
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polydopamine nanotubes-templated synthesis of TiO2 and its photocatalytic performance under visible light†
Zehuan Wang,
Jia Li,
Feng Tang,
Jun Lin and
Zhaoxia Jin *
*
Department of Chemistry, Renmin University of China, Beijing 100872, People's Republic of China. E-mail: jinzx@ruc.edu.cn
First published on 28th April 2017
Abstract
Herein, we demonstrated that an efficient TiO2 visible-light photocatalyst can be fabricated by templating mesoporous polydopamine (PDA) nanotubes. Because PDA is a nitrogen-doped carbon source, PDA nanotubes as templates elicited distinct advantages for obtained mesoporous TiO2. First, they introduced a codopant of carbon and nitrogen into anatase TiO2 that narrowed its band gap. Second, they could be graphitized in an optimized calcination process to form graphene-like carbon layers on TiO2 surfaces with high conductivity, which assisted the separation of photogenerated electron–holes of TiO2. These features endow TiO2@C-PDA (400-2) excellent photocatalytic performance for the degradation of methyl orange under visible-light irradiation (>420 nm).
1. Introduction
Porous scaffolds can provide catalysts with high accessibility and high diffusion rates of reactant and product. Also, catalysts can be stabilized through encapsulation in hollow nanostructures. Therefore, porous nanostructures have important applications as scaffolds for bio-, electro- and photocatalysts.1–5 Porous carbon nanostructures as catalyst carriers have attracted considerable attention because of their good thermal and chemical stability, low cost, large surface area, and versatile fabrication methods. In particular, carbon scaffolds can improve catalyst properties significantly. For example, hybrid TiO2@carbon has demonstrated promising photocatalytic ability under visible-light irradiation due to a narrowed band gap and accelerated charge transfer in hybrids.6–13 The band gap of TiO2 can be narrowed further by introducing a codopant for the TiO2 lattice (e.g. carbon, nitrogen-codoped TiO2). Thus, carbon nanomaterials doped with heteroatoms show superior features as scaffolds.14–16 Besides the doping effects mentioned above, a carbon scaffold can also modulate the structure and morphology of TiO2 and further influence its photocatalytic efficiency. Recent studies have shown that a highly conductive carbon layer on TiO2 is beneficial for fast electron transfer in a photocatalytic process of TiO2@carbon.15,17Polydopamine (PDA) is a promising precursor for nitrogen-doped carbon nanomaterials.18–20 The reactivity of functional groups on PDA gives it high absorbency of metal ions, which can be transformed further to useful metal oxides or metal nanoparticles depending on the conditions of carbonization or calcination. Previously, we reported successful fabrication of tubular PDA nanostructures,21 which could be templates for the production of various catalysts. Because tubular TiO2 has demonstrated very high efficiency as flow-through photocatalytic membranes,22 and has shown efficient solar water splitting after doping with carbon,7 we investigated the fabrication and properties of tubular TiO2 via templating PDA nanotubes (TiO2@C-PDA). Using it as a proof of concept, we hoped to reveal what effects PDA templates imparted for TiO2 as a photocatalyst. We observed that the photocatalytic ability of TiO2 could be improved significantly via scaffolding of PDA nanotubes. The latter, as TiO2-scaffolds, demonstrated versatile advantages through integration of morphological and structural control, multidoping and easily adjustable graphite content in the carbon layer.
2. Experimental sections
2.1 Materials
Dopamine hydrochloride (purity 98%) was obtained from Sigma-Aldrich. Analytical curcumin, ethanol, hydrochloric acid (12 M), terephthalic acid and titanium(IV) isopropoxide (TTIP) were purchased from Sinopharm Chemical Reagent Beijing Co. Ltd. Tris(hydroxymethyl) aminomethane (Tris)–HCl buffer (1.5 M, pH 8.8) was obtained from Shanghai Double-Helix Biotech. Co. Ltd. All chemicals were used without further purification. Deionized water (Millipore Q >18 MΩ cm at 25 °C) was used to prepare aqueous solutions or as non-solvent.2.2 Preparation of PDA nanotubes
According to our previous report,21 100 mg curcumin and 500 mg dopamine hydrochloride were dispersed in 100 mL ethanol under stirring, and then 400 mL deionized water was added. When precipitation was complete, 3 mL (Tris)–HCl buffer solution (1.5 M, pH 8.8) was added in this suspension. After gentle stirring for 24 h, the sediment was separated and cleaned by fresh water thrice, and collected by sedimentation or centrifugation. Then, the sediment was soaked in ethanol to dissolve curcumin crystals. When curcumin crystals were removed completely, the sediment was washed by deionized water and freeze-dried to obtain PDA nanotubes.2.3 Synthesis of TiO2@C-PDA samples
Titanium(IV) isopropoxide (0.1 g) was mixed with HCl (0.3 g, 12 M) under vigorous stirring for 5 min. Then, PDA nanotubes (0.1 g) were immersed in this mixture under stirring. Intensive stirring for 10 min enabled full absorption of titanium sol by PDA nanotubes. Then, the “mud-like” mixture was aged in a closed container at 70 °C for 24 h to further hydrolyze TTIP. The aged product was Ti(IV)–PDA gel. To carbonize PDA and crystallize TiO2, Ti(IV)–PDA gel was heated further in a tube furnace from room temperature to 500 °C at a heating rate of 5 °C min−1 under a continuous flow of argon (flow rate: 50 mL min−1), maintained at 500 °C for 3 h, and cooled to room temperature. The obtained product was named TiO2@PDA. Then, in air, the TiO2@PDA powder was heated in a muffle furnace from room temperature to 400 °C under a heating rate of 5 °C min−1 and maintained at 400 °C for 1, 2 and 3 h, respectively. Calcinated products were ground before further characterizations and experiments. Obtained samples were denoted as TiO2@C-PDA (400-1), TiO2@C-PDA (400-2) and TiO2@C-PDA (400-3) based on the temperature and duration of calcination. Besides, pristine TiO2 was prepared by directly heating the HCl-hydrolyzed TTIP gel at 500 °C for 3 h in a muffle furnace in air.2.4 Characterization and measurements
Morphological characterizations were conducted through scanning electron microscopy (SEM; SU8010, Hitachi) at 15 kV equipped with an energy-dispersive spectroscopy (EDS) detector. Transmission electron microscopy (TEM) was operated on an H-7650B device (Hitachi) at an accelerating voltage of 80 kV. High-resolution transmission electron microscopy (HRTEM) images were obtained on a FEI G2 F20 microscope (Tecnai) with an accelerating voltage of 200 kV.The chemical states and species of elements were verified using an X-ray photoelectron spectrometer (Axis Ultra; Kratos Analytical) with monochromatic Al Kα (hν = 1486.7 eV) radiation as the excitation and at an X-ray power of 150 W. All spectra were calibrated using the C 1s peak (284.7 eV). Fourier transform infrared (FTIR) spectroscopy was done using a Tensor 27 FTIR spectrophotometer (Bruker). Crystallization characterizations of calcinated products were performed with a XRD-7000 diffractometer (Shimadzu) in reflection mode using a Cu target (λ = 0.15418 nm) as the incident X-ray. Accelerating voltage of 40 kV, current of 40 mA, and 2θ-step of 0.013° were selected. The scan was collected between 20° and 80°. Raman spectra were acquired by a Raman spectrometer (XploRA PLUS; Horiba Scientific). The spot size of a 532 nm incident laser for signal collection was about 25 ×25 μm. Spectra were calibrated using the 520 cm−1 line of a silicon wafer, and the laser beam was focused by an optical microscope with a 10× objective lens, 1200 grooves per mm grating, and 100 μm slit. UV-visible absorption spectra of TiO2 and TiO2@C-PDA samples were collected using a UV-Vis spectrophotometer (U-3900; Hitachi) equipped with a diffuse reflectance accessory, and BaSO4 powder was used as the reflectance standard. Zeta potential values of TiO2@C-PDA in aqueous solution at different pH values were measured using a Zetasizer Nano ZS90 (Malvern) at 25 °C. Measurements were performed five times for each sample. Thermogravimetric analysis (TGA) was carried out on a TGA Q50 (TA Instruments) system from room temperature to 800 °C under air at a heating rate of 10 °C min−1. Brunauer–Emmett–Teller (BET) surface area was determined from N2 adsorption isotherm data collected at 77 K using a Belsorp-Mini II (MicrotracBEL) system. Before testing, ≈100 mg samples were dried under a N2 atmosphere at 300 °C for ≥12 h. The specific surface area was calculated using the BET method, and the pore volume and pore-size distribution were obtained using the Barrett–Joyner–Halenda (BJH) model.
2.5 Photocatalytic activity tests
The photocatalytic activity of various samples on degradation of methyl orange (MO) aqueous solution was investigated. A 300 W Xe arc lamp (CHFXM150, Beijing Trust Tech) equipped with a wavelength cutoff filter for λ > 420 nm and positioned ≈8 cm above the aqueous suspension. Briefly, 100 mg of sample was dispersed in 100 mL MO aqueous solution at 10 mg L−1. The solution was stirred for 1 h to ensure establishment of an adsorption–desorption equilibrium between TiO2 and MO in suspension before irradiation. At a given interval of 30 min, 3 mL of the suspension was sampled, and then the catalyst and MO solution were separated by centrifugation. The concentration of MO in the sample was determined by monitoring absorbance at 461 nm in a UV absorption spectrum with a UV-3600 spectrophotometer (Shimadzu).Photoluminescence (PL) spectra of TiO2@C-PDA aqueous suspensions were measured at room temperature using a fluorescence spectrophotometer (F-4600; Hitachi). The excitation wavelength was 420 nm.
To test the amount of hydroxyl radical (˙OH) generated over TiO2@C-PDA photocatalysts under visible-light irradiation, 40 mg of tested samples was suspended in 80 mL of aqueous solution containing NaOH (0.01 M) and terephthalic acid (3 mM).23 Before exposure to visible-light irradiation, the solution was stirred for 1 h in the dark to ensure establishment of an adsorption–desorption equilibrium. Then, the mixture was illuminated with a 300 W Xe arc lamp (CHFXM150, Beijing Trust Tech) equipped with a wavelength cut-off filter of λ > 420 nm. During illumination, at 30 min intervals, 3 mL of the mixture was removed and centrifuged. The fluorescence spectra of obtained supernatants were recorded at room temperature using a fluorescence spectrophotometer (F-4600; Hitachi) with a Xe lamp as the excitation source. The intensity of the emission peak at 426 nm of 2-hydroxyterepthalic acid (excitation, 315 nm) was used to evaluate the ability of photocatalysts to produce ˙OH radicals under light irradiation.
3. Results and discussion
3.1 Morphology and phase structure
Fig. 1 demonstrates images of original PDA nanotubes (Fig. 1a and d) as well as TiO2@C-PDA nanotubes before (Fig. 1b and e) and after calcination (Fig. 1c and f). The tubular morphology of original PDA nanotubes was maintained in TiO2@C-PDA even after calcination in air for several hours (Fig. 1c and f), whereas the carbon skeleton was almost “burned out”, leaving TiO2 as the main component in nanotubes. The average length of original PDA nanotubes was more than several tens of micrometers, but it decreased to several micrometers in hybrids after calcination. The average diameters of original PDA nanotubes, TiO2@PDA nanotubes and TiO2@C-PDA nanotubes were 465 ± 40, 366 ± 57, and 210 ± 30 nm, respectively. The thicknesses of tube walls for these PDA, TiO2@PDA and TiO2@C-PDA nanotubes were 107 ± 19, 99 ± 17, and 56 ± 12 nm, respectively, suggesting considerable shrinkage of TiO2@PDA after calcination in air due to TiO2 crystallization and burning out of carbon scaffolds. Element mapping of TiO2@C-PDA showed the smooth distribution of carbon, nitrogen on a single TiO2 nanotube (Fig. 1g–j).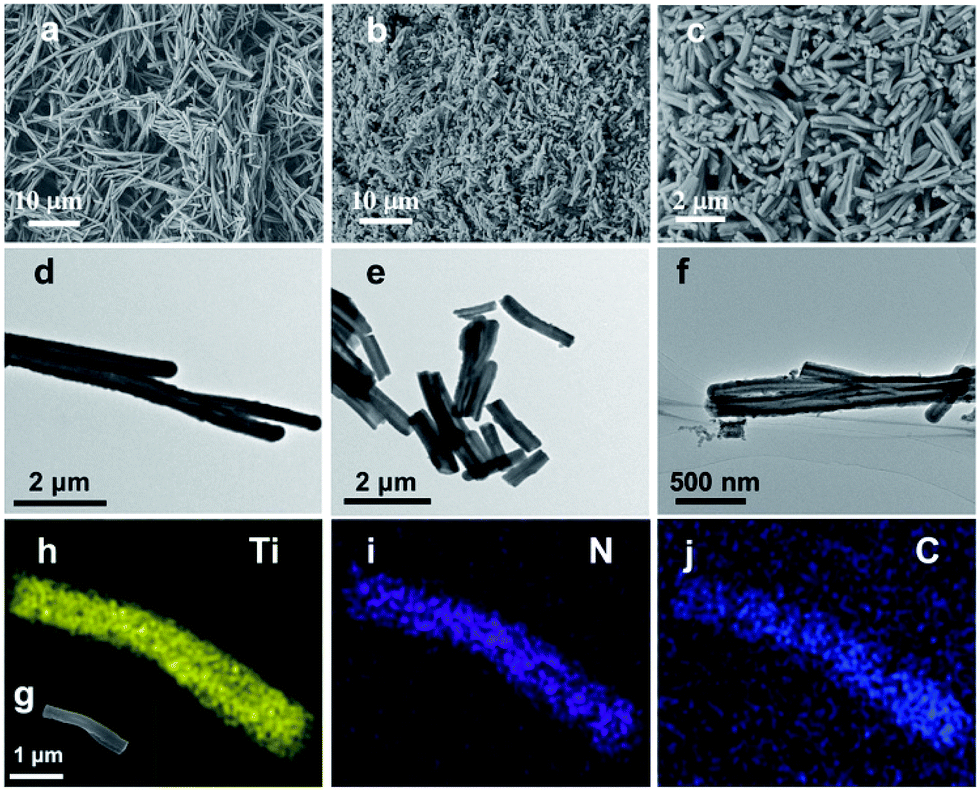 | ||
| Fig. 1 (a and d) SEM and TEM images of long PDA nanotubes. (b and e) SEM and TEM images of TiO2@PDA nanotubes. (c and f) SEM and TEM images of TiO2@C-PDA (400-2). (g) SEM image of a TiO2@C-PDA nanotube. Also shown are images of titanium (h), nitrogen (i) and carbon (j) mapping. | ||
To remove extra amorphous carbon in TiO2@PDA, calcination in air was conducted at 400 °C for 1, 2 and 3 h, respectively. Fig. 2 shows the TEM and HRTEM images of TiO2@C-PDA (400-2). Fig. 2a and b show the typical morphologies of a calcinated TiO2@C-PDA (400-2) nanostructure. The tube wall was mesoporous and composed of dark domains (Fig. 2b). All dark domain nanoparticles were well-crystallized (Fig. 2c), and the size distribution of domains was narrowed. The layer distance of an identical crystal was 0.35 nm, which is a typical feature of an anatase phase (101) lattice (Fig. 2e). Dark domains were TiO2 nanocrystals (Fig. 2c), whereas the light part around the TiO2 domain was a carbon layer (Fig. 2d). The carbon layer covering TiO2 nanocrystals was thick and amorphous in TiO2@C-PDA (400-1) (Fig. S1†) but was graphene-like after longer burning. Also, 0.34 nm was the distance between graphite (002) layers (Fig. 2f). The graphitization effect in calcination for carbonized PDA could be clearly observed even without TiO2. Fig. 3 shows HRTEM images of the residue of carbonized PDA nanotubes after calcination in air at 400 °C for 2 h. Graphite-like ordered carbon layers were concentrated in this residue. The measured lattice spacing of 0.18 nm corresponded to the (102) facet of graphitic carbon,24 and that of 0.21 nm belonged to the (1100) in-plane lattice fringes of graphene.25,26 Carbon with different degrees of graphitization shows different reactivity in burning: the one with a higher degree of graphitization survives longer during burning than its amorphous counterpart. Therefore, calcination in our experiments was not only a simple step to remove extra carbon, but also an optimal process to select a conductive graphite layer remaining on TiO2 nanoparticles. The presence of a graphite layer on TiO2@C-PDA (400-2) may be favorable for the efficient separation of photogenerated electron–hole pairs on TiO2.17
 | ||
| Fig. 2 (a and b) TEM images of TiO2@C-PDA (400-2), (c) SAED pattern of a TiO2 crystal, (d–f) HRTEM images of TiO2@C-PDA (400-2). Lattice fringes of anatase (e) and graphene-like carbon coverings (f). | ||
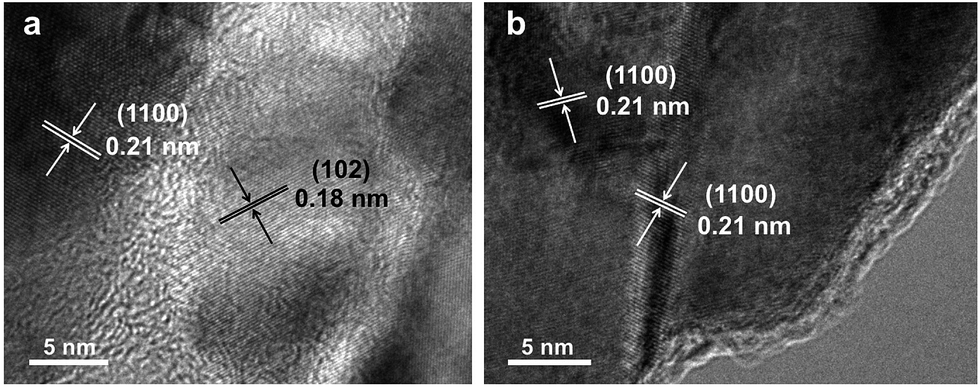 | ||
| Fig. 3 HRTEM images (a and b) of the residue of carbonized-PDA after calcination in air for 2 h at 400 °C. | ||
Interfacial interactions through chelated- and bidentate-binding modes27 and electron transfer in catechol/TiO2-anatase nanostructures have been confirmed.28,29 The advantage of PDA nanotubes as scaffolds for TiO2 is their inherent ability to chelate with Ti(IV) and TiO2.30–32 The strong interaction between Ti(IV) and the catechol groups of PDA promotes complete and tight contact between a carbon layer and TiO2 (Scheme 1). Further structural characterizations of TiO2@C-PDA were conducted. Fig. 4 presents X-ray diffraction (XRD) patterns of these hybrids. All samples showed pure anatase features that were in agreement with HRTEM observations. The (101) diffraction peaks in TiO2@C-PDA (400-1) and TiO2@C-PDA (400-2) showed clear shifts to smaller degrees as compared with that of pure TiO2. This result indicates expansion of the TiO2 lattice by introduction of foreign atoms after calcination. No obvious lattice expansion was observed in TiO2@C-PDA (400-3), suggesting that a prolonged calcination time decreases the content of foreign species in the host TiO2 lattice. The corresponding crystal sizes in different samples were calculated through application of the Scherrer equation to the peak at 25.3° (Table 1). The crystallite size decreased from 18.3 nm in pure TiO2 to 7.7 nm in TiO2@C-PDA (400-3). The precursor of Ti(IV) was absorbed and locked in these micropores on PDA tube walls. This spatial confinement was maintained even in its transformation to TiO2 crystals, which is similar to when TiO2 is generated in micelles.33 The bidentate bonding of catechol groups in PDA to TiO62− has been hypothesized to also contribute to the size and crystal control, which is similar in the case of SO42−.34 Because of this feature as well as inherent functional groups and microporous nanostructures, PDA nanotubes show better size-control compared with that of TiO2@C core–shell composites.35 No phase transformation from anatase to rutile occurred until the calcination temperature was increased to 800 °C, suggesting an extra-stabilized TiO2 crystal (Fig. S2†).
 | ||
| Scheme 1 Fabrication of TiO2@C-PDA nanotubes based on PDA nanotubes (schematic). | ||
 | ||
| Fig. 4 (a) XRD patterns of TiO2 and TiO2@C-PDA. (a) TiO2, (b) TiO2@PDA, (c) TiO2@C-PDA (400-1), (d) TiO2@C-PDA (400-2), (e) TiO2@C-PDA (400-3). (b) Partly magnified XRD patterns of (a). XRD patterns exhibit the diffraction peaks of anatase TiO2. | ||
| Samples name | Crystal size (nm) | BET surface area (m2 g−1) | Pore size (nm) | Band gap (eV) |
|---|---|---|---|---|
| Pristine TiO2 | 18.3 | 74.9 | 21.5 | 3.17 |
| TiO2@C-PDA (400-1) | 10.7 | 81.8 | 5.63 | 2.49 |
| TiO2@C-PDA (400-2) | 8.3 | 70.7 | 5.95 | 2.57 |
| TiO2@C-PDA (400-3) | 7.7 | 68.0 | 7.02 | 2.85 |
Raman spectroscopy can provide invaluable contributions towards understanding the material structure and its influence on the functional properties. In anatase TiO2 (Fig. 5), Raman peaks corresponding to the Eg mode (143 cm−1) became broader and significantly blue-shifted to 152 cm−1 for TiO2@C-PDA (400-1), and 147 cm−1 for TiO2@C-PDA (400-2). This effect is contributed mainly by a change in the crystal size of poorly crystalline materials36,37 and change in carrier density.17,38 Raman spectroscopy further confirmed the strong surface interaction between the carbon layer and TiO2 in TiO2@C-PDA.
 | ||
| Fig. 5 (a) Raman spectra of TiO2 and TiO2@C-PDA. (a) TiO2, (b) TiO2@C-PDA (400-1), (c) TiO2@C-PDA (400-2), (d) TiO2@C-PDA (400-3). (b) Partly magnified Raman spectra of (a). The original peak at 143 cm−1 in TiO2 was shifted to 152 cm−1 and 147 cm−1 in TiO2@C-PDA (400-1) and TiO2@C-PDA (400-2), respectively. | ||
3.2 Elemental analyses and optical properties
To investigate the composition and chemical state of the elements in TiO2@C-PDA after calcination, X-ray photoelectron spectroscopy (XPS) characterizations were conducted for all samples (Fig. 6, S3 and Table S1†). Calcination effectively removed large amounts of carbon in TiO2@PDA. Meanwhile, the amount of nitrogen was also sacrificed in calcination, from 8.93% in TiO2@PDA down to 1.54% in TiO2@C-PDA (400-1) (Table S1†). In the high-resolution XPS spectra of the N 1s core-level for three samples (Fig. 6a, c and e), two peaks, one located at 398.6 eV, and the other at 400.0 eV, were identified. The weak peak located at 398.6 eV was attributed to the N species in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N form from carbonized PDA,39 which is present on the surface of TiO2. The strong N 1s peak around 400.0 eV, widely observed in samples prepared via wet processes,40–43 could be assigned to interstitial N species bonding with the lattice O in the host TiO2. On the basis of the relative XPS area, the contents of the interstitial N species in TiO2@C-PDA (400-1), (400-2) and (400-3) were estimated to be 1.2, 0.98, and 0.49%, respectively. Given the relatively small space of the interstitial site in the host TiO2, occupation of the N species at the interstitial site would induce an expansion of the host TiO2 lattice, a notion that was well supported by XRD measurement given above.
N form from carbonized PDA,39 which is present on the surface of TiO2. The strong N 1s peak around 400.0 eV, widely observed in samples prepared via wet processes,40–43 could be assigned to interstitial N species bonding with the lattice O in the host TiO2. On the basis of the relative XPS area, the contents of the interstitial N species in TiO2@C-PDA (400-1), (400-2) and (400-3) were estimated to be 1.2, 0.98, and 0.49%, respectively. Given the relatively small space of the interstitial site in the host TiO2, occupation of the N species at the interstitial site would induce an expansion of the host TiO2 lattice, a notion that was well supported by XRD measurement given above.
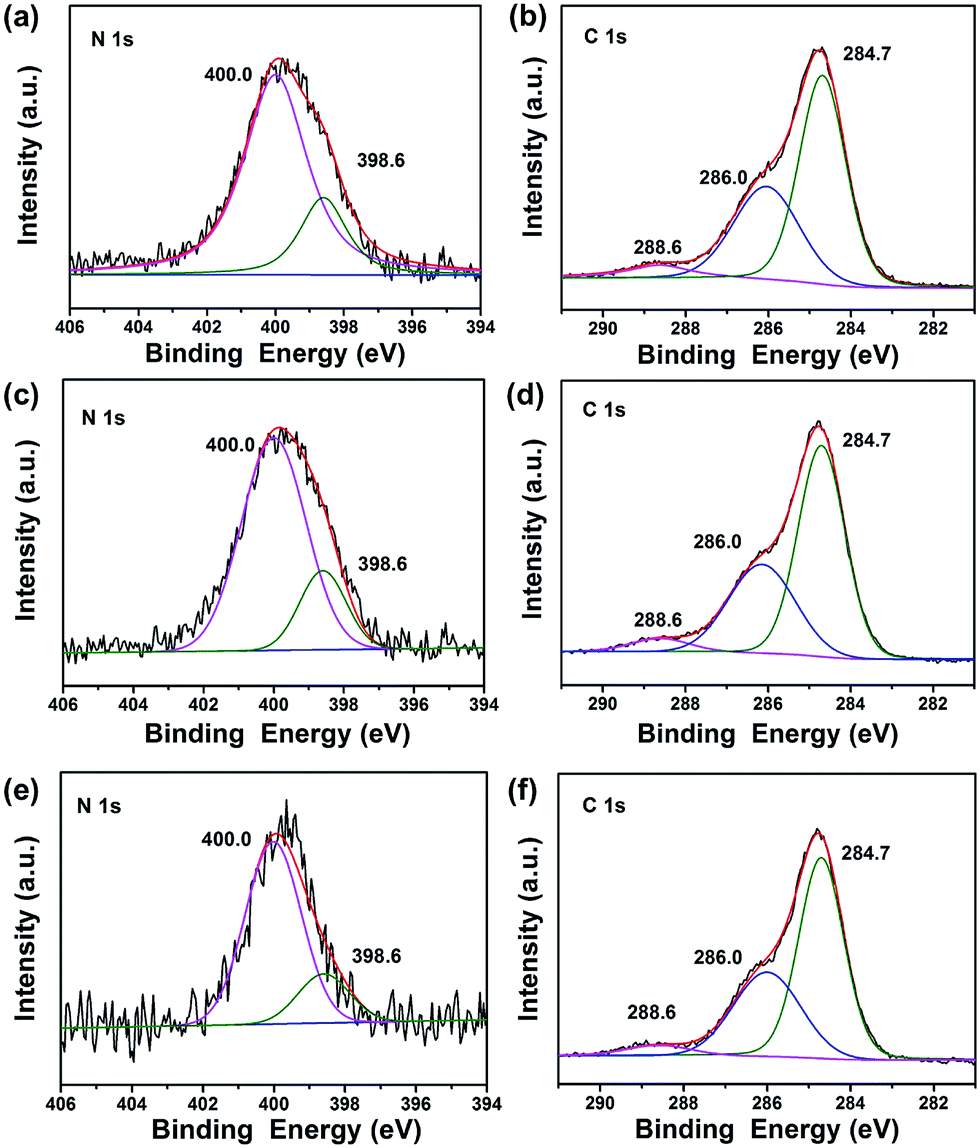 | ||
| Fig. 6 (a) N 1s XPS spectra of TiO2@C-PDA (400-1), (b) C 1s XPS spectra of TiO2@C-PDA (400-1). (c) N 1s XPS spectra of TiO2@C-PDA (400-2), (d) C 1s XPS spectra of TiO2@C-PDA (400-2). (e) N 1s XPS spectra of TiO2@C-PDA (400-3), (f) C 1s XPS spectra of TiO2@C-PDA (400-3). | ||
As shown in Fig. 6b, d and f, the high-resolution XPS spectra of C 1s for all samples consisted of three peaks located at 284.7, 286.0, and 288.6 eV, respectively. In combination with the HRTEM measurements given above, we believe that the main peak at 284.7 eV originated from both the surface graphene layer and adventitious carbon in XPS. The amount of surface carbon was calculated further based on the weight loss of samples in air (Fig. S4†). The carbon covered on TiO2 was 3.8, 1.6 and 1.1 wt% for TiO2@C-PDA (400-1), TiO2@C-PDA (400-2) and TiO2@C-PDA (400-3), respectively. The contribution at 286.0 eV was due to the C–N of carbonized PDA.39 The peak at 288.6 eV indicated formation of the Ti–O–C bond44 most likely by substitution of the titanium atom instead of the oxygen atom in the host TiO2 lattice. On the basis of the relative XPS area, the doping amounts of carbon in TiO2@C-PDA (400-1), TiO2@C-PDA (400-2) and TiO2@C-PDA (400-3) were 1.45, 0.84 and 0.82%, respectively. The content of the doped C species at the substitutional site and the N species at the interstitial site in TiO2 lattice decreased with increasing calcination time, whereas the amount of change of carbon cover was in accordance with them.
Fig. 7a presents the UV-vis diffuse reflectance spectra of various TiO2@C-PDA samples compared with pristine TiO2 without using PDA nanotubes as templates. From the Tauc plot (Fig. 7b), the corresponding band gap values of TiO2@C-PDA (400-1), (400-2) and (400-3) were estimated to be 2.49, 2.57, and 2.85 eV, respectively, as compared with the band gap (3.17 eV) of pure TiO2. Earlier investigations indicated that doping N at an interstitial site or carbon at a titanium site in a TiO2 lattice induces a localized level above the valence band (VB) or below the conduction band (CB) in the forbidden band, respectively, leading to extension of the host TiO2's absorption to the visible-light region.45–47 Thus, both the interstitial N and substitutional C dopants in the host TiO2, as evidenced by the XPS measurements given above, were responsible for the observed reduction in the band gap.
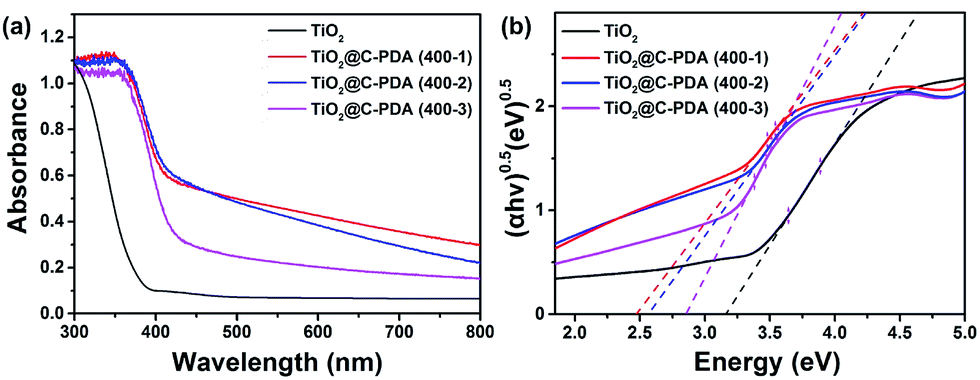 | ||
| Fig. 7 (a) UV-vis diffuse reflectance spectra of TiO2 and TiO2@C-PDA (400-1, 2, 3). (b) Band-gap evaluation from Tauc plots of TiO2 and TiO2@C-PDA (400-1, 2, 3). | ||
In addition, the surface area, pore size and surface charge of TiO2@C-PDA samples were measured (Fig. S5 and S6†). The surface charge of these samples showed no clear difference from that of pure TiO2 (Fig. S6†). The crystal sizes, BET surface area, pore size and band gaps of all samples are summarized in Table 1.
3.3 Photocatalytic performance of TiO2@C-PDA nanotubes
The photodegradation of MO was used to evaluate the photocatalytic activity of TiO2@C-PDA samples under visible light (>420 nm) (Fig. 8 and S7†). TiO2@C-PDA (400-2) showed remarkable photocatalytic activity, only 10% MO remained after 3 h. TiO2@C-PDA (400-1) and TiO2@C-PDA (400-3) had similar photocatalytic activities, leaving ≈43% and ≈53% MO in reaction systems after 3 h of irradiation under visible light (>420 nm). The photocatalytic oxidation of MO was a pseudo-zero-order reaction. Its apparent reaction rate constant was derived from the linear slope of the relationship between ln(c/c0) and kt, where c0 and c are the concentrations of initial solution and after t (h) of irradiation, respectively. TiO2@C-PDA (400-2) had the fastest reaction rate constant (0.738 h−1), whereas the constants of TiO2@C-PDA (400-1) and TiO2@C-PDA (400-3) were 0.284 h−1 and 0.214 h−1, respectively. The photocatalytic activity of TiO2@C-PDA (400-2) did not decrease after four cycles of photodegradation, demonstrating the good stability of this catalyst (Fig. S8†). A comparison of XRD patterns of this photocatalyst before and after photodegradation experiments also supported this finding (Fig. S9†). | ||
| Fig. 8 (a) Photocatalytic degradation of MO in the presence of TiO2 and TiO2@C-PDA (400-1, 2, 3) under visible-light irradiation (λ > 420 nm). (b) Linear extrapolations of TiO2@C-PDA (400-1, 2, 3) in photocatalytic degradation. | ||
The photocatalytic performance is strongly dependent on the extent of separation of electron–hole pairs over the excited semiconductor photocatalyst. Investigations have shown that a graphene-like carbon layer can promote remarkable separation of the electron–hole pairs over the excited TiO2 through an electron transfer from the CB to the graphene-like carbon layer. According to the HRTEM and XPS results shown above, graphene-like carbon layers were obviously present in TiO2@C-PDA (400-2) rather than in other samples. The effect of the graphene-like carbon layer in TiO2@C-PDA (400-2) on separation of the photoexcited electron–hole pairs was further evidenced by PL spectra (Fig. 9). The intensity of PL emission of TiO2@C-PDA (400-2) was decreased significantly compared with those of TiO2@C-PDA (400-1) and (400-3), suggesting efficient separation of the electron–hole pairs in the presence of the graphene-like carbon layer. Unsurprisingly, TiO2@C-PDA (400-2) exhibited the highest photocatalytic activity for MO degradation among these samples.
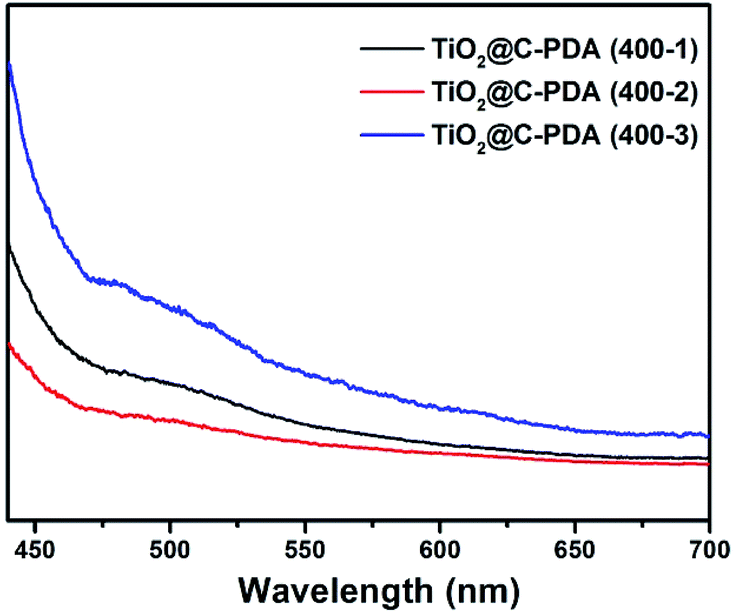 | ||
| Fig. 9 PL spectra of TiO2@C-PDA (400-1, 2, 3) using an excitation wavelength of 420 nm. | ||
Recently, an isotopic labeling study showed that the hole-driven oxidation of H2O in aqueous TiO2 suspensions is the main source of the free ˙OH radical,48 which plays an important part during the degradation of water pollutants in illuminated TiO2 photocatalysts,49 such as in case of MO degradation.50 Therefore, the rate of formation of ˙OH radicals is an important parameter to understand the reaction mechanism of illuminated photocatalysts. Quantitative detection of ˙OH radicals of TiO2@C-PDA under visible-light irradiation was conducted to investigate its photocatalytic mechanism. Terephthalic acid is an identical probe molecule for measuring ˙OH radicals.23 The change in the fluorescence intensity of supernatant solutions of various illuminated TiO2 photocatalysts is presented in Fig. 10. The amount of ˙OH radicals from TiO2@C-PDA (400-2) was highest and paralleled their best photocatalytical performance. The higher amount of ˙OH radicals from TiO2@C-PDA (400-2) under visible-light illumination revealed their increased ability to generate holes because of fast separation and transfer of electrons.
 | ||
| Fig. 10 Time dependence of the fluorescence intensity at 426 nm generated from 2-hydroxyterepthalic acid in the presence of TiO2@C-PDA (400-1, 2, 3) under visible-light irradiation (λ > 420 nm). | ||
The advantage of TiO2@C-PDA hybrids could be due to: (i) formation of Ti–O–C and interstitial N to vary the lattice structure of TiO2;7 (ii) surface contact of TiO2 and a carbon layer with a high degree of graphitization.17 Ti–O–C, similar to Ti–C, can extend light absorption to longer wavelengths.11,45 Conversely, electron transfer from a CB of TiO2 to a carbon layer (as a sensitizer) benefits charge separation.51,52 Optimization of charge separation requires a carbon layer on TiO2 of adequate thickness, better conductivity and close contact. Single-walled carbon nanotubes (SWCNTs) and graphene are highly conductive, so the photoexcitation charges from TiO2 can be rapidly separated and stabilized in TiO2@SWCNT53 and TiO2@graphene.10 However, the contact area between TiO2 and graphene or carbon nanotubes is limited. Zhang et al. reported an in situ polymer encapsulation–graphitization method to improve the contact between TiO2 and carbon coverings, and further programmed the thickness of a carbon shell on a TiO2 belt.17 However, the obtained TiO2@C showed an effect only under UV-light irradiation. Mesoporous PDA as a scaffold for TiO2 enlarged the contact area between a graphene-like carbon layer and TiO2, and provided a codopant for TiO2. Fig. 11 illustrates the electron transition and visible-light photocatalytic mechanism of TiO2@C-PDA. Moreover, theoretical calculations54 and experimental studies55 have indicated that codoping of non-metals and metals show synergistic effects on narrowing the band gap of TiO2. The catechol and amine groups on PDA nanotubes have strong interactions with various metal ions that can be transferred to multiple metal and non-metal dopants, thus showing the great versatility of PDA as a unique nitrogen-doped carbon scaffold for various catalysts. Visible light-induced photodecomposition is a green and renewable process for removing organic contaminants in water. Simple fabrication of TiO2@C-PDA photocatalysts endows it with great potential for industrial applications. It may have important and beneficial consequences on the environment and economy.
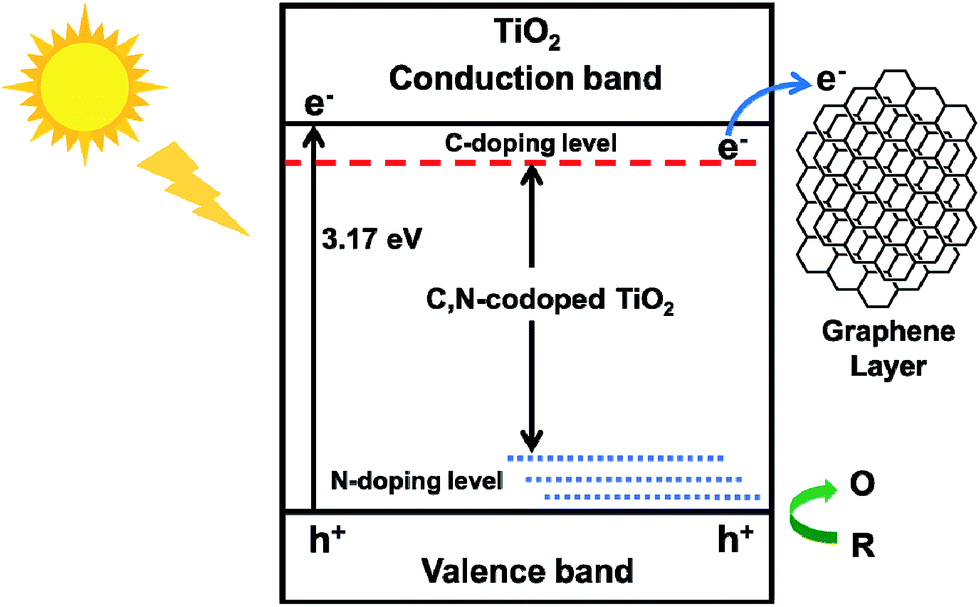 | ||
| Fig. 11 Electron transition and visible-light photocatalytic process of TiO2@C-PDA (schematic). | ||
4. Conclusions
Herein, tubular and mesoporous TiO2 were fabricated via sol–gel and calcination processes in the presence of PDA nanotubes. The obtained TiO2@C-PDA nanotubes were co-doped by carbon and nitrogen, which effectively narrowed the band gap of TiO2@C-PDA. The crystal size of TiO2 was confined through tightly covered PDA scaffolds, which efficiently suppressed crystal growth in calcination. Through optimized calcination, carbon cover on TiO2 was thinned and graphitized. Such thinning is convenient for taking advantage of a conductive carbon layer while avoiding the blocking effect from a thick carbon layer and, in turn, results in efficient separation of the photogenerated electron–holes of TiO2. The produced TiO2@C-PDA (400-2) with optimal structural and compositional advantages showed excellent photodegradation efficiency for MO under visible-light irradiation (>420 nm). PDA scaffolds can bring versatile benefits for TiO2 photocatalysts.Acknowledgements
The authors gratefully acknowledge the National Natural Science Foundation of China (Grant Numbers 21374132 and 21273281) for financial support.References
- X.-Y. Yang, L.-H. Chen, Y. Li, J. C. Rooke, C. Sanchez and B.-L. Su, Chem. Soc. Rev., 2017, 46, 481 RSC
.
- G. Prieto, H. Tüysüz, N. Duyckaerts, J. Knossalla, G.-H. Wang and F. Schüth, Chem. Rev., 2016, 116, 14056 CrossRef CAS PubMed
- S. A. Corr, D. P. Shoemaker, E. S. Toberer and R. Seshadri, J. Mater. Chem., 2010, 20, 1413 RSC
- S. Teixeira, P. M. Martins, S. Lanceros-Méndez, K. Kühn and G. Cuniberti, Appl. Surf. Sci., 2016, 384, 497 CrossRef CAS
- M. Zhu, C. Zhai, L. Qiu, C. Lu, A. S. Paton, Y. Du and M. C. Goh, ACS Sustainable Chem. Eng., 2015, 3, 3123 CrossRef CAS
- C. Zhai, M. Zhu, Y. Lu, F. Ren, C. Wang, Y. Du and P. Yang, Phys. Chem. Chem. Phys., 2014, 16, 14800 RSC
- J. H. Park, S. Kim and A. J. Bard, Nano Lett., 2006, 6, 24 CrossRef CAS PubMed
- J. S. Lee, K. H. You and C. B. Park, Adv. Mater., 2012, 24, 1084 CrossRef CAS PubMed
- J. Du, X. Lai, N. Yang, J. Zhai, D. Kisailus, F. Su, D. Wang and L. Jiang, ACS Nano, 2011, 5, 590 CrossRef CAS PubMed
- S. D. Perera, R. G. Mariano, K. Vu, N. Nour, O. Seitz, Y. Chabal and K. J. Balkus, ACS Catal., 2012, 2, 949 CrossRef CAS
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233 CrossRef CAS
- G. Wu, T. Nishikawa, B. Ohtani and A. Chen, Chem. Mater., 2007, 19, 4530 CrossRef CAS
- R. Leary and A. Westwood, Carbon, 2011, 49, 741 CrossRef CAS
- D. Chen, Z. Jiang, J. Geng, Q. Wang and D. Yang, Ind. Eng. Chem. Res., 2007, 46, 2741 CrossRef CAS
- L. W. Zhang, H. B. Fu and Y. F. Zhu, Adv. Funct. Mater., 2008, 18, 2180 CrossRef CAS
- H. Zhu, Y. Jing, M. Pal, Y. Liu, Y. Liu, J. Wang, F. Zhang and D. Zhao, Nanoscale, 2017, 9, 1539 RSC
- J. Zhang, M. Vasei, Y. Sang, H. Liu and J. P. Claverie, ACS Appl. Mater. Interfaces, 2016, 8, 1903 CAS
- R. Liu, S. M. Mahurin, C. Li, R. R. Unocic, J. C. Idrobo, H. Gao, S. J. Pennycook and S. Dai, Angew. Chem., Int. Ed., 2011, 123, 6931 CrossRef
- H. Zhu, Y. Jing, M. Pal, Y. Liu, Y. Liu, J. Wang, F. Zhang and D. Zhao, Nanoscale, 2017, 9, 1539 RSC
- J. Cao, C. J. Jafta, J. Gong, Q. Ran, X. Lin, R. Félix, R. G. Wilks, M. Bär, J. Yuan, M. Ballauff and Y. Lu, ACS Appl. Mater. Interfaces, 2016, 8, 29628 CAS
- J. H. Xue, W. C. Zheng, L. Wang and Z. X. Jin, ACS Biomater. Sci. Eng., 2016, 2, 489 CrossRef CAS
- S. P. Albu, A. Ghicov, J. M. Macak, R. Hahn and P. Schmuki, Nano Lett., 2007, 7, 1286 CrossRef CAS PubMed
- J. C. Barreto, G. S. Smith, N. H. Strobel, P. A. Mcquillin and T. A. Miller, Life Sci., 1995, 56, 89 Search PubMed
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Chem. Commun., 2012, 48, 8835 RSC
- K. Habiba, V. I. Makarov, J. Avalos, M. J. F. Guinel, B. R. Weiner and G. Morell, Carbon, 2013, 64, 341 CrossRef CAS PubMed
- J. Lee, K. Kim, W. I. Park, B.-H. Kim, J. H. Park, T.-H. Kim, S. Bong, C.-H. Kim, G. Chae, M. Jun, Y. Hwang, Y. S. Jung and S. Jeon, Nano Lett., 2012, 12, 6078 CrossRef CAS PubMed
- D. Finkelstein-Shapiro, S. K. Davidowski, P. B. Lee, C. Guo, G. P. Holland, T. Rajh, K. A. Gray, J. L. Yarger and M. Calatayud, J. Phys. Chem. C, 2016, 120, 23625 CAS
- P. Karthik, R. Vinoth, P. Selvam, E. Balaraman, M. Navaneethan, Y. Hayakawa and B. Neppolian, J. Mater. Chem. A, 2017, 5, 384 CAS
- L. G. C. Rego and V. S. Batista, J. Am. Chem. Soc., 2003, 125, 7989 CrossRef CAS PubMed
- S.-C. Li, L.-N. Chu, X.-Q. Gong and U. Diebold, Science, 2010, 328, 882 CrossRef CAS PubMed
- L.-M. Liu, S.-C. Li, H. Cheng, U. Diebold and A. Selloni, J. Am. Chem. Soc., 2011, 133, 7816 CrossRef CAS PubMed
- C. Creutz and M. H. Chou, Inorg. Chem., 2008, 47, 3509 CrossRef CAS PubMed
- J. Lin, Y. Lin, P. Liu, M. J. Meziani, L. F. Allard and Y.-P. Sun, J. Am. Chem. Soc., 2002, 124, 11514 CrossRef CAS PubMed
- M. Yan, F. Chen, J. Zhang and M. Anpo, J. Phys. Chem. B, 2005, 109, 8673 CrossRef CAS PubMed
- S. Shanmugam, A. Gabashvili, D. S. Jacob, J. C. Yu and A. Gedanken, Chem. Mater., 2006, 18, 2275 CrossRef CAS
- S. Sahoo, A. K. Arora and V. Sridharan, J. Phys. Chem. C, 2009, 113, 16927 CAS
- S. Balaji, Y. Djaoued and J. Robichaud, J. Raman Spectrosc., 2006, 37, 1416 CrossRef CAS
- P. Mazzolini, V. Russo, C. S. Casari, T. Hitosugi, S. Nakao, T. Hasegawa and A. L. Bassi, J. Phys. Chem. C, 2016, 120, 18878 CAS
- W. C. Zheng, H. L. Fan, L. Wang and Z. X. Jin, Langmuir, 2015, 31, 11671 CrossRef CAS PubMed
- F. Napoli, M. Chiesa, S. Livraghi, E. Giamello, S. Agnoli, G. Granozzi, G. Pacchioni and C. D. Valentin, Chem. Phys. Lett., 2009, 477, 135 CrossRef CAS
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed
- G. Wang, X. Xiao, W. Li, Z. Lin, Z. Zhao, C. Chen, C. Wang, Y. Li, X. Huang and L. Miao, Nano Lett., 2015, 15, 4692 CrossRef CAS PubMed
- R. Asahi, T. Morikawa, H. Irie and T. Ohwaki, Chem. Rev., 2014, 114, 9824 CrossRef CAS PubMed
- P. Ząbek, J. Eberl and H. Kisch, Photochem. Photobiol. Sci., 2009, 8, 264 Search PubMed
- C. D. Valentin, G. Pacchioni and A. Selloni, Chem. Mater., 2005, 17, 6656 CrossRef
- C. D. Valentin, G. Pacchioni and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 085116 CrossRef
- Y. Huang, W. Ho, S. Lee, L. Zhang, G. Li and J. C. Yu, Langmuir, 2008, 24, 3510 CrossRef CAS PubMed
- A. O. Kondrakov, A. N. Ignatev, V. V. Lunin, F. H. Frimmel, S. Bräse and H. Horn, Appl. Catal., B, 2016, 182, 424 CrossRef CAS
- C. S. Turchi and D. F. Ollis, J. Catal., 1990, 122, 178 CrossRef CAS
- J. Romão and G. Mul, ACS Catal., 2016, 6, 1254 CrossRef
- C. Lettmann, K. Hildenbrand, H. Kisch, W. Macyk and W. F. Maier, Appl. Catal., B, 2001, 32, 215 CrossRef CAS
- X. Xu, Y. Wang, R. Wang, J. Pan, J. Hu and H. Zeng, Adv. Mater. Interfaces, 2017, 4, 1600795 CrossRef
- Y. Yao, G. Li, S. Ciston, R. M. Lueptow and K. A. Gray, Environ. Sci. Technol., 2008, 42, 4952 CrossRef CAS PubMed
- R. Long and N. J. English, Chem. Mater., 2010, 22, 1616 CrossRef CAS
- X. Wu, S. Yin, Q. Dong, C. Guo, T. Kimura, J.-i. Matsushita and T. Sato, J. Phys. Chem. C, 2013, 117, 8345 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03063k |
| This journal is © The Royal Society of Chemistry 2017 |