
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and application of a MOF-derived Ni@C catalyst by the guidance from an in situ hot stage in TEM†
Dan Xu,
Ying Pan,
Mingyi Chen,
Qinying Pan,
Liangkui Zhu,
Ming Xue,
Daliang Zhang*,
Qianrong Fang and
Shilun Qiu*
and
Shilun Qiu*
State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University, Changchun, 130012, China. E-mail: sqiu@jlu.edu.cn
First published on 17th May 2017
Abstract
Metal–organic frameworks (MOFs) as a class of crystalline porous solids have attracted considerable attention due to their promising potential performance. MOFs have been recently proved to be ideal sacrificial templates for fabricating their respective derivatives by changing the thermal conditions. However, uncertainties still remain, and the direct observation of transition from MOF to metal nanoparticles (NPs) dispersed in carbon matrix is an important and crucial task for the development of MOF-derived materials. Here, transmission electron microscopy (TEM) combined with in situ hot stage technique was applied to directly observe the transition from MOF to metal NPs. Through in situ TEM experiment, the nanocrystals of Ni–ntca precursor (ntca = 1,4,5,8-naphthalenetetra carboxylic acid) are pyrolyzed under the temperature of 400, 500, or 600 °C to synthesize abundant Ni-NPs embedded in hierarchically porous carbon composites. Furthermore, the as-prepared samples show high catalytic activity and stability for the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) with NaBH4 in aqueous conditions. More importantly, Ni@C-600, which has nickel contents of 72.8%, shorten the reduction time to 3.5 min with high conversion of nearly 100%. When the catalyst is applied to recycle after being separated from the reaction by an extern magnet, it still keeps high conversion of 92% after 8 cycles, addressing the high stability of the composites. It is believed that these results will further facilitate the exploration of the technique of the TEM combined with in situ hot stage as a powerful tool in the carbonization of MOFs to obtain MOF-derived materials with different applications.
Introduction
Metal–organic frameworks (MOFs), also have been called porous coordination polymers (PCPs), consist of metal ions or metal-containing clusters in combination with a continuously expanding library of multi-functional organic ligands.1–6 Their fascinating feature of high porosity as well as large surface areas, made them useful for applications in gas storage and separation, ion exchange, catalysis, sensing, and electrochemical capacitance over the past two decades.7–12 Recently, MOFs have been proved to be ideal sacrificial templates for fabricating their respective derivatives by changing the thermal conditions in the different atmosphere.In 2008, Xu's group synthesized nanoporous carbon materials by pyrolyzing MOF-5 and furfuryl alcohol (FA) for the first time. They opened the gate of carbonizing MOFs and developed the applications of MOF-derived nanomaterials.13 After that, the use of MOFs as templates to create nanoporous carbon materials, nanostructured metal oxides and carbon–metal hybrids have been quickly developed.14–17 Liu's group prepared carbon–cobalt hybrid materials through the thermolysis of the Co–ZIF precursor at 750 °C which has been explored as a good catalyst for oxygen reduction reaction (ORR).18 The porous carbon-based nanomaterials prepared by this novel MOF-templated route possessed many advantages such as large internal surface area, high crystallinity, tailorable porous structure and tuned in modification, and more and more researchers focused on fabricating desired catalysts derived from MOFs precursors.19,20
There were various MOF-templated carbon-based nanomaterials synthesized via a choice of suitable MOF templates, pyrolysis atmosphere and temperature. Zhang's group employed zinc–nickel–terephthalate frameworks as precursors to prepare a carbon–nickel hybrid material. By adjusting the ratio of Zn/Ni, the catalysts with densely populated metal sites were applied in the converting of 4-nitrophenol.21 In addition, the use of other metal-based MOFs (Fe, Co, Cu etc.)17,22–25 as templates has promoted the development of the catalytic field. During the process of carbonization, the carbon generated from the organic linkers coated metal NPs and further prevented them from aggregating. Moreover, the in situ generation methods makes metal NPs more uniformly and stably dispersed in porous carbon matrix compared with other method of loading metal into carbon.
Transmission electron microscopy (TEM) with high resolution transmission electron microscopy (HRTEM) and electron diffraction (ED) was employed to characterize the microstructure and ultrastructure of materials.26–28 The TEM equipped with a heating holder and a hot stage controller allows in situ observations of phenomena, which accelerated the development of metallurgical applications. In 1970, the hot stage TEM was applied to study crystal nucleation in a lithia–silica glassy system.29 In 2007, a in situ TEM experiment was used as a guideline to synthesize as-grown ordered CoPt nanoparticles.30 So far, in situ TEM is a powerful method for the observation of micro structural phase changes,31 nucleation,32 growth and dissolution processes.33,34
Here, TEM combined with in situ hot stage technique was applied to directly observe the transition from MOF to metal NPs. Through in situ TEM experiment, the nanocrystals of Ni–ntca precursor (ntca = 1,4,5,8-naphthalenetetracarboxylic acid) are pyrolyzed under the temperature of 400, 500, or 600 °C to synthesize abundant Ni-NPs embedded in hierarchically porous carbon composites. Furthermore, the as-prepared samples show high catalytic activity and stability for the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) with NaBH4 in aqueous conditions. More importantly, Ni@C-600, which has nickel contents of 72.8%, shorten the reduction time to 3.5 min with high conversion of nearly 100%. When the catalyst is applied to recycle after being separated from the reaction by an extern magnet, it still keeps high conversion of 92% after 8 cycles, addressing the high stability of the composites.
Experimental
Synthesis of 1,4,5,8-naphthalenetetracarboxylic acid (NTCA)
1,4,5,8-Naphthalenetetracarboxylic dianhydride (NTCDA, 1 mmol) was dissolved in a aqueous solution of NaOH under stirring at 60–70 °C. After hydrolyzing for 1 h, 1 M HCl was added dropwise until a precipitate formed and pH was about 2–3. Then the precipitate was filtered and dried at room temperature.35Synthesis of Ni–ntca
Ni(NO3)2·6H2O (0.116 g, 0.4 mmol) was dissolved in a mixture solvent of 1.4 mL of dimethylformamide (DMF), 1.4 mL of ethanol, 1.4 mL of water. Then NTCA (0.015 g, 0.05 mmol) was added into the above solvent under sonicating. The resulting mixture was transferred into a Teflon autoclave and maintained at 100 °C for 3 days. The grown powders were washed with DMF and distilled water several times and dried at room temperature. The as-prepared powders were called as Ni–ntca.Synthesis of nickel@carbon hybrids
In a typical synthesis, the precursor Ni–ntca were paved in a ceramic boat and placed into a furnace. Carbonizing the powder under a nitrogen flow and heating to different temperatures with a heating rate of 5 °C min−1. After the pre-set temperature reached, the powders were annealed for 3 h. These products were donated as Ni@C-X (X is considered as pre-set temperature).Catalytic tests on reduction of 4-NP
In the quartz cell, pipetting 2.05 mL of deionized water, followed by the addition of 60 μL of 4-nitrophenol solution (5 mM), and the 0.65 mL of NaBH4 aqueous solution (0.2 M) was added to the mixture and the color changed to deep yellow solution. Then the catalyst suspension (50 μL, 6 mg mL−1) was added to the above mixture solution. During the reaction, the reaction was taken at 1 min intervals and analyzed by UV/vis spectroscopy, which were recorded in the scanning range of 250–500 nm at room temperature. For recycling, the catalyst was recovered using an external magnetic field for the next cycle.The in situ hot stage experiment
A small amount of Ni–ntca was dispersed in ethanol, after ultrasonic treatment, dripped onto a copper grid supporting holey carbon film. A single crystal of Ni–ntca was selected to observe the transition under beam irradiation. The TEM images and Selected Area Electron Diffraction (SAED) patterns of Ni–ntca were taken every 5 minutes. The parameters of this experiment were listed in Table 1.| Condition | Unit |
|---|---|
| High tension | 200 kV |
| Spot size | Spot 5 |
| Current density | 1.1–8.7 pA cm2 |
| Exposure time | 45 s M |
| Magnification | 50k |
Characterization
The X-ray diffraction patterns were recorded on a SHIMADZU LabX XRD-6000 diffractometer with Cu-Kα radiation (λ = 1.5418 Å) in the 2θ range of 10–80° with a step size of 0.02° and a time of 4 second per step. Raman spectra were measured on an inVia microscopic confocal laser Raman spectrometer. We investigated the morphology with a JEOS JSM-6510 and microstructure of the particles with a JEM-2100 TEM system. An in situ hot stage holder and heater control from GatanSmartSet Model 901 was used in this study. Thermogravimetric analysis (TGA) was performed using a SHIMADZU DTG-60 thermal analyzer system at a heating rate of 10 °C min−1 from 50 °C to 600 °C under dry air atmosphere with a flow rate of 30 mL min−1. Nitrogen adsorption–desorption measurements were measured with a Micro Meritics TriStar II 3020 surface area and pore size analyzer at 77 K. The absorbance of 4-NP was obtained on a UV-2450 UV/vis spectrophotometer.Results and discussion
The Ni–ntca crystals were synthesized by hydrothermal process. It can be easily obtained by adding nickel nitrate and NTCA into a mixture solvent, and then heated in a Teflon autoclave for 72 h. The light brown crystal powders were analyzed by X-ray diffraction (XRD) measurement (Fig. S1†). The relative intensities and peak positions in the XRD patterns were in good agreement with the predicted diffraction patterns, revealing the high crystalline of the synthesized Ni–ntca.36This moment, a hot stage was used for in situ experiments in TEM to directly observe the changes of the Ni–ntca. Lots of TEM images and the corresponding diffraction (ED) patterns of the Ni–ntca particles were taken as elevating the carbonization temperature in TEM (Fig. 1). The single crystal was displayed in Fig. 1a, and the ED pattern of crystal showed us high degree of crystallinity in Fig. 1b. Then, we raised the temperature in 100 °C increments, and stopped 5 minutes at each temperature. At a temperature of 300 °C, the nanoparticles (black dots) emerged in the carbon (Fig. 1c), which confirmed by the appearance of diffraction rings (Fig. 1d). The corresponding crystalline diffraction rings of (111) and (200) in the SAED pattern indicated the borning of Ni NPs. From 400 °C to 600 °C, the crystallization degree of the Ni NPs and the stability of the carbon gradually enhanced (Fig. 1e–h). When the temperature exceeded 600 °C, the Ni NPs were unstable and came out of the carbon to form large particles (Fig. 1h). When extending the holding time of 5 minutes to 20 minutes, we found that the results as shown in Fig. S2† were similar to those from 5 minutes. As it is well known the MOFs materials are sensitive to the beam irradiation, we tried to explain that the beam was not playing a leading role in the process of in situ experiment, and the following work was performed using TEM. The Ni–ntca was irradiated under beam for a long time using TEM. The TEM image and SAED patterns of the crystal were collected on time during irradiation and contrasted with Fig. 1d, there was no appearance of Ni NPs within 2 hours, addressing that beam didn't work for the transition from MOFs to metal NPs, while led to the collapse of MOFs structure (Fig. S3†). Following, these thermal phenomena became a reference for us in preparing Ni@C-X composite series by carbonizing the Ni–ntca at 300 °C, 400 °C, 500 °C, and 600 °C under a nitrogen flow.
 | ||
| Fig. 1 The hot stage TEM images at (a) room temperature, (b) Selected Area Electron Diffraction (SAED) pattern of Ni–ntca in [001] zone axe, (c) 300 °C, (d) SAED pattern of (c), (e) 400 °C, (f) 500 °C, (g) 600 °C, and (h) 700 °C. | ||
The XRD patterns of a series of samples after carbonization were showed in Fig. 2 and S4.† One low broad peak centered at 2θ = 25° assigned to amorphous carbon,37 the other three prominent peaks near at 2θ = 44.3°, 51.6° and 76.2°, being well assigned to the reflections of the (111), (200) and (220) crystalline planes of cubic-phased Ni (JCPDS no. 04-0850), respectively, were detected (Fig. 2). When pyrolyzing the Ni–ntca at 400 °C, 500 °C and 600 °C in a nitrogen flow, the Ni–ntca have been completely converted to amorphous carbon and cubic-phased Ni NPs, and there was no peaks showing Ni been oxidized to nickel monoxide (NiO). However, the peaks of Ni@C-300 were different from the cubic-phased Ni and were proven to be not pure Ni nanoparticles (Fig. S4†).
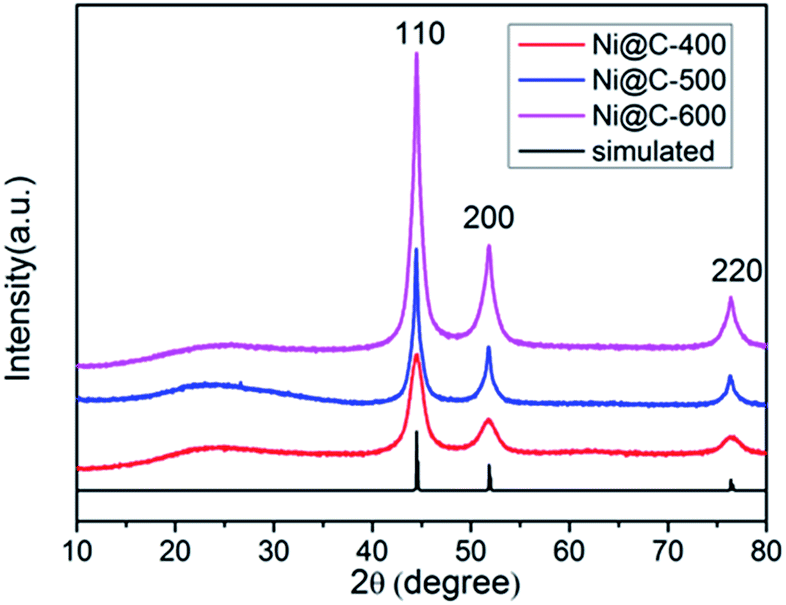 | ||
| Fig. 2 PXRD patterns of Ni@C-400, Ni@C-500, Ni@C-600 and the simulated data of cubic phased Ni (JCPDS no. 04-0850). | ||
As seen in Fig. 3, a broad D band appeared at 1340 cm−1 which ascribed to the disorder-induced mode, while a relatively sharp G band appeared at 1575 cm−1 which ascribed to the E2g mode from the sp2 carbon. The relative ratios of D bands to G bands (ID/IG) in Raman spectra, was 0.8, indicating the crystallization degree of graphitic carbon. This ID/IG ratio was comparable to those measured for GO and rGO (ID/IG = ∼1),38 indicating that high quality 3D graphite carbon was formed. Likewise, there was a 2D band at 2676 cm−1.
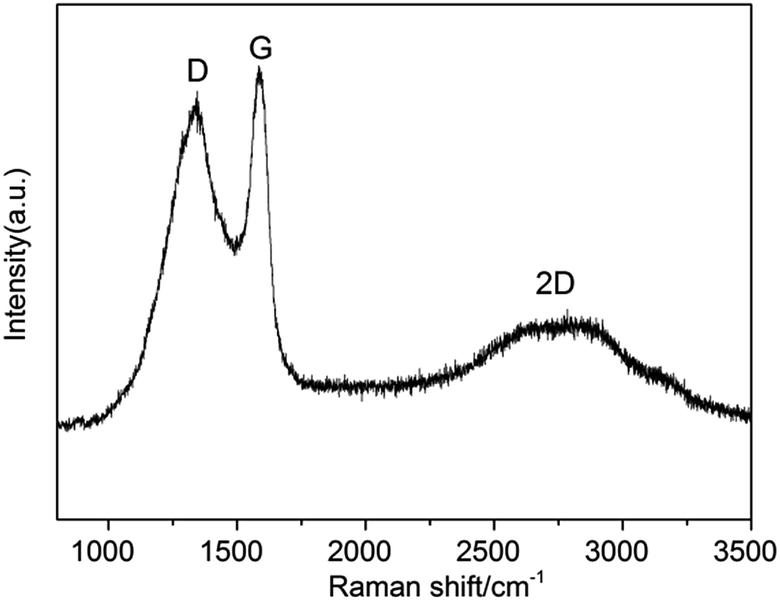 | ||
| Fig. 3 Raman pattern of Ni@C-600. | ||
To investigate the samples after carbonizing, we characterized the samples by TEM. From the TEM images (Fig. 4a–f), because of nanoscale morphology of Ni–ntca, they were all thermodynamically unstable and able to aggregate, and finally appeared irregularly shapes. After carbonizing, the composites still kept indefinite shapes (Fig. 4a and b). While further magnifying the edge at 300k, the Ni NP were uniformly deposited in the carbon matrix, almost all of the particles were wrapped by thin carbon, thus there was no reunion as well (Fig. 4c and d). The distribution range of Ni NPs was mainly distributed from 4 nm to 7 nm measured by TEM (Fig. S5†). To investigate crystallization degree of metal Ni, we continued to magnify the thin edge to 500k, and clearly observed metal lattice fringe, indicating that each particle had a single crystalline structure (Fig. 4e and f). The distance of the adjacent lattice fringes (marked by the white arrows) was calculated to be about 0.203 nm, corresponding well to the d111 spacing of cubic Ni. The corresponding fast Fourier transform (FFT) patterns were showed in the inset in Fig. 4e and f. Ni NPs were used as catalysts to induce the graphitization of carbon during the carbonization, thus three-dimensional graphite layers were readily produced around the metal Ni, which was in accordance with the results of Raman. The TEM images of Ni@C-400 and Ni@C-500 were placed in Fig. S6.† The images of scanning transmission electron microscope (STEM) were showed in Fig. S7,† further confirming the uniform distribution of Ni@C-X. From the TEM results, the synthetic Ni@C-X composites with a structure of thin 3D graphite carbon and plentiful Ni NPs will enhance the catalytic activity. The resultant samples were all composed of uniformly dispersed Ni NPs and carbon matrix.
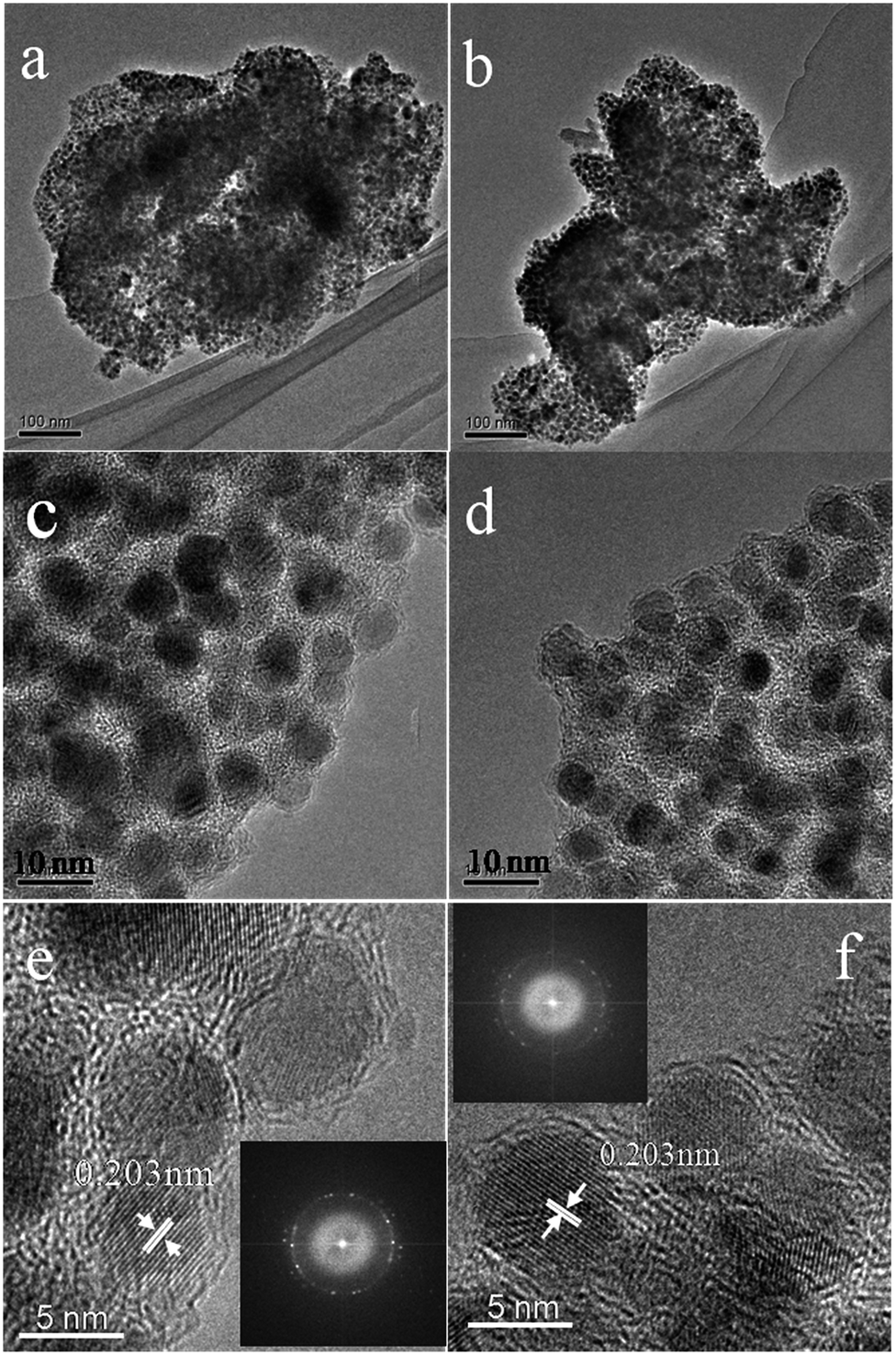 | ||
| Fig. 4 TEM images of Ni@C-600 at 100k (a and b), 300k (c and d), and 500k ((e) and (f), inset: FFT-ED pattern). | ||
To calculate the loading content of Ni NPs in the nanoporous carbon, the thermo gravimetric analysis (TGA) was carried out in the temperature range of 50–600 °C under dry air atmosphere at a heating rate of 10 °C min−1. From the TGA curves (Fig. 5), the nickel loading amounts of three samples were about 68.6%, 72.1% and 72.9% for Ni@C-400, Ni@C-500, Ni@C-600, respectively. Obviously, during carbonization, the metal contents increased while the carbon was generally declined as elevating the temperature. In the test of catalytic reduction of 4-nitrophenol (4-NP), the more Ni loading quality will acquire more excellent catalytic performance.
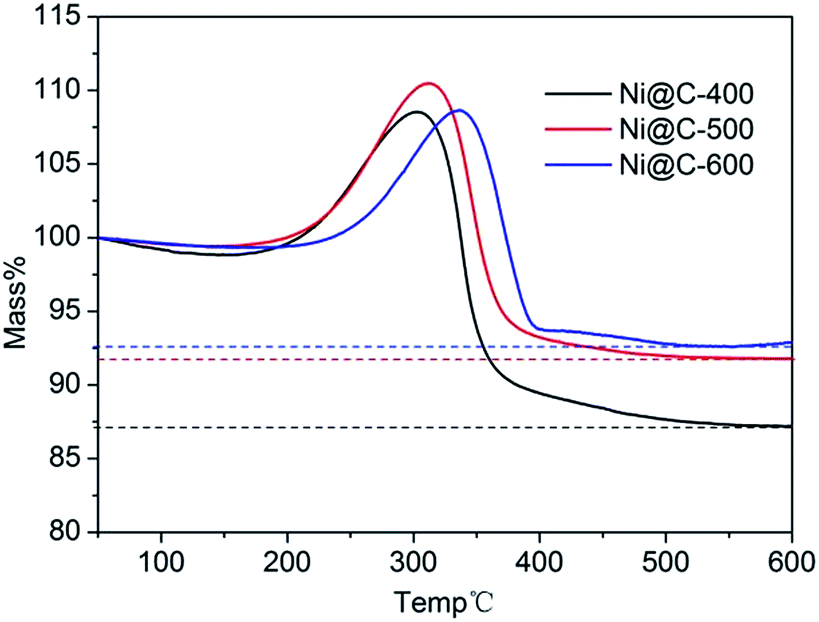 | ||
| Fig. 5 TGA curves of Ni@C-400 (black), Ni@C-500 (red), and Ni@C-600 (blue). | ||
The N2 adsorption/desorption isotherms of Ni@C-X were measured to explore porous structure (Fig. 6). Three samples exhibited typical type IV isotherms with a hysteresis loop, showing the existence of mesoporous (Fig. 6a). The pore size distribution curves (Fig. 6b) indicated that the mesoporous sizes were centered at around 9.0 nm, the microporous sizes were centered at about 1 nm, and the macroporous still existed in the samples, however, the dominating porous structure was mesopores. Brunauer–Emmett–Teller (BET) specific surface area of 138 m2 g−1, 142 m2 g−1 and 139 m2 g−1 were obtained for Ni@C-400, Ni@C-500, Ni@C-600, respectively. Furthermore, the N2 absorption isotherm of the Ni–ntca was also shown in Fig. S8.† The BET surface area of the Ni–ntca was 100 m2 g−1 and the pore size distribution showed it was microspores.
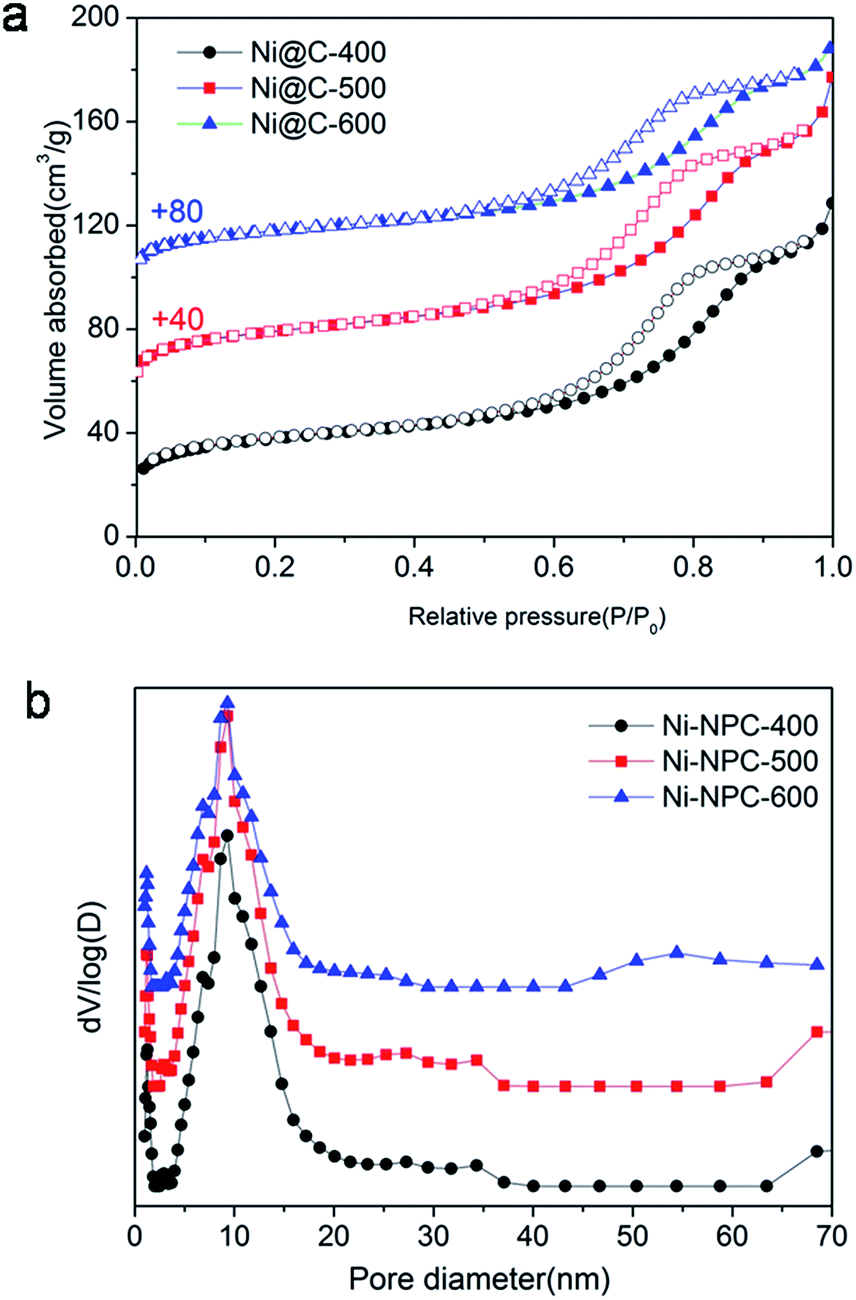 | ||
| Fig. 6 (a) N2 absorption–desorption isotherms and (b) their corresponding pore size distributions (N2 absorption–desorption isotherms for Ni@C-500 and Ni@C-600 are vertically offset by 40 and 80 cm3 g−1). | ||
At the end, we measured the catalytic activity of Ni@C-X for reduction of 4-NP. In recent years, the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) with NaBH4 in aqueous solution has become such a model reaction that many groups used to test catalytic performance. We can monitor accurately through UV/vis spectroscopy. It was clearly that 4-NP had a strong absorption peak at 400 nm in an aqueous solution of NaBH4 and the absorption peak shifted to 295 nm addressing the formation of 4-aminophenol (4-AP). The color of 4-NP solution was light yellow, with mixing NaBH4 into solution and turned into light yellow, with adding the catalyst composite and turned into colorless.39,40 The whole catalytic progress was measured regularly by UV/vis spectroscopy. Fig. 7a displayed the typical evolution of the UV/vis spectra with time for the sample. After the 0.3 mg catalyst is added to solution, the reaction immediately proceeded and we measured the absorbance curve every 1 minute. The intensity of peaks of the nitro compound at 400 nm decreased sharply, while the aminophenol at 295 nm increased. No other peak appeared, indicating no side reaction had taken place. When the peak belong to the nitro compound was no longer observed and the corresponding color changed from yellow to colorless, demonstrating the catalytic reduction of 4-NP completed. The reduction time of converting 4-NP to 4-AP over Ni@C-600 was 3.5 min (Fig. 7c), which was faster than Ni@C-400 of 6 min (Fig. 7a) and Ni@C-500 of 4 min (Fig. 7b).
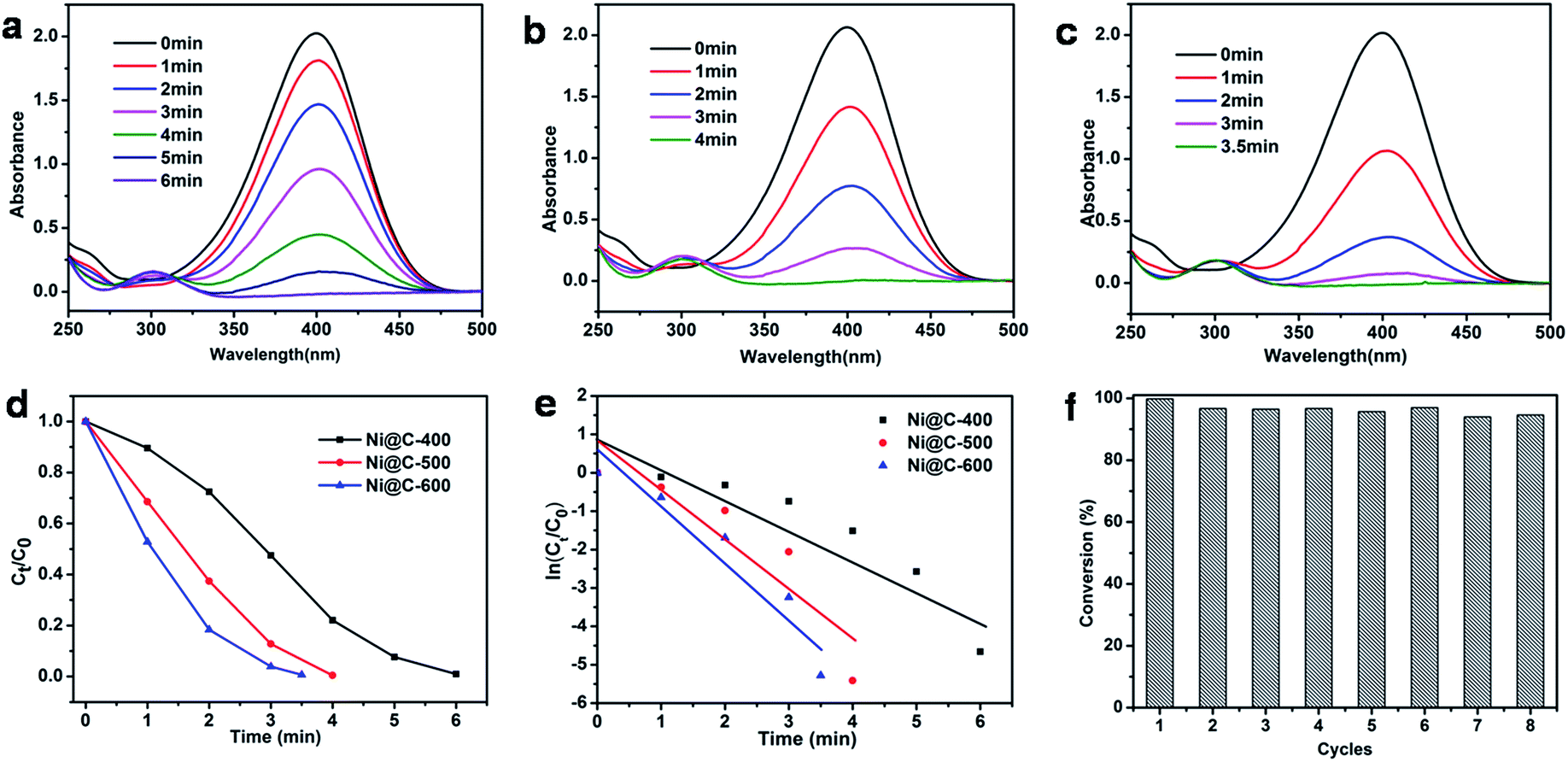 | ||
| Fig. 7 Successive UV-vis absorption spectra during the catalytic reduction of 4-NP using (a) Ni@C-400, (b) Ni@C-500, (c) Ni@C-600 as catalyst; (d) Ct/C0 versus reaction time for the reduction of 4-NP and (e) plot of ln(Ct/C0) against the reaction time of 4-NP reduction over Ni@C-400 (black line), Ni@C-500 (red line), Ni@C-600 (blue line); (f) the reusability of Ni@C-600 catalyst. | ||
According to the knowledge of physical chemistry principles, the above catalytic reduction reactions followed the Langmuir–Hinshelwood apparent first order kinetics mode without thinking about an amounts of NaBH4. The equation is described as dCt/dt = dAt/dt = kappCtt = kappAtt or ln(Ct/C0) = ln(At/A0) = kappt, Ct and At are the concentration and absorbance of 4-NP at 400 nm at time t, C0 and A0 are the initial concentration and absorbance of 4-NP at 400 nm, and kapp is the apparent rate constant.41 Fig. 7d showed the concentration of Ct (C at time t) to its initial value C0 (Ct/C0) versus reaction time for the reduction of 4-NP over the catalyst of Ni@C-400, Ni@C-500 and Ni@C-600. It can be found that the resultant catalysts were highly active for catalyzing 4-NP reduction with nearly 100% efficiency. Fig. 7e showed plots of ln(Ct/C0) versus reaction time. The respective rate constant was calculated to be 22.5, 20.8 and 12.0 × 10−3 s−1. The rate constant per gram of Ni@C-X (K) was then calculated, the activity factors K were calculated to be 40.0, 69.3 and 75.0 s−1 g−1 for Ni@C-400, Ni@C-500 and Ni@C-600, respectively. Obviously, Ni@C-600 exhibited high catalytic activity compared with Ni@C-500 and Ni@C-400, which benefited from its large nickel loading amounts and high crystallinity with quick accessibility in and out of the porous carbon matrix. The turnover frequency (TOF) was further evaluated, which was defined as the number of moles of 4-NP reduced per mole of nickel per hour (Table 2). The rate constant and TOF values of Ni@C-X were much bigger than the previously reported Ni-NPC-600 and other Ni-based catalysts21,39,42,43 (Table 2), which can be summarized to following factors: (1) the smaller Ni NPs, which supplied the catalytic reduction with a larger reaction area and more active sites for reduction; (2) the larger loading content of Ni NPs with more catalytic sites were beneficial to enhanced the catalytic performance; (3) the carbon surrounded Ni NPs were thinner and had a 3D structure, making the 4-nitrophenolate ion rapidly pass through and reach; (4) the easier hydrophilic characteristic due to a little amounts of carbon, which made it convenient to absorb borohydride anion on the surface of Ni NPs.
| Catalyst | Ni loading (wt%) | SBET (m2 g−1) | kapp × 10−3 (s−1) | Kb (s−1 g−1) | TOFc (h−1) | Reference |
|---|---|---|---|---|---|---|
| a Estimated by TGA.b The activity factors [K]: the rate constant per gram.c TOF (turnover frequency): moles of 4-NP converted per mole Ni per hour. | ||||||
| Ni@C-400 | 68.6a | 138 | 12.0 | 40.0 | 2.1 | This work |
| Ni@C-500 | 72.1a | 142 | 20.8 | 69.3 | 3.4 | This work |
| Ni@C-600 | 72.9a | 139 | 22.5 | 75.0 | 6.6 | This work |
| Ni–NPC-600 | — | 169 | 5.9 | 2.95 | 0.19 | 34 |
| Ni0.1/NPC | 46.5 | 389 | 2.0 | 6.7 | 0.468 | 21 |
| Ni1.0/NPC | 81.4 | 51 | 2.8 | 9.2 | 0.292 | 21 |
| Ni/m-CN | 48.3 | 134 | 9.1 | 9.1 | 1.2 | 42 |
| Ni0.52Sb0.48/SBA-15 | 2.85 | 416 | 3.7 | 140 | — | 43 |
| Ni/SiO2 | 14.6 | 185 | 4.5 | 1.5 | 12.2 | 39 |
To evaluate the reusability of our catalyst, the successive cycles of the catalytic reduction were carried out. As shown in Fig. 7f, the catalyst can be successfully used after being separated from reaction by an extern magnet, and still kept high activity and striking conversion of 92% after 8 cycles. The large conversion efficiency indicated the Ni NPs were stable and protected by the wrapped carbon. The TEM images of Fig. S9† told us there was no aggregation of Ni NPs, and structure and morphology were well maintained, fully demonstrating the good stability of the catalyst.
Conclusions
In conclusion, we selected a new Ni–ntca as the effective precursor to construct composites structure of magnetic nickel NPs embedded in hierarchically porous carbon matrix. The pyrolysis temperature was determined with transform of surface and the corresponding electron diffraction (ED) of the crystal during directly observing the transition of a Ni–ntca to Ni NPs. The product of 600 °C (Ni@C-600) exhibited excellent catalysis activity and remarkable durability for the reduction of 4-NP to 4-AP in mild condition. The nickel loading contents of Ni@C-600 were as high as 72.8%, which shortened catalysis reaction time to 3.5 min and possessed nearly 100% conversion. Moreover, the catalyst still remained large conversion efficiency of 92% after 8 cycles. It is believed that these results will further facilitate the exploration of the technique of the TEM combined with in situ hot stage as a powerful tool in the carbonization of MOFs to obtain derivations of MOFs with different applications.Acknowledgements
This work was supported by National Natural Science Foundation of China (21390394, 21571079, 21621001, 21571076, and 21571078), “111” project (B07016), and Guangdong Science and Technology Department Project (2013B090200052 and 2012D0501990028). Q. F. acknowledges the Thousand Talents program (China).Notes and references
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- Y. B. He, B. Li, M. O'Keeffe and B. L. Chen, Chem. Soc. Rev., 2014, 43, 5618–5656 RSC.
- J. W. Liu, L. F. Chen, H. Cui, J. Y. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- Q. R. Fang, G. S. Zhu, Z. Jin, Y. Y. Ji, J. W. Ye, M. Xue, H. Yang, Y. Wang and S. L. Qiu, Angew. Chem., 2007, 119, 6758–6762 CrossRef.
- S. L. Qiu, M. Xue and G. S. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- Z. J. Zhang, W. Shi, Z. Niu, H. H. Li, B. Zhao, P. Cheng, D. Z. Liao and S. P. Yan, Chem. Commun., 2011, 47, 6425–6427 RSC.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. B. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- H. Xu, F. Liu, Y. J. Cui, B. L. Chen and G. D. Qian, Chem. Commun., 2011, 47, 3153–3155 RSC.
- J. Tang, R. R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS PubMed.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed.
- S. R. Chen, M. Xue, Y. Q. Li, Y. Pan, L. K. Zhu and S. L. Qiu, J. Mater. Chem. A, 2015, 3, 20145–20152 CAS.
- Y. Pan, M. Xue, M. Y. Chen, Q. R. Fang, L. K. Zhu, V. Valtchev and S. L. Qiu, Inorg. Chem. Front., 2016, 3, 1112–1118 RSC.
- Y. Q. Li, T. Ben, B. Y. Zhang, Y. Fu and S. L. Qiu, Sci. Rep., 2013, 3, 2420–2426 CrossRef PubMed.
- X. Y. Li, C. M. Zeng, J. Jiang and L. H. Ai, J. Mater. Chem. A, 2016, 4, 7476–7482 CAS.
- S. Ma, G. A. Goenaga, A. V. Call and D. J. Liu, Chem.–Eur. J., 2011, 17, 2063–2067 CrossRef CAS PubMed.
- J. K. Sun and Q. Xu, Energy Environ. Sci., 2014, 7, 2071–2100 CAS.
- K. Shen, X. D. Chen, J. Y. Chen and Y. W. Li, ACS Catal., 2016, 6, 5887–5903 CrossRef CAS.
- Y. Yang, Y. Zhang, C. J. Sun, X. S. Li, W. Zhang, X. H. Ma, Y. Ren and X. Zhang, ChemCatChem, 2014, 6, 3084–3090 CrossRef CAS.
- W. Xia, R. Q. Zou, L. An, D. G. Xia and S. J. Guo, Energy Environ. Sci., 2015, 8, 568–576 CAS.
- H. Y. Niu, S. L. Liu, Y. Q. Cai, F. C. Wu and X. L. Zhao, Microporous Mesoporous Mater., 2016, 219, 48–53 CrossRef CAS.
- R. Q. Fang, R. Luque and Y. W. Li, Green Chem., 2016, 18, 3152–3157 RSC.
- F. Ke, J. F. Zhu, L. G. Qiu and X. Jiang, Chem. Commun., 2013, 49, 1267–1269 RSC.
- Z. Liu, T. Ohsuna, O. Terasaki, M. A. Camblor, M. Diaz-Cabaňas and K. Hiraga, J. Am. Chem. Soc., 2001, 123, 5370–5371 CrossRef CAS PubMed.
- Y. Sakamoto, M. Kaneda, O. Terasaki, D. Y. Zhao, J. M. Kim, G. Stuckyk, H. J. Shin and R. Ryoo, Nature, 2000, 408, 449–453 CrossRef CAS PubMed.
- Y. Han, D. L. Zhang, L. L. Chng, J. L. Sun, L. Zhao, X. D. Zou and J. Ying, Nat. Chem., 2009, 1, 123–127 CrossRef CAS PubMed.
- D. L. Kinser and L. L. Hench, J. Mater. Sci., 1970, 5, 369–373 CrossRef CAS.
- D. Alloyeau, C. Langlois, C. Ricolleau1, Y. Le Bouar and A. Loiseau, Nanotechnology, 2007, 18, 375301–375306 CrossRef.
- L. J. Bowen, R. J. Weston, T. G. Carruthers and R. J. Brook, J. Mater. Sci., 1978, 13, 341–350 CrossRef CAS.
- A. Garg and J. M. Howe, Acta Mater., 1991, 39, 1925–1937 CrossRef CAS.
- B. William and H. James, Philos. Mag. A, 1997, 75, 1641–1663 Search PubMed.
- D. Xu, D. L. Zhang, H. B. Zou, L. K. Zhu, Q. R. Fang and S. L. Qiu, Chem. Commun., 2016, 52, 10513–10516 RSC.
- U. Rana, K. Chakrabarti and S. Malik, J. Mater. Chem., 2011, 21, 11098–11100 RSC.
- L. F. Chen, J. Zhang, L. J. Song and Z. F. Ju, Inorg. Chem. Commun., 2005, 8, 555–558 CrossRef CAS.
- B. Liu, H. Shioyama, H. L. Jiang, X. B. Zhang and Q. Xu, Carbon, 2010, 48, 456–463 CrossRef CAS.
- J. Gong, J. Liu, L. Ma, X. Wen, X. C. Chen, D. Wan, H. O. Yu, Z. W. Jiang, E. Borowiak-Palen and T. Tang, Appl. Catal., B, 2012, 117–118, 185–193 CrossRef CAS.
- Z. Y. Niu, S. H. Zhang, Y. B. Sun, S. L. Gai, F. He, Y. L. Dai, L. Li and P. P. Yang, Dalton Trans., 2014, 43, 16911–16918 RSC.
- W. C. Pan, S. H. Zhang, F. He, S. L. Gai, Y. B. Sun and P. P. Yang, CrystEngComm, 2015, 17, 5744–5750 RSC.
- M. Schrinner, M. Ballauff, Y. Talmon, Y. Kauffmann, J. Thun, M. Möller and J. Breu, Science, 2009, 323, 617–620 CrossRef CAS PubMed.
- W. Zuo, G. Q. Yu and Z. P. Dong, RSC Adv., 2016, 6, 11749–11753 RSC.
- V. S. Marakatti and S. C. Peter, New J. Chem., 2016, 40, 5448–5457 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03162a |
| This journal is © The Royal Society of Chemistry 2017 |