
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCycling stability of Li metal in a mixed carbonate–ionic liquid electrolyte for lithium secondary batteries†
Ju-Sik Kim *a,
Hongsoo Choia,
Je-Nam Leeb,
Hyorang Kanga,
Dongmin Ima and
Hyunseok Kim*a
*a,
Hongsoo Choia,
Je-Nam Leeb,
Hyorang Kanga,
Dongmin Ima and
Hyunseok Kim*a
aSamsung Advanced Institute of Technology (SAIT), 130 Samsung-ro, Yeongtong-gu, Suwon-si, Gyeonggi-do 16678, Korea. E-mail: jusik.kim@samsung.com; hs13.kim@samsung.com
bAdvanced Batteries Research Center, Korea Electronics Technology Institute, 25 Saenari-ro, Bundang-gu, Seongnam 13509, Republic of Korea
First published on 8th May 2017
Abstract
Polymeric ionic liquids (PILs) containing a poly(ethylene glycol) methacrylate (POEM) coating layer significantly suppresses the reduction of the ionic liquid and of solvent molecules on Li metal anode in the Pyr14TFSI/carbonate electrolyte. Therefore, when Li metal was coated with the PILs–POEM, a cycling test performed with the electrolyte highlights an improved cycling stability.
The ever-increasing demands of electric vehicles, information technology (IT) devices, and other modern technologies require Li-ion rechargeable batteries with higher energy density per volume and per weight, compared to current batteries. In this context, Li metal has attracted significant interest as an anode material in Li-ion secondary batteries due to its intrinsically high energy density. Li metal anodes, however, have serious safety limitations associated with the formation and growth of Li dendrites and the decomposition of the electrolyte. These problems have prompted new investigations on the use of room-temperature ionic liquids (RTILs) as electrolytes, aimed at exploiting their high boiling and flash point temperatures.1–3
RTILs represent potential flame-retardant electrolytes for Li metal-based batteries;4–6 however, their viscosities are too high for practical applications. To decrease the viscosity of the electrolytes, RTILs can be used as additives or co-solvents in mixtures of conventional solvents with low viscosities, such as carbonate and ether.7–10 Lombardo et al. reported that the addition of an ionic liquid, N-methyl-N-propylpyrrolidinium bis(trifluoromethanesulfonyl) imide, to alkylcarbonate solvents led to improved thermal stability compared to the carbonate electrolyte alone.5
RTIL-containing carbonate electrolytes, however, are predicted to be electrochemically unstable at the interface of the Li metal anode due to solvent decomposition.11,12 The anionic and cationic components of the RTIL are reduced, forming a passivation layer on the anode that consumes active Li metal during the deposition and stripping processes.11–13 Using X-ray photoelectron spectroscopy (XPS), Howlett et al. found that, in the case of the ionic liquid N-methyl-N-alkylpyrrolidinium bis(trifluoromethanesulfonyl) imide (Pyr14TFSI), the reduction products of the TFSI anion were formed at the Li metal surface.11,12 The extent of reduction of RTILs in mixed solvents increases at high current rates due to the current density inhomogeneities at the anode–electrolyte interface, causing significant loss of capacity retention upon cycling. The application of a protective coating layer on the Li metal could represent an effective route to minimize side reactions affecting RTIL-containing electrolytes.
Therefore, in this work we investigate the interfacial electrochemical properties of a Li metal anode during Li deposition/stripping processes in an RTIL-containing carbonate electrolyte.
The electrolyte solvent was prepared by mixing diethyl carbonate (DEC), fluoroethylene carbonate (FEC), and Pyr14TFSI in 4.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.2:2 volume ratios. LiPF6 salt (1.2 M) was then dissolved in the solvent. The Li metal anode was coated with a composite layer consisting of a polymeric ionic liquid (PIL) and poly(ethylene glycol) methacrylate (POEM), both of which are compatible with RTILs, to stabilize the metal–electrolyte interface under electrochemical reaction conditions. The PIL was synthesized via anion exchange from a mixture of poly(diallyldimethylammonium) TFSI and N-methyl-N-butylpyrrolidinium TFSI.14,15 POEM and a LiTFSI salt were mixed with the PIL in 2:1:3 wt% ratios in a N,N-dimethylformamide solution. The mixed solution was coated on a 20 μm-thick Li metal foil using a bar coating method and dried in a vacuum chamber at 60 °C for 6 h.
:3.2:2 volume ratios. LiPF6 salt (1.2 M) was then dissolved in the solvent. The Li metal anode was coated with a composite layer consisting of a polymeric ionic liquid (PIL) and poly(ethylene glycol) methacrylate (POEM), both of which are compatible with RTILs, to stabilize the metal–electrolyte interface under electrochemical reaction conditions. The PIL was synthesized via anion exchange from a mixture of poly(diallyldimethylammonium) TFSI and N-methyl-N-butylpyrrolidinium TFSI.14,15 POEM and a LiTFSI salt were mixed with the PIL in 2:1:3 wt% ratios in a N,N-dimethylformamide solution. The mixed solution was coated on a 20 μm-thick Li metal foil using a bar coating method and dried in a vacuum chamber at 60 °C for 6 h.
Fig. 1 shows a scanning electron microscopy (SEM) image of the PIL–POEM composite layer coated on Li metal. The thickness of the coating layer was estimated to be approximately 5 μm.
 | ||
| Fig. 1 SEM images of the PIL–POEM coating layer ((a) lower resolution image, (b) higher resolution image). | ||
In order to compare the Li deposition/striping behavior of bare Li and PIL–POEM-coated Li metal, cyclic voltammograms (CVs) were obtained from the coin-type Cu/Li cells in the carbonate electrolyte (DEC/FEC = 6:4). The CVs measurements were performed in the potential range of −0.1 to 0.6 V (vs. Li/Li+) with a scan rate of 10 mV s−1, as shown in Fig. 2. For the coated Li cell, it is noted that the peak current densities for Li plating/striping decreased and the reduction potential for Li deposition was shifted in negative direction by comparison. This suggests that the coating layer possibly mitigate the reduction reaction of electrolyte with Li metal.
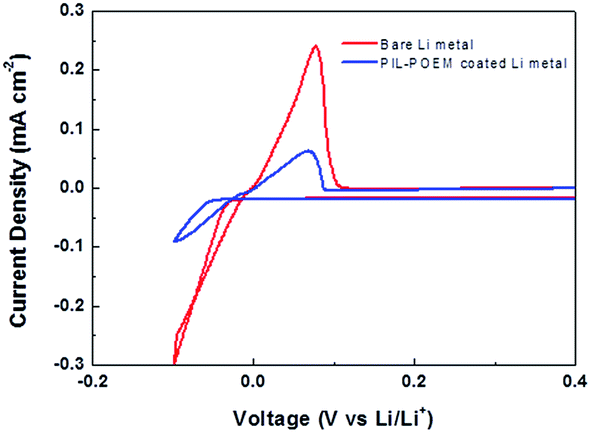 | ||
| Fig. 2 Cyclic voltammograms measured on the Cu/Li coin cells with and without the PIL–POEM coating layer in the carbonate electrolyte (DEC/FEC). | ||
Galvanostatic cycling was used to compare the electrochemical cycling stability of the coated and bare Li metal in the Pyr14TFSI-containing carbonate electrolyte during Li deposition and stripping. A Li symmetric cell containing a thin (20 μm thick) Li metal foil was used in these experiments. Galvanostatic deposition/stripping cycling was conducted at a current density of 0.8 mA cm−2 with a cut-off voltage at ±0.5 V (vs. Li/Li+). Fig. 3 shows the galvanostatic deposition/stripping curves for the Li symmetric cells with and without the PIL–POEM coating layer, measured in the mixed electrolyte. The cell containing the coated anode exhibited a relatively high overpotential for the Li deposition/stripping reactions, which indicates a sluggish interfacial reaction kinetics caused by the coating layer. Notably, after 130 cycles the electrode potential of the bare Li cell reached the cut-off voltage at a considerably faster rate compared to that of the coated Li cell. This indicates that the PIL–POEM coating on the Li metal resulted in a higher cycling stability of the cell.
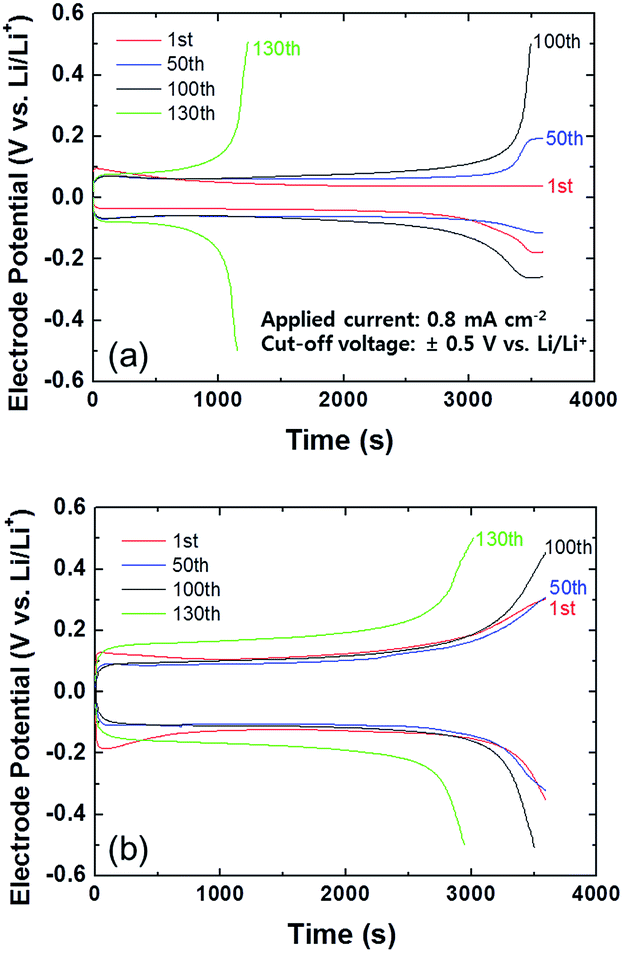 | ||
| Fig. 3 Charge/discharge voltage profiles for different Li symmetric cells at a constant current density of 0.8 mA cm−2. (a) Cell with bare Li metal anode; (b) cell with POEM–PIL-coated Li metal anode. | ||
Fig. 4 shows Nyquist plots of the impedance spectra measured after the first cycle for the Li symmetric cells both with and without the coating layer. All impedance spectra showed a depressed arc at high frequencies, which is related to the charge-transfer reactions at the Li metal/electrolyte interface.16 The values of the charge-transfer resistance, Rct, were quantitatively determined from complex non-linear least square (CNLS) fitting of the measured impedance spectra based on a simple resistance–capacitance (RC) circuit.17 Notably, the Rct value in Fig. 4(a) increased from 42.17 Ω cm2 for the bare Li metal cell to 224.4 Ω cm2 for the PIL–POEM-coated samples. Because a large polarization resistance favors a uniform distribution of nucleation and growth for Li deposition, the formation of Li dendrites can be suppressed by the coating PIL–POEM layer due to the relatively slow charge-transfer kinetics induced by this layer. After 130th cycling, the Rct value in both cases significantly decreased as compared to their initial values, probably due to the increased surface area resulting from the formation of Li dendrite.
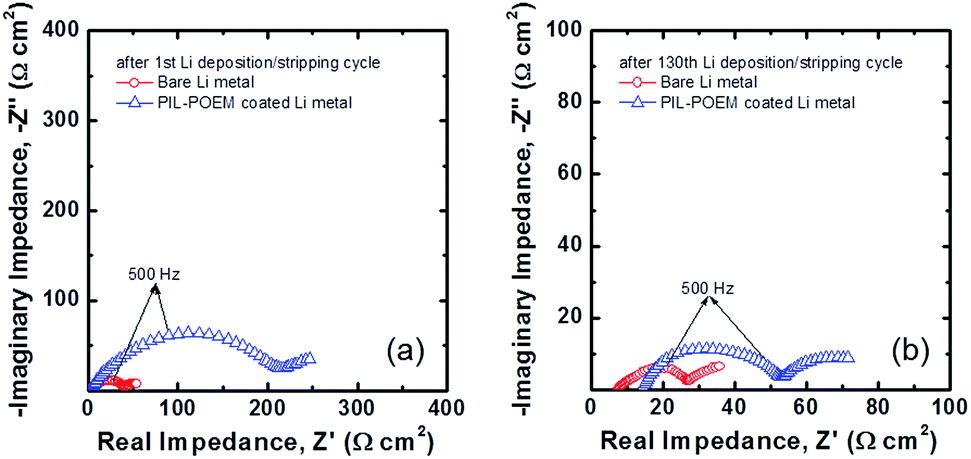 | ||
| Fig. 4 Nyquist plots of AC impedance spectra obtained from the bare Li and the POEM–PIL-coated Li symmetric cells (a) after the first deposition/stripping cycle and (b) after 130th cycling. | ||
The cycling performances of 2032 coin-type cells consisting of a high-loading LiCoO2 (LCO) cathode and the coated Li metal anode were assessed in the mixed electrolyte, and the results are shown in Fig. 5. The LCO electrode used in this measurement was fabricated from a mixture containing 97 wt% of the active material (Samsung SDI Co.) and 1.5 wt% of Denka black, which was added to a solution of N-methyl-2-pyrrolidene (NMP) containing 1.5 wt% polyvinylidene fluoride (PVDF). The resulting slurry was pasted onto an Al-foil current collector and dried at 120 °C for 2 h in a vacuum oven. After pressing, the dried paste was punched into a disc with a diameter of 1.2 cm. For reference, in addition to a cell containing the coated Li metal, an LCO/bare Li metal cell was also investigated.
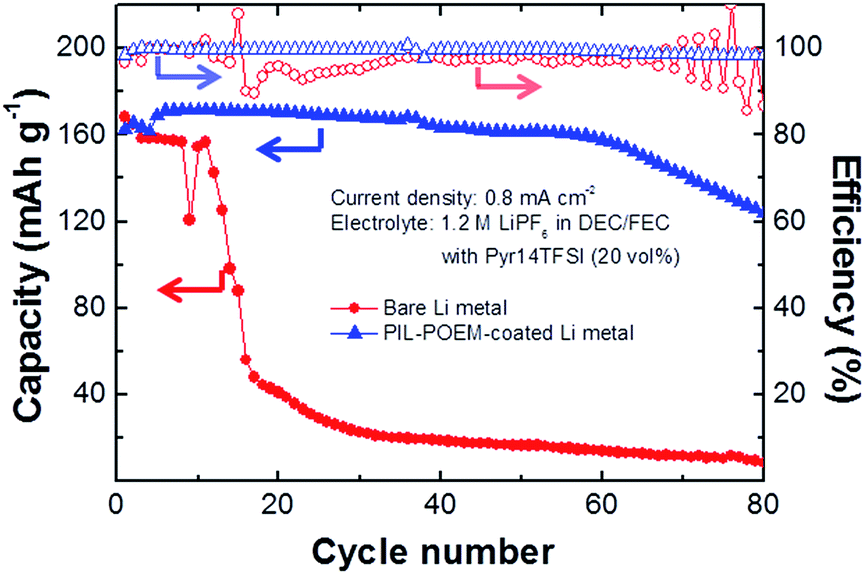 | ||
| Fig. 5 Cycling performance of coin-type cells. Discharging capacity and coulombic efficiency vs. cycle number in carbonate/Pyr14TFSI electrolyte for LCO/Li coin cells containing a bare Li metal anode and a PIL–POEM-coated Li metal anode. | ||
The cells were cycled at 25 °C by galvanostatically charging/discharging at potentials ranging from 3.0 to 4.3 V (vs. Li+/Li) with current densities of 0.8 mA cm−2. Fig. 5 shows the discharging capacities vs. cycle number measured for the cells containing the bare and coated Li metal anodes. In the case of the bare Li metal cell, the capacity drastically dropped after 20 cycles, and the retention was estimated to be below 0.5% after 80 cycles. Because LCO cathodes exhibit good electrochemical stability with ether and ionic liquid electrolytes,18,19 the poor performance observed for the bare Li metal cell is most likely associated with the loss of active Li metal due to side reactions with the electrolytes. In this cell, the carbonate solvent undergoes continuous decomposition at the Li metal interface at high charging/discharging cycle rates, resulting in the consumption of fresh Li metal.20–22 In addition, the cationic and anionic components of the RTILs are reduced at charged interfaces.12,13 Considering the oscillating potential pattern at the charged interfaces in RTIL-based electrolytes,23 the addition of RTILs to carbonate may cause an uneven electric field distribution at the Li metal surface. For the case of carbonate electrolyte without the addition of RTIL, in fact, a drastic capacity drop within 20 cycles was not observed even at higher current density, as shown in ESI Fig. 1.† Thus, addition of RTILs to carbonate solvents results in increased loss of Li.
The PIL–POEM-coated Li metal did not degrade significantly after 80 cycles: the capacity retention (75.7%) and coulombic efficiency (99.7%) of this cell were considerably higher than those of the cell containing a bare Li metal anode. This indicates that the coating layer effectively prevented the consumption of Li during cycling. The surface morphologies of the Li metal anodes of the cycled LCO/Li cells were characterized by SEM. The SEM images of the Li metal anodes with and without the PIL–POEM coating layer after 20 cycles, shown in Fig. 6, reveal rather different morphologies. Without the PIL–POEM coating, the interfacial reactions between the Li metal and the electrolyte during charging/discharging lead to a highly porous and dendritic morphology, shown in Fig. 6(a). In contrast, the SEM images for the system containing the coated Li metal show a relatively dense structure, indicating a significantly reduced dendrite formation on the coated Li metal anode during cycling.
 | ||
| Fig. 6 SEM images of the anode surfaces of LCO/Li coin cells after 20 cycles. (a) Bare Li metal anode; (b) PIL–POEM-coated Li metal anode. | ||
The chemical composition of the Li metal anode surface was characterized by XPS. In addition, the compositions at different depths were examined using Ar+ sputtering. Fig. 7 shows the XPS O 1s and N 1s spectra of the cycled Li metals with and without a PIL–POEM coating layer: decomposition products (Li2CO3, SOx) from Pyr14TFSI and DEC or FEC carbonate solvents were detected in both cases.
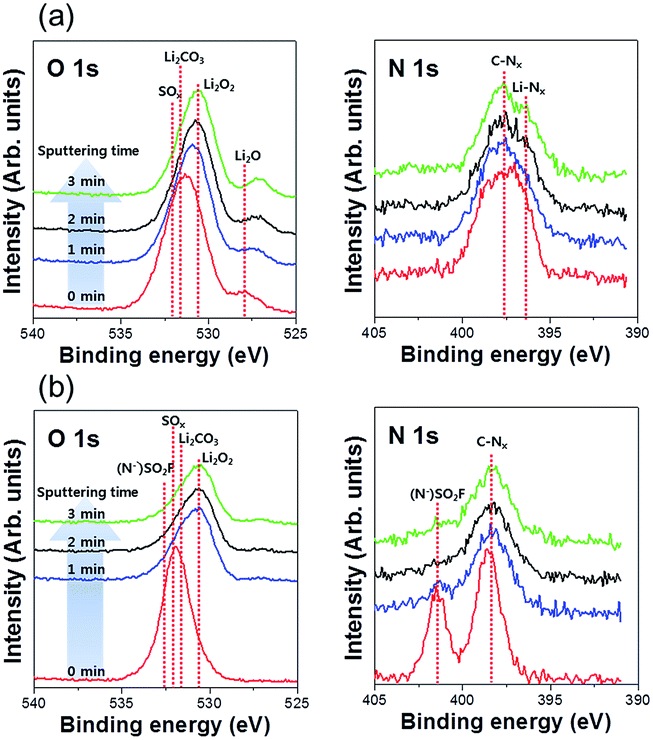 | ||
| Fig. 7 XPS O 1s and N 1s spectra obtained for LCO/Li coin cells after 20 cycles. (a) Cell containing bare Li metal anode; (b) cell containing PIL–POEM-coated Li metal anode. | ||
Notably, the (N−)SO2F peak originating from Pyr14TFSI is discernible only in the spectra of the systems containing the coated Li metal. The absence of the (N−)SO2F peak in the spectra of samples containing the uncoated Li metal (Fig. 7(a)) may be ascribed to the reduction of anions originating from Pyr14TFSI during the charging process. These results demonstrate that the PIL–POEM coating effectively suppresses the decomposition of the anionic and cationic components of the RTILs during the deposition/stripping of solvated Li complexes on Li metal. Because PIL and POEM exhibit good chemical/electrochemical compatibility with ionic liquids,24,25 we speculate that the coating layer can enhance the uniformity of the Li deposition/stripping reactions in RTIL-mixed electrolytes by preventing contact between the RTILs and the Li metal.
Conclusions
The work described in this study provides important insights on the poor cycling performance of cells with thin Li metal anodes in RTIL-containing carbonate electrolytes. Based on the XPS analysis, the rapid degradation of cells containing a Li metal anode can be attributed to the loss of excess Li metal caused by the reduction of the anionic components of the RTIL at the electrified Li metal interface. Although the identification of the exact mechanism by which the PIL–POEM coating layer affects the interfacial reactions during Li deposition/stripping represents a difficult task, the present results demonstrate that the coating layer effectively prevents the decomposition of electrolyte solvents at the Li metal interfaces. A coating layer thus represents a necessary requirement for achieving a high capacity retention in cells containing Li metal as the anode and RTIL-mixed carbonate solvents with low flammability as the electrolytes.Notes and references
- M. Armand, F. Endres, D. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS PubMed.
- A. Lewandowski and A. Świderska-Mocek, J. Power Sources, 2009, 194, 601 CrossRef CAS.
- G. T. Kim, S. S. Jeong, M. Z. Xue, A. Balducci, M. Winter, S. Passerini, F. Alessandrini and G. B. Appetecchi, J. Power Sources, 2012, 199, 239 CrossRef CAS.
- C. Arbizzani, G. Gabrielli and M. Mastragostino, J. Power Sources, 2011, 196, 4801 CrossRef CAS.
- L. Lombardo, S. Brutti, M. A. Navarra, S. Panero and P. Reale, J. Power Sources, 2013, 227, 8 CrossRef CAS.
- N. Wongittharom, T. C. Lee, C. H. Hsu, G. Ting-Kuo Fey, K. P. Huang and J. K. Chang, J. Power Sources, 2013, 240, 676 CrossRef CAS.
- M. Diaw, A. Chagnes, B. Carré, P. Willmann and D. Lemordant, J. Power Sources, 2005, 146, 682 CrossRef CAS.
- H. F. Xiang, B. Yin, H. Wang, H. W. Lin, X. W. Ge, S. Xie and C. H. Chen, Electrochim. Acta, 2010, 55, 5204 CrossRef CAS.
- G. H. Lane, A. S. Best, D. R. MacFarlane, M. Forsyth, P. M. Bayley and A. F. Hollenkamp, Electrochim. Acta, 2010, 55, 8947 CrossRef CAS.
- R. S. Kühnel, N. Böckenfeld, S. Passerini, M. Winter and A. Balducci, Electrochim. Acta, 2011, 56, 4092 CrossRef.
- P. C. Howlett, D. R. MacFarlane and A. F. Hollenkamp, Electrochem. Solid-State Lett., 2004, 7, A97 CrossRef CAS.
- P. C. Howlett, N. Brack, A. F. Hollenkamp, M. Forsyth and D. R. MacFarlane, J. Electrochem. Soc., 2006, 153, A595 CrossRef CAS.
- E. Markevich, R. Sharabi, V. Borgel, H. Gottlieb, G. Salitra, D. Aurbach, G. Semrau and M. A. Schmidt, Electrochim. Acta, 2010, 55, 2687 CrossRef CAS.
- R. Marcilla, F. Alcaide, H. Sardon, J. A. Pomposo, C. Pozo-Gonzalo and D. Mecerreyes, Electrochem. Commun., 2006, 8, 482 CrossRef CAS.
- A. L. Pont, R. Marcilla, I. De Meatza, H. Grande and D. Mecerreyes, J. Power Sources, 2009, 188, 558 CrossRef CAS.
- G. A. Umeda, E. Menke, M. Richard, K. L. Stamm, F. Wudl and B. Dunn, J. Mater. Chem., 2011, 21, 1593 RSC.
- J. R. Macdonald, Impedance Spectroscopy: Emphasizing Solid Materials and Systems, John Wiley & Sons, New York, 1987, p. 88 Search PubMed.
- A. Kraytsberg and Y. Ein-Eli, Adv. Energy Mater., 2012, 2, 922 CrossRef CAS.
- Z. Chen and J. R. Dahn, Electrochim. Acta, 2004, 49, 1079 CrossRef CAS.
- C. Wang, D. Wang and C. Dai, J. Electrochem. Soc., 2008, 155, A390 CrossRef CAS.
- H. Yoon, P. C. Howlett, A. S. Best, M. Forsyth and D. R. MacFarlane, J. Electrochem. Soc., 2013, 160, A1629 CrossRef CAS.
- D. Lv, Y. Shao, T. Lozano, W. D. Bennett, G. L. Graff, B. Polzin, J. Zhang, M. H. Engelhard, N. T. Saenz, W. A. Henderson, P. Bhattacharya, J. Liu and J. Xiao, Adv. Energy Mater., 2015, 5, 1402290 CrossRef.
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978 CrossRef CAS PubMed.
- Q. Hu, S. Osswald, R. Daniel, Y. Zhu, S. Wesel, L. Ortiz and D. R. Sadoway, J. Power Sources, 2011, 196, 5604 CrossRef CAS.
- Y. S. Ye, J. Rick and B. J. Hwang, J. Mater. Chem. A, 2013, 1, 2719 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03135a |
| This journal is © The Royal Society of Chemistry 2017 |