
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric transfer hydrogenation–Sonogashira coupling one-pot enantioselective tandem reaction catalysed by Pd(0)–Ru(III)/diamine-bifunctionalized periodic mesoporous organosilica†
Yuxi Zhao,
Ronghua Jin,
Yajie Chou,
Yilong Li,
Jingrong Lin and
Guohua Liu *
*
Key Laboratory of Resource Chemistry of Ministry of Education, Shanghai Key Laboratory of Rare Earth Functional Materials, Shanghai Normal University, Shanghai, 200234, China. E-mail: ghliu@shnu.edu.cn; Fax: +86-21-64322511; Tel: +86-21-64322280
First published on 25th April 2017
Abstract
Construction of a bifunctional heterogeneous catalyst through the immobilization of palladium nanoparticles within ethylene-bridged chiral ruthenium/diamine-functionalized periodic mesoporous organosilica is developed. Structural analyses and characterizations show its well-defined chiral single-site ruthenium species, and electron microscopy reveals its ordered mesostructure. As a bifunctional catalyst, it enables an efficient asymmetric transfer hydrogenation–Sonogashira coupling one-pot enantioselective tandem reaction, where Ru-catalysed asymmetric transfer hydrogenation followed by Pd-catalysed Sonogashira coupling affords various chiral conjugated alkynols in high yields with up to 97% enantioselectivity. As presented in this study, uniformly distributed palladium nanoparticles, high ethylene-bridged hydrophobicity, and single-site chiral ruthenium catalytic nature make a synergistic contribution to its catalytic performance. Furthermore, the high stability of palladium nanoparticles within its organosilicate network promotes high recyclability, and it could be used for the asymmetric transfer hydrogenation–Sonogashira coupling one-pot enantioselective tandem reaction of 4-iodoacetophenone and ethynylbenzene at least seven times without loss of its catalytic activity.
1. Introduction
Inspired by synergic multienzymatic reactions in biological systems, developement of multifunctional catalysts for multi-step sequential organic transformations is of practical interest but greatly challenging in asymmetric catalysis.1 Recently, by utilizing periodic mesoporous organosilicas (PMOs) as supports and incorporating multiple functionalities within their silicated networks, exploration of PMO-based heterogeneous catalysts for catalysis has made significant achievements.2 Some well-established PMO-based bifunctional heterogeneous catalysts, such as active site-isolated bifunctionalized catalysts, have realized various highly efficient tandem or sequential reactions.3 Despite great efforts made in construction of PMO-based bifunctional heterogeneous catalysts, most reports focus mainly on preparation of nonchiral molecules.4 Direct applications for construction of chiral molecules via an enantioselective tandem reaction are still rare.5 The main limitations are ascribed to two factors. One issue is that the cross-interference of two distinct organometallic complexes (active species) is difficult to avoid when they are anchored on a single material.6 Another issue is that a strict chiral configuration for catalytic mircoenvironment of active center is quite subtle. Therefore, exploration of alternative route to overcome intrinsic conflict of cross-interference and to retain originally chiral mircoenvironment of homogeneous catalysis is of great significance in asymmetric catalysis, which is beneficial to facilitate an enantioselective tandem reaction.Asymmetric transfer hydrogenation (ATH)7 and Sonogashira coupling reaction,8 as two types of classical catalytic reactions, are well-known to construct chiral conjugated acetylenic alcohols via ruthenium-catalysed ATH of ketones9 followed by palladium-catalysed Sonogashira coupling of arylalcohols and arylacetylenes. In particular, some of them can be used to prepare highly valuable natural products and chiral pharmaceuticals.10 However, among those highly efficient N-based Ru-complexes containing TsDPEN as ligands (TsDPEN: N-(2-amino-1,2-diphenylethyl)-4-methylbenzenesulfonamide)9 for ATH reductions and N- or P-based Pd-complexes with help of containing P and N ligands for Sonogashira coupling reactions,11 the cross-interference between metals and ligands often exists when both complexes are combined in one-pot manner. As a result, mismatched species between metals and ligands are unavoidable, which decrease greatly catalytic efficiency in a sequential organic transformation.
In order to eliminate the cross-interference of bifunctionality, our recent effort in the construction a yolk–shell-structured active site-isolated Pd(PPh3)/Ru(diamine)-bifunctionalized heterogeneous catalyst with PdCl2(PPh3)3-functionality into shell and chiral ruthenium/diamine-functionality onto core enables a Sonogashira coupling-ATH enantioselective tandem reaction.5a But, some inherent limitations, such as the complicated synthetic process of heterogeneous catalyst and the use of expensive phosphine ligands, still hinder greatly its practical application. In addition, the use of inorganosilica as a support also decreases its catalytic efficiency due to the inherent defect of slow mass diffusion during catalysis process. Interestingly, recent studies found that palladium nanoparticles immobilized on a solid-support without the addition of extra ligands also perform an highly efficiently Sonogashira reactions,8 which offers an alternative route to overcome this kind of cross-interference of bifunctionality through immobilization of palladium nanoparticles in hydrophobic chiral ruthenium/diamine-functionalized periodic mesoporous organosilica for this enantioselective tandem reaction. Benefit from palladium nanoparticles not only eliminates intrinsic conflict of cross-interference but also enhances heterogeneous catalyst' stability. As expected, assembly of the palladium nanoparticles in a chiral Ru/diamine-functionalized periodic mesoporous organosilica is synthetically simple, which enables an efficient ATH–Sonogashira coupling of iodoacetophenones and phenylacetylene.
2. Experimental
2.1. Characterization
Ru and Pd loading amounts in the catalyst was analysed using an inductively coupled plasma optical emission spectrometer (ICP, Varian VISTA-MPX). Fourier transform infrared (FTIR) spectra were collected on a Nicolet Magna 550 spectrometer using KBr method. X-ray powder diffraction (XRD) was carried out on a Rigaku D/Max-RB diffractometer with CuKα radiation. Scanning electron microscopy (SEM) images were obtained using a JEOL JSM-6380LV microscope operating at 20 kV. Transmission electron microscopy (TEM) images were performed on a JEOL JEM2010 electron microscope at an acceleration voltage of 220 kV. Nitrogen adsorption isotherms were measured at 77 K with a Quantachrome Nova 4000 analyser. The samples were measured after being outgassed at 423 K overnight. Pore size distributions were calculated by using the BJH model. The specific surface areas (SBET) of samples were determined from the linear parts of BET plots (p/p0 = 0.05–1.00). Solid state NMR experiments were explored on a Bruker AVANCE spectrometer at a magnetic field strength of 9.4 T with 1H frequency of 400.1 MHz, 13C frequency of 100.5 MHz and 29Si frequency of 79.4 MHz, and 19F frequency of 169.3 MHz with 4 mm rotor at two spinning frequency of 5.5 kHz and 8.0 kHz, TPPM decoupling is applied in the during acquisition period. 1H cross polarization in all solid state NMR experiments was employed using a contact time of 2 ms and the pulse lengths of 4 μs.2.2. Preparation of the heterogeneous catalyst 3
In a typical synthesis, 2.0 g of structure-directing agent, pluronic P123 ((CH2CH2O)20(CH2(CH3)CH2O)70(CH2CH2O)20), was completely dissolved in a mixture of 80 mL of hydrochloric acid (0.2 N) and 6.0 g of KCl. The mixture was stirred at room temperature for 1.0 h. Subsequently, 6.39 g (18.00 mmol) of the silica precursor 1,2-bis(triethoxysilyl)ethane was added at 40 °C. After a pre-hydrolysis period of 60 minute, 0.50 g (1.00 mmol) of (S,S)-DPEN-SO2Ph(CH2)2Si(OMe)3 (1) and 0.24 g (1.00 mmol) of 3-mercaptopropyltriethoxysilane were added. The reaction mixture was stirred at 40 °C for 24 h and then aged at 100 °C for 24 h. The resulting solid was filtered, rinsed with excess ethanol, and then dried overnight on a filter. The surfactant template was removed by refluxing in acidic ethanol (400 mL per gram) for 24 h. The solids were filtered, rinsed with ethanol again, and then dried at 60 °C under reduced pressure overnight to afford SH@ArDPEN@PMO (2) (3.62 g) in the form of a white powder. The part of collected solids (1.0 g) was suspended in 40 mL of dry ethanol, and 116.2 mg (0.66 mmol) of PdCl2 was added to the solution at ambient temperature. The resulting mixture was stirred for 12 h. After the reduction for 10 hours with NaBH4 (0.25 g, 6.60 mmol) at room temperature, the mixture was filtered through filter paper and then rinsed with excess water and CH2Cl2, and then dried at 60 °C under reduced pressure overnight to afford Pd@ArDPEN@PMO as in the form of a gray powder. The part of collected Pd@ArDPEN@PMO (0.50 g) was suspended in 20 mL of dry CH2Cl2 again, and 87.0 mg (0.15 mmol) of [RuCl2(mestylene)]2 was added to the solution at ambient temperature. The resulting mixture was stirred for 12 h. The mixture was filtered through filter paper and then rinsed with excess water and CH2Cl2. After Soxhlet extraction for 12 h in CH2Cl2 to remove homogeneous and unreacted starting materials, the solid was dried at ambient temperature under vacuum overnight to afford catalyst 3 (0.48 g) as a brown powder. ICP analysis showed that the Ru and Pd loadings were 10.98 mg (0.108 mmol) and 8.78 mg (0.0828 mmol) per gram of catalyst, respectively. 13C CP/MAS NMR (161.9 MHz): 136.3–116.6 (![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) of Ar and Ph groups), 102.1 ( of arene group), 72.4–68.4 ( of –NHPh), 60.8–54.2 ( of –OH2H2O– in P123 molecule), 31.3–23.8 ( of –H2Ar and of –SH2CH2CH2Si), 22.3–12.2 ( of arene H3, of –SCH2H2CH2Si, and of –CH2H3 in P123 molecule), 5.1 ( of –H2Si) ppm. 29Si MASNMR (79.4 MHz): T2 (δ = −58.7 ppm), T3 (δ = −65.2 ppm).
of Ar and Ph groups), 102.1 ( of arene group), 72.4–68.4 ( of –NHPh), 60.8–54.2 ( of –OH2H2O– in P123 molecule), 31.3–23.8 ( of –H2Ar and of –SH2CH2CH2Si), 22.3–12.2 ( of arene H3, of –SCH2H2CH2Si, and of –CH2H3 in P123 molecule), 5.1 ( of –H2Si) ppm. 29Si MASNMR (79.4 MHz): T2 (δ = −58.7 ppm), T3 (δ = −65.2 ppm).
2.3. General procedure for the ATH–Sonogashira coupling one-pot enantioselective tandem reaction
A typical procedure was as follows. Catalyst 3 (24.15 mg, 2.61 μmol of Ru, 2.0 μmol of Pd, based on ICP analysis), K2CO3 (27.6 mg, 0.20 mmol), HCO2Na (68.0 mg, 1.0 mmol), iodoacetophenone (0.10 mmol), aryne (0.11 mmol), and 4.0 mL of the mixed solvents (H2O/CH3OH v/v = 1/3) were added sequentially to a 10.0 mL round-bottom flask. The mixture was then stirred at 65 °C for 10–16 h. During this period, the reaction was monitored constantly by TLC. After completion of the reaction, the catalyst was separated by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm) for the recycling experiment. The aqueous solution was extracted with ethyl ether (3 × 3.0 mL). The combined ethyl ether extracts were washed with brine twice and then dehydrated with Na2SO4. After evaporation of ethyl ether, the residue was purified by silica gel flash column chromatography to afford the desired products. The ee values were determined by chiral HPLC analysis with a UV-vis detector using a Daicel chiralcel column (Φ 0.46 × 25 cm).
000 rpm) for the recycling experiment. The aqueous solution was extracted with ethyl ether (3 × 3.0 mL). The combined ethyl ether extracts were washed with brine twice and then dehydrated with Na2SO4. After evaporation of ethyl ether, the residue was purified by silica gel flash column chromatography to afford the desired products. The ee values were determined by chiral HPLC analysis with a UV-vis detector using a Daicel chiralcel column (Φ 0.46 × 25 cm).
3. Results and discussion
3.1. Synthesis and structural characterization of the heterogeneous catalyst
Immobilization of palladium nanoparticles within the ethylene-bridged chiral ruthenium/diamine-functionalized periodic mesoporous organosilica, abbreviated as Pd@mestyleneRuArDPEN@PMO (3) (MestyleneRuArDPEN:9 ArDPEN = (S,S)-4-((trimethoxysilyl)ethyl)phenylsulfonyl-1,2-diphenylethylene-diamine (1)), was performed through a co-condensation strategy. As outlined in Scheme 1, three-component co-condensation of 1, 3-(triethoxysilyl)propane-1-thiol, and 1,2-bis(triethoxysilyl)ethylane using the pluronic P123 as a structure-directing template gave mercapto- and chiral diamine-functionalized PMO, SH@ArDPEN@PMO (2), as a white powder. Anchor of PdCl2 within 2 followed by the reduction using NaBH4 as a reducing reagent gave the Pd@ArDPEN@PMO, which consecutive complexation with [RuCl2(mestylene)]2 led to catalyst 3 in the form of a light-yellow powder (see ESI in Fig. S1†). For comparison, an analogue 3′ of catalyst 3, (PdCl2@mestyleneRuArDPEN@PMO (3′)), was also prepared through a similar procedure without the reductive treatment (see ESI in Experimental Section†). | ||
| Scheme 1 Preparation of catalysts 3. | ||
Incorporation of well-defined chiral ruthenium/diamine species within the ethylene-bridged organosilicate network of 3 could be confirmed by solid-state 13C cross-polarization (CP)/magic angle spinning (MAS) NMR spectroscopy. As shown in Fig. 1, catalyst 3 produced the strong carbon signals of –SiH2H2Si– moiety around 5 ppm, corresponding to the ethylene-bridged organosilica. Characteristic peak around 102 ppm ascribed the carbon atoms of aromatic ring in the mestylene group, and those around 20 ppm were originated from the carbon atoms of the –H3 groups attached to the mestylene group. In addition, the peaks between 67 and 73 ppm were corresponded to carbon atoms of –NH groups connected to phenyl groups in ArDPEN moiety. All these observed carbon signals were similar to those of its homogeneous MestyleneRuTsDPEN, revealing that catalyst 3 had the same well-defined single-site active species as the MestyleneRuTsDPEN.9a Furthermore, the peak around 29 for the carbon atoms of –H2S– moiety could also be observed clearly. It was noteworthy that one groups of carbon signals were attributed to the carbon atoms at aromatic ring around 126 ppm, where the other two signals (about 176 and 76 ppm) denoted by asterisks were its rotational sidebands calculated by chemical shifts that often appeared in the MAS high speed rotation process. Two additional peaks around 17 and 59 ppm denoted by asterisks ascribed the carbon atoms of –OH2H2OH3 groups in P123 moiety left in the material. Solid-state 29Si MAS NMR spectrum of catalyst 3 (see ESI in Fig. S2†) produced one group of exclusive T signals, suggesting that all Si species were covalently attached to carbon atoms within its organosilica network. Two strong T signals around −59 and −65 ppm, corresponding to T2 {[R–Si(OSi)2(OH)]} and T3 [R–Si(OSi)3] (R = alkyl-linked MestyleneRuArDPEN groups or ethylene-bridged groups), demonstrated its organosilicate network with R–Si(OSi)2(OH) and R–Si(OSi)3 species as its main organosilicate wall that were similar to reported values.12
 | ||
| Fig. 1 Solid-state 29Si CP/MAS NMR spectra of 2 and catalyst 3. | ||
Ordered mesostructure and pore arrangement of 3 were further investigated by using nitrogen adsorption–desorption measurements, X-ray diffraction (XRD), and transmission electron microscopy (TEM). As shown in Fig. 2, the nitrogen adsorption–desorption isotherm of 3 was a typical type IV isotherm with an H1 hysteresis loop and a visible step at P/P0 = 0.45–0.85, indicating its mesostructure. The small-angle XRD pattern of catalyst 3 (see ESI in Fig. S3†) showed an intense d100 diffraction peak along with two similar weak diffraction peaks (d110, d200), suggesting its dimensional–hexagonal pore arrangement (p6mm).5b Its TEM image further confirmed its ordered dimensional–hexagonal pore arrangements (Fig. 3), where the palladium nanoparticles in its nanochannels could be observed clearly (also see ESI for the TEM viewed along the [001] direction in Fig. S4†).
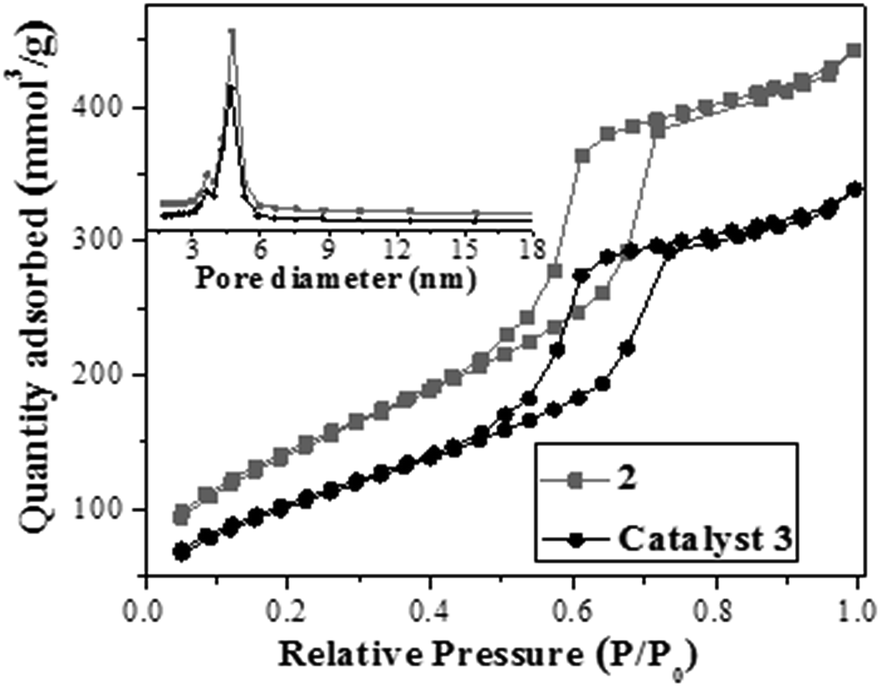 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms of 2 and catalyst 3. | ||
 | ||
| Fig. 3 TEM image of catalyst 3 viewed along the [100] direction. | ||
3.2. Catalytic properties of the heterogeneous catalyst
To obtain an efficient ATH–Sonogashira coupling one-pot enantioselective tandem reaction, the single-step Sonogashira reaction was optimized. In the this case, we investigated the Sonogashira coupling of 1-(4-iodophenyl)ethanone and ethynylbenzene catalyzed by 3 with 1.0 mol% of Pd-loading to compare its catalytic performance with those reported literatures at first.13 The results showed that this Sonogashira coupling reaction in 12 h could afford the 1-(4-(phenylethynyl)phenyl)ethanone in 95% yield, which was comparable to that of reported literature.13a Moreover, three different Pd-loadings of catalyst 3 in the Sonogashira coupling of (S)-1-(4-iodophenyl)ethanol and ethynylbenzene were further screened. Among three different contents of Pd-loadings (1.0 mol%, 2.0 mol% and 3.0 mol%), it was found that the Sonogashira reaction catalyzed by catalyst 3 with 2.0 mol% of Pd-loading gave the best result, which the reaction reached its catalytic complexion in 9 h with a clean product of (S)-1-(4-(phenylethynyl)phenyl)ethanol.On the basis of the optimized Sonogashira reaction, together with our previously well-established ATH of 4-iodoacetophenone,5 we then combined both reactions into one-pot process. In order to expatiate the catalytic nature and to examine the ATH–Sonogashira coupling catalytic sequence, we investigated its reaction time course in the ATH–Sonogashira coupling enantioselective tandem reaction of 4-iodoacetophenone and ethynylbenzene. As shown in Fig. 4, it was found that the ATH reaction occurs quickly concomitant with the Sonogashira coupling of C and phenylacetylene, where the tiny 1-(4-(phenylethynyl)phenyl)ethanone (B) was also observed. Subsequently, the Sonogashira coupling of C and phenylacetylene processes smoothly, reaching the reaction completion to provide (S)-1-(4-(phenylethynyl)phenyl)ethanol (D) in 10 h. This time course confirmed its catalytic nature in the ATH–Sonogashira coupling catalytic sequence.
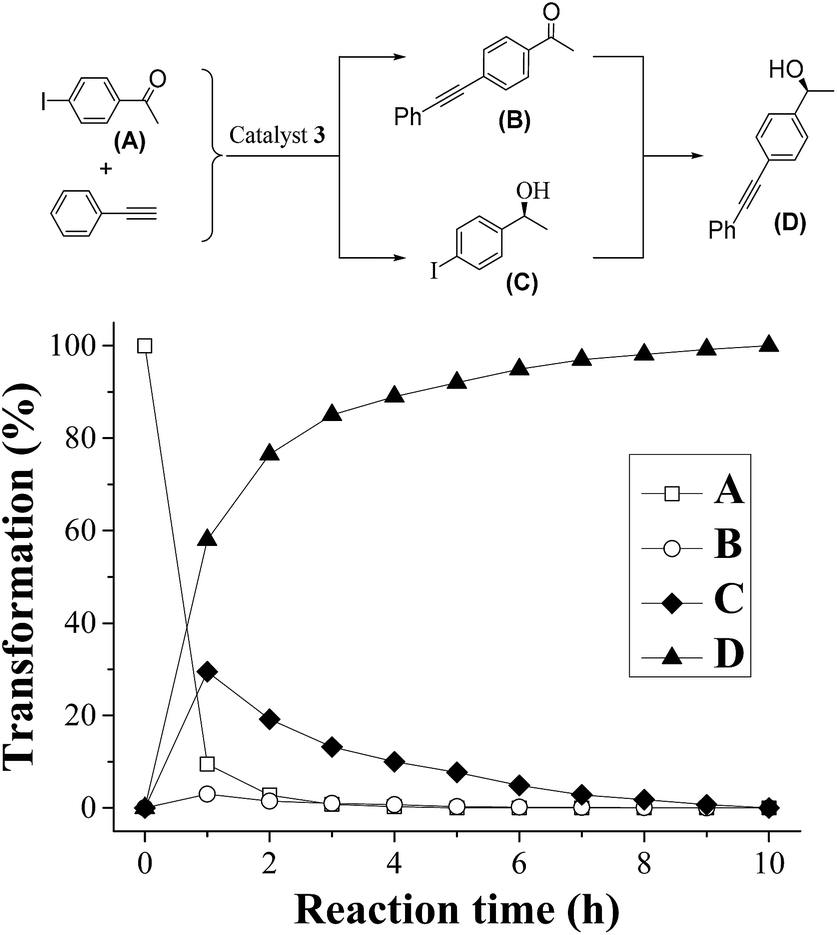 | ||
| Fig. 4 Time course for one-pot enantioselective tandem reaction of 4-iodoacetophenone and ethynylbenzene with catalyst 3 (65 °C, cat. = 2.6 mmol% of Ru and 2.0 mmol% of Pd, based on ICP analysis). | ||
It is worth mentioning that this ATH–Sonogashira coupling catalytic sequence was opposite of a Sonogashira coupling–ATH catalytic sequence.5a This finding demonstrated that the low active palladium nanoparticles (see ESI for XPS of catalyst 3 in Fig. S5†) relative to highly active PdCl2(PPh3)3 also enabled an efficiently enantioselective tandem reaction of 4-iodoacetophenone and ethynylbenzene. In order to confirm this ATH–Sonogashira coupling catalytic sequence that ascribed possibly the low active palladium nanoparticles in catalyst 3, a reaction time course in the enantioselective tandem reaction of 4-iodoacetophenone and ethynylbenzene catalysed by its analogue PdCl2@mestyleneRuArDPEN@PMO (3′) was also investigated. The result showed that this tandem reaction also presented a Sonogashira coupling–ATH catalytic sequence (see ESI for the time course in transformation of 4-iodoacetophenone and ethynylbenzene with 3′ in Fig. S6†), confirming that the highly active PdCl2 as catalytic species relative to the low active palladium nanoparticles was also an opposite catalytic sequence.
Furthermore, it is notable that such a clean ATH–Sonogashira coupling of 4-iodoacetophenone and ethynylbenzene was better than that obtained with its analogue 3′ because the latter afforded a low yield and ee value (Table 1, entry 1 versus entry 2). This finding indicated the existence of cross-interference between active species in 3′, suggesting the palladium nanoparticles in the designed catalyst 3 could be eliminated efficiently. The existance of cross-interference could be proved by using the mixed homogeneous PdCl2 plus the homogeneous AreneRuTsDPEN as the dual catalyst, where the enantioselective tandem reaction only afforded the target chiral products in 76% yield with 77% ee value (Table 1, entry 1 versus entry 1 in bracket 1). All these observations elucidate the benefit of the designed heterogenous catalyst 3 in the control of cross-interference.
|
||||
|---|---|---|---|---|
| Entry | X | Ar | Yield (%) | ee (%)b |
| a Reaction conditions: catalyst 3 (24.15 mg, 2.61 μmol of Ru, 2.0 μmol of Pd, based on ICP analysis), K2CO3 (27.6 mg, 0.20 mmol), HCO2Na (68.0 mg, 1.0 mmol), iodoacetophenone (0.10 mmol), aryne (0.11 mmol), and 4.0 mL of the mixed solvents (H2O/CH3OH v/v = 1/3) were added sequentially to a 10.0 mL round-bottom flask. The mixture was then stirred at 65 °C for 10–16 h.b Yields were determined by 1H-NMR analysis and ee values were determined by chiral HPLC analysis (see ESI in Fig. S7 and S91).c Data were obtained by the use of the mixed PdCl2 plus the homogeneous AreneRuTsDPEN as a combined catalyst.d Data was obtained by the use of its analogue 3′ as a catalyst. | ||||
| 1 | 4-I | Ph | 99(76) | 95(77)c |
| 2 | 4-I | Ph | 85 | 89d |
| 3 | 4-I | 4-FPh | 95 | 97 |
| 4 | 4-I | 3-FPh | 94 | 96 |
| 5 | 4-I | 4-ClPh | 93 | 96 |
| 6 | 4-I | 4-CF3Ph | 92 | 95 |
| 7 | 4-I | 4-OMePh | 93 | 93 |
| 8 | 4-I | 4-MePh | 96 | 96 |
| 9 | 4-I | 3-MePh | 95 | 96 |
| 10 | 3-I | Ph | 94 | 95 |
| 11 | 3-I | 4-FPh | 96 | 97 |
| 12 | 3-I | 3-FPh | 97 | 96 |
| 13 | 3-I | 4-ClPh | 97 | 96 |
| 14 | 3-I | 4-CF3Ph | 98 | 97 |
| 15 | 3-I | 4-OMePh | 94 | 96 |
| 16 | 3-I | 4-MePh | 93 | 96 |
| 17 | 3-I | 3-MePh | 92 | 96 |
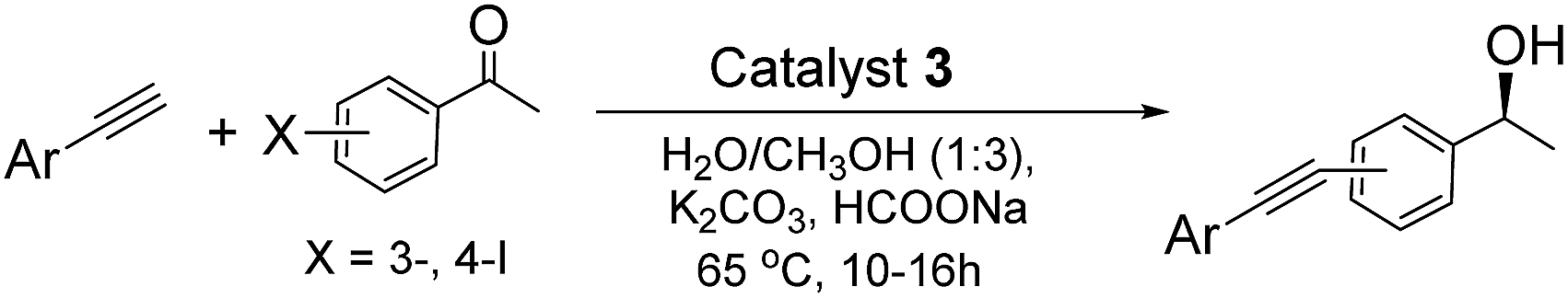
Having established an efficient ATH–Sonogashira coupling of 4-iodoacetophenone and ethynylbenzene, one-pot enantioselective tandem reactions with a series of substituted substrates were further tested for investigation of its general applicability. As shown in Table 1, the tandem reactions with all tested substrates catalysed by 3 could be converted smoothly into the corresponding chiral conjugated alkynols in high yields without intermediates under the same reaction conditions. Furthermore, these reactions were highly enantioselective, where the structures and electronic properties of the substituents on the aromatic ring did not significantly affect their enantioselectivities. Various electron-withdrawing and -donating substituents on the aromatic ring were equally efficient regardless of the reactions them with meta- or para-position iodoacetophenone (entries 3–17).
More importantly, this ATH–Sonogashira coupling one-pot enantioselective tandem reaction could also be applied in the preparation of chiral diols. As shown in Scheme 2, one representative distereocentered diols could be obtained, where the reaction of 1-(4-ethynylphenyl)ethanone and 3-iodoacetophenone catalysed by 3 afforded steadily the chiral products (S)-1-(3-((4-((S)-1-hydroxyethyl)phenyl)ethynyl)phenyl)ethanol with high diastereoselectivity.
 | ||
| Scheme 2 Extension of the ATH–Sonogashira coupling one-pot enantioselective tandem reaction. | ||
Another important aim in the design of the heterogeneous catalyst 3 is to expect that the heterogeneous catalyst 3 could be recovered easily and the recycled catalyst 3 could keep its catalytic activity and enantioselectivity after multiple cycles. As observed, catalyst 3 could be easily recovered by simple centrifugation and reused repeatedly. As shown in Table 2, in seven consecutive reactions, the ATH–Sonogashira one-pot tandem reaction of 4-iodoacetophenone and phenylacetylene still afforded the chiral products with 93% conversion and 93% ee value (see ESI in Fig. S8†). The obviously decreased conversion at eighth recycle was attributed mainly to the Pd-leaching in catalyst 3, where an inductively coupled plasma (ICP) optical emission spectrometer analysis found that the loss of Pd and Ru at eighth recycle were 11.5% and 7.2% relative to those of 6.9% and 6.7% at seventh recycle.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Run time | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| a Reaction conditions: catalyst 3 (241.5 mg), K2CO3 (276.0 mg, 2.0 mmol), HCO2Na (680 mg, 10.0 mmol), 4-iodoacetophenones (1.0 mmol), phenylacetylene (1.1 mmol), and 40.0 mL of the mixed solvents (H2O/MeOH v/v = 1/3), reaction temperature (65 °C), reaction time (10 h). | ||||||||
| Conversion [%] | 99 | 99 | 97 | 98 | 96 | 95 | 93 | 83 |
| ee [%] | 96 | 96 | 96 | 96 | 95 | 95 | 93 | 92 |

4. Conclusions
In conclusions, by assembling palladium nanoparticles within a chiral Ru/diamine-functionality periodic mesoporous organosilica, we construct a bifunctional heterogeneous catalyst. By utilizing the relatively low reactivity of palladium nanoparticles in Sonogashira coupling reaction, we perform an efficiently enantioselective tandem reactions of iodoacetophenone and arylacetylenes through the control of an ATH–Sonogashira coupling catalytic sequence. As presented in this study, the efficient catalytic performance is benefited from the combined contributions of the uniformly distributed small-size palladium nanoparticles, high ethylene-bridged hydrophobicity, and well-defined chiral single-site ruthenium active center. Furthermore, the highly stable palladium nanoparticles within a functionalized periodic mesoporous organosilica can result in a high recyclability in asymmetric transfer hydrogenation–Sonogashira coupling one-pot tandem reaction of 4-iodoacetophenone and ethynylbenzene, which the recycled catalyst could be used repeatedly for at least seven times without loss of its catalytic activity. This study described here also demonstrates that the combination of palladium nanoparticles within chiral Ru(III)/diamine-functionality periodic mesoporous organosilica overcomes efficiently the cross-interference of active species and performs a practical tandem reaction.Acknowledgements
We are grateful to China National Natural Science Foundation (21672149), Ministry of Education of China (PCSIRT-IRT-16R49), and Shanghai Municipal Education Commission (14YZ074) for financial support.References
- (a) D. E. Fogg, N. Eduardo and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS; (b) J. C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed; (c) M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2010, 111, 1072–1133 CrossRef PubMed; (d) M. J. Climent, A. Corma, S. Iborra and M. J. Sabater, ACS Catal., 2014, 4, 870–891 CrossRef CAS; (e) Y. M. He, Y. Feng and Q. H. Fan, Acc. Chem. Res., 2014, 47, 2894–2906 CrossRef CAS PubMed; (f) C. Li, H. D. Zhang, D. M. Jiang and Q. H. Yang, Chem. Commun., 2007, 547–558 RSC; (g) J. M. Fraile, J. I. García and J. A. Mayoral, Chem. Rev., 2009, 109, 360–417 CrossRef CAS PubMed; (h) M. Bartók, Chem. Rev., 2010, 110, 1663–1705 CrossRef PubMed; (i) C. G. Yu and J. He, Chem. Commun., 2012, 48, 4933–4940 RSC.
- (a) A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed; (b) N. Mizoshita, T. Taniab and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC; (c) F. Hoffmann and M. Fröba, Chem. Soc. Rev., 2011, 40, 608–620 RSC; (d) F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed; (e) M. Kruk, Acc. Chem. Res., 2012, 45, 1678–1687 CrossRef CAS PubMed; (f) J. A. Melero, R. van Grieken and G. Morales, Chem. Rev., 2006, 106, 3790–3812 CrossRef CAS PubMed; (g) Q. Yang, J. Liu, L. Zhang and C. Li, J. Mater. Chem., 2009, 19, 1945–1955 RSC; (h) P. van der Voort, D. Esquivel, E. De Canck, F. Goethals, I. van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC; (i) T. Y. Cheng, Q. K. Zhao, D. C. Zhang and G. H. Liu, Curr. Org. Chem., 2015, 19, 667 CrossRef CAS.
- (a) R. Srirambalaji, S. Hong, R. Natarajan, M. Yoon, R. Hota, Y. Kim, Y. H. Ko and K. Kim, Chem. Commun., 2012, 48, 11650–11652 RSC; (b) N. R. Shiju, A. H. Alberts, S. Khalid, D. R. Brown and G. Rothenberg, Angew. Chem., 2011, 123, 9789–9793 CrossRef.
- (a) Y. F. Lu, H. Y. Fan, N. Doke, D. A. Loy, R. A. Assink, D. A. Lavan and C. J. Brinker, J. Am. Chem. Soc., 2000, 122, 5258 CrossRef CAS; (b) B. D. Hatton, K. Landskron, W. Whitnall, D. D. Perovic and G. A. Ozin, Adv. Funct. Mater., 2005, 15, 823 CrossRef CAS; (c) M. P. Kapoor, Q. H. Yang and S. Inagaki, J. Am. Chem. Soc., 2002, 124, 15176 CrossRef CAS PubMed; (d) P. Borah, X. Ma, K. T. Nguyen and Y. Zhao, Angew. Chem., Int. Ed., 2012, 51, 7756 CrossRef CAS PubMed; (e) M. Waki, N. Mizoshita, T. Tani and S. Inagaki, Angew. Chem., Int. Ed., 2011, 50, 11667 CrossRef CAS PubMed; (f) M. Guan, W. Wang, E. J. Henderson, Ö. Dag, C. Kübel, V. S. K. Chakravadhanula, J. Rinck, I. L. Moudrakovski, J. Thomson, J. McDowell, A. K. Powell, H. Zhang and G. A. Ozin, J. Am. Chem. Soc., 2012, 134, 8439 CrossRef CAS PubMed; (g) M. Mandal and M. Kruk, Chem. Mater., 2012, 24, 123 CrossRef CAS; (h) B. X. Deng, W. Xiao, C. B. Li, F. Zhou, X. L. Xia, T. Y. Cheng and G. H. Liu, J. Catal., 2014, 320, 70 CrossRef CAS.
- (a) J. Y. Xu, T. Y. Cheng, K. Zhang, Z. Y. Wang and G. H. Liu, Chem. Commun., 2016, 52, 6005–6008 RSC; (b) D. C. Zhang, J. Y. Xu, Q. K. Zhao, T. Y. Cheng and G. H. Liu, ChemCatChem, 2014, 6, 2998–3003 CrossRef CAS; (c) L. Y. Kong, J. W. Zhao, T. Y. Cheng, J. R. Lin and G. H. Liu, ACS Catal., 2016, 6, 2244–2249 CrossRef CAS; (d) J. Z. An, T. Y. Cheng, X. Xiong, L. Wu, B. Han and G. H. Liu, Catal. Sci. Technol., 2016, 6, 5714–5720 RSC; (e) D. C. Zhang, X. S. Gao, T. Y. Cheng and G. H. Liu, Sci. Rep., 2014, 4, 5091–5097 CrossRef PubMed.
- H. U. Blaser and E. Schmidt, Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions, Wiley-VCH, Weinheim, 2010 Search PubMed.
- (a) R. Malacea, R. Poli and E. Manoury, Coord. Chem. Rev., 2010, 254, 729–752 CrossRef CAS; (b) X. F. Wu, J. K. Liu, D. D. Tommaso, J. O. Iggo, C. R. A. Catlow, J. Bacsa and J. L. Xiao, Chem.–Eur. J., 2008, 14, 7699–7715 CrossRef CAS PubMed; (c) K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef CAS; (d) S. Eagon, C. DeLieto, W. J. McDonald, D. Haddenham, J. Saavedra, J. Kim and B. Singaram, J. Org. Chem., 2010, 75, 7717–7725 CrossRef CAS PubMed; (e) E. T. Newcomb and E. M. Ferreira, Org. Lett., 2013, 15, 1772–1775 CrossRef CAS PubMed.
- L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS PubMed.
- (a) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS; (b) T. Ohkuma, K. Tsutsumi, N. Utsumi, N. Arai, R. Noyori and K. Murata, Org. Lett., 2007, 9, 255–257 CrossRef CAS PubMed.
- (a) H. Doucet and J. C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed; (b) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed; (c) K. Mori and H. Akao, Tetrahedron Lett., 1978, 43, 4127–4130 CrossRef; (d) A. F. Kluge, D. J. Kertesz, C. O. Yang and H. Y. Wu, J. Org. Chem., 1987, 52, 2860–2868 CrossRef CAS; (e) W. S. Johnson, R. S. Brinkmeyer, V. M. Kapoor and T. M. Yarnell, J. Am. Chem. Soc., 1977, 99, 8341–8343 CrossRef CAS PubMed; (f) L. E. Overman and K. L. Bell, J. Am. Chem. Soc., 1981, 103, 1851–1853 CrossRef CAS; (g) N. Cohen, R. J. Lopresti, C. Neukom and G. Saucy, J. Org. Chem., 1980, 45, 582–588 CrossRef CAS; (h) G. Stork and E. Nakamura, J. Am. Chem. Soc., 1983, 105, 5510–5512 CrossRef CAS; (i) R. A. Gibbs and W. H. Okamura, J. Am. Chem. Soc., 1988, 110, 4062–4063 CrossRef CAS; (j) H. Y. Kim, K. Stein and P. L. Toogood, Chem. Commun., 1996, 1683–1684 RSC.
- (a) R. Ciriminna, V. A. Pandarus, G. Gingras, F. Béland, P. D. Cará and M. Pagliaro, ACS Sustainable Chem. Eng., 2013, 1, 57–61 CrossRef CAS; (b) C. He, J. Ke, H. Xu and A. W. Lei, Angew. Chem., Int. Ed., 2013, 52, 1527–1530 CrossRef CAS PubMed; (c) M. García-Melchor, M. C. Pacheco, C. Nájera, A. Lledós and G. Ujaque, ACS Catal., 2012, 2, 135–144 CrossRef; (d) X. T. Pu, H. B. Li and T. J. Colacot, J. Org. Chem., 2013, 78, 568–581 CrossRef CAS PubMed; (e) C. W. D. Gallop, M. T. Chen and O. Navarro, Org. Lett., 2014, 16, 3724–3727 CrossRef CAS PubMed.
- (a) O. Krőcher, O. A. Kőppel, M. Frőba and A. Baiker, J. Catal., 1998, 178, 284–298 CrossRef; (b) N. Linares, A. E. Sepúlveda, M. C. Pacheco, J. R. Berenguer, E. Lalinde, C. Nájera and J. Garcia-Martinez, New J. Chem., 2011, 35, 225–234 RSC.
- (a) W. Xu, H. M. Sun, B. Yu, G. F. Zhang, W. Q. Zhang and Z. W. Gao, ACS Appl. Mater. Interfaces, 2014, 6, 20261–20268 CrossRef CAS PubMed; (b) Q. Cui, H. Zhao, G. S. Luo and J. H. Xu, Ind. Eng. Chem. Res., 2017, 56, 143–152 CrossRef CAS; (c) L. Yu, Z. Han and Y. H. Ding, Org. Process Res. Dev., 2016, 20, 2124–2129 CrossRef CAS; (d) P. Venkatesan and J. Santhanalakshmi, Langmuir, 2010, 26, 12225–12229 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03029k |
| This journal is © The Royal Society of Chemistry 2017 |