
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceYellow electrochromic polymer materials with fine tuning electrofluorescences by adjusting steric hindrance of side chains†
Jian Liu ,
Yuchen Shi,
Jingchuan Wu,
Mei Li,
Jianming Zheng and
Chunye Xu*
,
Yuchen Shi,
Jingchuan Wu,
Mei Li,
Jianming Zheng and
Chunye Xu*
Hefei National Laboratory for Physical Sciences at Microscale, CAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei 230026, P. R. China. E-mail: chunye@ustc.edu.cn; Tel: +86 551 63603459
First published on 11th May 2017
Abstract
A series of novel conducting conjugated yellow-to-transmissive electrochromic (EC) polymers were designed and synthesized to research their structure–property relationships, achieving electrofluorescent (EF) switching with applied external potential. These bifunctional materials are based on repeat units of bi-phenyl-1,3,4-oxadiazole and thiophene derivatives with different side chains. The polymers present a similar yellow color and emit various fluorescences with applied reduction potential, and they can turn to nearly transparent and non-fluorescent with oxidization potential. The optical contrast, electrochemical, electrochromic and electrofluorescent switching properties were also characterized in detail. A device containing these polymers was also fabricated that can achieve electrochromic and electrofluorescent switching simultaneously, making these polymers promising candidates for electro-bifunctional materials.
Introduction
Stimuli-responsive materials with optical change have gained much popularity in recent decades as they are promoted candidates for sensors, logical gates and military camouflage.1 Wherein, material for electrical stimuli attracted considerable research interest for display application, as the electrical stimuli can be applied rapidly and reversibly. Electrochromism and electrofluorescence are the most important parts in electrical stimuli material field.2–4 The material can change color or emission fluorescence quickly and reversibly in response to external potential so that they are widely used in smart windows, LED displays and mirrors.5–7From the aspect of electrochromic material, yellow-to-transmissive attaches importance to fulfill both of the subtractive primary color sets: cyan–magenta–yellow (CMY) and red–yellow–blue (RYB). A series of highly stable anodic yellow electrochromic polyamides with high contrast of optical transmittance change have synthesized by Guey-Sheng Liou's group.8 John R. Reynolds and his co-workers have also designed many yellow electrochromic polymers, which have excellent optical contrast and switching stability.9 These materials really provided strategies to explore variety of electrochromic materials, but presented either multicolored change or only simple electrochromic effect. In addition, Eunkyoung Kim's group have designed a series of π-conjugated polymer capable of electrofluorochromic switching and a device containing electrochromic polymer and fluorescent polymer to modulate color and fluorescence respectively.10,11 Above all, these reported materials are little to achieve the electrofluorescent and electrochromic effect synchronous by one material.
Thiophene derivatives with long side chains have been widely used in soluble electrochromic polymer materials, as they exhibited unique advantages such as tunable colors, facial processing procedure, high coloration efficiency and short response time.12–16 However, these thiophene derivatives polymers have strongly aggregation-caused quenching (ACQ) effect,17 which limits the fluorescent application in a state of aggregation, such as particles or film. In order to design the above mentioned bifunctional material, oxadiazole derivatives, excellent fluorescent functional molecule,18,19 was introduced in polymer backbone to act as electrofluorescent functional group. Additionally, color and emission fluorescence of conjugated polymers can be precisely tuned by controlling molecular structure. Some literatures have reported for tuning colors and emission fluorescences of polymers, such as M. Sassi et al. have reviewed the side chains functionalization approach to tune electrochromic colors and Atilla Cihaner successfully controlled the electrochromic colors by adjusting polymer backbone structure.20,21 Besides, Seogjae Seo' group have reported different electrofluorescent polymers with various fluorophores.22 It is observed that reports about fine tuning of color and fluorescence are really rare. As for solution-processable conducting polymer, the long side alkyl chains could bring obvious steric hindrance, and the resulted distortion of polymer backbone may cause the reduction of average conjugated length, further resulting change of color and fluorescence of functional material.23 Therefore, adjusting the steric hindrance by varying side chains is an effective way to control color and fluorescence of polymers. Hence similar compounds with different side chains were designed and synthesized to research for functional material with various colors and fluorescences.
In this article, combining fluorescent oxadiazole derivatives and electrochromic thiophene derivatives, a series of new conducting conjugated polymers with different side chains which can achieve bifunctions of electrochromic and electrofluorochromic switching were designed and synthesized, yielding insight for the future development of functional materials. The optical absorption, electrochemical, electrochromic and electrofluorescent properties of synthesized bifunctional polymer materials were characterized in detail as presented below. In addition, the device comprising these polymers was fabricated, and its electrochromic and electrofluorescent switching can be achieved synchronously by an applied external voltage.
Experimental section
Materials
All chemicals used in this paper were commercial products and used as received without further purification unless otherwise noted. Acetonitrile (ACN) and lithium perchlorate (LiClO4) were purchased from Sigma-Aldrich Chemical. HPLC tetrahydrofuran (THF) and N-methyl-2-pyrrolidone (NMP) were dried with 4A molecular sieves and stored in argon atmosphere for further use.Instruments
NMR spectra were measured by Bruker Avance AV400. The absorption spectra of films were measured with a UV-vis-NIR spectrophotometer (V-670; Jasco). Cyclic voltammetric (CV) measurements were carried out using CHI 660D electrochemical workstation in a three-electrode cell with working electrode (film coated on tin indium oxide (ITO) glasses, films area were approximately 0.7 cm × 3.0 cm), pseudo-reference electrode (Ag/Ag+), and counter electrode (platinum wire) in electrolyte of 0.1 M LiClO4/ACN. Spectroelectrochemical analysis was performed with electrolytic cells using 1 cm UV-cuvette, films (0.7 cm × 3.0 cm) on ITO glasses as working electrode, platinum wire as counter electrode, and silver wire as reference electrode. The fluorescent spectra and electrofluorescent behaviour of polymer film were characterized by OBIN YVON Fluorolog-3-TAV fluorescence spectrophotometer. The films were prepared by spraying chloroform solution of corresponding materials onto ITO glass. Photographs of the polymer films were collected with a Canon (IXUS 125 HS) digital camera.The synthesis of compound is as follows shown in Scheme 1:
 | ||
| Scheme 1 The synthesis route of compounds. | ||
Bromo-N′-(4-bromobenzoyl) benzohydrazide
4-Bromobenzoyl chloride and hydrazine monohydrate were mixed in THF at ice bath for 1 h, and then the reaction mixture was transferred to room temperature for another 3 h. The resulted precipitate was filtered, washed with saturated aqueous NaHCO3 solution and deionized water for three times, and dried to get white solid under vacuum condition. The crude product was directly used for next step without further purification.2,5-Bis(4-bromophenyl)-1,3,4-oxadiazole
The mixture of 4-bromo-N′-(4-bromobenzoyl)benzohydrazide and phosphorus oxychloride was refluxed for 6 h. After cooling to room temperature, the reaction mixture was poured slowly on ice. The resulted precipitate was filtered, washed with water, dried. Then the needle crystals were collected through recrystallization from ethanol. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.90 (d, 4H), 7.60 (d, 4H).General procedure of 1′–3′
3,4-Dimethoxythiophene (1 eq.), alkyl alcohol (3 eq.), p-toluene-sulfonic acid (0.1 eq.), and toluene were added in a three-necked flask equipped with Soxhlet extractor with 4A molecular sieves in cellulose thimble. The reaction mixture was refluxed for 24 h, after cooling to room temperature, the reaction solution was washed with deionized water and dried under vacuum. The resulted crude product was purified with column chromatography on silica gel with hexanes/methylene chloride (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
3,4-Dimethoxythiophene, dibromoneopentyl glycol, p-toluene-sulfonic acid, and toluene were added in three-necked flask equipped with Soxhlet extractor with 4A molecular sieves in cellulose thimble. The mixture was refluxed overnight, then it was cooled, and respectively extracted with saturated brine and anhydrous ether. The extracted solution was dried under vacuum, and crude product was purified by silica gel column chromatography, with petroleum ether/chloroform (10:1) as eluent, white powder was obtained as compound 4′′. 1H-NMR (400 MHz, CDCl3): δ (ppm) 6.49 (s, 2H), 4.12 (s, 4H), 3.65 (s, 4H).
A dried round-bottom flask filled with 100 mL of DMF, 12 mmol of 2-ethyl hexanol (4 eq.), and 18 mmol of NaH (6 eq.) was heated at 90 °C for 3 h. Then 3 mmol of compound 4′′ was added and continued to react for 24 h at 95 °C. After reaction, the reaction mixture was cooled and extracted with saturated brine and anhydrous ether twice. The organic phase was dried over magnesium sulfate, and the solvent was removed by rotary evaporation under reduced pressure. The obtained crude product was purified by column chromatography with petroleum to give colorless oil as compound 4′. 1H-NMR (400 MHz, CDCl3): δ (ppm) 6.45 (s, 2H), 4.0 (s, 4H), 3.47 (s, 4H), 3.28 (d, 4H), 1.2–1.6 (m, 20H), 0.8–1.0 (m, 12H).
General procedure of polymer 1′–4′
The compound was dissolved in a small amount of chloroform, then nitromethane solution dissolved with ferric chloride (5 eq.) was added. With the addition, the reaction solution gradually turned to green, the reaction was continued for 6 h, poured into large amount of methyl alcohol, filtered, and the filtrate was washed with Soxhlet extraction via methanol, hexane and chloroform respectively. The solvent was removed by rotary evaporation under reduced pressure to obtain the solid.General procedure of polymer 1–4
2,5-Bis(4-bromophenyl)-1,3,4-oxadiazole (1 eq.), thiophene derivative (1 eq.), potassium carbonate (2.6 eq.), palladium acetate (0.04 eq.), trimethylacetic acid (0.3 eq.) were added into dried two-necked flask in glove box. Then dried N-methyl-2-pyrrolidone (NMP) was injected to dissolve the mixture and the reaction mixture was heated to 120 °C for 1 h under agitation with N2 atmosphere. Upon cooling to the room temperature, the mixture was poured into methanol solution with vigorous stirring. The resulted precipitate was filtered through cellulose thimble and purified via Soxhlet extraction for 24 h with methanol and hexane respectively. The resulted solid was dissolved in chlorobenzene and heated to 60 °C. Diethyl dithiocarbamic acid dieth-ylammonium salt (Pd-scavenger, 4 eq. of Pd content) and 18-crown-6 were added and the solution was stirred for 4 h. The reaction mixture was cooled to room temperature, and precipitated into methanol. The precipitate was filtered, washed via Soxhlet extraction for 24 h with methanol and hexane respectively again and dried under vacuum overnight.Fabrication of device
A device (3.45 × 3.45 cm2) containing various polymers was fabricated, and its structural schematic diagram is shown in Fig. S11.† The polymer film sprayed onto the ITO glass was used as working electrode, NiO counter electrode was introduced in device, and electrolyte of LiClO4/ACN solution was bubbled with argon for 4 h and then injected into the device. The device was fabricated in a glove box under argon atmosphere in order to avoid any moisture and oxygen contamination. After gelation, the edges of device were sealed by UV-curable sealant.Results and discussion
Optical properties
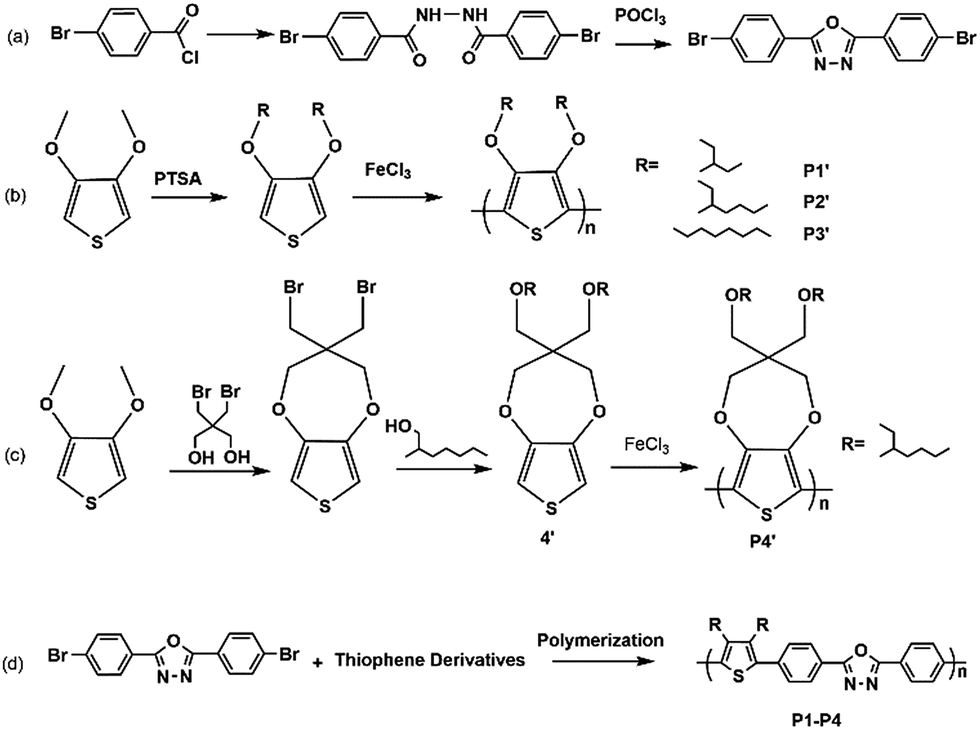
The polymer 1′–4′ were polymerized by oxidative polymerization, and color and absorbance spectra could be seen in Fig. 1(a), the polymer films present orange, yellow, red and purple respectively, correspondingly, the absorbance peak in spectra locates at the range of 450–600 nm. The diversity of absorbance spectra is what we based on to design the materials with fine tuning fluorescences (Fig. 1(b)).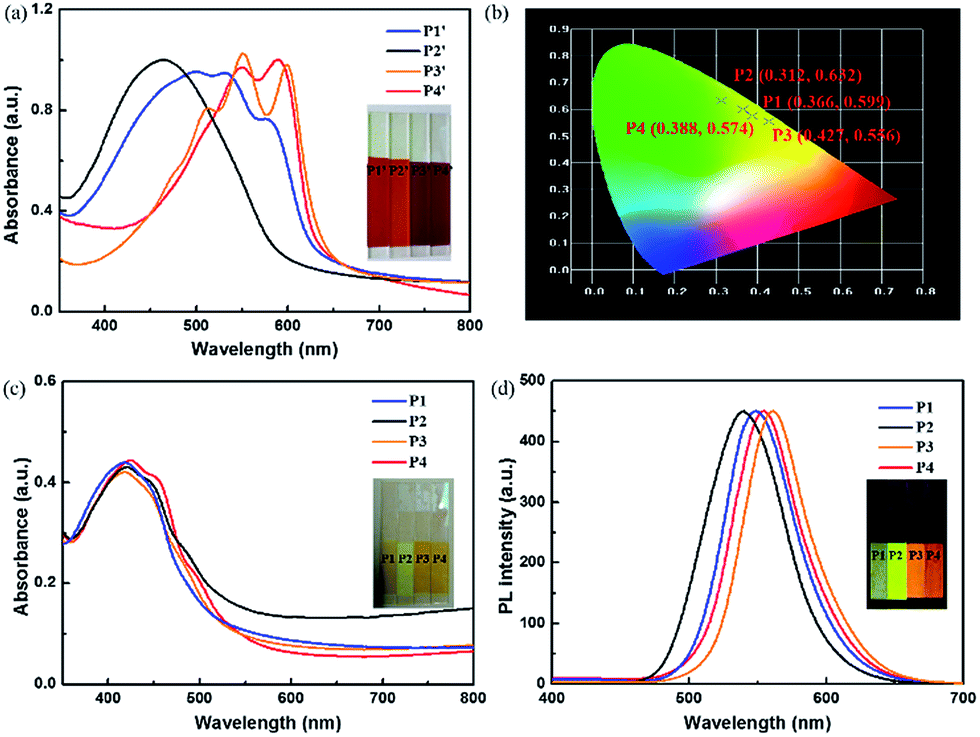 | ||
| Fig. 1 (a) The photos and absorbance spectra of the polymer 1′–4′ film. (b) CIE 1931 coordinates the fluorescence emission of different polymers. (c) The photos and absorbance spectra of polymers; (d) the photos and PL emission of polymers. | ||
The photos and spectra of polymer 1–4 films were displayed in Fig. 1(c) and (d). With introduction of oxadiazol derivative, all the films show a similar yellow color with the corresponding wavelength around 440 nm, as the higher electron delocalization energy of backbone benzene and oxadiazole than thiophene could cause a blue shift to yellow color region.24,25 As for PL emission spectra, polymer films show a slight change from yellow to orange bright fluorescence due to introducing excellent fluorescent group of oxadiazole derivative. The variety of fluorescences may be ascribed to steric hindrance of different side chains. As steric hindrance of branch chains is bigger than linear chain, P3 has a red shift compared with other three polymers. However, the emission peak of P4 owning branch chain occurs a red shift compared to that of P1 and P2. This may be caused by the ring attached to backbone structure, which can reduce the winding between side chains, further enhancing the conjugated degree of polymer backbone. Therefore, gradual red shift of emission spectra was observed with reduction of steric hindrance, resulting an obvious visible change from yellow to orange fluorescence under irradiation of 365 nm light. The trend is as follows: P2 < P1 < P4 < P3. The Commission Internationale de L'Eclairage (CIE) coordinates of various polymers are calculated to be (0.366, 0.599), (0.312, 0.632), (0.427, 0.556) and (0.388, 0.575) as shown in Fig. 1(b).
Electrochemical properties
To obtain a deep understanding of electrochemical behaviours of these polymers, the electrochemistry of polymers in 0.1 M LiClO4/ACN was explored by cyclic voltammetry (CV). The onset oxidization voltages (Eonset) of P1–P4 films were 1.08 V, 1.14 V, 1.04 V and 1.06 V respectively (Fig. 2), which were ascribed to the influence of different steric hindrances analyzed above. Obviously, weaker steric hindrance could induce conjugated length of polymers, further reducing oxidization voltage. This is also consistent with the optical results of polymers.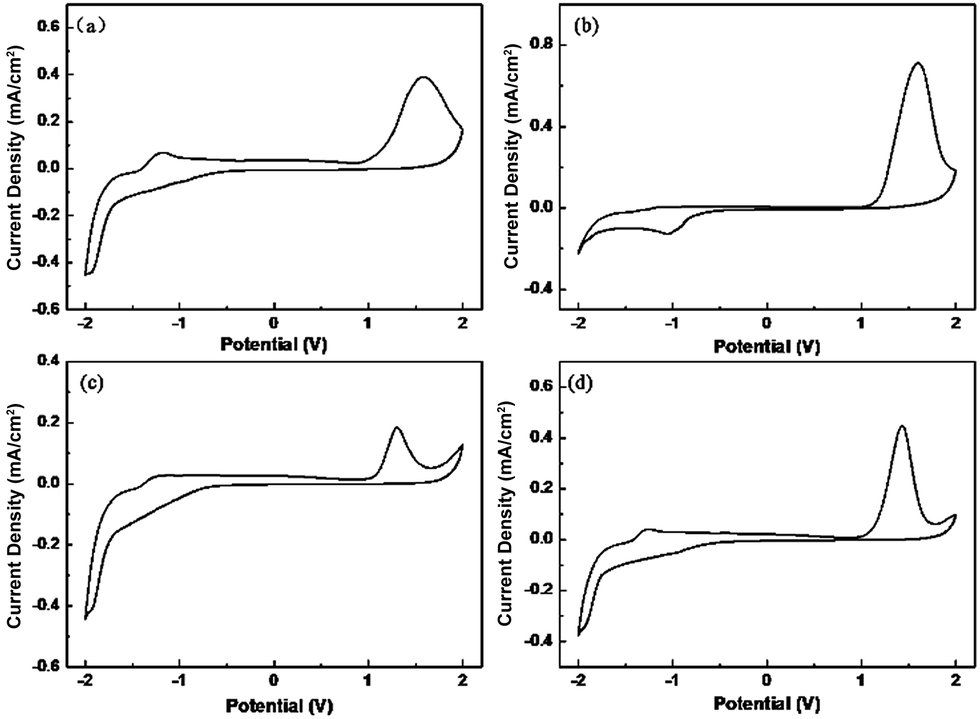 | ||
| Fig. 2 The CV curves of the P1 (a), P2 (b), P3 (c) and P4 (d) films in electrolyte of 0.1 M LiClO4/ACN. | ||
Spectroelectrochemistry
The electrochromic properties of polymer films were evaluated by spectroelectrochemistry. The changes in color and optical properties in details would be observed when different potentials were applied to films. Optical change, typical electrochromic absorbance spectra and photos of color change were displayed in Fig. 3. With increased applied potential, the absorption in wavelength of about 440 nm decreases indicating the formation of radical cations (polarons) and further dications (bipolarons) at the expense π–π* transition in a higher doping state.26,27 Additionally, all the polymers show similar yellow color in oxidization state and present approximate transparent color in reduction state.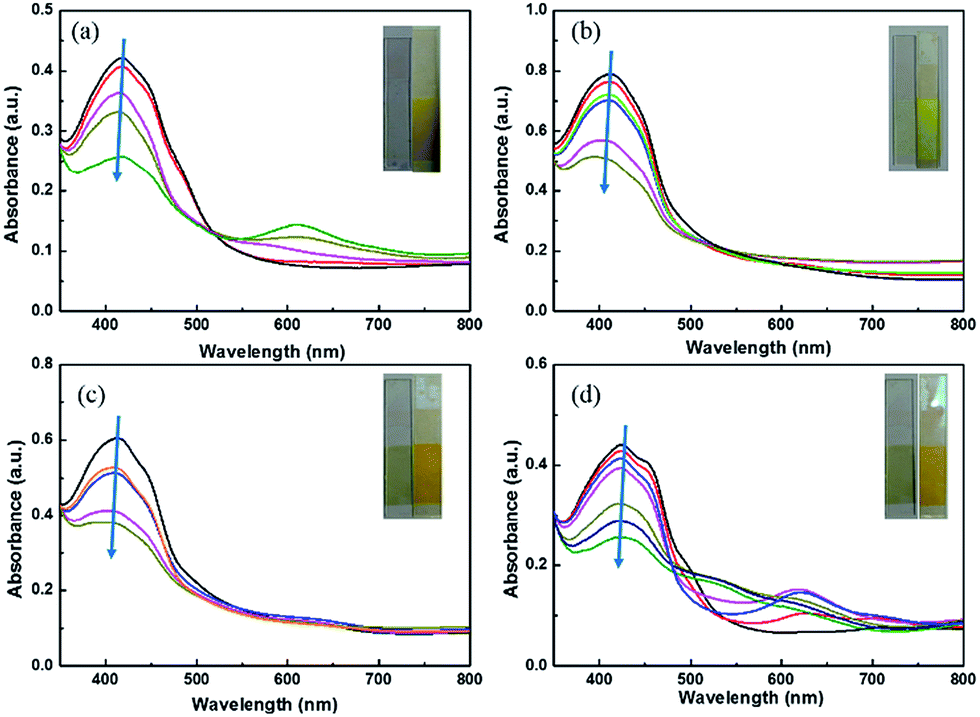 | ||
| Fig. 3 Spectroelectrochemistry of P1 (a), P2 (b), P3 (c) and P4 (d) films coated on ITO substrate from chloroform (2 mg mL−1) solution at different applied potentials between −1.6 V and 1.8 V, the potential increases as the direction of arrows. | ||
Electrochromic switching property
Transmittance change, a function of time at the maximum absorption wavelength achieving the largest change between reduction and oxidized state, and the switching time, the time needed to achieve 95% of transmittance changes, are crucial factors for electrochromic application. As shown in Fig. 4, P1–P4 films reveal transmittance change (ΔTmax) of 30%, 35%, 31% and 32% at the maximum absorption wavelength respectively, and all the polymer films possess similar switching time of 2 to 3.2 s.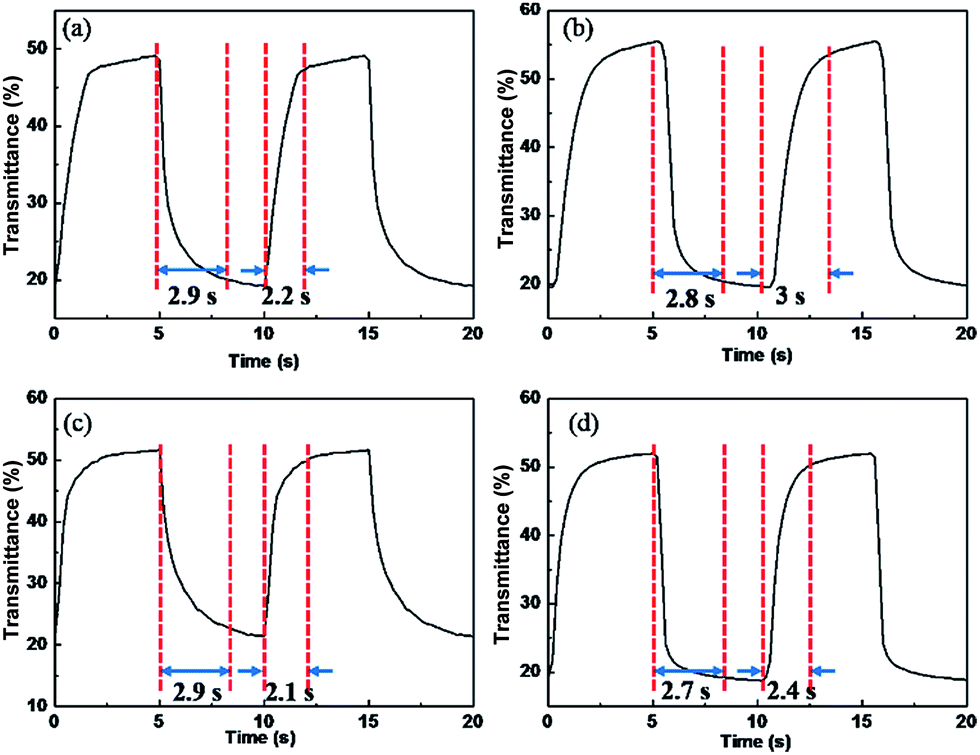 | ||
| Fig. 4 Electrochromic switching optical transmittance change of polymer films monitored at their λmax in electrolyte of 0.1 M LiClO4/ACN. (a) P1 at 416 nm, (b) P2 at 412 nm, (c) P3 at 418 nm and (d) P4 at 425 nm. | ||
Above all, typical electrochromic parameters such as maximum absorption wavelength, transmittance change and response time were displayed in Table 1.
Electrofluorescent property
The polymer films emit fluorescent colors by UV excitation and electrofluorescent switching properties of polymer films were characterized by monitoring their photoluminescent properties with different applied external potentials in a three-electrodes cell with electrochemical working station and fluorescence spectrophotometer combinedly (Fig. 5 and 6). All the polymers show similar electrofluorescent switching properties with different potentials. Taking P1 film for example, upon applying potential from −1.6 V to 1.8 V, the bright fluorescence was extinguished to nearly dark gradually. The fluorescence could reversibly return to its original state when applying potential of −1.6 V. Additionally, there is no obvious shift of emission peak (545 nm) of polymer film in potential change process, indicating that the quenching of fluorescence of polymer film originated from the produced cation radical in polymer backbone during redox process, rather than the production of side product.28,29 Furthermore, P1 film reveals a quenching time of 2.4 s and a recovery time of 2.3 s, which matches with the electrochromic response time. Other electrofluorescent properties of polymer films were depicted in Table 2.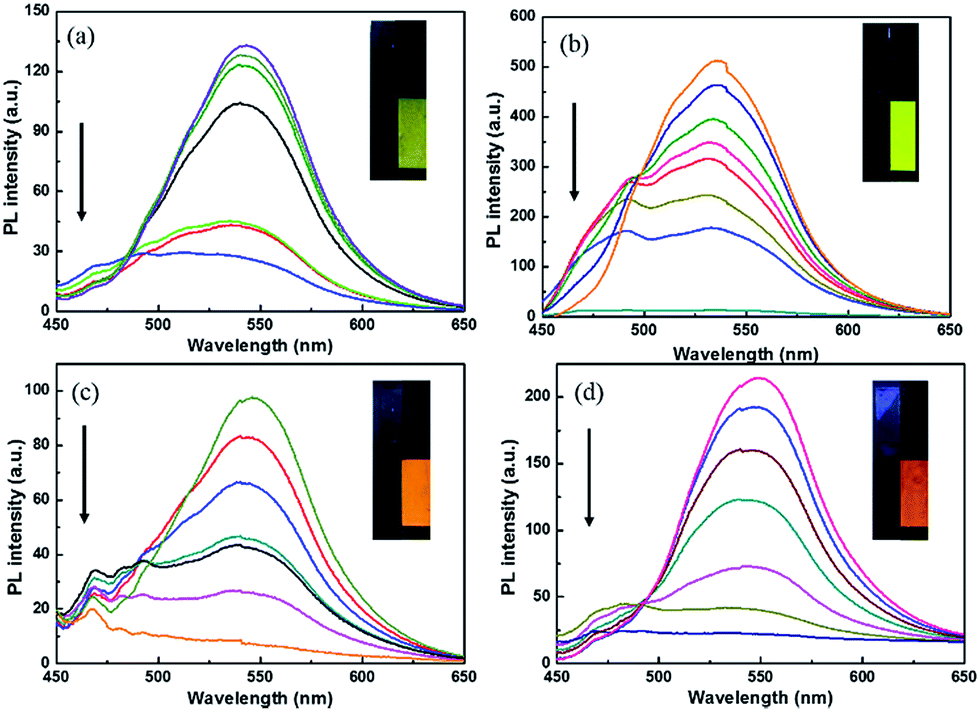 | ||
| Fig. 5 Electrofluorescent behaviours of polymer film between −1.6 V and 1.8 V and the insert is the photo of film in bleached state (left) and colored state (right). (a) P1; (b) P2; (c) P3; (d) P4. The potential increases as the direction of arrows. | ||
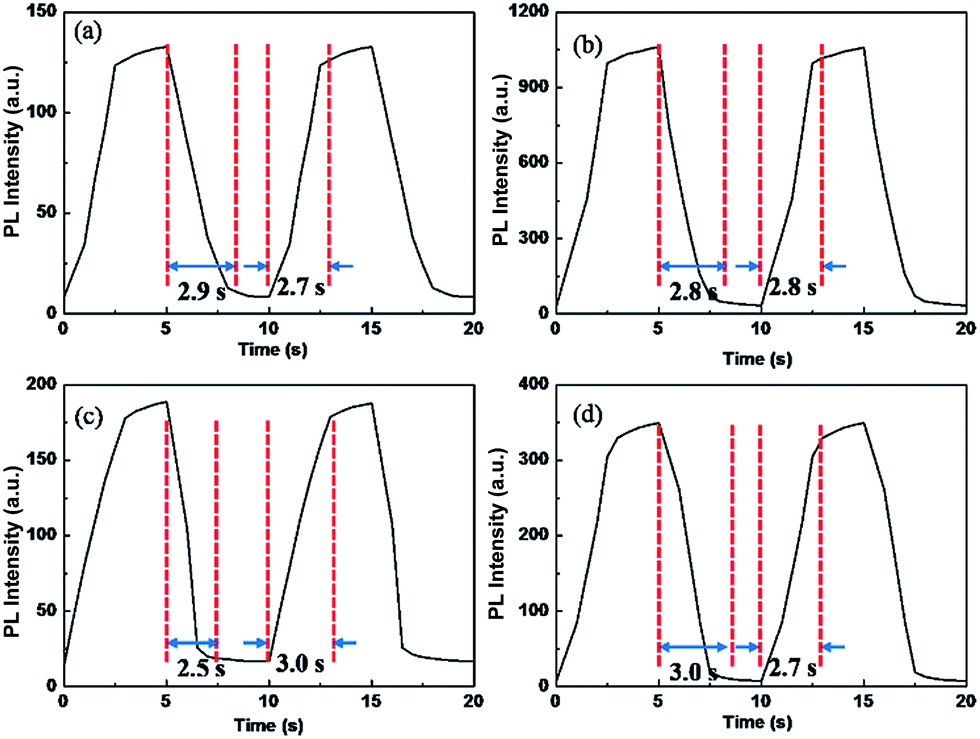 | ||
| Fig. 6 Electrofluorescent switching PL intensity of polymer films monitored at their λmax in electrolyte of 0.1 M LiClO4/ACN. (a) P1 at 547 nm, (b) P2 at 539 nm, (c) P3 at 561 nm and (d) P4 at 554 nm. | ||
| Polymer | λ (nm) | Quenching time (s) | Recovery time (s) |
|---|---|---|---|
| P1 | 545 | 2.9 | 2.7 |
| P2 | 539 | 2.8 | 2.8 |
| P3 | 561 | 2.5 | 3.0 |
| P4 | 554 | 3.0 | 2.7 |
Bifunctional effect of fabricated devices
The photos of devices containing different polymers were displayed in Fig. 7. Applying an external potential, the device turns to approximate transparent state, and the fluorescence of device was extinguished to nearly dark. The vanish of fluorescence and color is nearly synchronous, which may be caused by cation radical produced by applied external potential. It can decrease the absorption in visible region and also acts as strongly effective fluorescence quencher.22,30 With reverse potential, the color and fluorescence can return to its original state. After dozen cycles, the device couldn't work to achieve its bifunctional effect, similar devices with long service time are in research in our group. In addition, polymers show different fluorescences in one device, which provides a new way to design material with various fluorescences.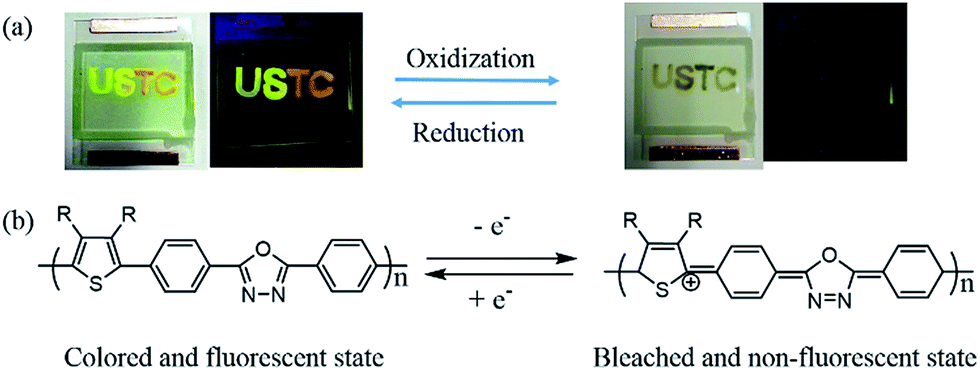 | ||
| Fig. 7 (a) The images of fabricated device of different doping state under sunlight and UV irradiation. (b) Electrochromic and electrofluorescent switching arise from reversible redox reactions in device. | ||
Conclusion
A series of novel conducting conjugated yellow-to-transmissive electrochromic and electrofluorescent polymers were designed and synthesized to investigate the structure–property relationship. With side chains of slight different steric hindrances, the polymers present similar yellow color and emit fining tuned fluorescence with applied reduction potential, and they can turn to nearly transparent state and non-fluorescent state with oxidization potential. The device containing various polymers was also fabricated which can achieve electrochromic and electrofluorescent switching simultaneously, making these polymers be promising candidate for electro-bifunctional material.Acknowledgements
We gratefully acknowledge supports of this work by the National Natural Science Foundation of China (51503193) and the Chinese Academy of Sciences (Integrated system of high efficiency building energy saving and its application, KFZD-SW-403).References
- Y.-K. Yang, K.-J. Yook and J. Tae, J. Am. Chem. Soc., 2005, 127, 16760–16761 CrossRef CAS PubMed.
- Z. Shi, W. T. Neo, T. T. Lin, H. Zhou and J. Xu, RSC Adv., 2015, 5, 96328–96335 RSC.
- S. Seo, Y. Kim, Q. Zhou, G. Clavier, P. Audebert and E. Kim, Adv. Funct. Mater., 2012, 22, 3556–3561 CrossRef CAS.
- J. Wu, C. Peng, H. Xiao, S. Bo, L. Qiu, Z. Zhen and X. Liu, Dyes Pigm., 2014, 104, 15–23 CrossRef CAS.
- P. Andersson Ersman, J. Kawahara and M. Berggren, Org. Electron., 2013, 14, 3371–3378 CrossRef CAS.
- S. V. Vasilyeva, P. M. Beaujuge, S. Wang, J. E. Babiarz, V. W. Ballarotto and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2011, 3, 1022–1032 CAS.
- C. Xu, L. Liu, S. E. Legenski, D. Ning and M. Taya, J. Mater. Res., 2011, 19, 2072–2080 CrossRef.
- G.-S. Liou and H.-Y. Lin, Macromolecules, 2009, 42, 125–134 CrossRef CAS.
- C. M. Amb, J. A. Kerszulis, E. J. Thompson, A. L. Dyer and J. R. Reynolds, Polym. Chem., 2011, 2, 812 RSC.
- Y. Kim, J. Kim, J. You and E. Kim, Mol. Cryst. Liq. Cryst., 2011, 538, 39–44 CrossRef CAS.
- S. Seo, Y. Kim, J. You, B. D. Sarwade, P. P. Wadgaonkar, S. K. Menon, A. S. More and E. Kim, Macromol. Rapid Commun., 2011, 32, 637–643 CrossRef CAS PubMed.
- M. Akbayrak and A. M. Önal, Polym. Chem., 2016, 7, 6110–6119 RSC.
- S. Govaerts, P. Verstappen, H. Penxten, M. Defour, B. Van Mele, L. Lutsen, D. Vanderzande and W. Maes, Macromolecules, 2016, 49, 6411–6419 CrossRef CAS.
- S. Ming, S. Zhen, X. Liu, K. Lin, H. Liu, Y. Zhao, B. Lu and J. Xu, Polym. Chem., 2015, 6, 8248–8258 RSC.
- Z. Xu, W. Wang, J. Wu, S. Mi, J. Zheng and C. Xu, New J. Chem., 2016, 40, 5231–5237 RSC.
- M. A. Invernale, J. G. Bokria, M. Ombaba, K.-R. Lee, D. M. D. Mamangun and G. A. Sotzing, Polymer, 2010, 51, 378–382 CrossRef CAS.
- G. He, W. Torres Delgado, D. J. Schatz, C. Merten, A. Mohammadpour, L. Mayr, M. J. Ferguson, R. McDonald, A. Brown, K. Shankar and E. Rivard, Angew. Chem., Int. Ed., 2014, 53, 4587–4591 CrossRef CAS PubMed.
- A. Baranczak, Y. Liu, S. Connelly, W. G. Du, E. R. Greiner, J. C. Genereux, R. L. Wiseman, Y. S. Eisele, N. C. Bradbury, J. Dong, L. Noodleman, K. B. Sharpless, I. A. Wilson, S. E. Encalada and J. W. Kelly, J. Am. Chem. Soc., 2015, 137, 7404–7414 CrossRef CAS PubMed.
- A. Paun, N. D. Hadade, C. C. Paraschivescu and M. Matache, J. Mater. Chem. C, 2016, 4, 8596–8610 RSC.
- L. Beverina, G. A. Pagani and M. Sassi, Chem. Commun., 2014, 50, 5413–5430 RSC.
- M. İçli, M. Pamuk, F. Algi, A. M. Önal and A. Cihaner, Chem. Mater., 2010, 22, 4034–4044 CrossRef.
- S. Seo, H. Shin, C. Park, H. Lim and E. Kim, Macromol. Res., 2013, 21, 284–289 CrossRef CAS.
- Z. Xu, X. Chen, S. Mi, J. Zheng and C. Xu, Org. Electron., 2015, 26, 129–136 CrossRef CAS.
- J. Wu, H. Xiao, L. Qiu, Z. Zhen, X. Liu and S. Bo, RSC Adv., 2014, 4, 49737–49744 RSC.
- J. A. Kerszulis, C. M. Amb, A. L. Dyer and J. R. Reynolds, Macromolecules, 2014, 47, 5462–5469 CrossRef CAS.
- J. Liu, S. Mi, Z. Xu, J. Wu, J. Zheng and C. Xu, Org. Electron., 2016, 37, 169–177 CrossRef.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS PubMed.
- J.-H. Wu and G.-S. Liou, Adv. Funct. Mater., 2014, 24, 6422–6429 CrossRef CAS.
- X. Yang, S. Seo, C. Park and E. Kim, Macromolecules, 2014, 47, 7043–7051 CrossRef CAS.
- S. Mi, J. Wu, J. Liu, Z. Xu, X. Wu, G. Luo, J. Zheng and C. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 27511–27517 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of target polymer and structural schematic diagram of device. See DOI: 10.1039/c7ra03097e |
| This journal is © The Royal Society of Chemistry 2017 |