
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic anion transporters 1 (OAT1) and OAT3 meditated the protective effect of rhein on methotrexate-induced nephrotoxicity
Zhihao Liu *ab,
Yongming Jiaa,
Changyuan Wangab,
Qiang Mengab,
Xiaokui Huoab,
Huijun Sunab,
Pengyuan Suna,
Xiaobo Yanga,
Xiaodong Maa,
Jinyong Pengab and
Kexin Liu*ab
*ab,
Yongming Jiaa,
Changyuan Wangab,
Qiang Mengab,
Xiaokui Huoab,
Huijun Sunab,
Pengyuan Suna,
Xiaobo Yanga,
Xiaodong Maa,
Jinyong Pengab and
Kexin Liu*ab
aDepartment of Clinical Pharmacology, College of Pharmacy, Dalian Medical University, 9 West Section, Lvshun South Road, Lvshunkou District, Dalian 116044, China. E-mail: liuzhihao12399@126.com; kexinliu@dlmedu.edu.cn; Fax: +86 411 8611 0407; Tel: +86 411 8611 0407
bProvincial Key Laboratory for Pharmacokinetics and Transport, Dalian Medical University, Liaoning, Dalian 116044, China
First published on 11th May 2017
Abstract
Rhein is identified as a major metabolite of diacerein, a prodrug used in the treatment of osteoarthritis. Methotrexate (MTX) is a highly toxic drug with a low therapeutic index. MTX and diacerein are used concomitantly in clinical practice for the treatment of rheumatoid arthritis. The purpose of this study was to investigate organic anion transporters (OAT)-mediated effect of rhein on the disposition of MTX, as well as to clarify the changes in nephrotoxicity of MTX when given rhein together. Concomitant administration of rhein could strongly increase the systemic exposure in rats. Similarly, MTX accumulation was markedly inhibited by rhein in rat kidney slices and hOAT1/3-HEK293 cell, indicating that OAT1 and OAT3 were involved in the drug–drug interaction (DDI) in the kidney. Additionally, concomitant administration of rhein attenuated cytotoxic effects of MTX in hOAT1/3-HEK293 cells, and alleviated nephrotoxicity caused by MTX in rats. In conclusion, rhein decreased renal elimination of MTX by inhibiting OAT1 and OAT3, and alleviated MTX-induced renal toxicity in vivo.
1. Introduction
Methotrexate (MTX), which is known as an antitumor drug through anti-metabolism, is extensively used in clinical in the treatment of rheumatoid arthritis and cancer chemotherapy.1,2 However, MTX is a highly toxic drug with a low therapeutic index. The side-effects of MTX include gastrointestinal disturbance, bone marrow suppression, serious infections, abnormal liver function and kidney damage, and, rarely, severe and life threatening pneumonitis.3–5 Given the narrow therapeutic index of MTX and the frequent requirement for careful individual patient dose titration, any DDI which significantly changes the pharmacokinetic profile of MTX is potentially clinically important.Rhubarb (Rheum officinale) constitutes an important food source and is consumed across the world in form of rhubarb pie, tarts and crumbles. It is also used in preparation of fruit wine and many baked food preparations. Rhein, a lipophilic anthraquinone, is one of the most important active components of rhubarb, which is widely used for its antidotal, anti-inflammatory, antipyretic and laxative properties in Asian countries including China, Korea, and Japan.6,7 This medicinal herb is also employed to treat diabetic nephropathy.8 It is identified as a major metabolite of diacerein, a prodrug used in the treatment of osteoarthritis.9,10 In vivo, diacerein is completely converted to rhein before entering the systemic circulation.11 In clinical practice, diacerein is utilized in polypharmacy therapies, e.g., a fixed dose of diacerein (50 mg) and aceclofenac (100 mg) has been approved in India to treat osteoarthritis.12 Recently, a phase II clinical trial has been completed in Thailand to investigate the safety and efficacy of the combined therapy of diacerein and MTX to treat early rheumatoid arthritis (http://clinicaltrials.gov/ct2/show/NCT01264211).
Transporters play an important role in the absorption, distribution, and excretion of many drugs.13–15 It is well known that transporter-mediated DDI has been shown to result in significant changes in drug disposition, efficacy, and even toxicity.16–18 Based on previous studies, hOAT1 and hOAT3, immunolocalized to the basolateral side of proximal tubules, were suggested to mediate the disposition of MTX.19–21 Furthermore, hOATs involved in the renal clearance of MTX, and would be a key factor in MTX therapy and nephrotoxicity.22 However, it was also showed that rhein was a potent inhibitor for hOAT1 and hOAT3, and could interfere with hOAT1- and hOAT3-mediated renal elimination in vivo, leading to unintended changes in pharmacokinetics, pharmacodynamics and toxicity of other drugs.23,24 As MTX and rhein are used together in clinic, the potential DDIs risk associated with the OAT transporters should be aware. In this study, we investigated the OAT transporters-mediated effect of rhein on the disposition of MTX, and to understand the changes in nephrotoxicity of MTX when the two drugs were co-administered.
2. Materials and methods
2.1. Chemicals
MTX was purchased from Sigma (ST. Louis, Mo, USA). Rhein and probenecid (PRO) were obtained from Dalian Meilun Biotech Co., Ltd. (China). MTT was provided by USB Corporation (Cleveland, OH, USA). All other reagents and solvents were of analytical grade and were commercially available.2.2. Cell culture
The stable transfected OAT1-human embryonic kidney (HEK) 293 cells, hOAT3-HEK293 cells and mock-HEK293 cells were a gift from Professor Gong Likun (Shanghai Institute of Materia Medica, Chinese Academy of Science, Shanghai, China). The cells were grown in low-glucose Dulbecco's Modified Eagle's Medium (DMEM) (Invitrogen, Carlsbad, CA) supplemented with 10% (v/v) fetal bovine serum (Invitrogen), 100 U ml−1 of penicillin, and 100 mg ml−1 of streptomycin at 37 °C with a 5% (v/v) CO2 atmosphere and 95% relative humidity. Culture medium was changed every other day, and the cells were passaged every 3–5 days by digesting with 0.5% trypsin and 0.02% EDTA at 37 °C.2.3. Animals
Male Wistar rats weighing 200–250 g, obtained from the Experimental Animal Center of Dalian Medical University (Dalian, China; permit number SCXK 2013-0003), were allowed free access to water and chow diet. The animals were fasted for 12 h before pharmacokinetic experiments.2.4. Live subject statement
All experiments were approved by the Animal Care and Use Committee of Dalian Medical University, and the experimental procedures were strictly carried out in accordance with Legislation Regarding the Use and Care of Laboratory Animals of China. No experiments involved human subjects.2.5. In vivo pharmacokinetic interaction in rats
In all cases, rats were fasted overnight but allowed free access to water prior to pharmacokinetic experiments and were anesthetized with pentobarbital (60 mg kg−1, i.p.) before the onset of each experiment. For oral study, rats received an oral administration of MTX (5 mg kg−1) dissolved in 2% sodium bicarbonate and brought up to volume with normal saline with or without rhein (10 mg kg−1, dissolved in 0.5% sodium carboxymethyl cellulose). Blood samples (0.2 ml) were collected via jugular vein at 5, 10, 20, 30 min, 1, 2, 4, 6, 8, 10, 12 and 24 h in heparin tubes for MTX determination as described before.25 For i.v. study, rats were received an i.v. administration of MTX (5 mg kg−1) and/or rhein (1 mg kg−1) dissolved in normal saline solution of hydroxypropyl-β-cyclodextrin via the jugular vein. Then blood samples (0.2 ml) were collected from the jugular vein at 1, 5, 10, 20, 30 min, 1, 2, 4, 6, 8, 10, 12 and 24 h.2.6. In vivo renal excretion in rats
Rats were fasted overnight but allowed free access to water prior to pharmacokinetic experiments and were anesthetized with pentobarbital (60 mg kg−1, i.p.) before the onset of each experiment. Then rats were received an i.v. administration of MTX (5 mg kg−1) and/or rhein (1 mg kg−1) according to the above process. Bladders were cannulated with polyethylene tubing for urine collection at 2, 4, 6, 8, 10, 12, 16, and 24 h after dosing. The concentrations of MTX in and urine were measured as described before.262.7. In vitro uptake in kidney slices
The kidneys of rats were incised, decapsulated, and immediately placed into oxygenated Krebs-bicarbonate slicing buffer (120 mM NaCl, 16.2 mM KCl, 1 mM CaCl2, 1.2 mM MgSO4, and 10 mM NaH2PO4/Na2HPO4, pH 7.4) at 4 °C and then rats' kidney cortical tissues were cut into slices as previously described.27,28 After pre-incubation for 3 min in 6-well culture plates with oxygenated buffer at 37 °C, kidney slices were transferred to 24-well culture plates containing fresh carbogen-saturated buffer with MTX (5 μM) or MTX and rhein (2 μM) for further incubation at 37 °C. The uptake was terminated by removing the buffer and washing the slices with ice-cold saline at 3, 5, 10, 15 and 30 min. After homogenization (IKA-T 10 homogenizer; IKA, Staufen, Germany), the accumulated concentrations of MTX in kidney slices were determined by LC-MS/MS.2.8. In vitro transporter uptake assays
Uptake experiments with hOAT1-HEK293 cells and hOAT3-HEK293 cells were performed as previously described.29 HEK293 cells were seeded in 24-well culture plates 48 h before uptake studies. After the cells were washed three times and incubated with transport buffer (118 mM NaCl, 23.8 mM NaHCO3, 4.8 mM KCl, 1.0 mM KH2PO4, 1.2 mM MgSO4, 12.5 mM HEPES, 5.0 mM glucose, 1.5 mM CaCl2, pH 7.4) at 37 °C for 15 min, uptake was initiated by adding 1 ml transport buffer containing MTX (5 μM) with or without rhein (0–2 μM). The uptake was terminated at 2 min upon removal of the medium and three washes. Then cell monolayers were lysed with 0.3 ml of 0.1% (v/v) Triton X-100, and then samples were analyzed by LC-MS/MS. Samples were then analyzed by LC-MS/MS as described before.2.9. MTT cytotoxicity assay
Cell viability was determined using an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Mock-HEK293, hOAT1-HEK293 and hOAT3-HEK293 cells were collected and seeded in 96-well plates (4 × 103 cells per well). After 24 h of culture, cells were exposed to a series of concentrations of MTX in the presence or absence of rhein (2 μM), and were cultured at 37 °C for 24 h. Following incubation with MTT (5 mg ml−1, 10 μl) for another 4 h, each well was added 100 μl per well of SDS-isobutanol-HCl solution (10% SDS, 5% isobutanol, and 12 μM HCl) and the plate was incubated overnight at 37 °C. Optical density in each well was measured at 570 nm with the microplate reader (Bio-Rad, San Diego, CA, USA). The IC50 values were calculated by Prism (Graphpad Software, La Jolla, CA, USA).2.10. Plasma markers of kidney damage
Rats were divided randomly into 4 groups (n = 3). Control groups received the vehicle alone via i.v. injection for 5 days. The MTX group received 5 consecutive daily i.v. injections of MTX at a dose of 5 mg kg−1 body weight. The rhein group received 5 consecutive daily i.v. injections of rhein (1 mg kg−1). The combination group received 5 consecutive daily i.v. injections of MTX (5 mg kg−1) and rhein (1 mg kg−1). Plasma markers of kidney function, blood urea nitrogen (BUN) and creatinine, were determined using a test kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).2.11. Histopathological staining and analysis
The rats were divided randomly into 4 groups as described above. Rat kidney cortex was removed for histopathological examination on the 6th day. Renal cortex was fixed in 10% neutral buffered formalin immediately after the application of ether and left in the same fixative solution for 24 h before being embedded in paraffin. Specimens (3 μm) were prepared for hematoxylin–eosin (H&E) for morphological evaluation.2.12. TUNEL assay
The rats were divided randomly into 4 groups as described above. Rat kidney cortex was removed for histopathological examination on the 6th day. Kidney tissue samples embedded in paraffin were prepared, and used for TUNEL assay. We utilized the apoptosis detection kit according to the manufacturer's instructions (in situ Cell Death Detection Kit, Fluorescein, Roche, Basle, Sweden). 10 microscopic fields within the view were randomly selected, and the positive cells (green spots) were counted. The extent of damage was evaluated using the average number of positive cells.2.13. LC-MS/MS analysis
An Agilent liquid chromatography system (Agilent HP1200; Agilent Technologies, Palo Alto, CA) and API 3200 triplequadrupole mass spectrometer (Applied Biosystems, Concord, Ont, USA) was used for LC-MS/MS analysis of MTX. Isocratic chromatographic separation was performed on a Hypersil BDS-C18 column (150 × 4.6 mm inner diameter, 5 μm; Dalian Elite Analytical Instruments Co., Ltd., Dalian, China). The mobile phase consisted of 20% (v/v) acetonitrile, 10% (v/v) methyl alcohol, and 70% (v/v) water with 0.1% (v/v) formic acid for MTX. The ionization was conducted using a TurboIonspray interface in positive ion mode. The selected transitions of m/z were m/z 455.2 → 308.2 for MTX. Data acquisition and analysis was performed with analyst software (Version 1.4.1).2.14. Statistical analysis
All statistical analyses were performed using the SPSS11.5 package (SPSS Inc, Chicago IL). Test results are expressed as means ± S.D. The statistical significance of differences between mean values was calculated using the non-paired t-test or one way ANOVA followed by the Newman–Keuls test among multiple treatments. Values of p < 0.05 were considered to statistically significant.3. Results
3.1. Effects of rhein on the pharmacokinetics of MTX in rats in vivo
To evaluate the nature and magnitude of DDI between rhein and MTX, in vivo pharmacokinetic studies in rats were conducted after intravenously or orally co-administration. When the two drugs were co-administered orally, the plasma exposure of MTX was increased significantly compared to that of control group (Fig. 1A). In addition, the Cmax and AUC of MTX were increased by 2.5-, and 4.4-fold, respectively (Table 1). The CL of MTX was decreased by 66.7%. Nevertheless, when the two drugs were co-administered intravenously, the plasma concentration of MTX and pharmacokinetic parameters were also altered significantly compared to control group (Fig. 1B, Table 1). The t1/2β of MTX was prolonged by 86.7% and CLP was decreased by 57.6% (Table 1). Furthermore, the AUC observed after combination was increased by 183.4%. Cumulative urinary excretions over 24 h were 73.7% and 35.8% in the absence and presence rhein, respectively (Fig. 2A). Cumulative urinary excretions and CLR of MTX decreased significantly when the two drugs were co-administered, indicating an interaction between MTX and rhein as measured by renal excretion. | ||
| Fig. 1 Effects of rhein on the mean plasma concentration-time curves of MTX by oral or intravenous administration in rats. (a) Plasma concentration of MTX after oral co-administration with MTX (5 mg kg−1) and rhein (10 mg kg−1). (b) Plasma concentration of MTX after intravenous administration with MTX (5 mg kg−1) and rhein (1 mg kg−1); data were expressed mean ± S.D. (*p < 0.05 vs. control; **p < 0.01 vs. control; n = 3–5). | ||
| Parameters | MTX | MTX + Rhein | |
|---|---|---|---|
| a p < 0.05 vs. MTX group. | |||
| p.o. | Cmax (μg ml−1) | 0.17 ± 0.04 | 0.59 ± 0.12a |
| AUC0→∞ (μg min ml−1) | 131.3 ± 53.1 | 703.7 ± 128.6a | |
| Tmax (min) | 73.3 ± 22.2 | 126.8 ± 53.5 | |
| CLp/F (ml kg−1 min−1) | 270.4 ± 53.2 | 90.6 ± 18.7a | |
| i.v. | AUC0→∞ (μg min ml−1) | 283 ± 20 | 802.4 ± 83.7a |
| t1/2 (min) | 60.3 ± 5.4 | 112.4 ± 13a | |
| CLp (ml kg−1 min−1) | 12.5 ± 0.6 | 5.3 ± 1.1a | |
| CLR (ml kg−1 min−1) | 11.24 ± 0.05 | 5.38 ± 0.14a | |
 | ||
| Fig. 2 Effects of rhein on the cumulative urinary excretion curves of MTX after intravenous injection (a) and concentration-time profile of MTX uptake into kidney slices (b). (a) Cumulative urinary excretion of MTX after intravenous administration with MTX (5 mg kg−1) and rhein (1 mg kg−1); (b) inhibition effects of rhein (2 μM) and PRO (probenecid, 100 μM) on MTX (5 μM) uptake in kidney slices. Data were expressed mean ± S.D. (*p < 0.05 vs. control; **p < 0.01 vs. control; n = 3). | ||
3.2. Effects of rhein on MTX uptake in rat kidney slices
To confirm the target of the DDI between MTX and rhein, fresh rat kidney slices were employed to investigate the effect of rhein on the uptake of MTX. The uptake of MTX in the co-administrated group decreased by 46.1% compared to the corresponding control group (Fig. 2B), indicating that rhein inhibited the uptake of MTX in the kidney. In the same manner, probenecid, a well-known inhibitor of OATs, inhibited MTX uptake in rat kidney slices (Fig. 2B). The in vitro results suggested that the target of the DDI between MTX and rhein is, at least in part, located in the kidney, and OATs might be involved in the DDI.3.3. Effects of rhein on MTX uptake in hOAT1/3-HEK293 cells
To confirm that the target transporters of the DDI between MTX and rhein are OATs in the kidney, we investigated the uptake interaction of MTX and rhein in hOAT1-/hOAT3-HEK293 cells. Rhein strongly blocked MTX accumulation in hOAT1-/hOAT3-HEK293 cells. Dose-response studies, applying increasing concentrations of rhein (0–2 μM), were performed to determine the IC50 value (Fig. 3). The IC50 values for rhein on hOAT1 and hOAT3 were estimated as 2 μM and 0.77 μM, respectively. The results indicated rhein showed much higher affinity for hOAT3 than for hOAT1. | ||
| Fig. 3 Effects of rhein on the concentration-time profiles of the uptake of MTX in hOAT1-/hOAT3-HEK293 cells. (a and b): Concentration-dependent inhibition of rhein (0–2 μM) on the uptake of MTX (5 μM) in hOAT1-/hOAT3-HEK293 cells. Data were expressed mean ± S.D. (*p < 0.05 vs. control; **p < 0.01 vs. control; n = 3). | ||
3.4. Effects of rhein on cytotoxic activities of MTX in hOAT1/3-HEK293 cells
To investigate the changes in the cytotoxic activities of MTX in the presence or absence of rhein in hOAT1-/hOAT3-HEK293 and mock-HEK293 cells, MTT assay was taken in the present study. Inhibition of proliferation in hOAT1/3-HEK293 cells was decreased significantly after co-administration of MTX with rhein (Fig. 4A and B). However, proliferation was almost unchanged in the mock-HEK293 cells after co-administration (Fig. 4C). IC50 values for MTX in hOAT1-HEK293 and hOAT3-HEK293 cells were increased by 1.84- and 8.29-fold, respectively (Table 2) in the combinated group compared to those in the corresponding single group. These results indicated that the cytotoxic activities of MTX were reduced when rhein was co-incubated with MTX. | ||
| Fig. 4 Effects of rhein on the cytotoxicity of MTX in mock-HEK293 cells (a), hOAT1-HEK293 cells (b) and hOAT3-HEK293 cells (c). After treating HEK293 cells with increasing concentrations of MTX in the presence or absence of rhein (2 μM), cell viability was measured by mtt assay. Data were expressed mean ± S.D. (*p < 0.05 vs. control; **p < 0.01 vs. control; n = 3). | ||
3.5. Effects of rhein on MTX-induced renal toxicity in vivo in rats
To investigate whether the changes of MTX pharmacokinetics caused by rhein could affect MTX-induced nephrotoxicity, we measured the levels of creatinine and BUN, which are considered as the main markers of renal function in the plasma of MTX-treated rats. After 5-day administration of MTX continuously, the plasma concentration levels of creatinine and BUN were increased by 74.8% and 285.4%, respectively, comparing to the control group (Fig. 5A and B). However, the levels of creatinine and BUN in the combination group were decreased by 25.6% and 34.2%, respectively, comparing to the MTX group. The results indicated that combination of rhein attenuated the nephrotoxicity caused by MTX in vivo. | ||
| Fig. 5 Effects of rhein on changes in creatinine (a) and bun (b) levels induced by MTX in rat plasma. The MTX group received 5 consecutive daily i.v. injections of MTX (5 mg kg−1). The rhein group received 5 consecutive daily i.v. injections of rhein (1 mg kg−1). The combination group received 5 consecutive daily i.v. injections of MTX (5 mg kg−1) and rhein (1 mg kg−1). Data were expressed mean ± S.D. (**p < 0.01 vs. control; ##p < 0.01 vs. MTX only group; n = 3). | ||
Histological damage was further evaluated in the study. Furthermore, tubular dilatation (arrow in Fig. 6A) with cellular casts composed mostly of detached cells, mono and polynuclear infiltrating cells and tubulitis were visible in the MTX treatment group. When rhein was co-administered with MTX, the histological damage was significantly alleviated (Fig. 6A). Compared to the normal group, the number of apoptotic cells stained by TUNEL in control group was significantly increased, indicating an increase in apoptotic degeneration of kidney cells as a result from MTX intoxication (Fig. 6B). In the combination group, there was a significant decrease in TUNEL-positive cells (Fig. 6B). All the results further showed that rhein alleviated the MTX-induced nephrotoxicity in rats.
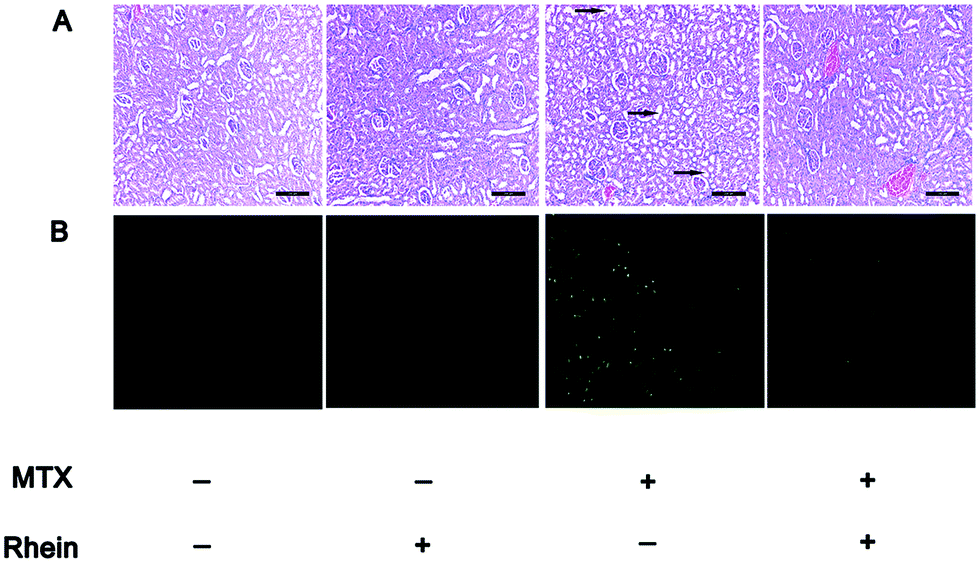 | ||
| Fig. 6 Effects of rhein on changes in kidney histopathological alteration induced by MTX in rats. (a): The protective effect of rhein against MTX-induced renal damage by hematoxylin and eosin (H&E) in rats (×200, magnification); (b): the protective effect of rhein against MTX-induced cell apoptosis though tunel method (×100, magnification). Green fluorescence indicates apoptotic cells. The MTX group received 5 consecutive daily i.v. injections of MTX (5 mg kg−1). The rhein group received 5 consecutive daily i.v. injections of rhein (1 mg kg−1). The combination group received 5 consecutive daily i.v. injections of MTX (5 mg kg−1) and rhein (1 mg kg−1). | ||
4. Discussion
Transporters play important roles in drug disposition and DDI, and can influence pharmacokinetics and pharmacodynamics (positively or negatively) by affecting drug disposition. In clinic, the ideal concomitant medication could be used to improve therapeutically effects while at the same time reducing potential toxicity. For instance, our previous study indicated that co-administration of resveratrol enhanced MTX exposure, and renal damage of MTX was attenuated by resveratrol mediated by OATs.25 In the present study, we show that OATs involved the DDI between rhein and MTX, which are co-administrated to treat rheumatoid arthritis in clinic, and the nephrotoxicity caused by MTX could be decreased by rhein.The in vivo pharmacokinetics data suggested the systemic exposure of MTX in rats was strongly increased when simultaneously given rhein. Both the AUC and t1/2β were increased in rats when MTX and rhein were co-administered intravenously (Table 1). Nevertheless, the pharmacokinetic parameters such as CLP and CLR of MTX were decreased. The results implied that elimination pathway was involved in the DDI between rhein and MTX in vivo. Indeed, the cumulative urinary excretion of MTX decreased significantly when given rhein together (Fig. 2A). Thus, we hypothesis that kidney might be the target organ for the DDI. To confirm this speculation, the DDI between MTX and rhein within the kidney was verified by uptake studies using fresh slices of rats' kidney. The results suggested that rhein suppressed MTX uptake in the rat kidney, and confirmed kidney was the target organ for the DDI (Fig. 2B). Rhein decreased the cumulative urinary excretion (Fig. 2A) and kidney uptake of MTX (Fig. 2B), and therefore resulted to the plasma concentration of MTX was increased (Fig. 1A and B).
It is well accepted that MTX is eliminated almost entirely in an unchanged form in urine, which involves glomerular filtration and active tubular secretion. The previous results suggest that hOATs mediate the transport of MTX in the basolateral side of the proximal tubule. It has been reported that concomitant administration of inhibitors for these transporters decreases MTX renal clearance, resulting in a higher plasma concentration and a longer t1/2.25 Furthermore, it was reported that rhein is the potent inhibitor of hOAT1 and hOAT3. Therefore, we predicted that hOATs would be a possible site involved in the interactions. To confirm this speculation, we also applied hOAT1/3-HEK293 cells to clarify the DDI mechanism, and we found that rhein inhibited the accumulation of MTX (Fig. 3). With this in mind, OAT1 and OAT3 would be the decisive factors influence the DDI between MTX and rhein in the kidney. These findings also suggested that compared with hOAT1, hOAT3 had an overwhelmingly greater role in the DDI between MTX and rhein. It should be noted that the affinity of different transporters to MTX may largely affect their overall contribution to the interaction of rhein with MTX. For example, MTX has a more than 26-fold greater affinity for OAT3 than OAT1.30 Besides, the affinity of rhein to OAT3 was 9.2 times higher than that of OAT1 as described before.23 Therefore, OAT3 would play a important role in the DDI between the two drugs.
Actual DDI in vivo is influenced by multiple extraneous factors and, as a consequence, the in vivo relevance of in vitro data is difficult to predict. Therefore, the OATs knockout mice would be a suitable model to verify the participation or contribution of OAT1/3 mediated the DDI. For instance, findings from pharmacokinetic studies indicate that MTX elimination or renal creatinine secretion is impaired in OAT3 knockout mice.31,32 Hence, the design of in vivo experiments in wild type and OAT3 knockout mice could directly examine the contribution of OAT3 in the elimination of MTX. However, due to the scarcity of OAT1/3 knockout animals, the experiment was not carried out to distinguish the contribution of the DDI mediated by OATs. Despite that, we think that the results from in vitro transport studies in cells that overexpress OAT1/3, together with results from studies in knockout animals or (tissues such as kidney slices), and rat experiments (e.g., after probenecid treatment) seem reasonably concordant with available human clinical data.33
The Food and Drug Administration guidance for industry recommends that DDI potency could be estimated using the DDI index, calculated as the [unbound] Cmax/Ki (or IC50) ratio.34 Based on clinical data and the equation, Li Wang et al. indicated that rhein expressed significant inhibition potential on hOAT1 (83%) and hOAT3 (98%) transport activity, resulting in reduced hOAT1- and hOAT3-mediated renal tubular secretion of co-administered therapeutics that are OAT substrates. Thus, the possibility of pharmacokinetics DDI between two drugs should be aware. It is worth noting that, besides elimination phase, the absorption phase of MTX was essentially influenced by rhein (Fig. 1A). The results suggested that other target organs might also involve in DDI between MTX and rhein except kidney when the two drugs were simultaneously administered orally. For several reasons we speculated that the small intestine would be also another target site for the DDI, which may in part explain the increased absorption phase of MTX caused by rhein. Chi-Sheng Shia et al. used MDCKII-multidrug resistance-associated protein 2 (MRP2) cells to evaluate the effect of rhein on the activity of MRP2. The results suggested that the activity of MRP2 could be inhibited by rhein.35 Nevertheless, it is well known that MTX is also a substrate of MRP2.36,37 Therefore, MRP2 would be another target site for the DDI between MTX and rhein. In fact, Chi-Sheng Shia et al. have found that the serum metabolite of rhubarb, the main component is rhein, significantly decreased the transepithelial efflux of MTX in Caco-2 cells, which are derived from human colorectal adenocarcinoma. Furthermore, the results suggested that co-administration of rhubarb increased the systemic exposure of oral MTX and prolonged the mean residence time, whereas Cmax was not affected. The results might contradict our hypothesis that absorption process involved in the DDI. The potential factors would be that the systemic exposure of rhein was different. Therefore, the serum concentration of rhein did not reach the Ki value. Nevertheless, further investigation should be taken to explain the phenomena. All in all, the hidden risks of interactions between rhein and MTX mediated by MRP2, warrant more investigations for ensuring drug safety.
As described above, concomitant administration of rhein could strongly increase the systemic exposure via transporter-mediated DDI. As we all know, nephrotoxicity is a major adverse effect of MTX and treatment at high doses may cause renal failure.38,39 The inhibitors of OATs may reduce the extent of OAT-induced nephrotoxicity of MTX. Indeed, we firstly conducted MTT assays using mock-HEK293 and hOAT1/3-HEK293 cells, and calculated the IC50 of the MTX in combination with rhein. When the two drugs were co-administered, the IC50 of MTX increased in both hOAT1-HEK293 and hOAT3-HEK293 cells (Fig. 4B and C). However, the IC50 showed no significant difference in the presence of rhein in mock-HEK293 cells (Fig. 4A). The results provide evidence that OAT1- and OAT3-mediated DDI could alleviate the cytotoxicity caused by MTX. In addition, pathological and histological damage, and an increase in plasma creatinine and BUN, both markers of renal damage, were observed in MTX-treated rats in our study (Fig. 5 and 6). Therefore, as expected, MTX could result in serious renal toxicity in vivo. However, the renal toxicity was alleviated via co-administration of rhein (Fig. 5 and 6). The results are rationalized by considering that OAT1 and OAT3, expressed in the proximal tubule basolateral membrane, contribute to the uptake of drugs into renal epithelial cells from the blood side.16 Efficient OAT-mediated basolateral uptake may result in high intracellular drug concentrations. The inhibition of OATs by rhein consequently decreases the cellular uptake and renal accumulation of MTX, resulting in reduced exposure of the kidney to MTX and a subsequent lowering of nephrotoxicity.40 However, attention should be paid to the increased exposure delayed elimination of MTX after combination with rhein, which would increase the risk of other adverse reactions. Further study should be taken to clarify whether the increased systemic exposure by rhein could result in other unexpected toxicity.
It was reported that diacerein, the prodrug of rhein, protected against doxorubicin-induced nephrotoxicity in rats most probably due to its antioxidant and anti-inflammatory activities.41 Similarly, Zhao et al. demented that protective effect of rhein against acetaminophen-induced kidney injuries might result from the amelioration of oxidative stress.42 Interestingly, Meng et al. also demonstrated that rhein significantly alleviated the symptoms of nephropathy through decreasing the production of proinflammatory cytokines, including interleukin 1β, prostaglandin E2, and TNFα.43 Therefore, the protective effects mediated by the signal pathway would be the influence factors mediated the DDI in the pharmacodynamic level. However, besides pharmacological activity, our results firstly suggested that rhein could also effectively attenuate the severity of nephrotoxicity, at least in part, through OATs-mediated DDI in the pharmacokinetic level. Further study should be taken to clarify whether the pharmacological functions of rhein involved the DDI between rhein and MTX. Nevertheless, rhein or its prodrug diacerein was optimal choice to use together with MTX in the treatment of early rheumatoid arthritis. Moreover, rhein was an effective drug employed to decrease the nephrotoxicity caused by the toxicities which were the substrates of OATs.
Taken together, our findings provided the evidence that a DDI mediated by OAT1/3 might occur when MTX and rhein were co-administered. Furthermore, concomitant administration of rhein alleviated MTX-induced renal damage. Therefore, diacerein, the prodrug of rhein, could be a rational combination drug for clinical application of MTX in the treatment of rheumatoid arthritis.
Conflict of interest
The authors declare that we have no conflicts of interest.Abbreviations
| MTX | Methotrexate |
| DMEM | Dulbecco's modified Eagle's medium |
| OATs | Organic anion transporters |
| MRP2 | Multidrug resistance-associated protein 2 |
| DDIs | Drug–drug interactions |
| BUN | Blood urea nitrogen |
| AUC | Area under the plasma concentration-time curve |
| CLp | Plasma clearance rate |
| MRP2 | Multidrug resistance-associated protein 2 |
Acknowledgements
This work was supported by a grant from the National Natural Science Foundation of China (No. 81603186, 81473280, U1608283). The authors thank Professor Yuichi Sugiyama (Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan) and Professor Gong Likun (Shanghai Institute of Materia Medica, Chinese Academy of Science, Shanghai, China) for providing hOAT1/3-HEK293 cells.References
- T. Pincus, Y. Yazici, T. Sokka, D. Aletaha and J. S. Smolen, Clin. Exp. Rheumatol., 2003, 21, S179–S185 CAS.
- W. M. Claudino, B. Gibson, W. Tse, M. Krem and J. Grewal, Am. J. Blood Res., 2016, 6, 1–5 Search PubMed.
- R. Elsby, L. Fox, D. Stresser, M. Layton, C. Butters, P. Sharma, V. Smith and D. Surry, E. J. Pharm. Sci.ci, 2011, 43, 41–49 CrossRef CAS PubMed.
- P. Jariwala, V. Kumar, K. Kothari, S. Thakkar and D. D. Umrigar, Case Reports in Dermatological Medicine, 2014, 2014, 946716 CrossRef PubMed.
- A. A. El-Sheikh, M. A. Morsy and A. H. Hamouda, Pharmacogn. Mag., 2016, 12, S76–S81 CrossRef PubMed.
- D. Bironaite and K. Ollinger, Chem.-Biol. Interact., 1997, 103, 35–50 CrossRef CAS PubMed.
- D. He, L. Lee, J. Yang and X. Wang, Biol. Pharm. Bull., 2011, 34, 1219–1226 CAS.
- C. C. Zeng, X. Liu, G. R. Chen, Q. J. Wu, W. W. Liu, H. Y. Luo and J. G. Cheng, J. Evidence-Based Complementary Altern. Med., 2014, 2014, 487097 Search PubMed.
- C. M. Spencer and M. I. Wilde, Drugs, 1997, 53, 98–106 CrossRef CAS PubMed , discussion 107–108.
- S. D. Mandawgade, S. Kulkarni, A. Pal, S. Srivastava, B. K. Padhi and R. S. Raghuvanshi, Curr. Drug Delivery, 2016, 13, 83–89 CrossRef CAS.
- P. Nicolas, M. Tod, C. Padoin and O. Petitjean, Clin. Pharmacokinet., 1998, 35, 347–359 CrossRef CAS PubMed.
- A. Ojha, R. Rathod and H. Padh, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1145–1148 CrossRef CAS PubMed.
- V. Vallon, T. Rieg, S. Y. Ahn, W. Wu, S. A. Eraly and S. K. Nigam, Am. J. Physiol. Ren. Physiol., 2008, 294, F867–F873 CrossRef CAS PubMed.
- Z. Liu and K. Liu, Curr. Drug Metab., 2016, 17, 456–468 CrossRef CAS PubMed.
- Z. Liu, J. Mi, S. Yang, M. Zhao, Y. Li and L. Sheng, Eur. J. Pharmacol., 2016, 791, 339–347 CrossRef CAS PubMed.
- Y. Hagos and N. A. Wolff, Toxins, 2010, 2, 2055–2082 CrossRef CAS PubMed.
- J. Zhang, C. Wang, Q. Liu, Q. Meng, J. Cang, H. Sun, Y. Gao, T. Kaku and K. Liu, Drug Met. Dispos.os, 2010, 38, 930–938 CrossRef CAS PubMed.
- K. L. Brouwer, D. Keppler, K. A. Hoffmaster, D. A. Bow, Y. Cheng, Y. Lai, J. E. Palm, B. Stieger and R. Evers, Clin. Pharmacol. Ther., 2013, 94, 95–112 CrossRef CAS PubMed.
- S. K. Nigam, K. T. Bush, G. Martovetsky, S. Y. Ahn, H. C. Liu, E. Richard, V. Bhatnagar and W. Wu, Physiol. Rev., 2015, 95, 83–123 CrossRef PubMed.
- A. Emami Riedmaier, A. T. Nies, E. Schaeffeler and M. Schwab, Pharmacol. Rev., 2012, 64, 421–449 CrossRef PubMed.
- R. Chioukh, M. S. Noel-Hudson, S. Ribes, N. Fournier, L. Becquemont and C. Verstuyft, Drug Metab. Dispos., 2014, 42, 2041–2048 CrossRef PubMed.
- K. Muhrez, I. Benz-de Bretagne, L. Nadal-Desbarats, H. Blasco, E. Gyan, S. Choquet, F. Montigny, P. Emond and C. Barin-Le Guellec, Pharmacol. Res., 2017, 118, 121–132 CrossRef CAS PubMed.
- L. Wang, X. Pan and D. H. Sweet, Biochem. Pharmacol., 2013, 86, 991–996 CrossRef CAS PubMed.
- L. Ma, L. Zhao, H. Hu, Y. Qin, Y. Bian, H. Jiang, H. Zhou, L. Yu and S. Zeng, J. Ethnopharmacol., 2014, 153, 864–871 CrossRef CAS PubMed.
- Y. Jia, Z. Liu, C. Wang, Q. Meng, X. Huo, Q. Liu, H. Sun, P. Sun, X. Yang, X. Ma and K. Liu, Toxicol. Appl. Pharmacol., 2016, 306, 27–35 CrossRef CAS PubMed.
- Y. Zhu, Q. Meng, C. Wang, Q. Liu, X. Huo, A. Zhang, P. Sun, H. Sun, H. Li and K. Liu, Int. J. Pharm., 2014, 465, 368–377 CrossRef CAS PubMed.
- Y. Nozaki, H. Kusuhara, T. Kondo, M. Hasegawa, Y. Shiroyanagi, H. Nakazawa, T. Okano and Y. Sugiyama, J. Pharmacol. Exp. Ther., 2007, 321, 362–369 CrossRef CAS PubMed.
- Y. Nozaki, H. Kusuhara, T. Kondo, M. Iwaki, Y. Shiroyanagi, H. Nakayama, S. Horita, H. Nakazawa, T. Okano and Y. Sugiyama, J. Pharmacol. Exp. Ther., 2007, 322, 1162–1170 CrossRef CAS PubMed.
- Q. Liu, C. Wang, Q. Meng, X. Huo, H. Sun, J. Peng, X. Ma, P. Sun and K. Liu, Pharm. Res., 2014, 31, 1120–1132 CrossRef CAS PubMed.
- M. Takeda, S. Khamdang, S. Narikawa, H. Kimura, M. Hosoyamada, S. H. Cha, T. Sekine and H. Endou, J. Pharmacol. Exp. Ther., 2002, 302, 666–671 CrossRef CAS PubMed.
- A. L. VanWert and D. H. Sweet, Pharm. Res., 2008, 25, 453–462 CrossRef CAS PubMed.
- V. Vallon, S. A. Eraly, S. R. Rao, M. Gerasimova, M. Rose, M. Nagle, N. Anzai, T. Smith, K. Sharma, S. K. Nigam and T. Rieg, Am. J. Physiol. Ren. Physiol., 2012, 302, F1293–F1299 CrossRef CAS PubMed.
- S. K. Nigam, W. Wu, K. T. Bush, M. P. Hoenig, R. C. Blantz and V. Bhatnagar, Clin. J. Am. Soc. Nephrol., 2015, 10, 2039–2049 CrossRef CAS PubMed.
- Food and Drug Administration, 2012.
- C. S. Shia, S. H. Juang, S. Y. Tsai, P. D. Lee Chao and Y. C. Hou, Am. J. Chin. Med., 2013, 41, 1427–1438 CrossRef CAS PubMed.
- M. L. Vlaming, Z. Pala, A. van Esch, E. Wagenaar, D. R. de Waart, K. van de Wetering, C. M. van der Kruijssen, R. P. Oude Elferink, O. van Tellingen and A. H. Schinkel, Clin. Cancer Res., 2009, 15, 3084–3093 CrossRef CAS PubMed.
- M. L. Vlaming, A. van Esch, Z. Pala, E. Wagenaar, K. van de Wetering, O. van Tellingen and A. H. Schinkel, Mol. Cancer Ther., 2009, 8, 3350–3359 CrossRef CAS PubMed.
- S. Iqbal, A. Armaghani, R. Aiyer and A. Kazory, J. Oncol. Pharm. Pract., 2013, 19, 373–376 CrossRef PubMed.
- T. Wiczer, E. Dotson, A. Tuten, G. Phillips and K. Maddocks, J. Oncol. Pharm. Pract., 2016, 22, 430–436 CrossRef CAS PubMed.
- K. M. Morrissey, S. L. Stocker, M. B. Wittwer, L. Xu and K. M. Giacomini, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 503–529 CrossRef CAS PubMed.
- M. M. Refaie, E. F. Amin, N. F. El-Tahawy and A. M. Abdelrahman, J. Toxicol., 2016, 9507563 Search PubMed.
- Y. L. Zhao, G. D. Zhou, H. B. Yang, J. B. Wang, L. M. Shan, R. S. Li and X. H. Xiao, Food Chem. Toxicol., 2011, 49, 1705–1710 CrossRef CAS PubMed.
- Z. Meng, Y. Yan, Z. Tang, C. Guo, N. Li, W. Huang, G. Ding, Z. Wang, W. Xiao and Z. Yang, Planta Med., 2015, 81, 279–285 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |