
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChroman-4-one and pyrano[4,3-b]chromenone derivatives from the mangrove endophytic fungus Diaporthe phaseolorum SKS019†
Hui Cui a,
Meng Dinga,
Dane Huangb,
Zhengrui Zhanga,
Huiting Liua,
Hongbo Huang*c and
Zhigang She*a
a,
Meng Dinga,
Dane Huangb,
Zhengrui Zhanga,
Huiting Liua,
Hongbo Huang*c and
Zhigang She*a
aSchool of Chemistry, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: cesshzhg@mail.sysu.edu.cn; Fax: +86-20-84113356; Tel: +86-20-84113356
bSchool of Pharmaceutical Sciences, Sun Yat-Sen University, Guangzhou 510080, China
cCAS Key Laboratory of Tropical Marine Bio-resources and Ecology, Guangdong Key Laboratory of Marine Materia Medica, South China Sea Institute of Oceanology, Chinese Academy of Sciences, 164 West Xingang Road, Guangzhou 510301, China. E-mail: huanghb@scsio.ac.cn; Fax: +86-20-34066449; Tel: +86-20-34066449
First published on 6th April 2017
Abstract
Seven new compounds: diaporchromanones A–D (1–4), (−)-phomopsichin A (5a), (+)-phomopsichin B (6a), and (±)-diaporchromone A (7), along with the known (+)-phomopsichin A (5b) and (−)-phomopsichin B (6b) were isolated from an endophytic fungus Diaporthe phaseolorum SKS019. The structures of the new compounds, including their absolute configurations, were determined on the basis of HRESIMS and NMR spectroscopic data, and experimental ECD and Rh2(OCOCF3)4-induced CD spectra analyses. Diaporchromanone A (1)/B (2), and C (3)/D (4) are two pairs of 3-epimers, and their structures possessing 3-substituted-chroman-4-one skeleton are rarely found in natural sources. (−)-Phomopsichin A (5a) and (+)-phomopsichin B (6a) are enantiomers of (+)-phomopsichin A (5b) and (−)-phomopsichin B (6b), respectively. All of the isolates were evaluated for their inhibitory effects against osteoclastogenesis in the RAW 264.7 cell line using luciferase reporter gene assays. Compounds 3–6b exhibited moderate inhibitory effects on osteoclastogenesis by suppressing the receptor activator of NF-κB by ligand-induced NF-κB activation.
Introduction
Chroman-4-one and 4H-chromen-4-one derivatives have been isolated from various natural sources, such as fungi, plants, and various marine organisms.1–3 These derivatives are reported to exhibit diverse and remarkable pharmacological activities, such as antiviral, antimicrobial, antifungal, and antitumor activities.3–8 However, 3-substituted-chroman-4-one and pyrano[4,3-b]chromenone derivatives were rarely described. Until now, only one 3-substituted-chroman-4-one compound and nine pyrano[4,3-b]chromenone analogues were found in nature.9,10Marine-derived fungi are widely recognized as prolific sources of biologically active and structurally unique natural products.3,11 As part of our ongoing investigation on bioactive metabolites from mangrove endophytic fungi,12–17 a chemical investigation of the mangrove endophytic fungus Diaporthe phaseolorum SKS019, isolated from a fresh branch of the mangrove plant Acanthus ilicifolius, was carried out. The CH2Cl2 extract of a fermentation broth of the fungus led to the isolation and identification of four chroman-4-one derivatives (1–4), and five pyrano[4,3-b]chromenone derivatives (5a–7). In bioactivity assays, compounds 3–6b exhibited moderate inhibitory effects on osteoclastogenesis by suppressing RANKL-induced NF-κB activation. Herein, details of the isolation, structure elucidation, and bioactivity of these compounds (Fig. 1) are described.
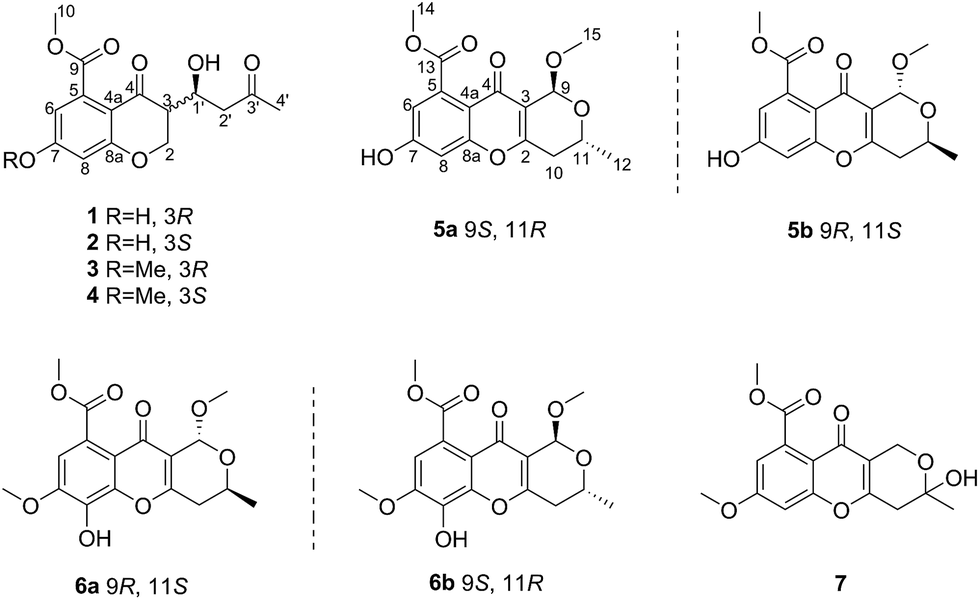 | ||
| Fig. 1 Structures of the isolated compounds 1–7. | ||
Results and discussion
The mangrove endophytic fungus D. phaseolorum SKS019 was cultured on solid rice medium with seawater for 28 days. After incubation, the medium were extracted with CH2Cl2, and then the organic layers were dried under vacuum to yield solid residue. The residue was purified by various column chromatographic separations to obtain the compounds 1–7. Compound 5b and 6b were identified as (+)-phomopsichin A (5b) and (−)-phomopsichin B (6b), respectively, by comparisons of the MS and NMR data with those previously reported.9Compound 1 was isolated as a brown powder. Its molecular formula was assigned as C15H16O7 on the basis of (−)-HRESIMS at m/z 307.0818 [M − H]− (calcd for C15H15O7−, 307.0818), indicating eight degrees of unsaturation. The IR spectrum of 1 exhibited absorption bands for hydroxyl (3367 cm−1) and conjugated carbonyl (1732 and 1620 cm−1) groups. In the 1H NMR spectrum (Table 1), the signals for two meta-coupled aromatic protons at δH 6.46 (1H, d, J = 2.3 Hz, H-6) and 6.38 (1H, d, J = 2.3 Hz, H-8), two methylene groups [δH 4.55 (1H, dd, J = 11.5, 5.1 Hz, H-2b), 4.42 (1H, dd, J = 11.5, 9.6 Hz, H-2a); δH 2.81 (1H, dd, J = 17.3, 3.6 Hz, H-2′b), 2.74 (1H, dd, J = 17.3, 8.7 Hz, H-2′a)], two methine groups [δH 4.49 (1H, ddd, J = 8.7, 5.1, 3.6 Hz, H-1′), δH 2.90 (1H, ddd, J = 9.6, 5.1, 5.1 Hz, H-3)], one methoxy group at δH 3.93 (3H, s, H3-10), and one methyl group at δH 2.19 (3H, s, H3-4′) were observed. The 13C NMR and DEPT spectra showed 15 carbon resonances corresponding to two sp2 methine (δC 110.4, 104.7), two sp3 methine (δC 66.5, 49.6), two sp3 methylene (δC 68.6, 46.9), seven sp2 quaternary (δC 209.2, 191.0, 170.5, 164.0, 163.0, 136.3, 112.0), one methoxyl (δC 53.4), and one methyl (δC 31.0) carbons. These spectroscopic features illustrated that 1 belonged to the family of chromanone. A literature survey suggested that the 1H and 13C NMR data of 1 were very similar to the known polivione isolated from fungus Penicillium frequentans.10 The presence of a 1,2,3,5-tetrasubstituted aromatic ring was confirmed by the meta-coupled aromatic protons of H-6 and H-8. The COSY spectrum showed the 1H–1H spin system of H2-2/H-3/H-1′/H2-2′, assigned the fragment of –CH2–CH–CH–CH2–. The HMBC correlations from H2-2 to C-3, C-4, and C-8a, and from H-3 to C-4 established the core structure of a chroman-4-one. The HMBC correlations (Fig. 2) from the aromatic proton H-6 and from the methoxy protons H3-10 to the carbonyl C-9 indicated the existence of a methyl benzoate moiety. The attachment of 7-OH group was confirmed by the HMBC correlations from H-6 to C-7 and from H-8 to C-7. The substitution of a butan-2-one fragment at C-3 was inferred from the cross peaks of H3-4′/C-3′, C-2′, and of H-1′/C-2, C-4, and C-3′ in the HMBC spectrum. The above mentioned data established the planar structure of 1.
| Position | 1 (CDCl3) | 2 (CDCl3) | ||
|---|---|---|---|---|
| δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | |
| 2 | 4.55, dd (11.5, 5.1) | 68.6, CH2 | 4.70, dd (11.6, 6.3) | 68.0, CH2 |
| 4.42, dd (11.5, 9.6) | 4.52, dd (11.6, 3.9) | |||
| 3 | 2.90, ddd (9.6, 5.1, 5.1) | 49.6, CH | 2.64, ddd (8.5, 6.3, 3.9) | 50.6, CH |
| 4 | 191.0, C | 190.3, C | ||
| 4a | 112.0, C | 111.6, C | ||
| 5 | 136.3, C | 136.4, C | ||
| 6 | 6.46, d (2.3) | 110.4, CH | 6.44, d (2.3) | 110.3, CH |
| 7 | 163.0, C | 162.8, C | ||
| 8 | 6.38, d (2.3) | 104.7, CH | 6.39, d (2.3) | 104.6, CH |
| 8a | 164.0, C | 163.9, C | ||
| 9 | 170.5, C | 170.4, C | ||
| 1′ | 4.49, ddd (8.7, 5.1, 3.6) | 66.5, CH | 4.38, ddd (8.5, 8.5, 2.9) | 64.1, CH |
| 2′ | 2.81, dd (17.3, 3.6) | 46.9, CH2 | 2.85, dd (18.1, 2.9) | 47.5, CH2 |
| 2.74, dd (17.3, 8.7) | 2.72, dd (18.1, 8.5) | |||
| 3′ | 209.2, C | 210.2, C | ||
| 4′ | 2.19, s | 31.0, CH3 | 2.19, s | 30.9, CH3 |
| 10-CH3O | 3.93, s | 53.4, CH3 | 3.94, s | 53.3, CH3 |
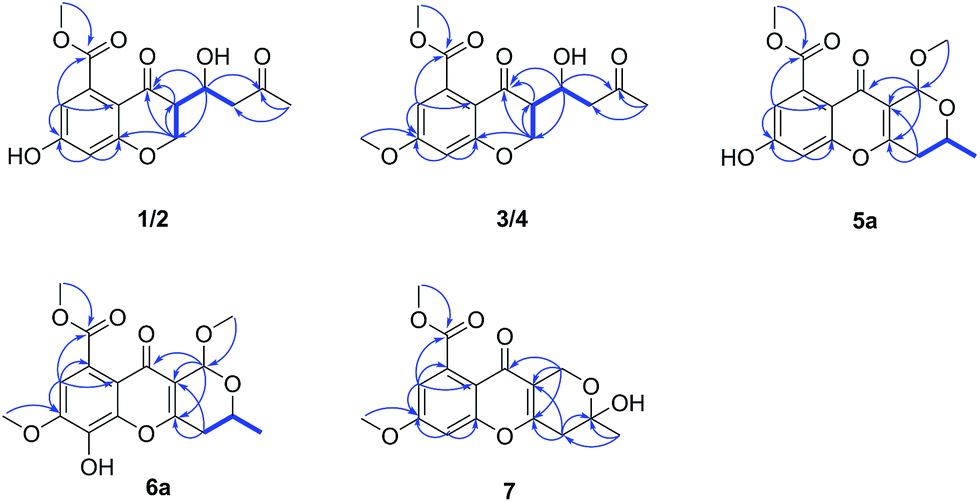 | ||
| Fig. 2 Selected 1H–1H COSY (bold line) and HMBC (arrow) correlations of compounds 1–7. | ||
There are two stereogenic centers (C-3 and C-1′) in 1. The absolute configuration at C-3 in 1 was determined by analysis of electronic circular dichroism (ECD) spectrum using ketone octant rule.18 For cyclic aryl–ketone compounds, a modified octant rule demonstrated that the positive cotton effect (CE) for the n → π* carbonyl transition indicate 3R absolute configuration, while the negative CE for the n → π* carbonyl transition indicate 3S absolute configuration.18,19 Compound 1 showed UV absorption bands at 279 nm (π → π*) and 320 nm (n → π*). The ECD spectrum of 1 displayed a positive CE at 329 nm (Fig. 3, left) and confirmed the absolute configuration of the stereogenic center C-3 as R. The absolute configuration of the C-1′ was assigned to be S on the basis of the CD spectrum for the complex of compound 1 with [Rh2(OCOCF3)4], which showed a positive E band (Fig. 5).20–22 In addition, the predicted ECD curves of 1 were calculated by a quantum chemical method at the CAM-B3LYP/6-311+G(2d,p) level, and the theoretical ECD curve for 3R1′S-1 showed an excellent fit with the experimental one. Therefore, the absolute configuration of the stereogenic centers for 1 was assigned as 3R and 1′S. Thus, compound 1 was named diaporchromanone A. Since the first 3-substituted-chroman-4-one derivative polivione was described in 1985,10 this was the second example to report the 3-substituted-chroman-4-one analogues.
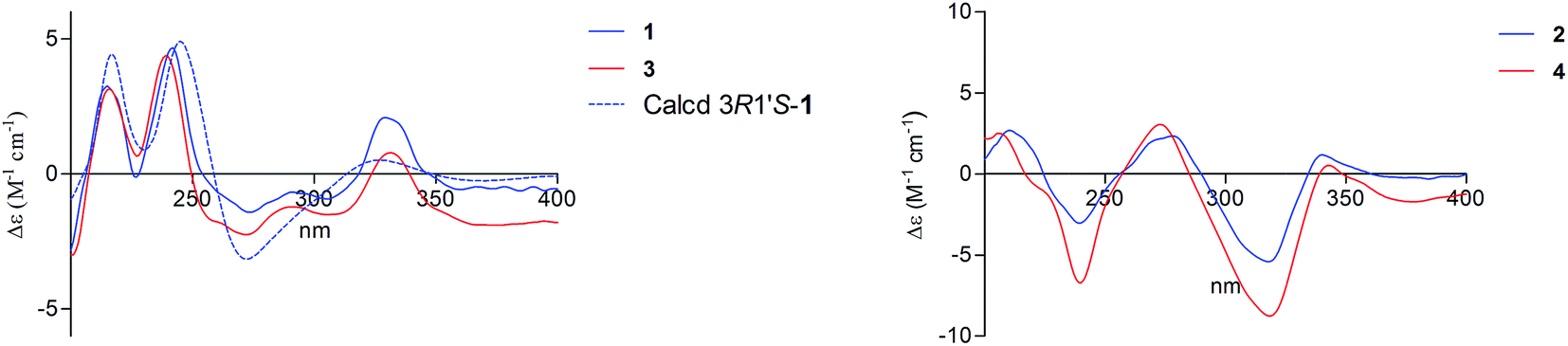 | ||
| Fig. 3 Experimental and calculated ECD spectra of compounds 1/3 (left) and 2/4 (right) in methanol. | ||
Compound 2 was isolated as a brown powder, possessing the same molecular formula C15H16O7 as 1. The 1H and 13C NMR spectroscopic data (Table 1) of 2 were very similar to those of 1, except that the 13C NMR chemical shifts at C-2, C-3, C-1′, C-2′ (δC 68.0, 50.6, 64.1, and 47.5) were different from those in 1 (δC 68.6, 49.6, 66.5, and 46.9), respectively. Further investigations of the 1H–1H COSY and HMBC experiments (Fig. 2) indicated that 2 shared the same planar structure with 1. In the 1H NMR spectrum, although the resonance value for H-3 of 2 (δH 2.64) was up-shifted slightly than that in 1 (δH 2.90), the coupling constants of H-3/H2-2 (J = 6.3, 3.9 Hz) and of H-3/H-1′ (J = 8.5 Hz) were obviously different with those in 1 (JH-3/H-2 = 9.6, 5.1 Hz, JH-3/H-1′ = 5.1 Hz). Compound 2 displayed a negative CE at the n → π* carbonyl transition (318 nm) in the ECD spectrum (Fig. 3, right), which confirmed 3S absolute configuration via applying the octant rule.18 Furthermore, the Rh-complex of 2 displayed a positive E band (Fig. 5), indicating 1′S absolute configuration for 2, which was the same with that in 1. Therefore, compound 2 was identified as 3-epimer of 1, and was named diaporchromanone B.
The molecular formula of compounds 3 and 4 were determined to be C16H18O7 by (−)-HRESIMS peak at m/z 321.0970 [M − H]−, which was one CH2 unit greater than that for 1 and 2. The 1H and 13C NMR spectroscopic data (Table 2) of 3 and 4 resembled those of 1 and 2, respectively, except that the additional signals for a methoxy group (δH/C 3.85/56.1 in 3; δH/C 3.84/56.1 in 4) were observed. The methoxy group C-11 was connected to C-7 on the basis of the HMBC correlation (Fig. 2) from the methoxy protons to C-7 (δC 164.1) in 3 and 4. The absolute configurations of stereogenic centers for 3 and 4 were assigned as 3R, 1′S, and 3S, 1′S on the basis of the same ECD absorption curves as 1 and 2 (Fig. 3), respectively. Compounds 3 and 4, a pair of 3-epimers, were named diaporchromanones C and D, accordingly.
| Position | 3 (CDCl3) | 4 (CDCl3) | ||
|---|---|---|---|---|
| δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | |
| 2 | 4.60, dd (11.5, 5.1) | 68.6, CH2 | 4.71, dd (11.6, 6.3) | 68.2, CH2 |
| 4.47, dd (11.5, 9.6) | 4.57, dd (11.6, 3.9) | |||
| 3 | 2.96, ddd (9.6, 5.1, 5.1) | 49.6, CH | 2.66, ddd (8.5, 6.3, 3.9) | 50.6, CH |
| 4 | 190.9, C | 190.2, C | ||
| 4a | 112.3, C | 111.7, C | ||
| 5 | 136.3, C | 136.3, C | ||
| 6 | 6.56, d (2.4) | 109.8, CH | 6.54, d (2.4) | 109.8, CH |
| 7 | 164.1, C | 164.0, C | ||
| 8 | 6.46, d (2.4) | 102.2, CH | 6.46, d (2.4) | 102.0, CH |
| 8a | 165.6, C | 165.5, C | ||
| 9 | 169.7, C | 169.7, C | ||
| 1′ | 4.51, ddd (8.7, 5.1, 3.6) | 66.4, CH | 4.38, ddd (8.5, 8.5, 2.9) | 64.0, CH |
| 2′ | 2.80, dd (17.3, 8.7) | 46.8, CH2 | 2.88, dd (18.1, 2.9) | 47.7, CH2 |
| 2.74, dd (17.3, 3.6) | 2.71, dd (18.1, 8.5) | |||
| 3′ | 208.8, C | 210.0, C | ||
| 4′ | 2.18, s | 31.0, CH3 | 2.18, s | 30.9, CH3 |
| 10-CH3O | 3.93, s | 53.1, CH3 | 3.94, s | 53.1, CH3 |
| 11-CH3O | 3.85, s | 56.1, CH3 | 3.84, s | 56.1, CH3 |
Compound 5a was obtained as a white powder. The molecular formula of C16H16O7 was determined on the basis of (−)-HRESIMS m/z 319.0818 [M − H]−, representing nine degrees of unsaturation. The 1H and 13C NMR data (Table 3) of 5a were the same as the known phomopsichin A (5b),9 which was previously isolated from mangrove endophytic fungus phomopsis sp. 33#. However, compound 5a showed a negative optical rotation value [α]25D −12 (c 0.3, MeOH), which was opposite to that of 5b [α]25D +12.5. Furthermore, compounds 5a and 5b presented reverse absorption curves in the ECD spectra (Fig. 4, left). Based on the above data, it was deduced that 5a and 5b were enantiomers, and the absolute configuration of stereogenic centers for 5a was assigned as 9S, 11R. Finally, compound 5a was named (−)-phomopsichin A. To the best of our knowledge, compound 5a is the tenth example of pyrano[4,3-b]chromenone derivatives from natural sources.
| Position | 5a (acetone-d6) | 6a (CDCl3) | 7 (CDCl3) | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | |
| 2 | 164.1, C | 163.5, C | 158.4, C | |||
| 3 | 117.4, C | 116.2, C | 115.5, C | |||
| 4 | 173.2, C | 173.5, C | 173.7, C | |||
| 4a | 114.8, C | 116.7, C | 114.4, C | |||
| 5 | 136.4, C | 124.7, C | 134.7, C | |||
| 6 | 6.85, d (2.3) | 113.9, CH | 6.93, s | 107.9, CH | 6.89, d (2.3) | 113.0, CH |
| 7 | 162.5, C | 148.9, C | 163.2, C | |||
| 8 | 6.93, d (2.3) | 104.4, CH | 134.7, C | 6.87, d (2.3) | 101.7, CH | |
| 8a | 158.4, C | 144.3, C | 158.0, C | |||
| 9 | 5.40, s | 95.3, CH | 5.58, s | 94.6, CH | 4.72, m | 57.5, CH2 |
| 10 | 2.67, dd (17.7, 3.8) | 34.6, CH2 | 2.65, m | 34.5, CH2 | 2.86, dd (17.4, 2.8) | 37.1, CH2 |
| 2.58, dd (17.7, 10.9) | 2.76, dd (17.4, 9.8) | |||||
| 11 | 4.34, m | 62.8, CH | 4.42, m | 62.1, CH | 95.4, C | |
| 12 | 1.34, d (6.3) | 21.0, CH3 | 1.40, d (6.3) | 21.0, CH3 | 1.63, s | 29.5, CH3 |
| 13 | 169.5, C | 169.7, C | 169.5, C | |||
| 14-CH3O | 3.86, s | 52.8, CH3 | 3.97, s | 53.2, CH3 | 3.98, s | 53.2, CH3 |
| 15-CH3O | 3.43, s | 55.6, CH3 | 3.55, s | 55.9, CH3 | ||
| 16-CH3O | 4.01, s | 56.9, CH3 | 3.90, s | 56.2, CH3 | ||
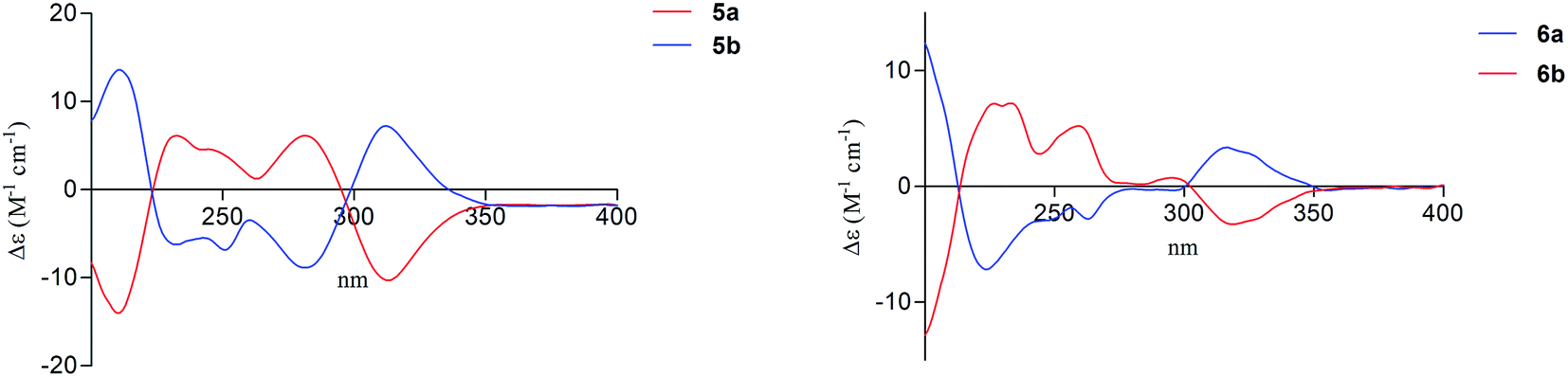 | ||
| Fig. 4 Experimental ECD spectra of compounds 5a/5b (left) and 6a/6b (right) in methanol. | ||
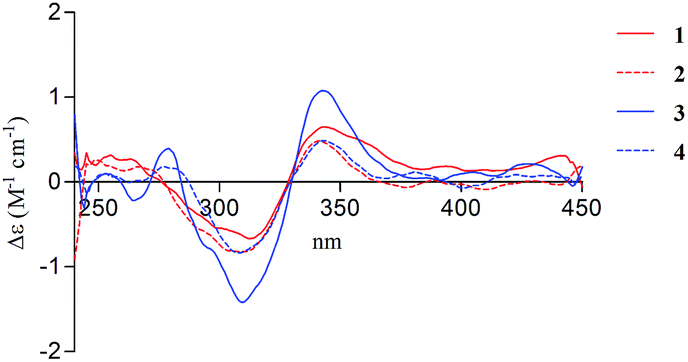 | ||
| Fig. 5 CD spectrum of Rh-complex of 1–4 with the inherent CD spectrum subtracted. | ||
The molecular formula of compound 6a was established as C17H18O8 on the basis of (−)-HRESIMS at m/z 349.0921 [M − H]− (calcd for 349.0923), indicating nine degrees of unsaturation. Detailed analysis of the 1H and 13C NMR data (Table 3) of 6a indicated that the 1H and 13C NMR data of 6a were the same as phomopsichin B (6b).9 As seen in compounds 5a and 5b, the optical rotation value [α]25D +16 (c 0.4, MeOH) of 6a was opposite to that of phomopsichin B (6b). Moreover, compounds 6a and 6b presented the reverse ECD absorption curves (Fig. 4, right). Thus, compound 6a was identified to be enantiomer of 6b, and the absolute configuration of stereogenic centers for 6a was determined as 9R, 11S. Finally, compound 6a was named as (+)-phomopsichin B.
Compound 7 had a molecular formula of C16H16O7, as established by the (−)-HRESIMS m/z 319.0811 [M − H]−, implying nine degrees of unsaturation. The 13C NMR data showed 16 carbons (Table 3), including two sp2 methine (δC 113.0, 101.7), two sp3 methylene (δC 57.5, 37.1), eight sp2 quaternary (δC 173.7, 169.5, 163.2, 158.4, 158.0, 134.7, 115.5, 114.4), one sp3 quaternary (δC 95.4), two methoxyl (δC 56.2, 53.2), and one methyl (δC 29.5) carbons. Comparing the 1H and 13C NMR data of 7 with those of 5a revealed that two sp3 methine (C-9 and C-11) in 5a were replaced by one sp3 methylene and one sp3 quaternary carbons in 7. These changes were proved by the HMBC correlations from H2-10 to C-11, from H3-12 to C-10 and C-11, and from H2-9 to C-3, C-4 and C-11. In addition, the methoxy group C-16 was linked to C-7 established by the HMBC correlation (Fig. 2) from the methoxy protons (δH 3.90, H3-16) to C-7 (δC 163.2). Thus, the planar structure was elucidated. Because the optical rotation value of 7 was nearly zero and the ECD spectrum of 7 didn't show absorption curves, it was deduced that diaporchromone A (7) was obtained as racemic mixture. Unfortunately, further chiral separations of 7 by HPLC using S-Chiral A or chiralcel OD columns were failed. Finally, compound 7 was named (±) diaporchromone A.
Compounds (1–6b) were tested for their inhibitory activity of osteoclastogenesis by suppressing RANKL-induced NF-κB activation using RAW 264.7 cell line. The results (Table 4) showed that compounds 3–6b exhibited moderate activities with IC50 values from 28 to 46 μM, in contrast to positive control (JSH-23, IC50 = 10.8 μM). Interestingly, the different configurations in 5a/5b and 6a/6b almost didn't affect their activities. However, compounds 1 and 2 showed no inhibitory effects at a concentration of 50 μM. Compounds (1–6b) were tested for their cytotoxicity using MDA-MB-435 and MCF10A human cell lines. None of these compounds showed growth inhibitory activity against MDA-MB-435 and MCF10A cell lines at a concentration of 50 μM.
| No. | % inhibition (50 μM) | IC50a (μM) | CC50b (μM) |
|---|---|---|---|
| a IC50: 50% inhibitory concentration.b CC50: 50% cytotoxic concentration.c Positive control; compound 7 was not evaluated for their bioactivity because of sample limitations. | |||
| 1 | 15 | — | >50 |
| 2 | 20 | — | >50 |
| 3 | 51 | 43 ± 1.3 | >50 |
| 4 | 60 | 41 ± 0.3 | >50 |
| 5a | 46 | 46 ± 0.8 | >50 |
| 5b | 50 | 43 ± 2.1 | >50 |
| 6a | 65 | 30 ± 1.3 | >50 |
| 6b | 70 | 28 ± 0.5 | >50 |
| JSH-23c | 10.8 ± 1.3 | ||
Experimental section
General experimental procedures
IR data were recorded on a Nicolet 5DX-FTIR (Thermo Fisher Scientific, Inc., Hudson, NH, USA), in KBr discs. UV data were recorded on a Shimadzu UV-240 spectrophotometer (Shimadzu, Kyoto, Japan). Optical rotations were measured on a Bellingham-Stanley ADP 440+ polarimeter at 25 °C. The 1H NMR (500 MHz), 13C NMR (125 MHz), and 2D NMR spectra were obtained on a Bruker AVANCE-500 (Bruker BioSpin Corporation, Billerica, MA, USA) using TMS as an internal reference. ESIMS were acquired on a Thermofisher LTQ Orbitrp Elite LC-MS spectrometer (Thermo Fisher Scientific, Inc., Hudson, NH, USA), and the HRESIMS data were measured on a Micro Mass Q-TOF spectrometer (Waters Corporation, Milford, MA, USA). TLC analysis was carried out on silica gel plates (Marine Chemical Ltd., Qingdao, China). The chiral HPLC separation was accomplished over an S-Chiral A column (column size: 10 × 250 mm 5 μm; Acchrom Technologies Co., Ltd., Beijing, China). RP-C18 silica gel (Fuji, 40–75 μm, Fuji Silysia Chemical Ltd., Kasugai, Japan), silica gel (200–300 mesh, Marine Chemical Ltd., Qingdao, China), high silica gel (H, Marine Chemical Ltd., Qingdao, China), and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Stockholm, Sweden) were used for column chromatography (CC). RAW264.7 cells were gifted from Professor Jiake Xu from the School of Pathology and laboratory Medicine, the University of Western Australia. Human breast cancer cell line MDA-MB-435 and immortalized non-cancer breast epithelial cells MCF10A were purchased from the cell bank of Chinese Academy of Sciences (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), 2 mM L−1 glutamine, 100 units mL−1 penicillin, and 100 μg mL−1 streptomycin were obtained from Invitrogen-Gibco (Grand Island, NY, USA). RANKL was purchased from Peprotech EC Ltd. (London, UK). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reagent was purchased from Genview (Houston, TX, USA). 4-Methyl-N1-(3-phenylpropyl)-1,2-benzenediamine (JSH-23, >98%) was purchased from Sigma (Buchs, Switzerland), and all other chemicals were obtained from Sigma (St. Louis, USA).Fungal material
The fungal strain SKS019 was isolated from the branches of the mangrove plant Acanthus ilicifolius collected from Shankou in Guangxi province, China. The fungus was identified by our team as Diaporthe phaseolorum SKS019, according to a molecular biological protocol by DNA amplification and sequencing of the ITS region (deposited in GenBank, accession no. KX815357). A voucher strain was deposited in School of Chemistry, Sun Yat-Sen University, Guangzhou, China, with the access code, 2016-SKS019.Fermentation, extraction and isolation
The fungus Diaporthe phaseolorum SKS019 was grown on a solid autoclaved rice substrate medium (sixty 500 mL Erlenmeyer flasks, each containing 50 g of rice and 50 mL of 0.3% of saline water) for 28 days at room temperature. The mycelia and solid rice medium were extracted with CH2Cl2 for three times. The organic solvents were evaporated under reduced pressure to yield a crude extract (26.3 g), then chromatographed on silica gel column (100 g, 100–200 mesh, 6 × 50 cm) eluting with a step gradient of petroleum ether (PE)–ethyl acetate (EA) (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0; 9:1; 8:2; 7:3; 6:4; 5:5; 4:6; 3:7; 0:100, v/v, each 500 mL) to give nine fractions (F1–F9). F4 (1.5 g) was subjected to a silica gel column (5.0 × 30 cm), eluting with gradient of CH2Cl2/MeOH (100:1) to yield five subfractions (F401–F405). F401 (0.5 g) was further fractionated on Sephadex LH-20 (110 g, 110 × 3 cm) eluting with CH2Cl2–MeOH (1 L, 1:1, v/v) to give two sets of mixtures F401a (13 mg) and F401b (9 mg). After evaluation by the 13C NMR analysis, F401a and F401b both contained two sets of diastereoisomers. And then F401a was purified by HPLC on an S-Chiral A column, using hexane/2-propanol (70:30, v/v, flow rate: 1.5 mL min−1) as the solvent system, to yield 1 (5 mg, tR = 11.5 min) and 2 (3 mg, tR = 13.0 min), and F401b was also purified by HPLC on an S-Chiral A column, using hexane/2-propanol (90:10, v/v, flow rate: 1.5 mL min−1) as the solvent system, to obtain 3 (4 mg, tR = 34.9 min) and 4 (3 mg, tR = 38.8 min). F402 (200 mg) was purified by HPLC on a semipreparative SB-C18 column, using MeOH–H2O (45:55, v/v, flow rate: 1.0 mL min−1) as the solvent system, to afford F402-3 (5a/5b, 25 mg), F402-4 (6a/6b, 20 mg), and 7 (4 mg). F402-3 and F402-4 were further separated by chiral HPLC using an S-Chiral A column to yield 5a (8 mg) and 5b (10 mg), 6a (9 mg) and 6b (7 mg), respectively.
ε) 216 (4.4), 240 (4.1), 279 (4.1), 320 (4.0) nm; ECD (MeOH) λmax (Δε) 215 (+3.3), 242 (+4.2), 329 (+2.0) nm; IR (KBr) νmax 3367, 2936, 1732, 1620, 1306, 1166 cm−1; 1H and 13C NMR data, Table 1; (−)-ESIMS m/z 307.1 [M − H]−; (−)-HRESIMS at m/z 307.0818 [M − H]− (calcd for C15H15O7 307.0818).ε) 216 (4.4), 240 (4.1), 279 (4.1), 320 (4.0) nm; ECD (MeOH) λmax (Δε) 240 (−2.8), 318 (−5.2) nm; IR (KBr) νmax 3356, 2948, 1716, 1604, 1300, 1160 cm−1; 1H and 13C NMR data, Table 1; (−)-ESIMS m/z 307.1 [M − H]−; (−)-HRESIMS at m/z 307.0810 [M − H]− (calcd for C15H15O7 307.0818).ε) 216 (4.4), 240 (4.1), 279 (4.1), 320 (4.0) nm; ECD (MeOH) λmax (Δε) 215 (+3.3), 242 (+4.2), 329 (+0.7) nm; IR (KBr) νmax 3503, 2925, 1725, 1600, 1292, 1150 cm−1; 1H and 13C NMR data, Table 2; (−)-ESIMS m/z 321.1 [M − H]−; (−)-HRESIMS at m/z 321.0970 [M − H]− (calcd for C16H17O7 321.0974).ε) 216 (4.4), 240 (4.1), 279 (4.1), 320 (4.0) nm; ECD (MeOH) λmax (Δε) 240 (−6.0), 318 (−8.6) nm; IR (KBr) νmax 3458, 2950, 1739, 1610, 1298, 1158 cm−1; 1H and 13C NMR data, Table 2; (−)-ESIMS m/z 321.1 [M − H]−; (−)-HRESIMS at m/z 321.0973 [M − H]− (calcd for C16H17O7 321.0974).ε) 218 (4.4), 290 (4.0) nm; ECD (MeOH) λmax (Δε) 211 (−8.6), 233 (+6.3), 248 (+4.3), 284 (+6.1), 314 (−10.5) nm; IR (KBr) νmax 3410, 2925, 1744, 1655, 1287, 1225 cm−1; 1H and 13C NMR data, Table 3; (−)-ESIMS m/z 319.1 [M − H]−; (−)-HRESIMS at m/z 319.0818 [M − H]− (calcd for C16H15O7 319.0818).ε) 238 (4.4), 299 (3.8) nm; ECD (MeOH) λmax (Δε) 224 (−6.6), 250 (−3.0), 265 (−2.6), 318 (+3.1) nm; IR (KBr) νmax 3421, 2944, 1726, 1658, 1429, 1357, 1110 cm−1; 1H and 13C NMR data, Table 3; (−)-ESIMS m/z 349.1 [M − H]−; (−)-HRESIMS at m/z 349.0921 [M − H]− (calcd for C17H17O8 349.0923).ε) 220 (4.4), 290 (4.0) nm; IR (KBr) νmax 3420, 2923, 1736, 1615, 1292, 1151 cm−1; 1H and 13C NMR data, Table 3; (−)-ESIMS m/z 319.1 [M − H]−; (−)-HRESIMS at m/z 319.0811 [M − H]− (calcd for C16H15O7 319.0818).
:0; 9:1; 8:2; 7:3; 6:4; 5:5; 4:6; 3:7; 0:100, v/v, each 500 mL) to give nine fractions (F1–F9). F4 (1.5 g) was subjected to a silica gel column (5.0 × 30 cm), eluting with gradient of CH2Cl2/MeOH (100:1) to yield five subfractions (F401–F405). F401 (0.5 g) was further fractionated on Sephadex LH-20 (110 g, 110 × 3 cm) eluting with CH2Cl2–MeOH (1 L, 1:1, v/v) to give two sets of mixtures F401a (13 mg) and F401b (9 mg). After evaluation by the 13C NMR analysis, F401a and F401b both contained two sets of diastereoisomers. And then F401a was purified by HPLC on an S-Chiral A column, using hexane/2-propanol (70:30, v/v, flow rate: 1.5 mL min−1) as the solvent system, to yield 1 (5 mg, tR = 11.5 min) and 2 (3 mg, tR = 13.0 min), and F401b was also purified by HPLC on an S-Chiral A column, using hexane/2-propanol (90:10, v/v, flow rate: 1.5 mL min−1) as the solvent system, to obtain 3 (4 mg, tR = 34.9 min) and 4 (3 mg, tR = 38.8 min). F402 (200 mg) was purified by HPLC on a semipreparative SB-C18 column, using MeOH–H2O (45:55, v/v, flow rate: 1.0 mL min−1) as the solvent system, to afford F402-3 (5a/5b, 25 mg), F402-4 (6a/6b, 20 mg), and 7 (4 mg). F402-3 and F402-4 were further separated by chiral HPLC using an S-Chiral A column to yield 5a (8 mg) and 5b (10 mg), 6a (9 mg) and 6b (7 mg), respectively.
ε) 216 (4.4), 240 (4.1), 279 (4.1), 320 (4.0) nm; ECD (MeOH) λmax (Δε) 215 (+3.3), 242 (+4.2), 329 (+2.0) nm; IR (KBr) νmax 3367, 2936, 1732, 1620, 1306, 1166 cm−1; 1H and 13C NMR data, Table 1; (−)-ESIMS m/z 307.1 [M − H]−; (−)-HRESIMS at m/z 307.0818 [M − H]− (calcd for C15H15O7 307.0818).ε) 216 (4.4), 240 (4.1), 279 (4.1), 320 (4.0) nm; ECD (MeOH) λmax (Δε) 240 (−2.8), 318 (−5.2) nm; IR (KBr) νmax 3356, 2948, 1716, 1604, 1300, 1160 cm−1; 1H and 13C NMR data, Table 1; (−)-ESIMS m/z 307.1 [M − H]−; (−)-HRESIMS at m/z 307.0810 [M − H]− (calcd for C15H15O7 307.0818).ε) 216 (4.4), 240 (4.1), 279 (4.1), 320 (4.0) nm; ECD (MeOH) λmax (Δε) 215 (+3.3), 242 (+4.2), 329 (+0.7) nm; IR (KBr) νmax 3503, 2925, 1725, 1600, 1292, 1150 cm−1; 1H and 13C NMR data, Table 2; (−)-ESIMS m/z 321.1 [M − H]−; (−)-HRESIMS at m/z 321.0970 [M − H]− (calcd for C16H17O7 321.0974).ε) 216 (4.4), 240 (4.1), 279 (4.1), 320 (4.0) nm; ECD (MeOH) λmax (Δε) 240 (−6.0), 318 (−8.6) nm; IR (KBr) νmax 3458, 2950, 1739, 1610, 1298, 1158 cm−1; 1H and 13C NMR data, Table 2; (−)-ESIMS m/z 321.1 [M − H]−; (−)-HRESIMS at m/z 321.0973 [M − H]− (calcd for C16H17O7 321.0974).ε) 218 (4.4), 290 (4.0) nm; ECD (MeOH) λmax (Δε) 211 (−8.6), 233 (+6.3), 248 (+4.3), 284 (+6.1), 314 (−10.5) nm; IR (KBr) νmax 3410, 2925, 1744, 1655, 1287, 1225 cm−1; 1H and 13C NMR data, Table 3; (−)-ESIMS m/z 319.1 [M − H]−; (−)-HRESIMS at m/z 319.0818 [M − H]− (calcd for C16H15O7 319.0818).ε) 238 (4.4), 299 (3.8) nm; ECD (MeOH) λmax (Δε) 224 (−6.6), 250 (−3.0), 265 (−2.6), 318 (+3.1) nm; IR (KBr) νmax 3421, 2944, 1726, 1658, 1429, 1357, 1110 cm−1; 1H and 13C NMR data, Table 3; (−)-ESIMS m/z 349.1 [M − H]−; (−)-HRESIMS at m/z 349.0921 [M − H]− (calcd for C17H17O8 349.0923).ε) 220 (4.4), 290 (4.0) nm; IR (KBr) νmax 3420, 2923, 1736, 1615, 1292, 1151 cm−1; 1H and 13C NMR data, Table 3; (−)-ESIMS m/z 319.1 [M − H]−; (−)-HRESIMS at m/z 319.0811 [M − H]− (calcd for C16H15O7 319.0818).Determination of absolute configurations of the secondary alcohol in compounds 1–4
According to the published procedure,20,22 compounds 1–4 (0.5 mg) were dissolved in a dry solution of the stock [Rh2(OCOCF3)4] complex (1.5 mg) in CH2Cl2 (200 μL). The first CD spectrum was recorded immediately after mixing, and its time evolution was monitored until stationary (about 10 min after mixing). The inherent CD was subtracted. The observed sign of the E band at 350 nm in the induced CD spectrum was correlated to the absolute configuration of the C-1′ secondary alcohol.Cytotoxicity assay
As our previous reported method.12 Human breast cancer cell line MDA-MB-435 and immortalized non-cancer breast epithelial cells MCF10A were cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 5% fetal bovine serum (Hyclone, Logan, UT, USA), 100 μg mL−1 streptomycin, 2 mM L−1 glutamine and 100 units mL−1 penicillin (Invitrogen). The cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2.Cells were seeded in 96-well flat-bottom plates at a density of 1 × 104 cells per well and cultured in a humidified incubator for 24 h, followed by exposure to various concentrations of compounds for 48 h. Subsequently, 20 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reagent (Genview, Houston, TX, USA) dissolved in phosphate-buffered saline (PBS) (pH 7.4) at a concentration of 5 mg mL−1 was added to each well, and the cells were incubated for additional 4 h. The MTT-formazan crystals formed were dissolved in 150 μL DMSO (Sangon Biotech, Shanghai, China), and the absorbance was measured at 570 nm with a reference wavelength of 630 nm using a microplate reader. Cell growth inhibition was determined using the following formula according to a previously published method: growth inhibition (%) = (1 − OD of treated cells/OD of control cells) × 100%. The half maximal inhibitory concentration (IC50) was calculated from the dose-response curves with GraphPad Prism software.
Luciferase reporter gene activity assay
As previous reported method with slight modifications,23 the effect of isolated compounds on RANKL-induced NF-κB activation was conducted using RAW 264.7 cells stably transfected with an NF-κB driven luciferase reporter gene construct (3kB-Luc-SV40). Various concentrations of isolated compounds (1–6b) were treated for 1 h followed by 100 ng mL−1 RANKL for 6 h on RAW 264.7 cells stably transfected with an NF-κB-driven luciferase reporter gene. Luciferase activities were measured using a Promega Luciferase Assay System (Promega, Madison, WI, USA), and normalized to that of the vehicle control.Conclusions
In summary, seven new (1–4, 5a, 6a, and 7) and two known metabolites (5b, 6b) were isolated and identified from the culture of the endophytic fungus D. phaseolorum SKS019. Diaporchromanones A–D (1–4) possessing 3-substituted-chroman-4-one nucleus are rarely found in natural sources. To the best of our knowledge, the first 3-substituted-chroman-4-one derivative, polivione, was described in 1985.10 Diaporchromanones A–D (1–4) descripted in this paper were the second example of the 3-substituted-chroman-4-one analogues. Furthermore, chromanone components are reported for the first time from the Diaporthe sp. Our findings would enrich chemical context of the genus Diaporthe and expand the chemical and biological diversity of chromanones. In bioactivity assays, compounds 3–6b exhibited moderate inhibitory effects by suppressing RANKL-induced NF-κB activation using RAW 264.7 cell line.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the National Natural Science Foundation of China (21472251, 41276146), the Science & Technology Plan Project of Guangdong Province of China (2013B021100011), the Key Project of Natural Science Foundation of Guangdong Province (2016A040403091), Special Financial Fund of Innovative Development of Marine Economic Demonstration Project (GD2012-D01-001), China Postdoctoral Science Foundation Funded Project (2013M542223), and China's Marine Commonwealth Research Project (201305017) for generous support.Notes and references
- S. Saengchantara and T. Wallace, Nat. Prod. Rep., 1986, 3, 465–475 RSC.
- W. Y. Tsui and G. D. Brown, Phytochemistry, 1996, 43, 871–876 CrossRef CAS.
- D. L. Zhao, C. L. Shao, L. S. Gan, M. Wang and C. Y. Wang, J. Nat. Prod., 2015, 78, 286–293 CrossRef CAS PubMed.
- T. Ishikawa, Y. Oku, T. Tanaka and T. Kumamoto, Tetrahedron Lett., 1999, 40, 3777–3780 CrossRef CAS.
- F. Cottiglia, B. Dhanapal, O. Sticher and J. Heilmann, J. Nat. Prod., 2004, 67, 537–541 CrossRef CAS PubMed.
- D. G. Wei, G. F. Yang, J. Wan and C. G. Zhan, J. Agric. Food Chem., 2005, 53, 1604–1611 CrossRef CAS PubMed.
- G. Wu, G. Yu, T. Kurtán, A. Mándi, J. Peng, X. Mo, M. Liu, H. Li, X. Sun and J. Li, J. Nat. Prod., 2015, 78, 2691–2698 CrossRef CAS PubMed.
- D. J. Payne, J. A. Hueso-Rodríguez, H. Boyd, N. O. Concha, C. A. Janson, M. Gilpin, J. H. Bateson, C. Cheever, N. L. Niconovich and S. Pearson, Antimicrob. Agents Chemother., 2002, 46, 1880–1886 CrossRef CAS PubMed.
- M. Huang, J. Li, L. Liu, S. Yin, J. Wang and Y. Lin, Mar. Drugs, 2016, 14, 215 CrossRef PubMed.
- A. K. Demetriadou, E. D. Laue, F. J. Leeper and J. Staunton, J. Chem. Soc., Chem. Commun., 1985, 762–764 RSC.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2014, 31, 160–258 RSC.
- H. Cui, J. Yu, S. Chen, M. Ding, X. Huang, J. Yuan and Z. She, Bioorg. Med. Chem. Lett., 2017, 27, 803–807 CrossRef CAS PubMed.
- Y. Liu, Q. Yang, G. Xia, H. Huang, H. Li, L. Ma, Y. Lu, L. He, X. Xia and Z. She, J. Nat. Prod., 2015, 78, 1816–1822 CrossRef CAS PubMed.
- H. Cui, Y. Liu, Y. Nie, Z. Liu, S. Chen, Z. Zhang, Y. Lu, L. He, X. Huang and Z. She, Mar. Drugs, 2016, 14, 86 CrossRef PubMed.
- Z. Liu, Y. Chen, S. Chen, Y. Liu, Y. Lu, D. Chen, Y. Lin, X. Huang and Z. She, Org. Lett., 2016, 18, 1406–1409 CrossRef CAS PubMed.
- Y. Liu, G. Xia, H. Li, L. Ma, B. Ding, Y. Lu, L. He, X. Xia and Z. She, Planta Med., 2014, 80, 912–917 CrossRef CAS PubMed.
- S. Chen, Y. Liu, Z. Liu, R. Cai, Y. Lu, X. Huang and Z. She, RSC Adv., 2016, 6, 26412–26420 RSC.
- S. A. Ahmed, S. A. Ross, D. Slade, M. M. Radwan, I. A. Khan and M. A. Elsohly, Tetrahedron Lett., 2008, 49, 6050–6053 CrossRef CAS PubMed.
- D. Slade, D. Ferreira and J. P. J. Marais, Phytochemistry, 2005, 66, 2177–2215 CrossRef CAS PubMed.
- J. Frelek and W. J. Szczepek, Tetrahedron: Asymmetry, 1999, 10, 1507–1520 CrossRef CAS.
- M. Gerards and G. Snatzke, Tetrahedron: Asymmetry, 1990, 1, 221–236 CrossRef CAS.
- Y. Wang, Z. Zheng, S. Liu, H. Zhang, E. Li, L. Guo and Y. Che, J. Nat. Prod., 2010, 73, 920–924 CrossRef CAS PubMed.
- S. Yang, X. Li, L. Cheng, H. Wu, C. Zhang and K. Li, Biochem. Biophys. Res. Commun., 2015, 466, 615–621 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra of all new compounds (1H NMR, 13C NMR, 2D NMR, and HRESIMS). See DOI: 10.1039/c7ra03032k |
| This journal is © The Royal Society of Chemistry 2017 |