| CuO/K2CO3/MgAl2O4 | 350 | 75.6 | 1.09 | 24.1 |
| 400 | 97.7 | 1.31 | 32.8 | |
| 450 | 99.9 | 1.56 | 34.0 | |
| 500 | 99.0 | 1.48 | 24.3 | |
| 550 | 94.7 | 1.42 | 19.6 | |
| CuO/K2CO3/Al2O3 | 350 | 23.6 | 0.80 | 14.1 |
| 400 | 80.8 | 0.88 | 33.3 | |
| 450 | 78.6 | 0.57 | 32.7 | |
| 500 | 71.6 | 0.49 | 23.8 | |
| 550 | 37.5 | 0.38 | 18.9 |
Differently from the CuO/K2CO3/MgAl2O4 catalyst, the CuO/K2CO3/Al2O3 catalyst nearly presents no “lean trap” period in the NOx storage profiles. Additionally, the CuO/K2CO3/MgAl2O4 catalyst achieves maximum NSC value at 450 °C, which is 50 °C higher than that of the CuO/K2CO3/Al2O3 catalyst. With further increasing the reaction temperature above 400 °C, the NSC value of the CuO/K2CO3/Al2O3 catalyst decreases seriously, whereas, the CuO/K2CO3/MgAl2O4 catalyst can maintain the large NSC values, making it suitable for the NSR reaction at high temperatures. The NO to NO2 conversion of the CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts was also measured, and the results were calculated and summarized in Table 1. The NO to NO2 conversions of the two catalysts are very similar because the oxidation of NO to NO2 is thermodynamically limited at high temperatures.5,37,38
The distinct difference of the NOx storage behavior at high temperature between the CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts probably results from the different thermal stability of the formed nitrate during the NOx storage reaction. To investigate the thermal stability of the trapped NOx on the catalysts, the NOx-TPD experiments were conducted after the saturated adsorption of NOx at 450 °C. Fig. 2 shows the NOx-TPD profiles of the CuO/K2CO3/MgAl2O4, CuO/K2CO3/Al2O3 and CuO/MgAl2O4 catalysts. In the profile of the CuO/MgAl2O4, a single NOx desorption peak is observed at 365 °C, which is attributed to the decomposition of nitrates species formed on the MgAl2O4 support, confirming that the support offers additional sites for NOx adsorption. It is likely caused by the strong basicity of MgAl2O4. However, the MgAl2O4 support is not the main NOx storage sites at the high temperatures as indicated by its small desorption amount and poor thermal stability. The desorption peak located at 481 °C in the profile of the CuO/K2CO3/Al2O3 catalyst can be ascribed to the decomposition of KNO3 on it. Two desorption peaks are clearly observed in the profile of the CuO/K2CO3/MgAl2O4 catalyst. The first peak at 365 °C is assigned to the NOx desorption from the MgAl2O4 support as mentioned above. The latter peak appearing at 649 °C can be attributed to the desorption of the stored NOx species on K2CO3. Thus, the K2CO3 is identified as the main NOx storage sites according to the large desorption amount and high thermal stability. Meanwhile, a distinct difference in the decomposition temperatures of KNO3 between the CuO/K2CO3/Al2O3 (481 °C) and CuO/K2CO3/MgAl2O4 catalysts (649 °C) is observed, suggesting the higher thermal stability of the nitrates on the K sites of the CuO/K2CO3/MgAl2O4 catalyst, which is the main reason of its higher NOx storage ability at high temperatures.
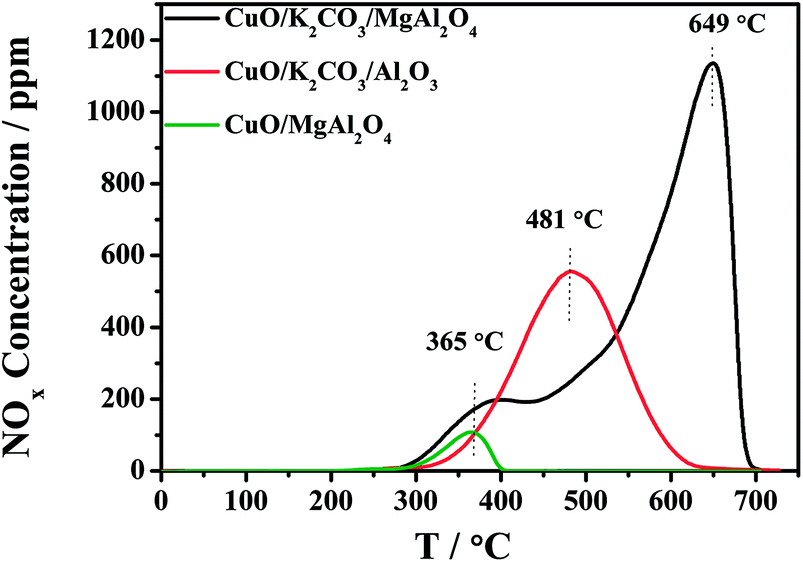 | ||
| Fig. 2 NOx-TPD profiles of the catalysts. | ||
The components of CuO and K2CO3 in the catalyst are designed to act as the NOx oxidation sites and storage sites, respectively. To clarify the roles of these two components, the stationary NOx storage tests were also performed over the K2CO3-free or CuO-free catalysts. In Fig. S1,† the K2CO3-free catalyst of CuO/MgAl2O4 and CuO-free catalysts of K2CO3/Al2O3 and K2CO3/MgAl2O4 show the poor NOx adsorbability. The NSC of the CuO/MgAl2O4 sample is only 0.11 mmol g−1, because of the absence of K2CO3, suggesting that K2CO3 is the main NOx storage sites. The NSC values of the K2CO3/Al2O3 and K2CO3/MgAl2O4 catalysts are 0.43 mmol g−1 and 0.60 mmol g−1, respectively. The decline in the NSC values suggests that the NO oxidation ability of CuO plays a significant role in the NOx storage process, because NO2 oxidized from NO is considered to be more easily captured than NO over NSR catalysts.4,8,39 The result above indicates that both the CuO and K2CO3 play the important roles in the NOx storage process.
3.2. NOx storage/reduction in the lean–rich cycles
The NOx storage and reduction tests in the periodical lean/rich cyclic atmospheres (50 s/10 s) were carried out in the temperature range of 350–550 °C. The evolution of outlet NOx is depicted in Fig. 3. The NRP was calculated and summarized in Table 1. Fig. 3a shows the concentration of outlet NOx for the CuO/K2CO3/MgAl2O4 catalyst during the lean–rich cycles. The NSR performance of the CuO/K2CO3/MgAl2O4 catalyst behaves differently depending on the reaction temperatures. At 350 °C, the NRP of the catalyst is 75.6%. The escaping NOx increases with the prolonged operating period because the NOx can hardly be reduced. When the reaction temperature is above 400 °C, the NSR performance of the CuO/K2CO3/MgAl2O4 catalyst improves greatly. As the profiles of the NOx concentration during the lean–rich cycles at 400–550 °C show, NOx in the feeding gas is captured completely in the lean period firstly; once switched to the rich period, the outlet NOx concentration is quite low, suggesting that most of the NOx can be reduced and nearly no NOx escapes. At 450 °C, the highest NRP of 99.9% is achieved. As the temperature continues to rise, the NRP deceases slightly. The NRP of CuO/K2CO3/MgAl2O4 catalyst can remain above 90% over a wide temperature range (400–550 °C). During the 20 lean–rich cycles, little N2O is produced as byproduct (Fig. S2†). The selectivity of 99.7% is obtained on the CuO/K2CO3/MgAl2O4 catalyst at 450 °C. Fig. 3b shows the concentration of outlet NOx for the CuO/K2CO3/Al2O3 catalyst during the lean–rich cycles. The NOx reduction ability of the CuO/K2CO3/Al2O3 catalyst at 350 °C is only 23.6%. At 400 °C, the CuO/K2CO3/Al2O3 catalyst obtains the highest NRP (80.8%). As the profile of the NOx concentration during the lean–rich cycles on the CuO/K2CO3/Al2O3 catalyst at 400 °C shows, the catalyst exhibits outstanding NSR performance at the beginning, however, with the prolonged operating time, the storage sites can not be fully regenerated and the NOx removal activity decreases from cycle to cycle. With further increasing the reaction temperature, the NRP of the CuO/K2CO3/Al2O3 catalyst decreased gradually. At 550 °C, its NRP decreases sharply to 37.5%.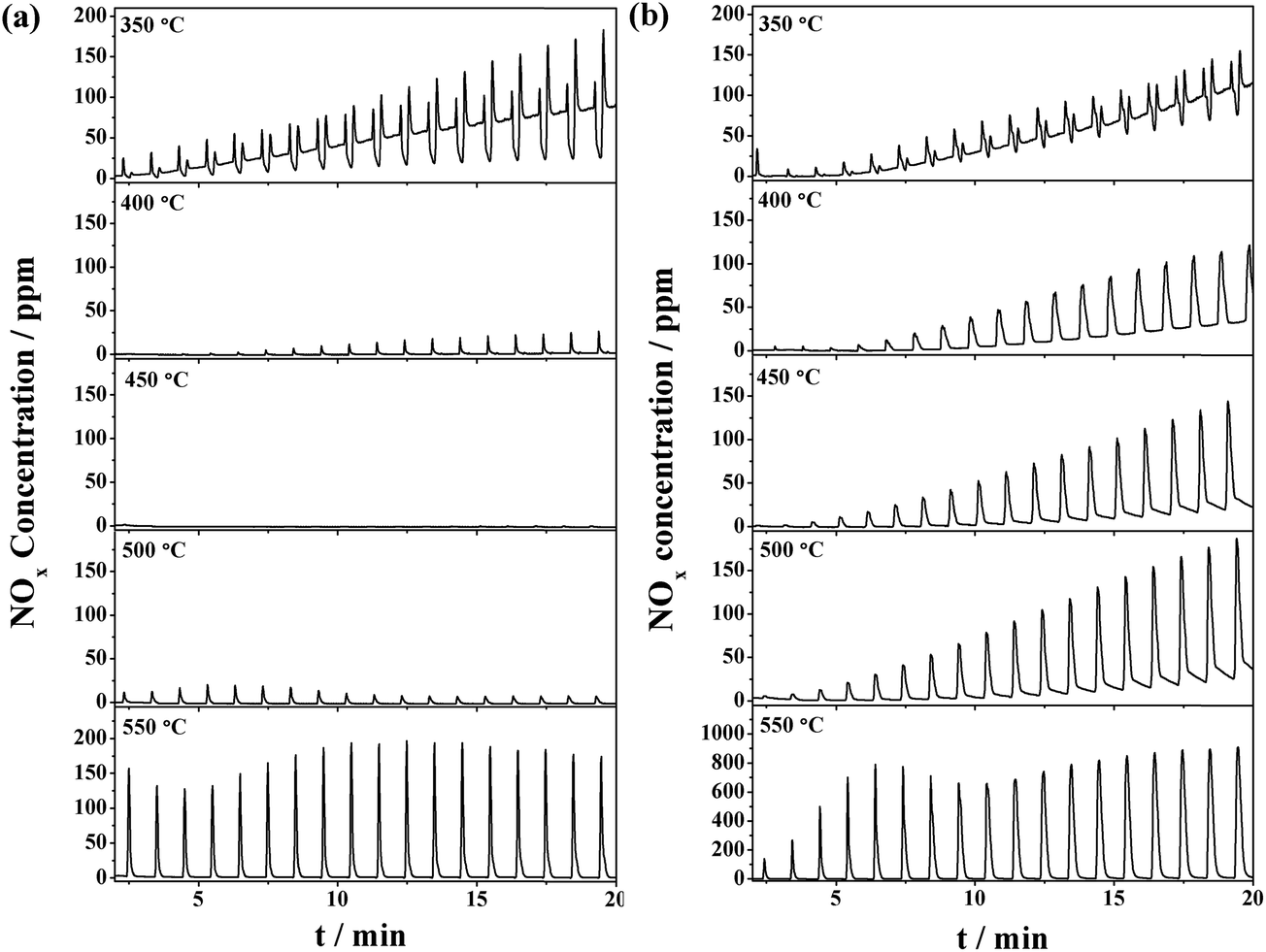 | ||
| Fig. 3 NOx concentration curves during the lean/rich cycles over the (a) CuO/K2CO3/MgAl2O4 and (b) CuO/K2CO3/Al2O3 catalysts. | ||
Compared with the CuO/K2CO3/Al2O3 catalyst, the NSR performance of the CuO/K2CO3/MgAl2O4 catalyst is consistently more effective and steady in the successive 20 cycles in the whole high-temperature region. Additionally, the NRP results show that the NOx reduction efficiency is poor at 350 °C for both catalysts. It indicates that CuO is only active above 400 °C, which coincides with the NSC results.
3.3. Structure of the catalysts
XRD is employed to investigate the structure of the catalysts. Fig. 4 shows the XRD patterns of the fresh catalysts and the catalysts after NOx storage at 450 °C. All of the diffraction peaks in Fig. 4a are well matched with spinel-type MgAl2O4 phase (JCPDS 21-1152). It indicates that the MgAl2O4 support is successfully synthesized after calcination at 800 °C with no other phase detected. Crystallized CuO phase (JCPDS 48-1548) with 2θ at 35.6°, 38.7° and 48.7° can be clearly identified on the CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts. No characteristic peak of K-related species is detected on the pattern of the fresh CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts in Fig. 4b and c, suggesting that the K-related species may be highly dispersed or in amorphous state.40 Additionally, the mean crystallite size of CuO was calculated on the basis of the XRD data by using Scherrer equation. The mean crystallite size of CuO on the fresh CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts is 24.4 and 22.4 nm, respectively. | ||
| Fig. 4 XRD patterns of the catalysts: (a) fresh MgAl2O4, (b) fresh CuO/K2CO3/MgAl2O4, (c) fresh CuO/K2CO3/Al2O3, (d) CuO/K2CO3/MgAl2O4 after NOx storage at 450 °C and (e) CuO/K2CO3/Al2O3 after NOx storage at 450 °C. | ||
The phase structure of the catalysts after NOx storage was also characterized as shown in Fig. 4d and e. After the NOx storage treatment at 450 °C, new diffraction peaks at 27.2°, 29.6° and 32.9° assigned to KNO3 phase (JCPDS 32-0824) emerge in the XRD patterns of the CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts. The transformation of carbonates to nitrates is revealed on the K-related storage sites. Stronger peaks of KNO3 are observed in the XRD pattern of the CuO/K2CO3/MgAl2O4 catalyst than those of the CuO/K2CO3/Al2O3 catalyst, implying that more NOx was trapped on the CuO/K2CO3/MgAl2O4 catalyst. It is in good agreement with the results of NSC. Additionally, after the NOx storage, the crystallite size of CuO on the two catalysts is 25.0 and 24.6 nm, respectively, which is similar to the fresh catalysts. Probably, the CuO phase is the active site in the NSR reaction.29,41
We measured the specific surface areas of the catalysts by N2 physisorption. The MgAl2O4 support has a specific surface area of 100.2 m2 g−1, lower than the commercial-Al2O3 support (179.8 m2 g−1). After loading the same amount of Cu and K, the surface areas of the CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts drop to 60.5 and 109.8 m2 g−1, respectively. Combined with the NRP and NSC results, the CuO/K2CO3/MgAl2O4 catalyst owns the higher NSR activity in spite of the smaller specific surface area compared with the CuO/K2CO3/Al2O3 catalyst, suggesting that the specific surface area is not the key factor to determine the catalytic activity of the catalysts.
Fig. S3† shows the SEM image of the fresh CuO/K2CO3/MgAl2O4 catalyst to determine its morphology. The SEM image clearly indicates that the catalyst possesses dense lamellar and needle-like structure, which may be attributed to MgAl2O4 and K2CO3.42
3.4. Chemical states of Cu- and K-species
To investigate the chemical states of the Cu- and K-species on the surface of the CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts, the XPS characterization was carried out, as shown in Fig. 5. Fig. 5a displays the XPS spectra in the Cu 2p region of the fresh CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts. For the two catalysts, the Cu 2p spectra are both composed of two typical peaks of Cu2+ at about 933.5 eV (Cu 2p3/2) and 952.9 eV (Cu 2p1/2) with a satellite shakeup at around 942.3 eV.43–46 No Cu-related species other than CuO is observed, suggesting the NOx oxidation and reduction center of the two catalysts is CuO. The above result is in agreement with the XRD result in Fig. 4. | ||
| Fig. 5 XPS spectra of the fresh catalysts: (a) Cu 2p, (b) K 2p and C 1s and (c) O 1s. | ||
Fig. 5b shows the XPS spectra in the C 1s and K 2p region of the fresh CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts. An apparent peak centering at about 296.1 eV (K 2p3/2) accompanied by a less intense peak at about 293.0 eV (K 2p1/2) can be assigned to K+. Contamination carbon was taken as a reference at 284.6 eV. The C 1s peaks in the region of 278–287 eV are ascribed to the impurities or adventitious in the fresh catalysts.47 The weak peak locating at 289.7 eV is related to surface carbonate species.48 In Fig. 5c, the catalysts present a single asymmetric peak of O 1s at about 531.1 eV. It was reported that the C 1s and O 1s peaks of pure K2CO3 were at 288.1 eV and 531.1 eV.47 Therefore, the C 1s at 289.7 eV and O 1s at 531.1 eV may be assigned to K2CO3 on the surface of the catalysts.49 In the preparation process of the catalysts, a part of the K2CO3 precursor on the catalysts has decomposed into K2O and CO2 during the calcination at 600 °C. However, the K2O is easy to react with CO2 and form K2CO3 again when the samples are exposed in air. So, the detected K2CO3 by the XPS measurement probably results from the reaction of K2O with CO2 in air.50
The binding energies and elemental compositions from the XPS data are given in Table 2. The surface atomic ratios of Cu/K of the fresh CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts are much lower than their theoretical composition (Cu/K = 0.62). It indicates that the K2CO3 may partially cover the CuO phase on the surface of the catalysts. The surface Cu/K atomic ratio of the CuO/K2CO3/MgAl2O4 catalyst is 0.29, larger than that of the CuO/K2CO3/Al2O3 catalyst (0.24). As we discussed in Fig. S1,† the NOx storage process will be substantially hindered without the aid of CuO. The higher surface Cu/K ratio of the MgAl2O4-supported catalyst reveals that there is more CuO existing and uncovered on the surface of CuO/K2CO3/MgAl2O4 catalyst, which can improve the accessibility of the active CuO sites, and then enhance the catalytic activity.
| Catalysts | Cu 2p3/2 (eV) | K 2p3/2 (eV) | O 1s (eV) | Cu/K (atomic%) |
|---|---|---|---|---|
| CuO/K2CO3/MgAl2O4 | 933.5 | 295.9 | 531.0 | 0.29 |
| CuO/K2CO3/Al2O3 | 933.8 | 296.1 | 531.1 | 0.24 |
To further identify the chemical state of Cu species, Fig. 6 shows the XANES spectra of Cu K-edge of the fresh CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts and the catalysts after NOx storage at 450 °C. The reference samples are Cu, Cu2O and CuO. Both of the two fresh catalysts show the similar shape and location of the adsorption edge to that of CuO.41 There is no Cu and Cu2O detected on the two catalysts. Therefore, the Cu species on the CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts is in the form of CuO. After NOx storage, the Cu K-edge of the spent catalysts are also the same as that for the fresh ones, suggesting that little change has taken place for the Cu species before and after reaction. It coincides with the XRD results in Fig. 4. Therefore, CuO is the active component of the CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts.29,41,51
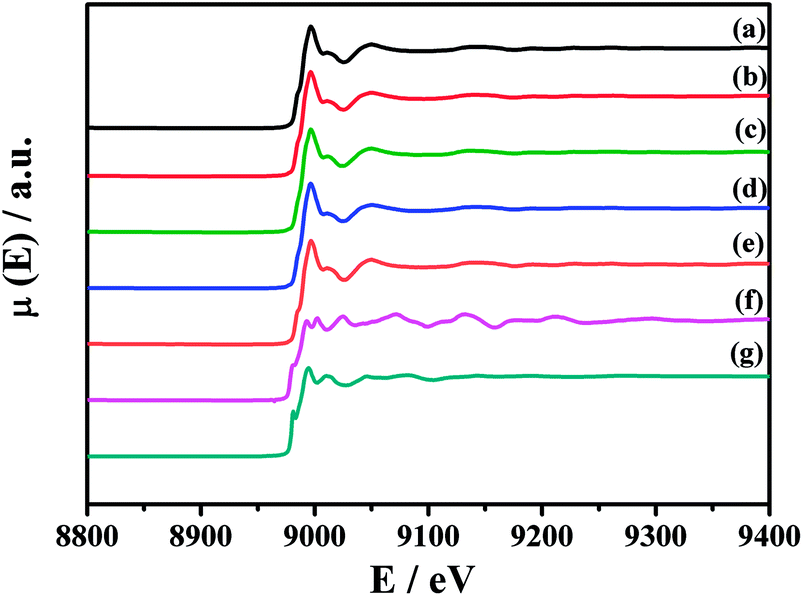 | ||
| Fig. 6 XANES spectra of Cu K-edge of the samples: (a) fresh CuO/K2CO3/MgAl2O4, (b) CuO/K2CO3/MgAl2O4 after NOx storage at 450 °C, (c) fresh CuO/K2CO3/Al2O3, (d) CuO/K2CO3/Al2O3 after NOx storage at 450 °C, (e) CuO, (f) Cu and (g) Cu2O. | ||
The H2-TPR experiment was conducted to elucidate the reducibility of the fresh catalysts, as shown in Fig. 7. No reduction reaction happens in the profile of the inert MgAl2O4 support. For contrast, pure CuO shows a H2 consumption peak at 350 °C. CuO particles on the MgAl2O4 support and Al2O3 support can be reduced at much lower temperature of 240 °C and 238 °C, respectively. After 10% K loading on the CuO/MgAl2O4 sample, the main reduction peak shifts to 327 °C, which is close to the unsupported-bulk CuO. Notably, a weak reduction peak at 264 °C is observed, which is assignable to the uncovered CuO on the CuO/K2CO3/MgAl2O4 catalyst. While after 10% K loading on the CuO/Al2O3 sample, the reduction of CuO occurs at above 300 °C. Herein, only the uncovered CuO on the CuO/K2CO3/MgAl2O4 catalyst can be easy to contact to H2 and results in the similar reducibility of CuO as the CuO/MgAl2O4 catalyst. The K2CO3 loading on the catalysts may cover the CuO, which can hinder the diffusion of H2 to CuO. Based on the H2-TPR result, some of the uncovered CuO by K2CO3 exists on the CuO/K2CO3/MgAl2O4 catalyst, while the CuO on the CuO/K2CO3/Al2O3 catalyst is totally covered. Thus, the CuO/K2CO3/MgAl2O4 catalyst shows a better NSR performance than the CuO/K2CO3/Al2O3 catalyst.
 | ||
| Fig. 7 H2-TPR profiles of the samples: (a) CuO/MgAl2O4, (b) CuO/K2CO3/MgAl2O4, (c) CuO/Al2O3, (d) CuO/K2CO3/Al2O3, (e) CuO and (f) MgAl2O4. | ||
Additionally, the released CO2 via the decomposition of K2CO3 during the TPR process can dilute the feed gas and generate pseudo H2 consumption peaks. The high temperature peaks at 724 °C and 763 °C can be assigned to the decomposition of bulk-like K2CO3.
3.5. Property of the supported potassium carbonates on the catalysts
To further investigate the thermal decomposition of K2CO3 species, the CO2-TPD experiments of the fresh CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts were implemented, and the evolution profiles of CO2 are presented in Fig. 8. As reported in the literature, two kinds of K2CO3 with different thermal stability can be distinguished as a function of decomposition temperature.41 The reaction occurs as the following expression: K2CO3 → K2O + CO2. For the CuO/K2CO3/MgAl2O4 catalyst, the desorption of CO2 at 304–640 °C is regard to the decomposition of unstable surface K2CO3 on the catalyst. The second desorption stage within 640–840 °C can be attributed to bulk/bulk-like K2CO3 species with the high thermal stability. For the CuO/K2CO3/Al2O3 catalyst, the desorption of CO2 initiates at 245 °C. The unstable surface K2CO3 decomposes from 245 to 763 °C, and the bulk/bulk-like K2CO3 decomposes from 763 to 840 °C. By the integration of the CO2-TPD curves area, we find that the CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts have similar amount of CO2 desorption, suggesting that the amount of the desorbed K2CO3 is similar on the two catalysts. However, the amount of bulk/bulk-like K2CO3 on the CuO/K2CO3/MgAl2O4 catalyst is much larger than that on the CuO/K2CO3/Al2O3 catalyst, suggesting the higher thermal stability of the K2CO3 on the CuO/K2CO3/MgAl2O4 catalyst.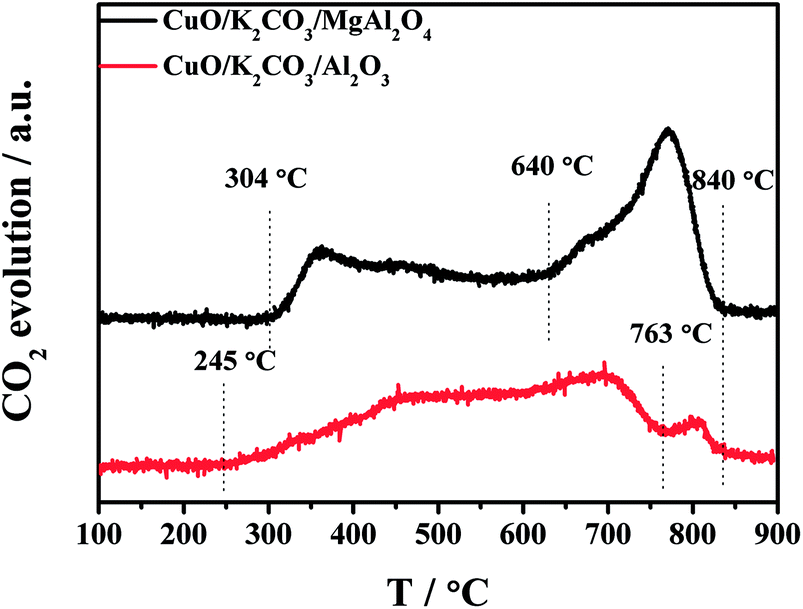 | ||
| Fig. 8 CO2-TPD profiles of the fresh catalysts. | ||
In order to explicit the nature of K2CO3 species, the FT-IR technique was carried out for the fresh CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts, as well as the references of the bulk K2CO3 and MgAl2O4. Fig. 9 shows their FT-IR spectra. The characteristic peaks of the reference bulk K2CO3 appear at 1652 cm−1, 1427 cm−1, 1386 cm−1 and 1110 cm−1. The bands at 695 cm−1 and 518 cm−1 are assignable to typical MgAl2O4 spinel structure. Compared with the bulk K2CO3, the FT-IR spectra of the loaded K2CO3 on the fresh CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts shows an additional IR band at 1530 cm−1, belonging to chelating bidentate carbonates.52 Thus, three kinds of K2CO3 species, including bridging bidentate carbonates (1652 cm−1),53 free ionic carbonate CO32− (1410 cm−1 and 1110 cm−1)52 and chelating bidentate carbonates (1530 m−1), co-exist on the fresh catalysts.
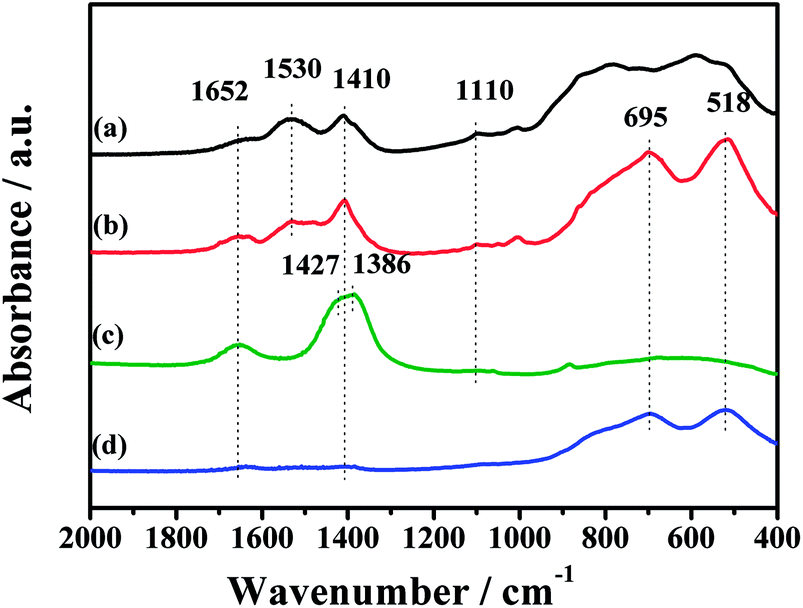 | ||
| Fig. 9 FT-IR spectra of the samples: (a) CuO/K2CO3/Al2O3, (b) CuO/K2CO3/MgAl2O4, (c) pure K2CO3 and (d) MgAl2O4. | ||
The IR spectra of the CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts after the NOx storage reaction at different temperatures were also collected to investigate to the reactivity of the different types of K2CO3. Fig. 10a(2–6) shows the IR spectra of the CuO/K2CO3/MgAl2O4 catalyst after the NOx storage reaction at different temperatures. As reported, both the ionic and bidentate nitrates presented on a series of K2O/Al2O3 catalysts after NO2 dosing.54 The most intense peak at 1380 cm−1 can be identified as ionic nitrates after the NOx storage.52 The intensity of this peak varies with the reaction temperature of CuO/K2CO3/MgAl2O4 catalyst, the tendency of which is consistent with the NSC order. The intensity of this peak becomes strongest when the CuO/K2CO3/MgAl2O4 catalyst reacts at 450 °C. Differently from the fresh CuO/K2CO3/MgAl2O4 catalyst in Fig. 9, the IR bands of bridging bidentate carbonate and chelating bidentate carbonate almost disappear leaving a tiny amount of carbonate residue on the CuO/K2CO3/MgAl2O4 catalyst, suggesting most of carbonates on the CuO/K2CO3/MgAl2O4 catalyst participate in the NOx storage. Fig. 10b(2–6) shows the IR spectra of the CuO/K2CO3/Al2O3 catalyst after the NOx storage reaction at different temperatures. The peak at 1380 cm−1 attributed to ionic nitrates can also be detected. However, the peaks at 1652 cm−1 and 1530 cm−1 do not completely disappear after the NOx storage process, suggesting that a part of the bridging bidentate carbonate and chelating bidentate carbonate in the CuO/K2CO3/Al2O3 catalyst can not completely store NOx at high temperatures. Comparing the IR spectra of the CuO/K2CO3/MgAl2O4 and CuO/K2CO3/Al2O3 catalysts after the NOx storage, there is more K2CO3 on CuO/K2CO3/MgAl2O4 transformed to nitrate. It suggests that the MgAl2O4 can improve the NOx storage efficiency of K2CO3 at high operating temperatures. Additionally, after NOx storage at 450 °C, the two catalysts were heated in the N2 flow at 450 °C for 10 min. The IR spectra of the two catalysts after NOx-TPD are shown in Fig. 10a(7) and b(7). For the CuO/K2CO3/MgAl2O4 catalyst, the intensity of the peak at 1380 cm−1 in Fig. 10a(7) has little change compared with Fig. 10a(4). For the CuO/K2CO3/Al2O3 catalyst, a distinct decrease of peak at 1380 cm−1 in Fig. 10b(7) is observed compared with Fig. 10b(4). The different behaviors at 1380 cm−1 on the two catalysts reveal that the nitrates on the CuO/K2CO3/MgAl2O4 catalyst exhibit higher thermal stability.
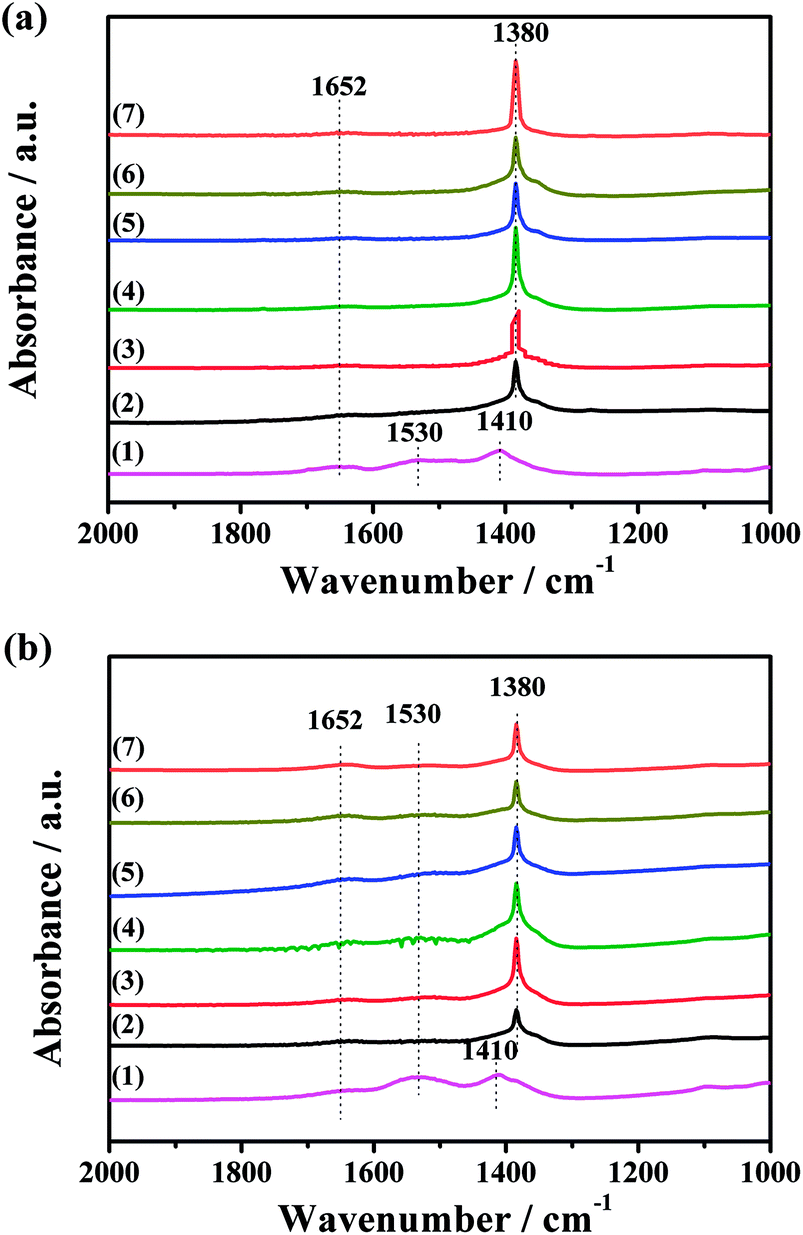 | ||
| Fig. 10 FT-IR spectra of the (a) CuO/K2CO3/MgAl2O4 and (b) CuO/K2CO3/Al2O3 catalysts: (1) fresh catalyst, and the catalyst after NOx storage reaction at (2) 350 °C, (3) 400 °C, (4) 450 °C, (5) 500 °C and (6) 550 °C, and (7) the catalyst (4) heated at 450 °C in N2 flow for 10 min. | ||
3.6. Thermal stability of the stored nitrates
The low thermal stability of the nitrate formed on NSR catalysts in the NOx storage reaction will limit their application at high operating temperatures.16 In Fig. 2, the thermal stability of the nitrates on the K sites of the CuO/K2CO3/MgAl2O4 catalyst is higher compared with the CuO/K2CO3/Al2O3 catalyst. Based on the previous report, the strength of N–O bond in nitrate is sensitive to the metal cations exposed from the support because of electronic polarization.17 The charge density of Al3+ and Mg2+ is 4.8 × 10−3 e nm−3 and 7.5 × 10−4 e nm−3. Lower extent of polarization of Mg2+ makes N–O bond less prone to breakage, and thus the nitrates on the CuO/K2CO3/MgAl2O4 show the higher thermal stability.Additionally, in our study, the temperatures of NOx desorption correlate to the temperatures of K2CO3 decomposition as mentioned in Fig. 8. Thus, the thermal stability of nitrates is related to the thermal stability of K2CO3. In other word, the nitrates with high thermal stability may be transformed from the thermal stable K2CO3. As discussed in Fig. 10, there is more K2CO3 with high thermal stability on the CuO/K2CO3/MgAl2O4 catalyst than that on the CuO/K2CO3/Al2O3, and this part of K2CO3 is the main storage sites in the high-temperature NSR reaction. Thus, the trapped nitrate on the CuO/K2CO3/MgAl2O4 catalyst shows the high thermal stability.
Through the above discussion, the CuO/K2CO3/MgAl2O4 catalyst can store much more NOx than CuO/K2CO3/Al2O3 catalyst above 400 °C, resulting from the former's higher thermal stability of nitrates. The high thermal stability of nitrate is the main reason to induce the high NOx storage capacity of the CuO/K2CO3/MgAl2O4 catalyst at the high temperatures.
Therefore, with the high thermal stability of nitrates and the high redox capacity of CuO above 400 °C, the CuO/K2CO3/MgAl2O4 catalyst exhibits the prominent NRP performance at high operating temperatures.
4. Conclusion
Herein, we report the high NSR activity of the newly designed CuO/K2CO3/MgAl2O4 catalyst at high operating temperatures. As concerns of the NSR activity in successive 20 lean/rich cycles between 350–550 °C, the CuO/K2CO3/MgAl2O4 catalyst performs more effectively and steadily on De-NOx in alternative lean/rich atmospheres than the Al2O3-supported catalyst. Especially, the NOx reduction efficiency of the CuO/K2CO3/MgAl2O4 catalyst achieves a maximum value of 99.9% with a large NOx uptake of 1.56 mmol g−1 at 450 °C. Meanwhile, a high N2 selectivity of 99.7% was also obtained. The NRP of the CuO/K2CO3/MgAl2O4 catalyst maintains above 90% at 400–550 °C. While the NRP of the CuO/K2CO3/Al2O3 catalyst decreases sharply with the increased reaction temperatures.The states of CuO and K2CO3 species have an apparent difference on the MgAl2O4 and Al2O3 supported catalysts. After K loading, CuO is probably covered by K2CO3. There is still a part of the uncovered CuO on the MgAl2O4 supported catalyst, but none for the Al2O3 supported catalyst. This part of CuO is beneficial for the oxidation and reduction of NOx in alterative lean/rich cycles. CuO exhibits the high redox ability at the temperatures above 400 °C. Based on the FT-IR results, the carbonates on the two catalysts can be classified into three types: free ionic carbonate, bridging bidentate carbonate and chelating bidentate carbonate. Most of the K2CO3 on the CuO/K2CO3/MgAl2O4 catalyst can convert to nitrates, but only a part of them participates on the CuO/K2CO3/Al2O3 catalyst. Moreover, our results show that the thermal stability of nitrate is consistent with the thermal stability of K2CO3. The formed nitrates during the lean operation on the CuO/K2CO3/MgAl2O4 catalyst have the high thermal stability, because of the high thermal stability of K2CO3 supported on the catalyst. Accordingly, the CuO/K2CO3/MgAl2O4 catalyst exhibits a high NSR activity at high operating temperatures and is a promising high-temperature NSR catalyst.
Acknowledgements
We are grateful to the National Natural Science Foundation of China [No. 21476159, U1232118], the Program of Introducing Talents of Discipline to China Universities [No. B06006], the 973 program [2014CB932403], and the Natural Science Foundation of Tianjin, PR China [15JCZDJC37400, 15JCYBJC23000]. We gratefully acknowledge Shanghai Synchrotron Radiation Facility (SSRF) for the assistance in the XAFS experiments.References
- R. Burch, Catal. Rev.: Sci. Eng., 2004, 46, 271–334 CAS.
- T. Kreuzer, E. S. Lox, D. Lindner and J. Leyrer, Catal. Today, 1996, 29, 17–27 CrossRef CAS.
- U. Alkemade and B. Schumann, Solid State Ionics, 2006, 177, 2291–2296 CrossRef CAS.
- S. Roy and A. Baiker, Chem. Rev., 2009, 109, 4054–4091 CrossRef CAS PubMed.
- C. H. Kim, G. Qi, K. Dahlberg and W. Li, Science, 2010, 327, 1624–1627 CrossRef CAS PubMed.
- N. Takahashi, H. Shinjoh, T. Iijima, T. Suzuki, K. Yamazaki, K. Yokota, H. Suzuki, N. Miyoshi, S. Matsumoto, T. Tanizawa, T. Tanaka, S. Tateishi and K. Kasahara, Catal. Today, 1996, 27, 63–69 CrossRef CAS.
- C. Sedlmair, J. Catal., 2003, 214, 308–316 CrossRef CAS.
- W. S. Epling, L. E. Campbell, A. Yezerets, N. W. Currier and J. E. Parks, Catal. Rev.: Sci. Eng., 2004, 46, 163–245 Search PubMed.
- M. Piacentini, M. Maciejewski and A. Baiker, Appl. Catal., B, 2005, 59, 187–195 CrossRef CAS.
- P. Forzatti, L. Lietti and I. Nova, Energy Environ. Sci., 2008, 1, 236–247 CAS.
- L. Masdrag, X. Courtois, F. Can and D. Duprez, Appl. Catal., B, 2014, 146, 12–23 CrossRef CAS.
- B. Pereda-Ayo, J. R. González-Velasco, R. Burch, C. Hardacre and S. Chansai, J. Catal., 2012, 285, 177–186 CrossRef CAS.
- J. Dupré, P. Bazin, O. Marie, M. Daturi, X. Jeandel and F. Meunier, Appl. Catal., B, 2016, 181, 534–541 CrossRef.
- R. D. Clayton, M. P. Harold, V. Balakotaiah and C. Z. Wan, Appl. Catal., B, 2009, 90, 662–676 CrossRef CAS.
- M. Takeuchi and S. Matsumoto, Top. Catal., 2004, 28, 151–156 CrossRef CAS.
- N. Takahashi, S. i. Matsunaga, T. Tanaka, H. Sobukawa and H. Shinjoh, Appl. Catal., B, 2007, 77, 73–78 CrossRef CAS.
- J. Luo, F. Gao, A. M. Karim, P. Xu, N. D. Browning and C. H. F. Peden, ACS Catal., 2015, 5, 4680–4689 CrossRef CAS.
- J. H. Kwak, D. H. Kim, J. Szanyi, S. J. Cho and C. H. F. Peden, Top. Catal., 2012, 55, 70–77 CrossRef CAS.
- M. Hatanaka, N. Takahashi, N. Takahashi, T. Tanabe, Y. Nagai, A. Suda and H. Shinjoh, J. Catal., 2009, 266, 182–190 CrossRef CAS.
- T. Tanabe, Y. Nagai, K. Dohmae, H. Sobukawa and H. Shinjoh, J. Catal., 2008, 257, 117–124 CrossRef CAS.
- L. Lietti, P. Forzatti, I. Nova and E. Tronconi, J. Catal., 2001, 204, 175–191 CrossRef CAS.
- S. Roy, N. van Vegten and A. Baiker, J. Catal., 2010, 271, 125–131 CrossRef CAS.
- H. Xian, X. Zhang, X. Li, H. Zou, M. Meng, Z. Zou, L. Guo and N. Tsubaki, Catal. Today, 2010, 158, 215–219 CrossRef CAS.
- W. Wen, X. Wang, S. Jin and R. Wang, RSC Adv., 2016, 6, 74046–74052 RSC.
- X. G. Li, Y. H. Dong, H. Xian, W. Y. Hernández, M. Meng, H. H. Zou, A. J. Ma, T. Y. Zhang, Z. Jiang, N. Tsubaki and P. Vernoux, Energy Environ. Sci., 2011, 4, 3351–3354 CAS.
- X. Wang, X. Qi, Z. Chen, L. Jiang, R. Wang and K. Wei, J. Phys. Chem. C, 2014, 118, 13743–13751 CAS.
- R. Vijay, R. J. Hendershot, S. M. Rivera-Jiménez, W. B. Rogers, B. J. Feist, C. M. Snively and J. Lauterbach, Catal. Commun., 2005, 6, 167–171 CrossRef CAS.
- Z. S. Zhang, M. Crocker, B. B. Chen, Z. F. Bai, X. K. Wang and C. Shi, Catal. Today, 2015, 256, 115–123 CrossRef CAS.
- F. Fan, M. Meng and Y. Tian, Acta Phys.-Chim. Sin., 2015, 31, 1761–1770 CAS.
- T. Tanabe, Y. Nagai, K. Dohmae, H. Sobukawa and H. Shinjoh, J. Catal., 2008, 257, 117–124 CrossRef CAS.
- Q. Wang and J. S. Chung, Appl. Catal., A, 2009, 358, 59–64 CrossRef CAS.
- M. Takeuchi and S. Matsumoto, Top. Catal., 2004, 28, 151–156 CrossRef CAS.
- A. J. Ma, S. Z. Wang, C. Liu, H. Xian, Q. Ding, L. Guo, M. Meng, Y. S. Tan, N. Tsubaki, J. Zhang, L. R. Zheng and X. G. Li, Appl. Catal., B, 2014, 146, 24–34 CrossRef CAS.
- S. Bennici and A. Gervasini, Appl. Catal., B, 2006, 62, 336–344 CrossRef CAS.
- A. Patel, T. Rufford, V. Rudolph and Z. Zhu, Catal. Today, 2010, 166, 188–193 CrossRef.
- Q. Wang, J. H. Sohn and J. S. Chung, Appl. Catal., B, 2009, 89, 97–103 CrossRef CAS.
- J. Despres, M. Elsener, M. Koebel, O. Krocher, B. Schnyder and A. Wokaun, Appl. Catal., B, 2004, 50, 73–82 CrossRef CAS.
- M. Yung, E. Holmgreen and U. Ozkan, J. Catal., 2007, 247, 356–367 CrossRef CAS.
- S. Salasc, M. Skoglundh and E. Fridell, Appl. Catal., B, 2002, 36, 145–160 CrossRef CAS.
- N. Hou, Y. Zhang and M. Meng, J. Phys. Chem. C, 2013, 117, 4089–4097 CAS.
- Y. Zhang, R. You, D. Liu, C. Liu, X. Li, Y. Tian, Z. Jiang, S. Zhang, Y. Huang, Y. Zha and M. Meng, Appl. Surf. Sci., 2015, 357, 2260–2276 CrossRef CAS.
- M. Galvez, S. Ascaso, R. Moliner and M. Lazaro, Chem. Eng. Sci., 2013, 87, 75–90 CrossRef CAS.
- J. O. Shim, H. S. Na, A. Jha, W. J. Jang, D. W. Jeong, I. W. Nah, B. H. Jeon and H. S. Roh, Chem. Eng. J., 2016, 306, 908–915 CrossRef CAS.
- K. I. Shimizu, H. Maeshima, H. Yoshida, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2000, 2, 2435–2439 RSC.
- E. Moretti, M. Lenarda, L. Storaro, A. Talon, T. Montanari, G. Busca, E. Rodríguez-Castellón, A. Jiménez-López, M. Turco, G. Bagnasco and R. Frattini, Appl. Catal., A, 2008, 335, 46–55 CrossRef CAS.
- F. Márquez, A. E. Palomares, F. Rey and A. Corma, J. Mater. Chem., 2001, 11, 1675–1680 RSC.
- J. M. Moggia, V. G. Milt, M. A. Ulla and L. M. Cornaglia, Surf. Interface Anal., 2003, 35, 216–225 CrossRef CAS.
- X. Deng, A. Verdaguer, T. Herranz, C. Weis, H. Bluhm and M. Salmeron, Langmuir, 2008, 24, 9474–9478 CrossRef CAS PubMed.
- Y. Zhang, M. Meng, F. Dai, T. Ding and R. You, J. Phys. Chem. C, 2013, 117, 23691–23700 CAS.
- Z. Zou, M. Meng and J. He, Mater. Chem. Phys., 2010, 124, 987–993 CrossRef CAS.
- X. Wang, Z. Chen, Y. Luo, L. Jiang and R. Wang, Sci. Rep., 2013, 3, 1559 CrossRef PubMed.
- T. J. Toops, D. B. Smith and W. P. Partridge, Appl. Catal., B, 2005, 58, 245–254 CrossRef CAS.
- R. You, Y. Zhang, D. Liu, M. Meng, L. Zheng, J. Zhang and T. Hu, J. Phys. Chem. C, 2014, 118, 25403–25420 CAS.
- D. H. Kim, K. Mudiyanselage, J. Szanyi, J. C. Hanson and C. H. F. Peden, J. Phys. Chem. C, 2014, 118, 4189–4197 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03200e |
| This journal is © The Royal Society of Chemistry 2017 |