
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrospinning synthesis of Co3O4@C nanofibers as a high-performance anode for sodium ion batteries†
Zhenwei Mao,
Min Zhou*,
Kangli Wang,
Wei Wang,
Hongwei Tao and
Kai Jiang *
*
State Key Laboratory of Advanced Electromagnetic Engineering and Technology, School of Electrical and Electronic Engineering, State Key Laboratory of Materials Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, Wuhan 430074, P. R. China. E-mail: kjiang@hust.edu.cn; minzhou0729@hust.edu.cn; Tel: +86-27-87559524
First published on 26th April 2017
Abstract
A unique 1D structure of Co3O4 nanoparticles encapsulated in carbon nanofibers has been fabricated by a facile, cost-effective and scalable electrospinning method followed by a two-step heat-treatment. When tested as an anode for sodium ion batteries, the composite exhibits an initial discharge and charge capacity of 768.4 mA h g−1 and 422.4 mA h g−1 at 50 mA g−1, and retains a reversible specific capacity of ∼300 mA h g−1 up to 100 cycles. At a higher current density of 500 mA g−1, a reversible capacity of 251.7 mA h g−1 is still obtained. Meanwhile, compared to the pure Co3O4 nanofibers, Co3O4@C nanofibers demonstrate much improved capacity retention of 84.3% over 500 cycles. The good electrochemical performance is mainly attributed to the unique 1D nanofiber structure with stable structural integrity and improved electrical conductivity rendered by the carbon nanofiber framework, which is more convenient for the electron transport and Na+ insertion/extraction of the active materials.
Introduction
Large-scale energy storage technologies play a crucial role in the integration of renewable clean energy which is important to address the increasing concerns in relation to depletion of fossil-based resources, global warming and environmental pollution.1–3 Among the various available energy storage technologies, lithium ion batteries (LIBs) have conquered the energy storage market of portable electronics for decades, and are currently considered as the most promising technology for electric vehicle and large storage energy applications due to their advantages of high energy density, long cycling life, and environmental benignity.4,5 Despite the widespread use of LIBs, the rising cost of lithium resources and their geographical limitation create a barrier for their applications in the large-scale storage field.6,7 As sodium is a naturally abundant alkali element widely distributed around the world, sodium ion batteries (SIBs) seem to be an appealing choice as a low cost alternative to LIBs, especially for large-scale energy storage applications.8However, it remains a great challenge to develop suitable electrode materials for SIBs as Na ions are much larger than Li ions. During the past few years, much progress has been made in the Na-storage cathode field, varieties of Na storage materials have been reported to demonstrate certain Na-storage capacity and cycling performances, such as layered transition metal oxides, phosphates, Prussian blue analogues, organic cathodes and so on.9–14 In the case of anode materials, transitional metal oxides (TMOs) are considered to be promising anode materials for sodium ion batteries due to their resource abundance, low cost and high theoretical capacity (1005, 889, 718, 674 and 661 mA h g−1 for Fe2O3, Co3O4, NiO, CuO and ZnO, respectively). Nevertheless, transitional metal oxides usually suffer from serious volume expansion during cycling. In addition, the poor conductivity of TMOs lead to inferior capacity utilization, poor cycling stability and poor rate capability. Recent researches show that the electrochemical performance of TMOs electrode strongly depend on the structure and pore size of the particles.15–17 As a result, great efforts have been devoted to design specific structures, such as hollow spheres, core–shell structure, nanotubes and so on.18–23 Lu et al. synthesized micro-nanostructured CuO/C spheres by aerosol spray pyrolysis, delivering a capacity of 402 mA h g−1 after 600 cycles at a current density of 200 mA g−1.21 Another effective strategy is to incorporate various conductive matrix to the nanostructured TMOs, thus improving the electronic conductivity as well as relieving the volume expansion during repeated cycling.18–23 Jian et al. synthesized Fe2O3 nanocrystals@GNS by coating graphene nanosheets (GNS) on Fe2O3 nanocrystals. The discharge–charge capacity delivered by Fe2O3@GNS reaches 400 mA h g−1 over 200 cycles and no capacity decay can be observed.18
One dimensional nanostructures such as nanotubes, nanowires, nanorods and nanofibers are considered as ideal structure for Na-storage due to their uniform structure which possess high electronic and ionic conductivities as well as strong tolerance to resist structural pulverization, leading to great enhancement of the electrochemical performances.21,22 Electrospinning is a versatile, low-cost and scalable method to fabricate 1D nanofibers. Co3O4 is widely considered as promising anode materials for both LIBs and SIBs based on its relatively high conductivity and high theoretical capacity (889 mA h g−1). In this work, we fabricated a unique 1D structure carbon coated Co3O4 with the structure of Co3O4 nanoparticles uniformly wrapped in carbon nanofibers (denoted as Co3O4@CNFs) by a facile electrospinning technique followed by a two-step heat-treatment. The unique 1D carbon framework can not only buffer the volume variation of Co3O4 nanoparticles, but also serves as the electron and ion pathways to improve the conductivity of the composite. The as prepared Co3O4@CNFs composites demonstrate great enhancement in electrochemical performances with high reversible capacity of 422.4 mA h g−1 and cycling stability over 500 cycles.
Experimental
Synthesis of Co3O4@C nanofibers composite
Polyacrylonitrile (PAN, MW = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Sigma-Aldrich. Cobalt(II) acetate tetrahydrate (CoAc, >99.9%) and N,N-dimethylformamide (DMF, 99.8%) were purchased from Sinopharm and used as received without any purified.
000) was purchased from Sigma-Aldrich. Cobalt(II) acetate tetrahydrate (CoAc, >99.9%) and N,N-dimethylformamide (DMF, 99.8%) were purchased from Sinopharm and used as received without any purified.
In a typical process, 0.6 g CoAc was added into 10 mL DMF and stirred for a few minutes to dissolve. After that, 1 g PAN was slowly added into the above solution and stirred continuously at 80 °C for 6 h to yield a homogeneous solution. The prepared homogeneous solution was then loaded into a plastic syringes (10 mL) with a needle of 20 G and then placed on a commercial electrospinning setup (Ucalery, Beijing). The collector to the needle distance was kept constant at 15 cm to collect a light-pink film on an aluminium foil wrapped around a drum collector rotating at a speed of 0.6 m min−1. A positive high-voltage power of 12 kV was applied to the needle tip, and a negative high-voltage power of 3 kV was applied to the collecting drum. The flow rate of fluid was set to be 0.8 mL h−1. The humidity level inside the electrospinning chamber was 40%, and the temperature was around 30 °C. The as-spun CoAc–PAN nanofibers composite film was first stabilized at 280 °C for 90 min in air. Then the as-stabilized film was carbonized at 650 °C for 1 h in argon atmosphere. Finally, the carbonized film was oxidized at 300 °C for 30 min. The heating rate was set to be 3 °C min−1. Other different amount of CoAc was added to the precursor solution respectively to determine the optimized carbon content of the composite, and the obtained composites were labelled as Co3O4@CNFs-2 and Co3O4@CNFs-3. In order to identify the optimized carbonization temperature, different carbonized temperature were set, namely, 550 °C, 600 °C, 650 °C and 750 °C. The obtained composites were denoted as Co3O4@CNF-550, Co3O4@CNF-600, Co3O4@CNF-650 and Co3O4@CNF-750. In this paper, Co3O4@CNFs without suffix correspond to the composite carbonized at 650 °C.
Synthesis of Co3O4 nanofibers
Pure Co3O4 nanofibers (denoted as Co3O4 NFs) were also prepared by electrospinning for comparison. The synthesis process was almost the same with Co3O4@C nanofibers composite except the 650 °C heat-treatment in air for 2 h after stabilizing the film in air to burn the carbon.Synthesis of carbon nanofibers
Pure carbon nanofibers (denoted as CNFs) were synthesized by electrospinning 10 wt% PAN solution without adding CoAc. The electrospinning process and heat-treatment condition were the same with those of Co3O4@C nanofibers composite.Characterization
The crystal structure of the composites was characterized by X-ray diffraction system (XRD-7000S) equipped with Cu Kα radiation between 10° and 80°. The morphological features were examined with a field-emission scanning electron microscope (FE-SEM, JEOL-7600F). Particle morphology of the synthesized composites was observed with transmission electron microscopy (TEM, JEOL-2100). X-Ray photoelectron spectroscopy (XPS, AXIS-ULTRA DLD-600W) was employed to evaluate the surface chemistry of materials. Thermogravimetric analysis (TGA) was conducted (TGA/DSC, Netzsch-STA 449F5) in air over a temperature range of 30–800 °C at a heating rate of 10 °C min−1. The surface area was determined by nitrogen adsorption/desorption using the Brunauer–Emmett–Teller method (BET, Micromeritics Tristar 2000). The sample was degassed under nitrogen gas at 180 °C for a whole night prior to BET measurements.Electrochemical measurements
All the electrochemical studies were conducted using CR2025 coin cells. The anodes were prepared by mixing the active materials (Co3O4@CNFs, Co3O4 NFs, CNFs), the conductive additives (super P) and the binder (carboxymethyl cellulose) in a mass ratio of 80:10:10. The mixture was prepared as slurry and spread onto copper foil. The electrode was dried under vacuum at 120 °C for a whole night to remove the solvent before pressing. Then the electrodes were cut into disks (11 mm in diameters) to be used as the working electrodes, sodium metal foil as a counter electrode, and a glass microporous fiber filter (Whatman, cat. no. 1825-047) as a separator, 1 M NaClO4 in propylene carbonate (PC) with 2% FEC (fluoroethylene carbonate) additive as the electrolyte. Assembled cells were allowed to soak overnight. The cyclic voltammetry (CV) tests were performed between 0.01 V and 3.0 V at a scan rate of 0.1 mV s−1 on an electrochemical workstation (CHI660c), and the galvanostatic discharge–charge cycling of the cells was carried out at different current densities between potentials of 0.01 V and 3.0 V on a battery tester (LAND 2001 CT) at room temperature. Electrochemical impedance spectroscopy (EIS) was carried out by applying an AC voltage of 5 mV in the frequency range of 100 kHz to 0.1 Hz on an electrochemical workstation (CHI660c).
Results and discussions
Carbon coated Co3O4 nanofibers were synthesized via a single-nozzle electrospinning technique using PAN as the carbon source. For comparison, pure Co3O4 nanofibers (denoted as Co3O4 NFs) were prepared in the same way except for annealing in air to burn the carbon. During the followed stabilization process, PAN was converted to aromatic cyclized ladder type structure by cyclization, dehydrogenation, aromatization and crosslinking.24,25 After carbonization, the stabilized composite was transformed to Co3O4@CNFs. The Co3O4@CNFs composites carbonized at different temperatures are denoted as Co3O4@CNF-550, Co3O4@CNF-600, Co3O4@CNF-650 and Co3O4@CNF-750. In the following text, the samples denoted as Co3O4@CNFs correspond to the composite carbonized at 650 °C.The crystal structure and phase characterizations of both the Co3O4@CNFs and Co3O4 NFs were conducted by XRD as shown in Fig. 1(a). All the diffraction peaks in Fig. 1(a) can be assigned to (111), (220), (311), (222), (400), (311), (422), (511) and (440) planes of cubic Co3O4 phase [(JCPDS no. 78-1970), space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (no. 227)]. The high peak intensity indicates that the material was highly crystallized after annealing at 650 °C. No peaks of any other phases were detected, demonstrating the high purity of the as-prepared sample. N2 adsorption and desorption isotherm measurements were carried out to examine the specific surface area and pore size of Co3O4@CNFs and Co3O4 NFs. As shown in Fig. 1(b), the BET surface area and average pore size of Co3O4@CNFs are calculated to be 14.19 m2 g−1 and 5.6 nm, respectively. In addition, the specific surface area and pore size of Co3O4 NFs are 5.80 m2 g−1 and 4.4 nm, respectively.
m (no. 227)]. The high peak intensity indicates that the material was highly crystallized after annealing at 650 °C. No peaks of any other phases were detected, demonstrating the high purity of the as-prepared sample. N2 adsorption and desorption isotherm measurements were carried out to examine the specific surface area and pore size of Co3O4@CNFs and Co3O4 NFs. As shown in Fig. 1(b), the BET surface area and average pore size of Co3O4@CNFs are calculated to be 14.19 m2 g−1 and 5.6 nm, respectively. In addition, the specific surface area and pore size of Co3O4 NFs are 5.80 m2 g−1 and 4.4 nm, respectively.
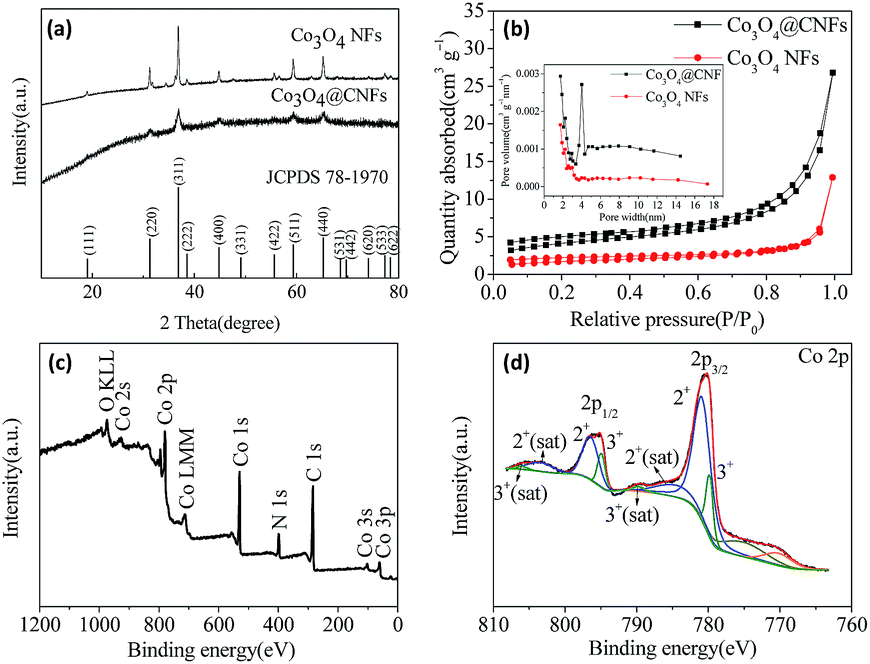 | ||
| Fig. 1 (a) XRD patterns of Co3O4 NFs and Co3O4@CNFs; (b) nitrogen adsorption–desorption isotherm for Co3O4@CNFs and Co3O4 NFs; (c) XPS survey spectra of Co3O4@CNFs composite; (d) deconvoluted XPS spectrum of Co 2p. | ||
The chemical state and molecular environment of the as prepared Co3O4@CNFs was evaluated by X-ray photoelectron spectroscopy (XPS). The survey spectra of the Co3O4@CNFs (Fig. 1(c)) confirms the existence of Co, O, C and N. Fig. 1(d) displays the high resolution spectrum of Co. There are two binding energy peaks located at 795.6 eV and 780.2 eV corresponding to the electronic states of Co 2p1/2 and Co 2p3/2 respectively with a spin-energy separation of 15.4 eV, which is the characteristic of the Co3O4 phase.23,26–28
The morphology of the as prepared Co3O4@CNFs and Co3O4 NFs were characterized by FESEM. As shown in Fig. 2(a and b), the as prepared Co3O4@CNFs composite emerges as smooth nanofibers with uniformly distributed diameters ranging from 150–350 nm. In contrast, the Co3O4 NFs shows much rougher surface with the aggregation of the Co3O4 nanoparticles (Fig. 2(c and d)). The microstructure of Co3O4@CNFs composite was further examined by TEM. Fig. 2(e) displays the microstructure of a single Co3O4@C nanofiber. As can be seen from the Fig. 2(e), the Co3O4 nanoparticles are uniformly embedded in the carbon nanofiber matrix. The high-resolution TEM image reveals the lattice fringes with an interplanar spacing of 0.21 nm, corresponding to the (222) plane of Co3O4 nanoparticles, which are well consistent with the XRD results.
 | ||
| Fig. 2 (a and b) SEM images of the Co3O4 NFs nanofibers; (c and d) SEM images of the Co3O4@CNFs; (e) TEM image of Co3O4@CNFs; (f) HRTEM image of Co3O4@CNFs. | ||
Cyclic voltammograms (CVs) of the electrodes were measured at a scan rate of 0.1 mV s−1 in the potential range of 0.01–3.0 V (vs. Na+/Na) at room temperature. Fig. 3(a) displays the CV curves of the first three cycles for Co3O4@CNFs. In the first cycle, there is a sharp cathodic peak at ∼0.40 V, which can be ascribed to the electrochemical reduction (sodiation) reaction of Co3O4 with Na to metallic Co, the formation of Na2O and a partially irreversible solid electrolyte interphase (SEI) layer. In the anodic scan, the anodic peak at around 0.87 V and 1.25 V is ascribed to the oxidation (desodiation) reaction of Co3O4. The formation of Co and Na2O and the reformation of Co3O4 can be described by the following electrochemical conversion reaction.23 In the subsequent cycles, the cathodic peak shifts to 0.70 V. The peaks intensity and the integral areas of the next cycle is very close to those of the second cycle. These results show that the electrochemical reversibility of the Co3O4@CNFs is gradually improved after the first cycle. The CV curves of the Co3O4 NFs (Fig. S5†) are very similar to those of Co3O4@CNFs, except for a little shift of the peak location, suggesting the similar reaction mechanism as below.
| Co3O4 + 8Na+ + 8e− ↔ 4Na2O + 3Co |
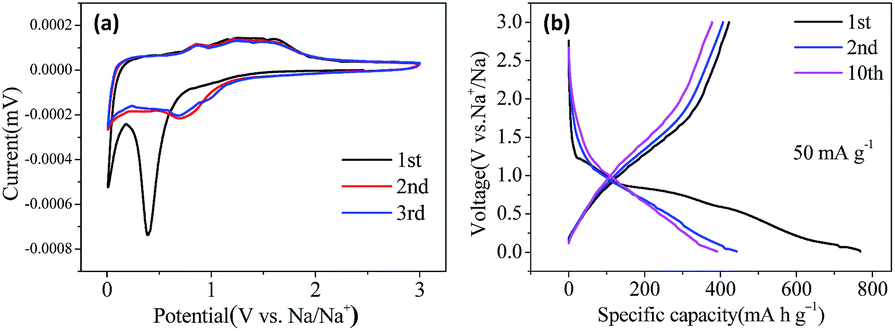 | ||
| Fig. 3 (a) Cyclic voltammogram (CV) of Co3O4@CNFs at a scan rate of 0.1 mV s−1 between 0.01 V and 3.0 V for the first three cycles; (b) galvanostatic charge/discharge curves of Co3O4@CNFs for the 1st, 2nd and 10th cycles between 0.01–3.0 V versus Na+/Na at a current density of 50 mA g−1. | ||
To evaluate the electrochemical performance of the Co3O4@CNFs composite for SIBs applications, the galvanostatic charge and discharge measurements of the assembled cells were performed at the current density of 50 mA g−1 in the voltage range of 0.01–3.0 V (versus Na+/Na). As shown in Fig. 3(b), in accordance with the CV curves, the Co3O4@CNFs demonstrate a charge/discharge plateau at about 0.75 V in the first cycle, which gradually disappears in the subsequent cycles. The initial discharge and charge capacities are 768.4 mA h g−1 and 422.4 mA h g−1, corresponding to the initial coulombic efficiency of about 55%. The irreversible capacity loss arising during the first cycle is due to the incomplete conversion reaction and the formation of SEI layer caused by the irreversible decomposition of the electrolyte and other side effects. The reversible capacities of the Co3O4@CNFs remain at about 400 mA h g−1 in the subsequent cycles with the coulombic efficiency quickly increasing to over 95%.
In order to evaluate the electrochemical performances of the Co3O4@CNFs carbonized at different temperatures, the cycling performance of the Co3O4@CNF-550, Co3O4@CNF-600, Co3O4@CNF-650 and Co3O4@CNF-750 was conducted at a current density of 100 mA g−1, as shown in Fig. S6.† The initial discharge and charge capacities of the above-mentioned composites are 492.6/294.7 mA h g−1, 533.9/324.6 mA h g−1, 768.4/422.4 mA h g−1 and 651.1/331.5 mA h g−1, respectively. After 50 cycles, the composite carbonized at 650 °C presents the highest discharge specific capacity of 314.3 mA h g−1, demonstrating the best cycling performances.
Fig. 4(a) shows the comparison of cycling performance of Co3O4@CNFs, Co3O4 NFs and CNFs at a rate of 50 mA g−1 in the voltage range of 0.01–3.0 V (versus Na+/Na) up to 100 cycles. The reversible capacity of Co3O4@CNFs composite remains ∼300 mA h g−1 with a coulombic efficiency over 99% up to 100 cycles. However, the reversible capacity of Co3O4 NFs is only 46.9 mA h g−1 after 100 cycles. The much improved electrochemical performance of Co3O4@CNFs may be attributed to the unique 1D structure with Co3O4 nanoparticles uniformly embedded in the carbon framework, enabling stable structural integrity to accommodate the volume change during the repeated cycling. The volume expansion of both the Co3O4@CNFs and Co3O4 NFs during the cycling process (before and after 50 cycles) were evaluated by the SEM imagine. As we can see from Fig. S7,† the structure of Co3O4@CNFs electrode after 50 cycles shows negligible changes, indicating the attractive morphological stability. In contrast, the cycled Co3O4 NFs electrode shows obvious collapse and aggregation, demonstrating that the carbon nanofibers can effectively relieve the volume expansion and guarantee the structure stability of the electrode.
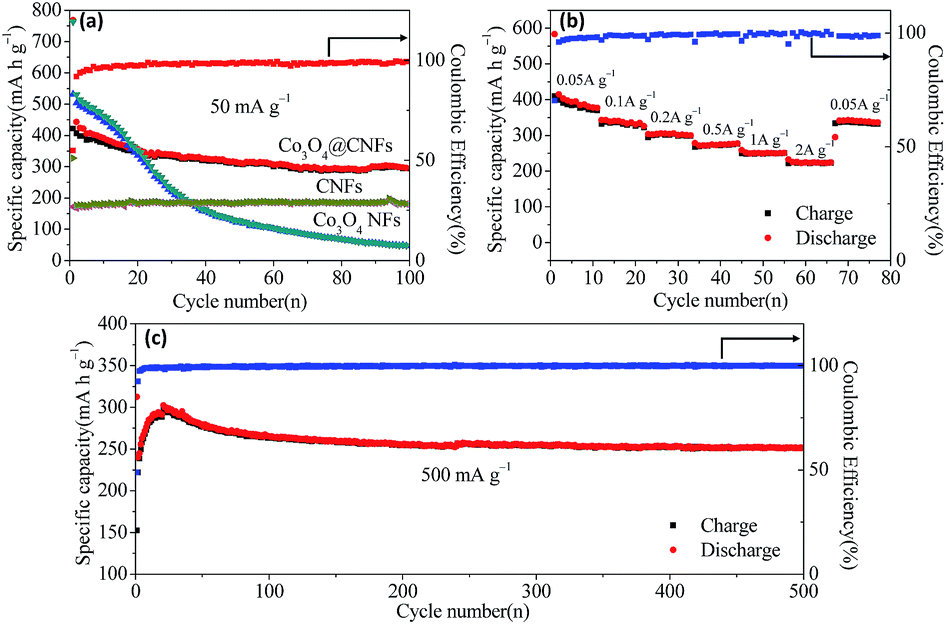 | ||
| Fig. 4 (a) The cycling performance of Co3O4@CNFs (red), Co3O4 NFs (green) and CNFs (yellow); (b) rate capability of the Co3O4@CNFs at various current rates between 0.05 A g−1 to 2 A g−1; (c) high current cycling performance of Co3O4@CNFs at a current density of 500 mA g−1. | ||
Rate capability measurement was carried out on the Co3O4@CNFs composite cells at various current densities (Fig. 4(b)). The reversible capacity of Co3O4@CNFs is 394.8 mA h g−1 at the current density of 50 mA g−1 for 10 cycles. When the current density was increased to 100, 200, 500, 1000 and 2000 mA g−1, the corresponding capacity values are 343, 305.5, 278.1, 258.5 and 225.1 mA h g−1, respectively. Moreover, the reversible capacity of 342.3 mA h g−1 can still be retained when the current density was returned to 50 mA g−1. Besides, the coulombic efficiency quickly increases to over 96% after the initial few cycles and remains stable in the subsequent cycles. This excellent rate capability can be ascribed to the improved electrical conductivity rendered by the carbon nanofiber framework, which is more convenient for electron transport. Moreover, the unique 1D nanofiber is favourable for the Na+ insertion/extraction, leading to fast interfacial charge transfer and Na+ diffusion in the composite materials.
In addition to the high rate capability, the Co3O4@CNFs also exhibit superior long cycling performances. Fig. 4(c) shows the long-cycling stability of Co3O4@CNFs at 500 mA g−1 over 500 cycles. The reversible capacity has been increasing during the initial 20 cycles and reaches the highest specific capacity value of 298.5 mA h g−1, which may be caused by the gradual activation of the composite materials in the first few cycles. After 500 cycles, the Co3O4@CNFs composite material still remains 251.7 mA h g−1 with a capacity retention of 84.3%. In addition, the coulombic efficiency rapidly rises up to 99.6% in the subsequent cycles, indicating stable reversibility.
The electrochemical impedance spectroscopy (EIS) of Co3O4@CNFs and Co3O4 NFs were conducted to further confirm the mechanism of the electrochemical behavior. The Nyquist plots of Co3O4@CNFs and Co3O4 NFs electrodes after 50 cycles as well as the equivalent circuit are presented in Fig. S8.† Apparently, the Co3O4@CNFs electrode exhibits much lower SEI film resistance (Rsf, 2.1 Ω) and charge transfer resistance (Rct, 41.4 Ω) than those of the Co3O4 NFs electrode (3.4 Ω and 223.4 Ω) based on the equivalent circuit simulation, revealing the high electronic and ionic conductivity of the Co3O4@CNFs electrodes, resulting in great enhancement in the rate capability and cycling stability.
Conclusions
A unique 1D structure of Co3O4@CNFs composite has been synthesized by a facile, cost-effective and scalable electrospinning method and applied as anode materials of SIBs. Compared to the pure Co3O4 NFs, the Co3O4@CNFs exhibit great enhancement in electrochemical performances of high reversible capacity of 422.4 mA h g−1 with good rate capability and cycling stability over 500 cycles. The outstanding electrochemical performances of Co3O4@CNFs can be ascribed to the unique 1D structure with Co3O4 nanoparticles uniformly distributed in the carbon nanofiber frameworks. The 1D carbon nanofiber can not only increase the electronic and ionic conductivity of the composite electrodes, but also serve as a buffering matrix to accommodate the volume change and protect the composite electrode from pulverization during the repeated Na+ insertion/extraction, enabling Co3O4@CNFs to be promising anode materials for SIBs. Furthermore, the nanostructure design and synthesis described in this work may provide new insights for development of better host materials for practical energy storage applications.Acknowledgements
This work was supported by the Natural Foundation of China (Grant 21503084, 51622703), 973 Program (2015CB258400) and the National Thousand Talents Program of China. The authors thank Analytical and Testing Center of HUST for XRD, SEM and FETEM measurements.Notes and references
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed.
- C. J. Barnhart and S. M. Benson, Energy Environ. Sci., 2013, 6, 1083 CAS.
- A. L. Mohana Reddy, S. R. Gowda, M. M. Shaijumon and P. M. Ajayan, Adv. Mater., 2012, 24, 5045–5064 CrossRef PubMed.
- J. B. Goodenough, Energy Environ. Sci., 2014, 7, 14–18 CAS.
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- B. L. Ellis and L. F. Nazar, Curr. Opin. Solid State Mater. Sci., 2012, 16, 168–177 CrossRef CAS.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338 CAS.
- C. Fang, Y. Huang, W. Zhang, J. Han, Z. Deng, Y. Cao and H. Yang, Adv. Energy Mater., 2016, 6, 1501727 CrossRef.
- D. Kim, S.-H. Kang, M. Slater, S. Rood, J. T. Vaughey, N. Karan, M. Balasubramanian and C. S. Johnson, Adv. Energy Mater., 2011, 1, 333–336 CrossRef CAS.
- H. Kim, H. Kim, Z. Ding, M. H. Lee, K. Lim, G. Yoon and K. Kang, Adv. Energy Mater., 2016, 1600943, DOI:10.1002/aenm.201600943.
- Q. Zhang, W. Wang, Y. Wang, P. Feng, K. Wang, S. Cheng and K. Jiang, Nano Energy, 2016, 20, 11–19 CrossRef CAS.
- M. Zhou, W. Li, T. Gu, K. Wang, S. Cheng and K. Jiang, Chem. Commun., 2015, 51, 14354–14356 RSC.
- Y. You, X. Yu, Y. Yin, K.-W. Nam and Y.-G. Guo, Nano Res., 2014, 8, 117–128 CrossRef.
- I. Hasa, R. Verrelli and J. Hassoun, Electrochim. Acta, 2015, 173, 613–618 CrossRef CAS.
- Z. L. Brown, S. Smith and M. N. Obrovac, J. Electrochem. Soc., 2014, 162, A15–A20 CrossRef.
- Y. Jiang, M. Hu, D. Zhang, T. Yuan, W. Sun, B. Xu and M. Yan, Nano Energy, 2014, 5, 60–66 CrossRef CAS.
- Z. Jian, B. Zhao, P. Liu, F. Li, M. Zheng, M. Chen, Y. Shi and H. Zhou, Chem. Commun., 2014, 50, 1215–1217 RSC.
- X. Liu, T. Chen, H. Chu, L. Niu, Z. Sun, L. Pan and C. Q. Sun, Electrochim. Acta, 2015, 166, 12–16 CrossRef CAS.
- F. Zou, Y. M. Chen, K. Liu, Z. Yu, W. Liang, S. M. Bhaway, M. Gao and Y. Zhu, ACS Nano, 2016, 10, 377–386 CrossRef CAS PubMed.
- Y. Lu, N. Zhang, Q. Zhao, J. Liang and J. Chen, Nanoscale, 2015, 7, 2770–2776 RSC.
- Z. Jian, P. Liu, F. Li, M. Chen and H. Zhou, J. Mater. Chem. A, 2014, 2, 13805 CAS.
- Y. Liu, Z. Cheng, H. Sun, H. Arandiyan, J. Li and M. Ahmad, J. Power Sources, 2015, 273, 878–884 CrossRef CAS.
- C. T. Cherian, J. Sundaramurthy, M. Kalaivani, P. Ragupathy, P. S. Kumar, V. Thavasi, M. V. Reddy, C. H. Sow, S. G. Mhaisalkar, S. Ramakrishna and B. V. R. Chowdari, J. Mater. Chem., 2012, 22, 12198 RSC.
- X. Zhang, H. Liu, S. Petnikota, S. Ramakrishna and H. J. Fan, J. Mater. Chem. A, 2014, 2, 10835 CAS.
- S. Abouali, M. Akbari Garakani, B. Zhang, H. Luo, Z.-L. Xu, J.-Q. Huang, J. Huang and J.-K. Kim, J. Mater. Chem. A, 2014, 2, 16939–16944 CAS.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- S. Abouali, M. A. Garakani, B. Zhang, Z. L. Xu, E. K. Heidari, J. Q. Huang, J. Huang and J. K. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 13503–13511 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02965a |
| This journal is © The Royal Society of Chemistry 2017 |