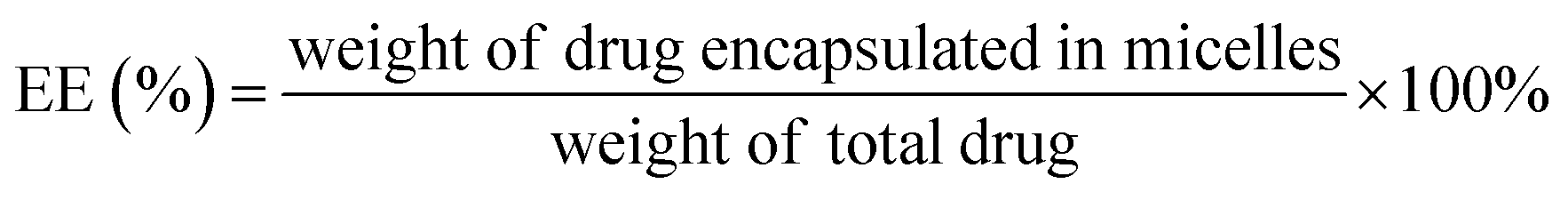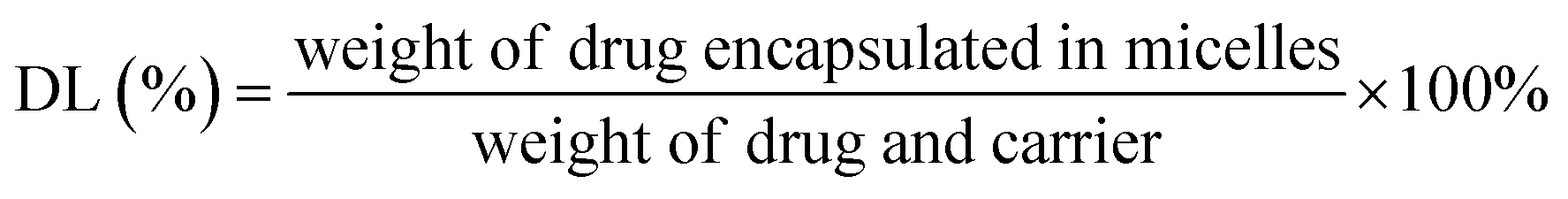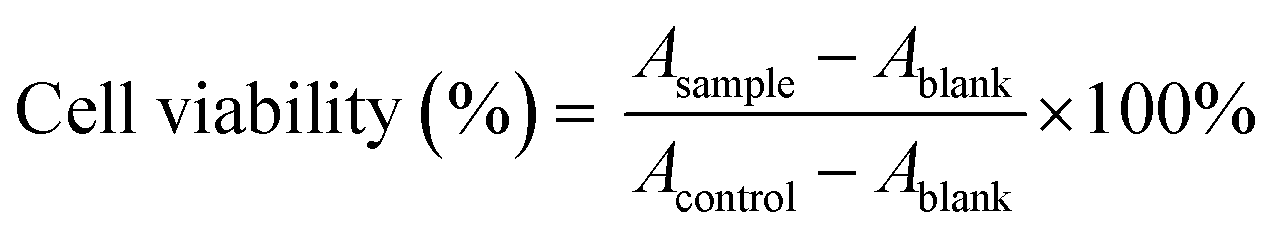DOI:
10.1039/C7RA02909H
(Paper)
RSC Adv., 2017,
7, 24030-24039
Co-delivery of hydrophilic gemcitabine and hydrophobic paclitaxel into novel polymeric micelles for cancer treatment†
Received
10th March 2017
, Accepted 22nd April 2017
First published on 3rd May 2017
Abstract
This study was carried out to investigate an effective method for the co-delivery of Gemcitabine (GEM) and paclitaxel (PTX) into tumor cells. GEM and PTX were modified with functional (+)-α-tocopherol (VE) to obtain similar water solubility. Folic acid-poly(ethylene glycol)–(+)-α-tocopherol (FA–PEG–VE) was designed to co-encapsulate the modified GEM and PTX. Methoxy poly(ethylene glycol)–poly(lactide-co-glycolide) (MPEG–PLGA) was used as a control. The characterizations of micelles were examined by DLS and TEM. It was found that two drugs-loaded FA–PEG–VE micelles, (GPF) and MPEG–PLGA micelles (GPM), had a spherical morphology with an average diameter of 127 nm and 118.9 nm, respectively. GEM–VE and PTX–VE encapsulation efficiencies of GPF were 91.09 ± 0.03%, 92.46 ± 0.02% (88.60 ± 0.03%, 89.32 ± 0.04% of GPM). In vitro release of GPF, 2.73% of GEM–VE and 2.88% of PTX–VE, were accumulatively released in 72 h (4.04% of GEM–VE and 3.88% of PTX–VE from GPM). Furthermore, comparisons of cytotoxicity were made with different fomulations. The IC50 of GPF after 72 h incubation was lowest. FA–PEG–VE micelle showed higher uptake efficiency than that of MPEG–PLGA micelle. Clathrin-mediated and energy-dependent endocytosis was involved in uptake mechanisms. These results demonstrated that GEM–VE and PTX–VE loaded FA–PEG–VE micelles would be a potentially useful prodrug-based nano-drug delivery system for cancer treatment.
Introduction
Cancer, one of the leading causes of mortality worldwide, is called “the leading killer”.1 At present, the most common treatment of cancer is surgery, chemotherapy, and radiotherapy. Chemotherapeutic drugs play an essential role in tumor treatment but are far from perfect because of their undesirable toxic side-effects and limited bioavailability, which is due to low targeting specificity and poor water solubility of anticancer agents, as well as multidrug resistance (MDR) of cancer cells. In addition, cancer is becoming a more and more complex disease so that a single drug or even a stand-alone molecularly targeted therapeutic agent may not be sufficient, and this has become the major limitation of anti-tumor clinical treatment.2–4
Combination chemotherapy has attracted more and more attention from pharmaceutical researchers; this generally involves either co-administering two or more therapeutic agents or combining different types of therapy (e.g., chemotherapy and radiotherapy).5 The hope is that synergistic anticancer effects can be obtained by focusing on different signaling pathways in tumor cells. This approach holds enormous interest for reducing toxicity and undesirable severe side effects, overcoming multidrug resistance, reducing dosage of each agent, and improving therapeutic profiles.6,7
For example, simultaneously administering hydrophilic gemcitabine (GEM) and hydrophobic paclitaxel (PTX) has shown distinct mechanisms of action, synergistic activity in vitro, and non-overlapping toxicity; this has now become a standard clinical treatment method and has proven to be an effective way to suppress proliferation of cancer cells.6,8 However, combining GEM and PTX has several limitations. This drug combination, after injection, is distributed and eliminated independently due to their different chemical and physical properties. For example, free GEM is not stable in the bloodstream and exhibits low permeability across cellular membranes, while free PTX is hindered by its low water solubility.9,10 Therefore, different pharmacokinetics, biodistribution, and membrane transport exhibited by GEM and PTX result in differences in uptake of the two drugs by the same tumor cells or non-specific cellular sites, which consequently can induce relapse or tumor metastases due to low accumulation in patients with malignancy.11
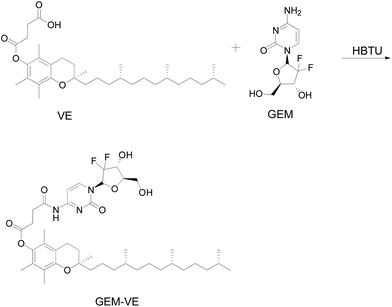
Accordingly, in order to develop an effective method to co-deliver GEM and PTX into target cells, nanocarrier systems were investigated to solve this problem because pharmacokinetic characteristics of the individual drugs depend on the nanocarriers. Nevertheless, it is still a challenge to co-deliver two drugs with different chemical and physical characteristics. For example, liposomes are capable of carrying hydrophilic and hydrophobic drugs in the inner core and bilayer of membranes, respectively. However, there are a number of disadvantages, such as lower encapsulation efficiency of hydrophobic drugs in the lipid bilayer and a higher initial burst release, as well as thermodynamic instability.12 Polymeric micelles have been designed to deliver hydrophobic drugs due to their hydrophobic core, but this limits their application for combining hydrophobic and hydrophilic anticancer drugs.13 In addition, there are problems involving poor stability and inconsistency in drug loading and release kinetics. Recently, application of pro-drugs was extended to combination therapy. After chemical modification, the drugs had a number of advantages compared with parent free drugs including solubility changes in water or the lipid membrane, a reduction in adverse effects, and increased cellular uptake.14,15 In the present study, GEM and PTX were first conjugated with (+)-α-tocopherol (VE), which is thought to be an inhibitor of P-gp,16,17 an efflux transporter active against a variety of structurally unrelated anticancer drugs.18,19 After modification, GEM–VE became hydrophobic and could be easily assembled into a micelle system through hydrophobic interactions. Meanwhile, applications targeting self-assembling block copolymers have opened unprecedented opportunities for controlled drug delivery and novel combination therapy strategies. To improve compatibility of GEM–VE, PTX–VE, and polymeric carriers, we designed a novel polymeric material, FA–PEG–VE, which has the same hydrophobic blocks as GEM–VE and PTX–VE. Thus, co-delivering modified drugs using polymeric micelles was employed to load drugs with different physiochemical properties to target tumor cells with an optimal ratio of the co-delivered drugs; this results in minimizing drug dose and achieving a synergistic therapeutic effect after a single injection.20–22
To co-encapsulate GEM and PTX with the same carrier and deliver them to the same cancer cell simultaneously, GEM and PTX were conjugated with VE. Amphiphilic copolymer FA–PEG–VE was designed to co-encapsulate two modified drugs. Amphiphilic copolymer methoxy poly(ethylene glycol)–poly(lactide-co-glycolide) (MPEG–PLGA) was chosen as a control material, which can be self-assembled into micelles. The hydrophilic block PEG and hydrophobic block PLGA have met with FDA's approval for clinical use as drug delivery carriers.2 The micelles were characterized by their particle size, morphology, and in vitro release. In vitro cell studies were carried out to confirm combination efficacy of the two drugs co-loaded FA–PEG–VE micelles.
Experimental section
Materials
Gemcitabine was obtained from Fujian Vanke Pharmaceutical Co. Ltd (China), while PTX–VE and sulforhodamine B-(+)-α-tocopherol (SRB–VE) were synthesized in our lab (ESI Fig. S1†).7,23
Folic acid (FA) was obtained from Zhengzhou Longsheng Chemical Products Co., Ltd. (China). PEG2000 was obtained from Jiangsu Haian Petro Chemical Plant (China), while (+)-α-tocopherol (VE) was obtained from Hubei Prosperity Galaxy Chemical Co., Ltd. (China). 3-(Ethyliminomethylideneamino)-N,N-dimethylpropan-1-amine (EDCI) was obtained from GL Biochem Ltd, and di(imidazol-1-yl)methanone (CDI) was obtained from Tianjin Weiyi Chemical Reagent Co., Ltd. (China). 2,2,2-Trifluoroacetic acid (TEA) and N,N-dimethylpyridin-4-amine (DMAP) were obtained from Tianjin Kemiou Chemical Reagent Co., Ltd. and N,N-dimethylformamide (DMF) was obtained from Shanghai Energy Chemical (China). O-Benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) was obtained from Shanghai Ziyi Reagent Co. Ltd. and methylsulfonylmethane (DMSO), dichloromethane and methanol were obtained from Concord Technology (Tianjian) Co., Ltd. (China). Acetone was obtained from the Chemical Reagent Factory of Shenyang Pharmaceutical Company (China).
Synthesis of FA–PEG–VE
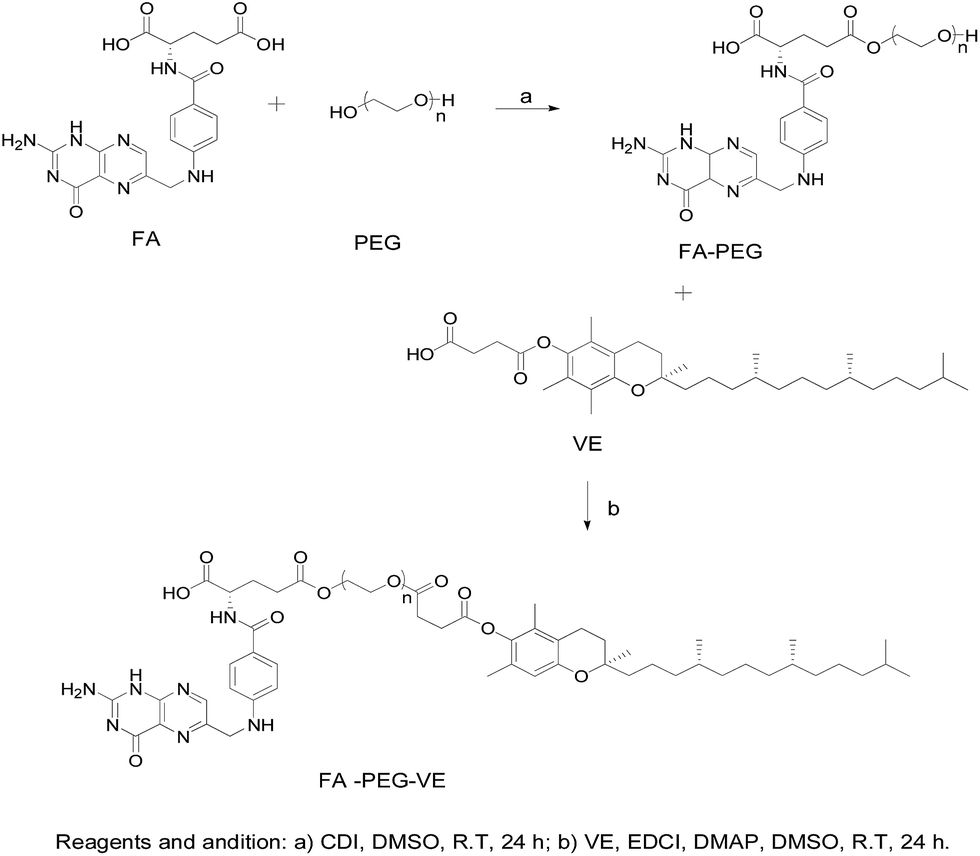
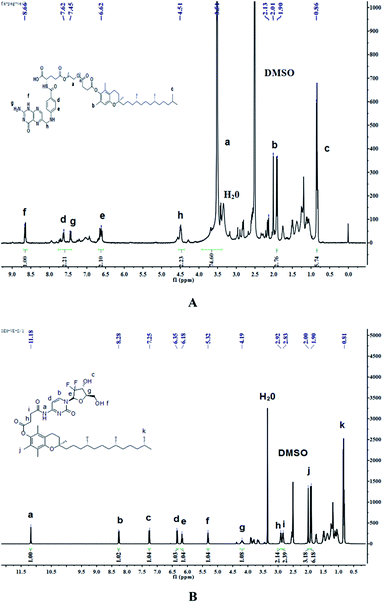
Synthesis of FA-PEG. The synthetic route of FA–PEG–VE is shown in Fig. 1. For this, 2.207 g FA, and 0.892 g CDI were dissolved in 80 mL dry DMSO. The reaction was carried out at room temperature in the dark for 4 h under a nitrogen atmosphere. Then, 37.500 g PEG2000 was dissolved in 170 mL dry DMSO and added to the above solution. The reaction was allowed to proceed for 24 h in the dark. Next, the sediment was allowed to separate after adding 2000 mL dichloromethane to the reaction mixture. This sediment was collected by filtration and dissolved in water. The solution was transferred to a dialysis tube (MWCO 1000) and dialyzed in the dark for 2 days against distilled water. Finally, the product was obtained after vacuum drying in an oven for 2 days. The 1H-NMR spectrum of FA-PEG (ESI Fig. S2†): peaks at 11.71 ppm(a), 8.65 ppm(b), 8.10 ppm(c), 7.66 ppm(d), 7.03 ppm(e), 6.66 ppm(f), 4.49 ppm(g), 4.33 ppm(h) belong to the H of the FA fragment; the peak at 3.51 ppm(i) belongs to the H of the PEG fragment.
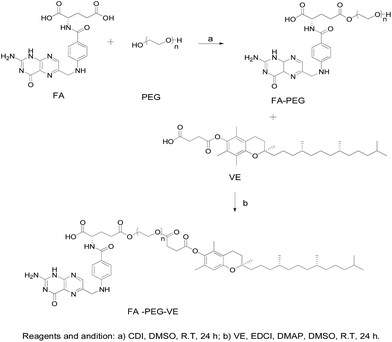 |
| | Fig. 1 The synthetic route for FA–PEG–VE. | |
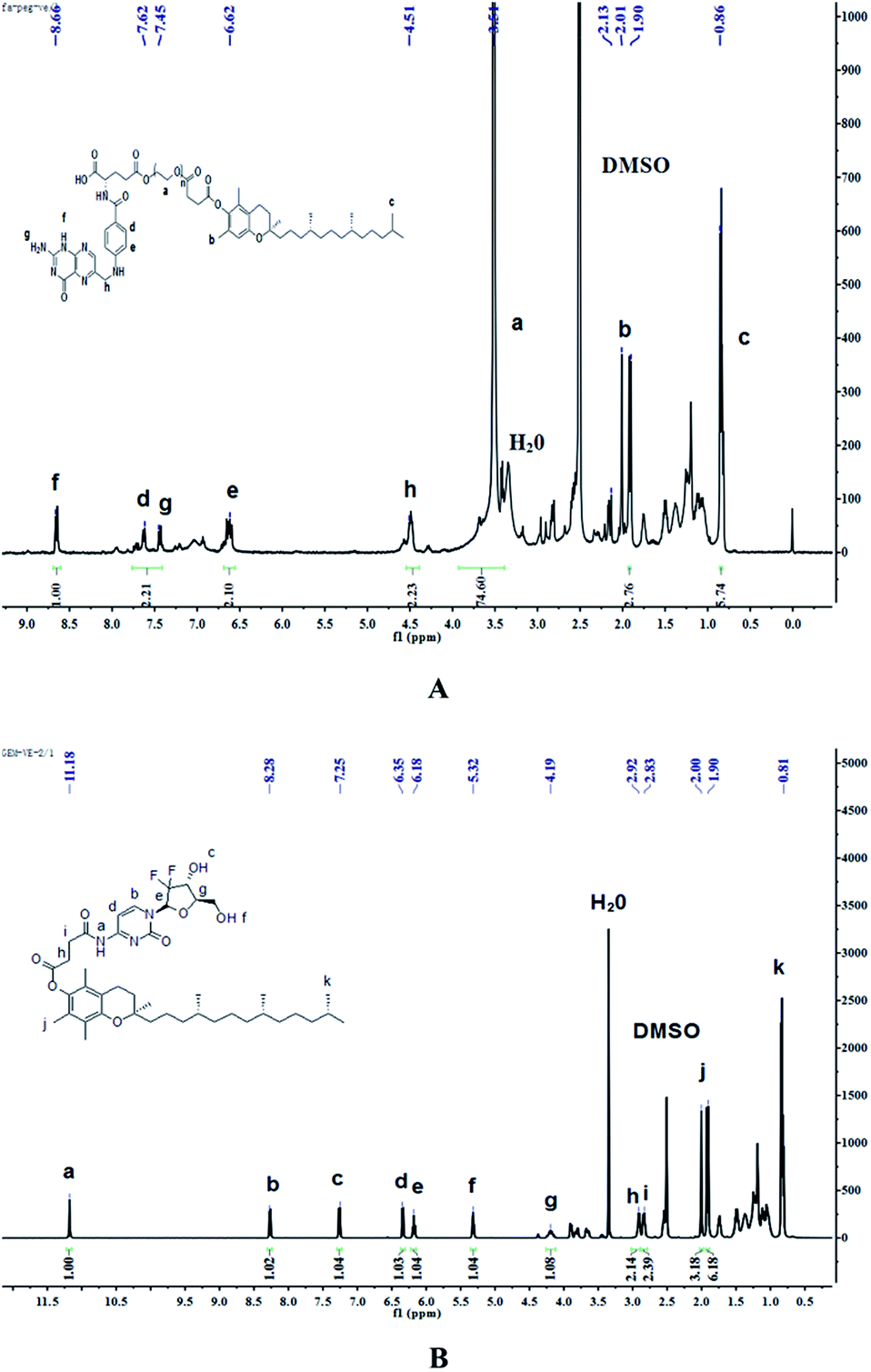
Synthesis of FA–PEG–VE. The product of the last step, FA-PEG, was dissolved in 70 mL dry DMSO. Then, 1.2 mol eq. VE, 3 mol eq. EDCI and 0.5 mol eq. DMAP were dissolved in 10 mL dry DMSO, and the solution was stirred for 30 min at room temperature before adding it to the FA-PEG solution. Next, the reaction was allowed to proceed for 24 h in the dark at room temperature and then the product was purified by dialyzing (MWCO 2000) against deionized water for 3 days in the dark. Finally, the water was removed by reduced pressure distillation and the product was dried under vacuum at 45 °C for 2 days. The 1H-NMR spectrum of FA–PEG–VE is shown in Fig. 3A: peaks at 4.51 ppm(h), 7.45 ppm(g), 6.62 ppm(e), 7.62 ppm(d), 8.66 ppm(f) belong to the H of the FA fragment; the peak at 3.51 ppm(a) belongs to the H of the PEG fragment, and peaks at 1.90–2.13 ppm(b) and 0.86 ppm(c) belong to the H of the VE fragment. The FTIR analysis of the spectrums for FA, PEG-VE was reported.24,25 The FT-IR spectrum is shown in Fig. S3;† 2892.43 (C–H), 1728.87, 1688.87 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1060.38(C–O).
O), 1060.38(C–O).
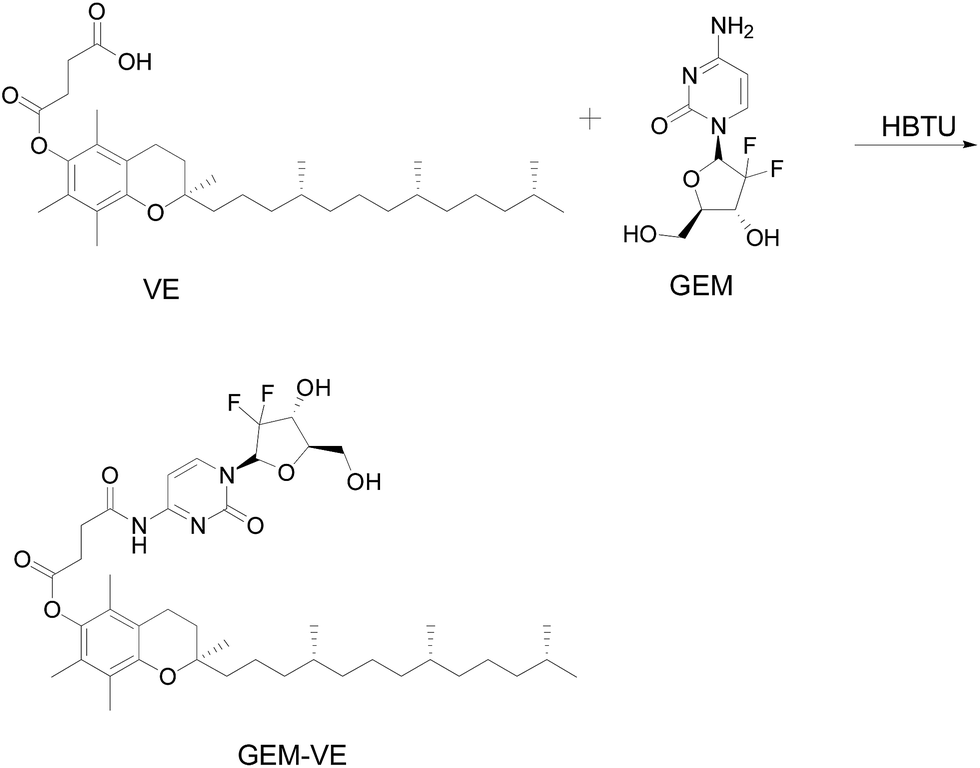
Synthesis of GEM–VE. The synthetic route of GEM–VE is shown in Fig. 2. For this, 0.5 g VE, 0.248 g GEM, and 0.714 g HBTU were dissolved in 15 mL DMF. Then 5 drops of TEA were added to the solution and the reaction was carried out at room temperature for 24 h under a nitrogen atmosphere. Next, the solution was mixed with 60 mL ethyl acetate and successively washed with a small quantity of HCl, deionized water, saturated NaHCO3 solution, and saturated NaCl solution. Next, the ethyl acetate layer was collected and separated by silica-gel column chromatography. The crude product was re-crystallized from ethyl acetate and petroleum ether and, after pulping in a solution of ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (1:2), the pure product was obtained. The 1H-NMR spectrum of GEM–VE in Fig. 3B shows peaks at 11.18 ppm(a), 8.28 ppm(b), 7.25 ppm(c), 6.35 ppm(d), 6.18 ppm(e), 5.32 ppm(f), 4.19 ppm(g), 2.92 ppm(h), and 2.83 ppm(i) belonging to the H of the GEM fragment, and peaks at 0.81 ppm(k), and 1.90–2.00 ppm(j) belong to the H of the VE fragment. The FTIR analysis with the spectra for VE, GEM was reported,12,25 and our FTIR spectrum is shown in Fig. S4;† 3484.65(–OH), 1664.78(CO), 1077.31(C–O). MS (ESI): m/z 798.4 (M + Na).
:water (1:2), the pure product was obtained. The 1H-NMR spectrum of GEM–VE in Fig. 3B shows peaks at 11.18 ppm(a), 8.28 ppm(b), 7.25 ppm(c), 6.35 ppm(d), 6.18 ppm(e), 5.32 ppm(f), 4.19 ppm(g), 2.92 ppm(h), and 2.83 ppm(i) belonging to the H of the GEM fragment, and peaks at 0.81 ppm(k), and 1.90–2.00 ppm(j) belong to the H of the VE fragment. The FTIR analysis with the spectra for VE, GEM was reported,12,25 and our FTIR spectrum is shown in Fig. S4;† 3484.65(–OH), 1664.78(CO), 1077.31(C–O). MS (ESI): m/z 798.4 (M + Na).
 |
| | Fig. 2 The synthetic route for GEM–VE. | |
 |
| | Fig. 3 The 1H-NMR spectrum of FA–PEG–VE (A) and GEM–VE (B). | |
Determination of the critical micelle concentration (CMC)
The CMC values of micelle solutions are generally determined by fluorescence measurements using pyrene as a probe,26 Briefly, aliquots of the pyrene solution (2.4 × 10−6 mol L−1, 500 μL) in acetone were added to a volumetric flask and acetone was removed by vacuum drying, followed by adding blank micelles solutions with concentrations ranging from 1 × 10−6 mg mL−1 to 1 × 10−3 mg mL−1. The final pyrene concentration in the copolymer solution was fixed at 1.2 × 10−7 mol L−1. The mixture was then sonicated for 30 min, and kept in the dark for 24 h at room temperature to reach soluble equilibrium. Fluorescence of soluble pyrene was measured at an excitation wavelength of 335 nm and emission wavelengths of 373 nm (I1) and 384 nm (I3), respectively, using a microplate reader (Thermo Scientific, U.S.). The CMC was determined by plotting intensity ratio of (I1/I3) versus the logarithm of the micelles solution concentration.
Preparation of FA–PEG–VE and MPEG–PLGA polymeric micelles
The GEM–VE- and PTX–VE-loaded FA–PEG–VE micelles (GPF) were prepared by an organic solvent evaporation method.13,27 Briefly, moderate amounts of GEM–VE and PTX–VE were dissolved in 3 mL acetone, while 2 mg FA–PEG–VE was dissolved in 6 mL aqueous solution containing 0.1% (w/v) Na2CO3. Then, the organic phase was added dropwise to the water phase under gentle stirring at 60 °C in a water bath until the organic reagent had evaporated completely. Following this, the micelle solution was obtained by passing through a 0.45 μm syringe filter to remove aggregates and free drugs. MPEG–PLGA was used as a control. As shown in Table 1, blank FA–PEG–VE micelle (BF), blank MPEG–PLGA micelle (BM), GEM–VE loaded FA–PEG–VE micelle (GF), GEM–VE loaded MPEG–PLGA micelle (GM), PTX–VE loaded FA–PEG–VE micelle (PF), PTX–VE loaded MPEG–PLGA micelle (PM) and dual drugs loaded MPEG–PLGA micelle (GPM), SRB–VE, GEM–VE and PTX–VE loaded FA–PEG–VE micelle (SGPF) and MPEG–PLGA micelle (SGPM) were prepared by a similar method.
Table 1 Abbreviations of different formulations
| Abbreviations |
Formulations |
| GEM–VE |
Free GEM–VE solution |
| PTX–VE |
Free PTX–VE solution |
| GEM–VE + PTX–VE |
GEM–VE and PTX–VE solution |
| BM |
Blank MPEG–PLGA micelle |
| BF |
Blank FA–PEG–VE micelle |
| PM |
PTX–VE loaded MPEG–PLGA micelle |
| PF |
PTX–VE loaded FA–PEG–VE micelle |
| GM |
GEM–VE loaded MPEG–PLGA micelle |
| GF |
GEM–VE loaded FA–PEG–VE micelle |
| GPM |
GEM–VE and PTX–VE loaded MPEG–PLGA micelle |
| GPF |
GEM–VE and PTX–VE loaded FA–PEG–VE micelle |
| SGPM |
SRB–VE GEM–VE and PTX–VE loaded MPEG–PLGA micelle |
| SGPF |
SRB–VE GEM–VE and PTX–VE loaded FA–PEG–VE micelle |
Characterization of micelles
Measurements of particle size and zeta potential. Particle size, size distribution, and zeta potential of different formulations were determined by DLS using a Zetasizer Nano instrument (ZS90 Malvern Instruments Ltd., UK). Particle size was determined at a fixed scattering angle of 90° at 25 °C, and the zeta potential was measured at 25 °C.
Determination of encapsulation efficiency and drug loading capacity (EE and DL). For the measurement of entrapment efficiency (EE) and drug loading (DL) of drug-loaded micelle, the amount of free drug (non-encapsulated in micelle) was determined by ultrafiltration centrifugation. 0.5 mL of micelle solution in an ultrafiltration tube was centrifuged at 10000 rpm for 15 min to separate non-encapsulated drug from the micelle solution. The total amount of drug was determined by mixing drug-loaded micelles with methanol and sonicating for 30 min. After centrifuging for 10 min at 5000 rpm, the contents of drugs were calculated using a validated HPLC method involving an LC-ATVP pump and SPD-10AVP ultraviolet light detector (Shimadzu, Kyoto, Japan). The HPLC conditions were as follows: a C18 column (4.6 mm × 150 mm, 5 μm, Dikma, Tianjin, China) was eluted with methanol/water (97:3, v/v) at a flow rate of 1.0 mL min−1. The detection wavelength of GEM–VE and PTX–VE was set at 248 nm and 227 nm, respectively. And the sample injection volume was kept constant at 20 μL. EE(%) and DL(%) of the drug-loaded micelles were calculated according to the following eqn (1) and (2):| |
 | (1) |
| |
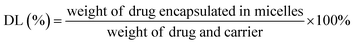 | (2) |
Transmission electron microscopy (TEM). The morphologies of BF, BM, PF, PM, GF, GM, GPF, and GPM were observed under TEM using a JEM-100SX electron microscope (JEOL, Tokyo, Japan). A drop of aqueous micelle solution with a selected concentration was placed onto a 300-mesh film-coated copper grid. After air-drying, negative staining was performed using a droplet of 1% (w/v) phosphotungstic acid for 1 min. Then, excess liquid was removed with a filter paper, and dried prior to imaging.28
In vitro release. The in vitro release behaviors of GEM–VE and PTX–VE from polymeric micelles were tested by a dialysis method. GF, GM, PF, PM, GPF, and GPM solutions were transferred into a dialysis tube (molecular weight cutoff: 8–14 kDa), and then dialyzed against 100 mL pH 7.4 PBS containing 0.5% (w/v) SDS and stirred at 100 rpm and 37 °C. At predetermined times, 1 mL of release medium was withdrawn for further determination and equal fresh release medium was added. The contents of GEM–VE and PTX–VE were assayed by HPLC and accumulative drug release was plotted as a function of time.
Cell tests in vitro
Cell cultures. Human lung carcinoma cells (A549) were used as a model cell line and obtained from Nanjing Keygen Biotech Co., Ltd. (Nanjing, China). They were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, penicillin (100 units per mL), and streptomycin (100 μg mL−1) at 37 °C and 5% CO2. Cells in the exponential phase of growth were used in the experiments.
In vitro cytotoxicity. In vitro cytotoxicity of different formulations was carried out in A549 cells. First, comparisons of cytotoxicity of GEM and GEM–VE, PTX, and PTX–VE were performed. Briefly, cell suspension at a density of 5 × 103 cells per mL was seeded in a 96-well plate and subsequently cultured for 24 h. After attachment, the culture medium was replaced with GEM and GEM–VE, PTX and PTX–VE at various concentrations and incubated for another 24 h. Then, comparisons of cytotoxicity of PTX–VE, GEM–VE, a mixture of GEM–VE, and PTX–VE, BF, BM, PF, PM, GF, GM, GPF, and GPM were also conducted and incubated with A549 cells for 72 h. Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay. For this, 20 μL MTT stock solutions (5 mg mL−1) were added to each well, and then the cells were incubated for another 4 h at 37 °C. The medium was then removed and 150 μL DMSO was added to dissolve formazan crystals formed in the live cells. Absorbance of each well was measured at 490 nm after the plates were incubated at 37 °C for 10 min to confirm that all the crystals had dissolved, using a microplate reader. Cell viability was calculated using the following eqn (3):| |
 | (3) |
In vitro cellular uptake. SRB–VE was chosen as the fluorescent material which was encapsulated with the drugs into GPF and GPM micelles to further estimate cellular uptake. The cells were seeded in a 24-well plate at a concentration of 1 × 105 cells per mL and incubated for 24 h. After attachment, the culture medium was replaced with different concentrations of micelle solutions (SGPF, SGPM) (SRB–VE concentrations: 0.07, 0.13, 0.27, 0.50, 1.21, and 1.39 μg mL−1) diluted by FBS free culture medium. After incubation for 2 h at 37 °C and 5% CO2, the culture medium was removed and the cells were washed three times with cold PBS, and then 150 μL RIPA Lysis Buffer (Beyotime, China) was added to digest the cells. Following this, 100 μL Lysis Buffer was transferred to an opaque 96-well plate. Fluorescence intensity of the cellular uptake was measured using a Microplate Reader and uptake was expressed as the amount (ng) of SRB–VE. In addition, cellular uptake of the micelles (SRB–VE concentration, 1.21 μg mL−1) at different incubation times (0.5 h, 1 h, 2 h, 4 h, and 6 h) was also investigated in the same way.Furthermore, the cellular uptake mechanism was investigated by adding inhibitors to prevent potential internalization pathways, such as chlorpromazine (10 μg mL−1), colchicine (40 μg mL−1), indomethacin (15 μg mL−1), and sodium azide (30 μg mL−1). Inhibitors were added to each well and incubated with A549 cells for 1 h. Then, the medium was replaced by SGPM and SGPF solutions (SRB–VE concentration, 1.21 μg mL−1); synergism with different internalization inhibitors are described above. Cellular uptake at 4 °C and room temperature was also studied. After further incubation for another 2 h, the treatment was the same as that described above.29
Confocal laser scanning microscopy (CLSM) observations. Cellular uptake of different micelles was observed by CLSM (Carl Zeiss LSM 710, Germany). A549 cells were seeded on a sterile cover slip in a 24-well plate and incubated until attachment. Then, the culture medium was replaced with fresh FBS free culture medium containing SGPM and SGPF with a SRB–VE concentration of 1.21 μg mL−1. After incubation for 0.5 h, 1 h, 2 h, 4 h, and 6 h, the cells were washed with cold PBS 3 times and fixed with 4% paraformaldehyde for 30 min at room temperature. The nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) for 5 min before observation.
Results and discussion
Critical micelle concentration (CMC)
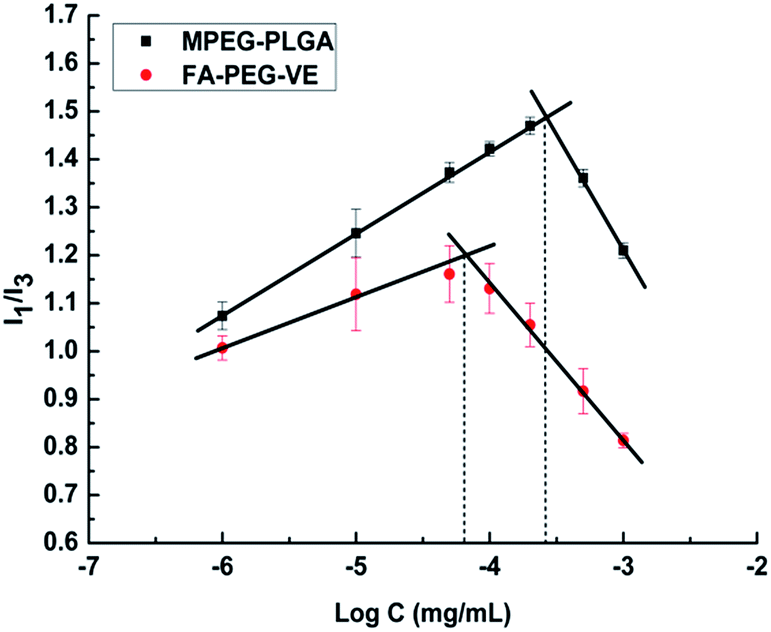
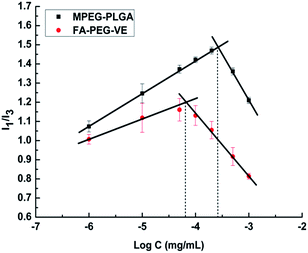
The CMC value is the concentration of amphiphilic copolymers under which the copolymer is present in monomer form or a large aggregate, above which core–shell micelles are formed by hydrophobic interactions.30 The CMC value has a great significance in a drug delivery system. The concentration of copolymers will be greatly diluted by blood following intravenous administration. If the diluted concentration is higher than the CMC, then the micelle solution will be stable until it reaches the tumor site. Otherwise, there will be depolymerization of the micelles, wrapped hydrophobic drug precipitation, and adverse reactions. In this study, as shown in Fig. 4, the CMC value of FA–PEG–VE was 0.0637 μg mL−1, much lower than that of MPEG–PLGA (0.272 μg mL−1). This relatively lower CMC value indicated that copolymer micelles would be stable in the blood stream after intravenous injection. Theoretically, a higher drug loading capacity of micelle will be obtained due to the lower CMC. Therefore, synthetic FA–PEG–VE as an amphiphilic copolymer for self-assembly micelles has potential.
 |
| | Fig. 4 Fluorescence intensity (I1/I3) ratio of pyrene versus the logarithm of the polymer concentration. | |
Preparation and characterization of polymeric micelles
According to the properties of amphiphilic copolymers, drug-loaded micelles GPF and GPM were successfully prepared by an organic solvent evaporation method. To examine advantages of the FA–PEG–VE micelles, a comparison with MPEG–PLGA micelles was conducted.
Particle size, size distribution, and zeta potential are the main influence factors of physical stability and transfer behaviors in vitro and in vivo.31,32 As shown in Table 2, the average particle size of a blank MPEG–PLGA micelle was 64.0 nm; after encapsulating a drug into a MPEG–PLGA micelle, a slight increase particle size was observed (PM 102.1 nm, GM 109.1 nm, and GPM 118.9 nm). The average particle size of a blank FA–PEG–VE micelle was 175.6 nm, while after encapsulating a drug into a FA–PEG–VE micelle, decreased particle sizes were observed (PF 141.8 nm, GF 125.8 nm, and GPF 127 nm). This might be due to GEM–VE, PTX–VE, and FA–PEG–VE because with the same hydrophobic core VE, the binding force between drugs and copolymer become intensely stronger after drug encapsulation. Therefore, a smaller particle size of drug-loaded FA–PEG–VE micelles was obtained. Furthermore, the prepared micelles all had a unimodal particle size distribution and a negative charge, which could prevent aggregation and increase the stability of micelles.
Table 2 Physicochemical characterizations of micelle formulations (mean ± SD, n = 3, each experiment repeated 3 times)
| Micelles |
Particle size (nm) |
PDI |
Zeta potential (mv) |
| BM |
64.0 ± 1.6 |
0.268 ± 0.01 |
−7.6 ± 0.55 |
| PM |
102.1 ± 9.6 |
0.267 ± 0.02 |
−9.6 ± 0.47 |
| GM |
109.1 ± 2.6 |
0.079 ± 0.05 |
−10.9 ± 0.54 |
| GPM |
118.9 ± 1.0 |
0.200 ± 0.006 |
−11.4 ± 0.52 |
| BF |
175.6 ± 2.1 |
0.227 ± 0.008 |
−23.6 ± 0.55 |
| PF |
141.8 ± 5.8 |
0.128 ± 0.028 |
−35.7 ± 0.44 |
| GF |
125.8 ± 5.9 |
0.189 ± 0.02 |
−47.9 ± 0.63 |
| GPF |
127 ± 1.1 |
0.245 ± 0.003 |
−47.6 ± 0.91 |
As shown in Table 3, in comparison with drug-loaded MPEG–PLGA micelles, FA–PEG–VE micelles showed better drug loading capacity. Furthermore, dual drugs-loaded micelles displayed better drug loading capacity than single drug-loaded micelles. This may be due to an interaction between drugs. GEM–VE and PTX–VE EE% of GPF was 91.09 ± 0.03% and 92.46 ± 0.02% and the DL% was 36.64 ± 0.01% and 2.04 ± 0.01%, respectively. However, the GEM–VE and PTX–VE EE% of GPM was 88.60 ± 0.03% and 89.32 ± 0.04%, and the DL% was 4.14 ± 0.02% and 0.35 ± 0.02%, respectively. Obviously, the GEM–VE loading efficiency of FA–PEG–VE micelles was approximately 9 times higher than that of MPEG–PLGA micelles (PTX–VE DL% was 6 times). This may be attributed to the lower critical micelle concentration of FA–PEG–VE. When the equivalent amount of drug was encapsulated, less FA–PEG–VE material was used compared with MPEG–PLGA (weight ratio 2:15).
Table 3 Encapsulation efficiency (EE) and drug loading capacity (DL) (mean ± SD, n = 3, each experiment repeated 3 times)
| Micelles |
EE (% ± SD) |
DL (% ± SD) |
| GEM–VE |
PTX–VE |
GEM–VE |
PTX–VE |
| PM |
— |
86.87 ± 0.12 |
— |
0.32 ± 0.01 |
| GM |
84.70 ± 0.20 |
— |
2.17 ± 0.05 |
— |
| GPM |
88.60 ± 0.03 |
89.32 ± 0.04 |
4.14 ± 0.02 |
0.35 ± 0.02 |
| PF |
— |
88.15 ± 1.18 |
— |
2.03 ± 0.15 |
| GF |
89.30 ± 0.69 |
— |
18.99 ± 0.58 |
— |
| GPF |
91.09 ± 0.03 |
92.46 ± 0.02 |
36.64 ± 0.01 |
2.04 ± 0.01 |
In addition, the morphologies of BF, BM, PF, PM, GF, GM, GPF, and GPM were investigated by transmission electron microscopy (TEM). We can see from Fig. 5A that all formulations were spherical and homogeneous. However, the size observed by TEM was little smaller than that measured by DLS. This may be due to a shrinkage of the micelles during the drying process when TEM samples were made.33
 |
| | Fig. 5 TEM images of BF, BM, GPF, GPM, PF, PM, GF and GM. | |
In vitro release
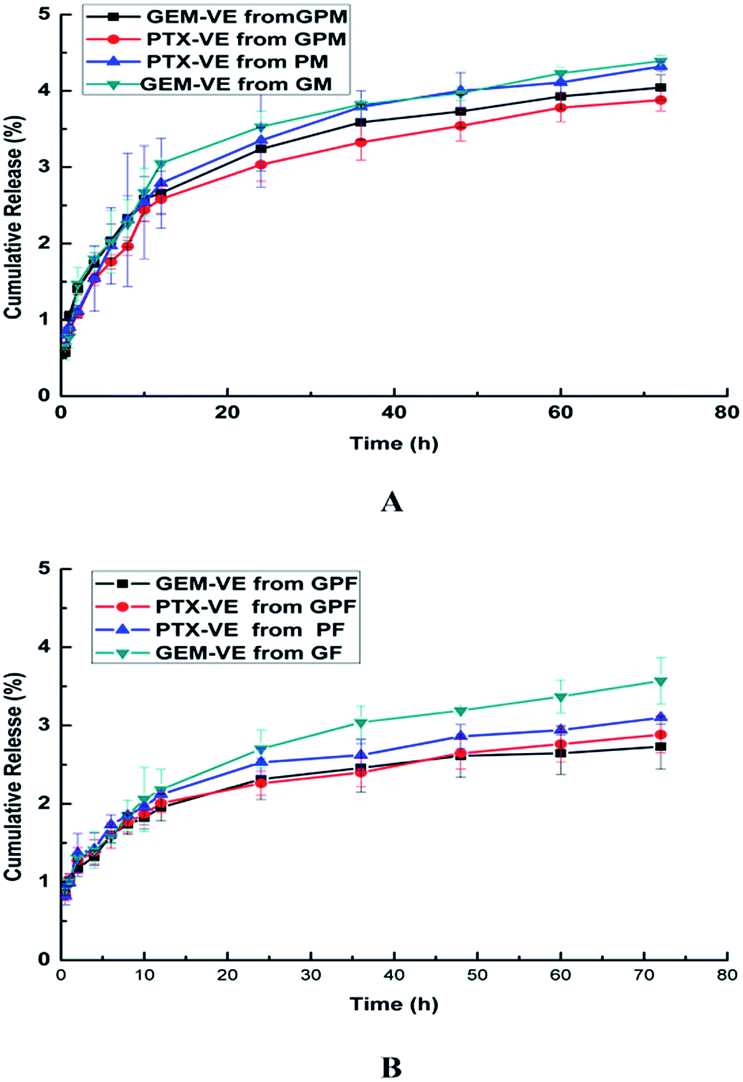
The in vitro drug release profiles of GF, PF, GM, PM, GPF, and GPM were examined at pH 7.4. As shown in Fig. 6, the release of GEM–VE and PTX–VE was sustained, and the release from single drug loaded micelle was a little faster than that from dual drug-loaded micelles. This may be affected by the interaction between drugs. Furthermore, 2.73% of GEM–VE and 2.88% of PTX–VE from GPF were accumulatively released within 72 h; 4.04% of GEM–VE and 3.88% of PTX–VE from GPM were accumulatively released within 72 h. The obtained similar profiles of GEM–VE and PTX–VE released from both GPF and GPM indicated that the co-delivery system provided a possibility of their synergistic effect. It has been mentioned in previous reports that synchronous release of hydrophilic and hydrophobic drugs from a drug delivery system is necessary.2,4 As for hydrophilic GEM, after conjugating with VE it became hydrophobic, after which it could be co-encapsulated into micelles and released from micelles synchronously.
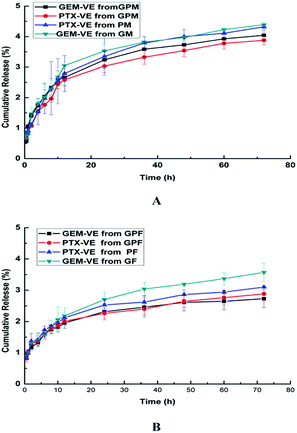 |
| | Fig. 6 Release profiles of GEM–VE or PTX–VE from GM, PM, GPM (A); GF, PF, GPF (B) in the PBS (pH 7.4) at 37 °C. | |
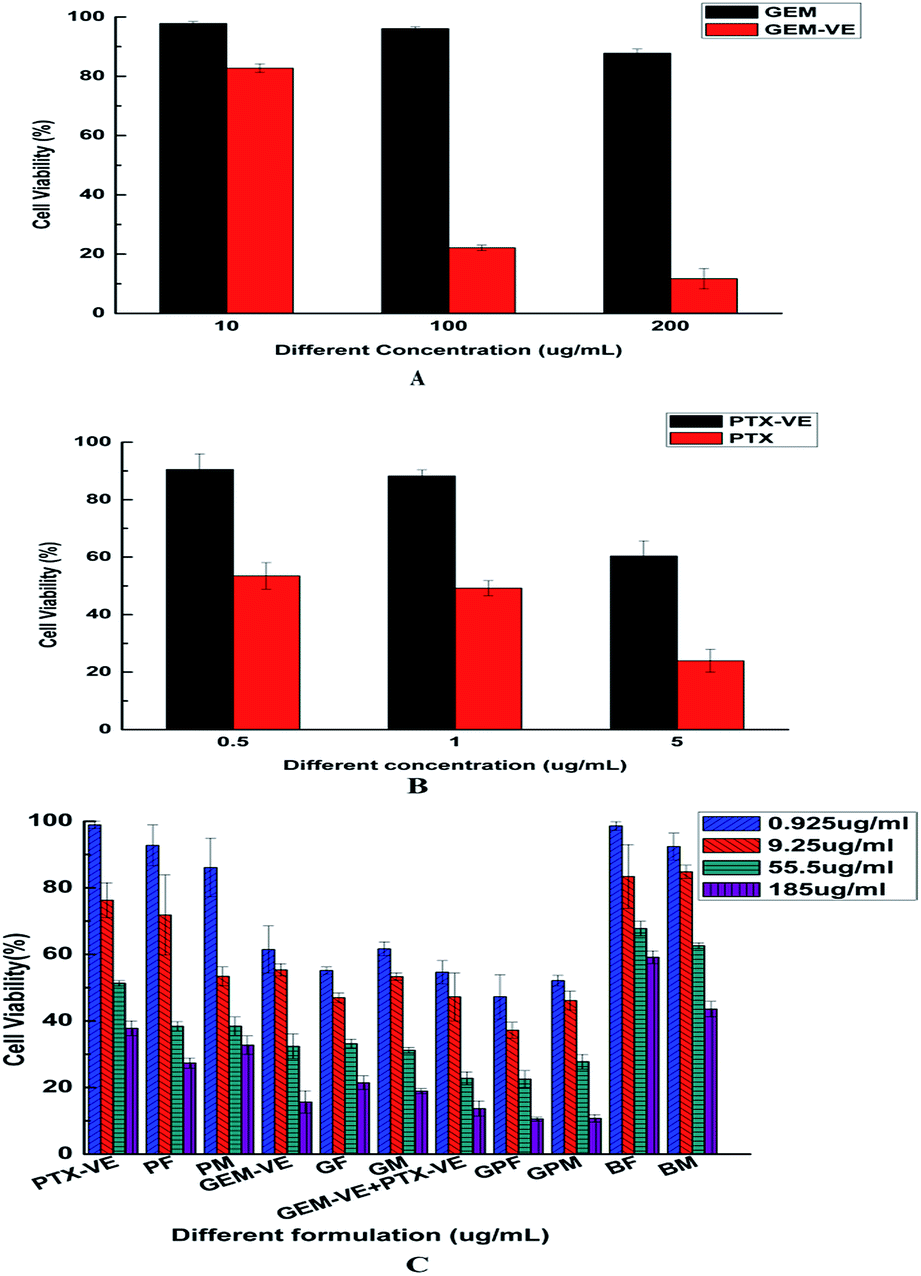
In vitro cytotoxicity
In this study, we investigated the cytotoxicity of different formulations in A549 cells using an MTT assay. The viability of A549 cells after incubation with GEM and GEM–VE for 24 h at three different concentrations (10, 100, and 200 μg mL−1) is shown in Fig. 7A; GEM–VE showed higher tumor cell growth inhibition efficiency than that of GEM. This was due to the low permeability of free GEM across the cellular membrane.12 The viability of A549 cells after incubation with PTX and PTX–VE for 24 h at three different concentrations (0.5, 1, and 5 μg mL−1) is shown in Fig. 7B; PTX–VE showed lower tumor cell growth inhibition efficiency. Despite this, as mentioned before, PTX–VE exhibited higher average blood concentration than that of PTX, and longer half-lives and retention times in vivo.23,34
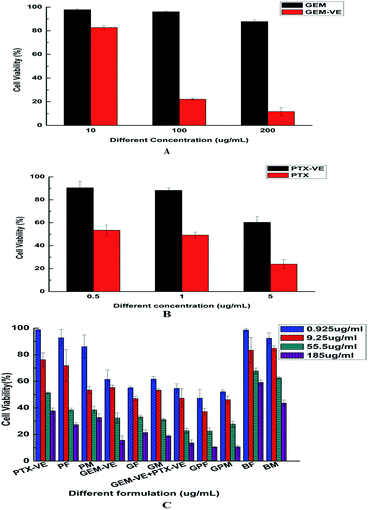 |
| | Fig. 7 In vitro cytotoxicity of GEM and GEM–VE in A549 cells for 24 h (A), PTX and PTX–VE for 24 h (B), different formulations for 72 h (C) (mean ± SD, n = 3. The legend in C shows the concentration of GEM–VE). | |
Furthermore, we evaluated the advantages of co-delivery drugs into novel micelles. The viability of A549 cells after incubation with free conjugated drug solutions, blank micelles, and drug-loaded micelles for 72 h at four different concentrations (GEM–VE concentrations: 0.925, 9.25, 55.5, and 185 μg mL−1; corresponding PTX–VE concentrations: 0.0775, 0.775, 4.65, and 15.5 μg mL−1) is shown in Fig. 7C. We observed that the cell toxicity of BF and BM on A549 cells was minimal, while the free drug solution groups (GEM–VE, PTX–VE, GEM–VE, and PTX–VE) and the drug-loaded micelle groups (GF, PF, GPF; GM, PM, and GPM) showed inhibitory effects on A549 cells. Therefore, we confirmed that MPEG–PLGA and synthetic FA–PEG–VE had no significant cytotoxic effects on cancerous cells. What's more, with increasing drug concentrations, a marked inhibitory effect on cell growth was obtained. In addition, according to the IC50 values (72 h) in Table 4, the cytotoxicity of formulations can be ranked as follows: GPF > GEM–VE + PTX–VE > GF > GEM–VE > PF > PTX–VE > BF; GPM > GEM–VE + PTX–VE > GM > PM > GEM–VE > PTX–VE > BM. No matter which drug-loaded FA–PEG–VE or MPEG–PLGA micelles were used, the cytotoxicity of micelle solutions was higher than those of the corresponding drug-free solutions. Furthermore, GEM–VE and PTX–VE loaded micelles showed higher cytotoxicity than those of single drug loaded micelles. In addition, no matter whether single drug-loaded micelles or two drug-loaded micelles were used, the cytotoxicity of FA–PEG–VE micelles was greater than those of MPEG–PLGA micelles.
Table 4 Cytotoxicity of different formulations in A549 cell lines for 72 h, expressed as IC50 values (GEM–VE concentration, μg mL−1). Significant differences are indicated as follows: comparisons in the same raw *P < 0.05 and **P < 0.01. Comparisons in the same column, different letters (a, b, c, d, e, and f) represent P < 0.05
| MPEG–PLGA |
IC50(μg mL−1) |
FA–PEG–VE |
IC50(μg mL−1) |
Comparison |
| GEM–VE |
19.3 ± 6.1 b |
GEM–VE |
19.3 ± 6.1 c |
— |
| PTX–VE |
74.3 ± 0.6 a |
PTX–VE |
74.3 ± 0.6 a |
— |
| GEM–VE + PTX–VE |
3.9 ± 0.5 d |
GEM–VE + PTX–VE |
3.9 ± 0.5 d |
— |
| GM |
4.2 ± 0.5 d |
GF |
2.1 ± 0.3 e |
** |
| PM |
13.6 ± 2.6 c |
PF |
24.9 ± 6.2 b |
* |
| GPM |
2.5 ± 0.1 e |
GPF |
0.5 ± 0.3 f |
** |
To sum up all the comparisons, GEM and PTX after modifying with VE can be synergistically encapsulated into the same copolymer to produce co-delivery drug micelles, and can significantly improve the toxicity of A549 cells compared with a single drug delivery nano-system. Also, micelles prepared using the synthetic FA–PEG–VE copolymer exhibited an excellent inhibitory effect on tumor cell growth in comparison with the commercially available copolymer MPEG–PLGA. This may be attributed to folic acid being able to target tumor cells, which will be investigated in future work.
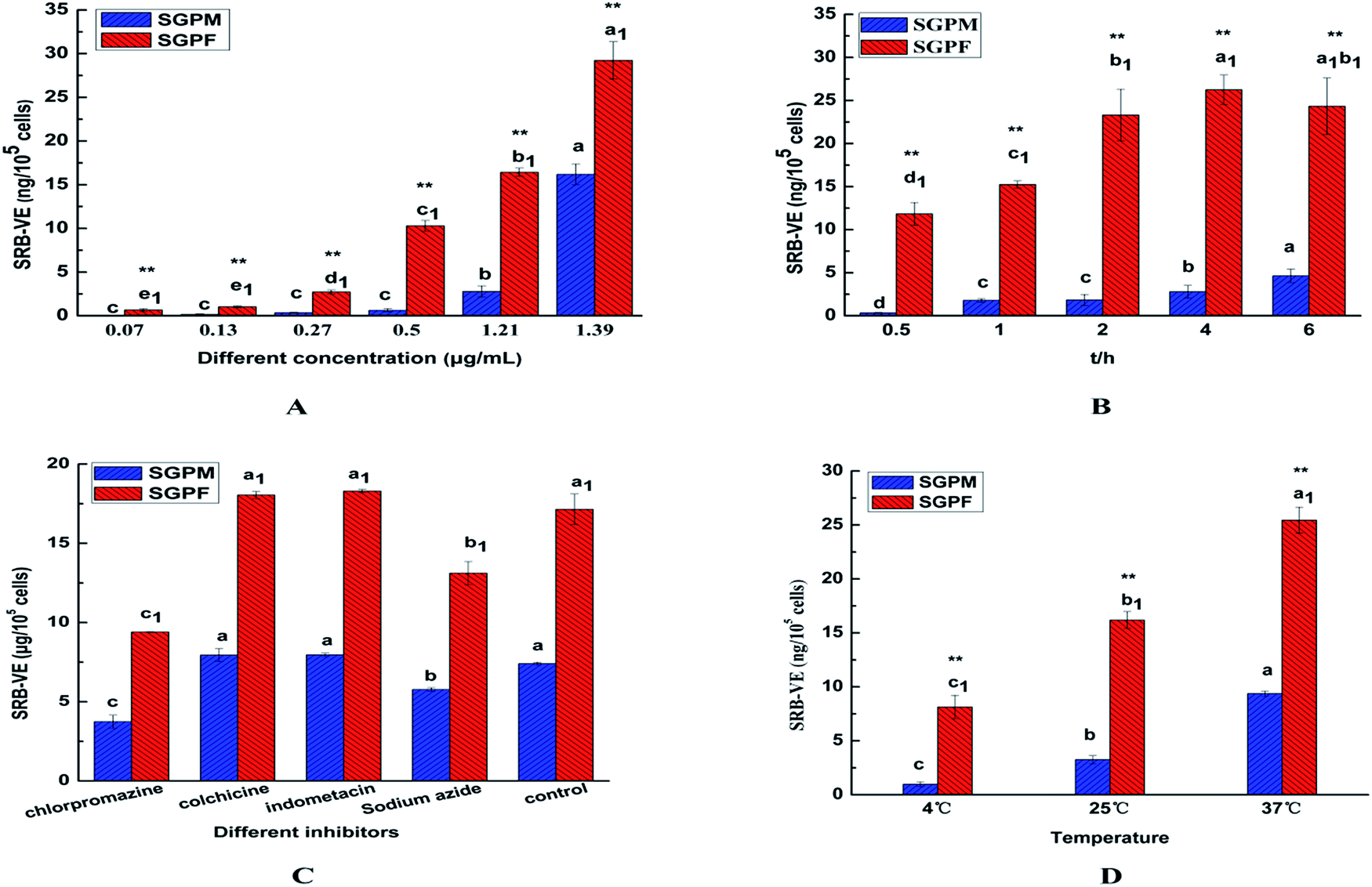
Cell uptake studies
In the present work, A549 cells were incubated with SRB–VE-loaded micelles at different concentrations and different incubation periods. As shown in Fig. 8A, the cell uptake of SGPF showed no difference below a concentration of 0.13 μg mL−1, and above this cell uptake was concentration-dependent. However, up to 0.50 μg mL−1, the cellular uptake of SGPM gradually appeared to exhibit concentration-dependence. Furthermore, the uptake of SGPF was much greater than that of SGPM at all concentrations.
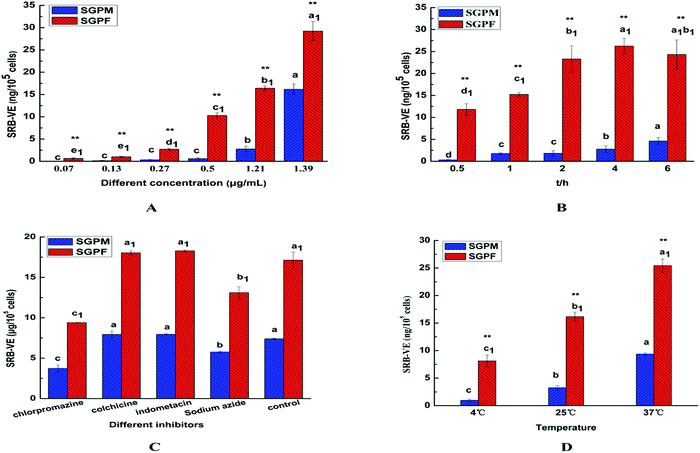 |
| | Fig. 8 In vitro cell uptake of SRB–VE and drug-loaded micelles (SGPM, SGPF) at different concentrations (A); different incubation times (B); different inhibitors (C); different temperatures (D). (Mean ± SD, n = 4. Significant differences are indicated as follows: a, b, c mean comparisons in SGPM, a1, b1, c1 mean comparisons in SGPF P < 0.05, ** means comparisons between SGPM and SGPF P < 0.01). | |
In addition, as shown in Fig. 8B, with an increase of time, the uptake of SGPM increased gradually, and reached a maximum at 6 h. However, SGPF uptake reached a maximum at 4 h, then gradually decreased, becoming equal to that at 2 h. This may be because the uptake of SGPM was originally lower, and the endocytosis protein on the surface was partly used and after 4 h the cell uptake continued to increase. On the contrary, the uptake of SGPF was higher at first and endocytosis protein on the surface of cells was fully used; excretion played an important role when the endocytosis carrier protein on the surface reached saturation. After a certain period of time, the efflux phenomenon is more obvious and results in reduced uptake.
Different endocytosis inhibitors were used to study the SGPM and SGPF cellular internalization mechanisms. Chlorpromazine can interact with clathrin from the coated pits and lead to their loss from the surface membrane,35 so it was used to learn if clathrin-dependent endocytosis was involved in the two kinds of micelles uptake; colchicine can block the assembly of the microtubes, as a macropinocytic route which is microtube-dependent, and it can be inhibited by colchicine.36 Indomethacin is an inhibitor of blood cell membrane caveolae-mediated endocytosis, and sodium azide can inhibit cytochrome C oxidase in the mitochondrial electron transport chain and reduce intracellular ATP production,37–39 which can produce a cell energy barrier. Using sodium azide as an energy inhibitor, it is possible to examine whether uptake of the two micelles is energy-dependent. As shown in Fig. 8C, we found that chlorpromazine and sodium azide significantly inhibited cellular internalization of the two kinds of micelles. However, following pre-incubation with other inhibitors, cellular uptake was not significantly different from the control group. In addition, incubation of the two kinds of micelles with A549 cells at 4 °C and room temperature were also conducted. As shown in Fig. 8D, when incubated at 4 °C, uptake was markedly reduced, which is consistent with the results obtained with sodium azide. Therefore, uptake of these two kind micelles by A549 cells is clathrin-mediated endocytosis and energy-dependent.
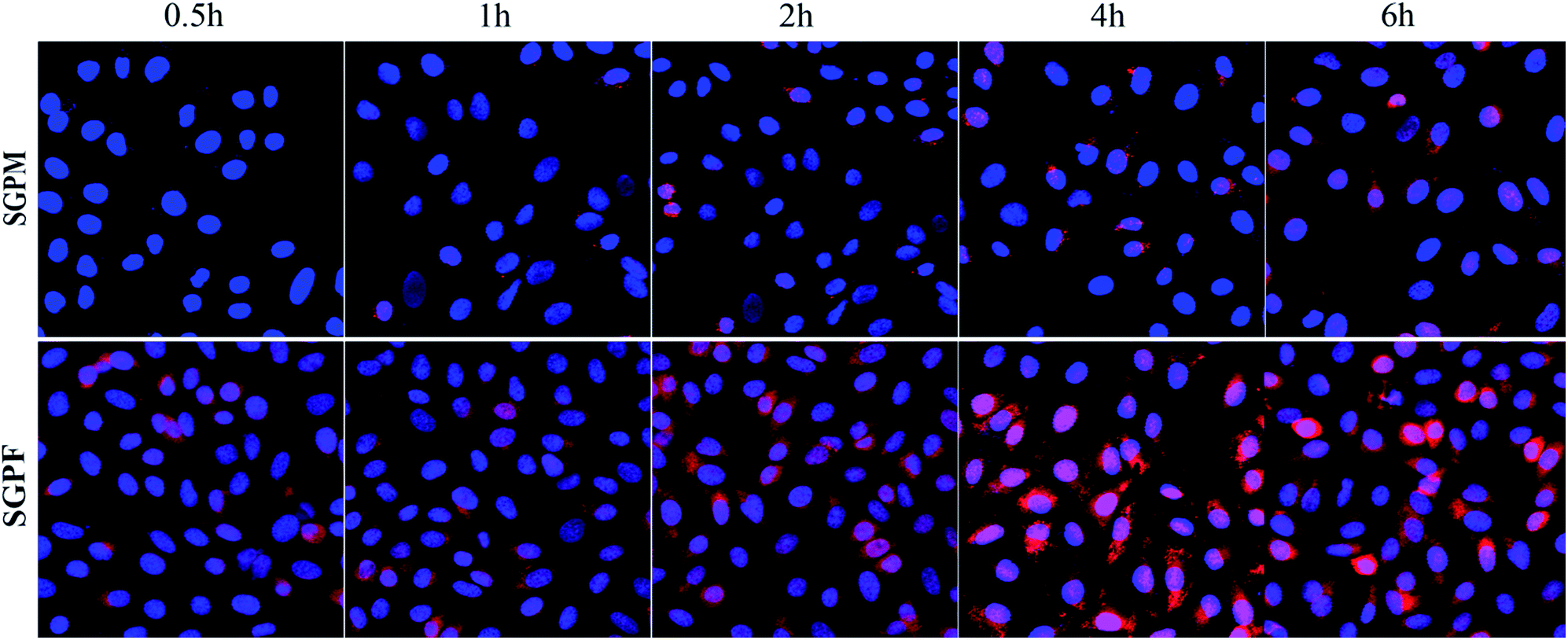
The CLSM images were used to help us qualitatively examine intracellular distribution after merger of the DAPI and SRB channels. Blue represents DAPI stained cell nuclei and red represents the SRB in cytoplasm. As shown in Fig. 9, SGPM exhibited time-dependence. Over time, uptake of SGPM gradually increased, while the uptake of SGPF reached a maximum at 4 h, and then gradually decreased, which is in accordance with above results.
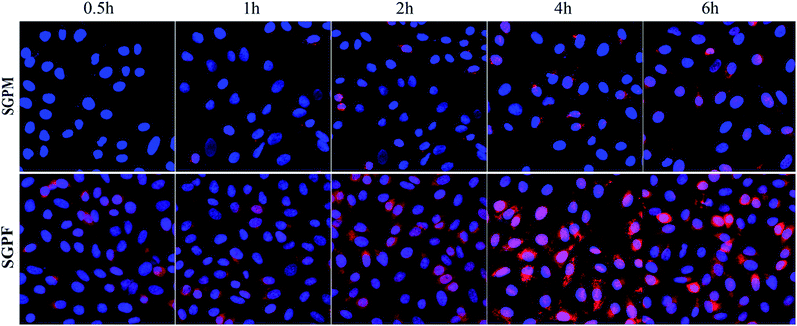 |
| | Fig. 9 The CLSM images of A549 cells after incubation with SGPM and SGPF micelle for 0.5 h, 1 h, 2 h, 4 h, and 6 h (blue: fluorescence of DAPI stained nuclei, red: fluorescence of SRB–VE internalized by A549 cells, and pink: merged blue and red fluorescence). | |
Conclusions
In this study, we successfully synthesized amphiphilic block copolymer FA–PEG–VE to construct a novel polymeric micelle system to co-deliver GEM and PTX; VE modified GEM and PTX were co-encapsulated into the cores of micelles with a good particle size and high drug-loading capacity. Similar in vitro release profiles of GEM–VE and PTX–VE were obtained. With the in vitro cytotoxicity study, the drug-loaded FA–PEG–VE micelles exhibited a dramatic inhibitory effect on A549 cells. In addition, the co-delivery micelle system exhibited superior toxicity compared with the single drug-loaded micelle system, and confirmed the synergistic combinational efficiency of GEM and PTX. Meanwhile, FA–PEG–VE micelles showed higher uptake efficiency than that of MPEG–PLGA micelles. Clathrin-mediated and energy-dependent endocytosis was involved in uptake mechanisms.
Overall, co-delivery of GEM and PTX into a FA–PEG–VE micelle system can lead to pronounced synergy in inhibiting growth of cancer cells. Furthermore, in vivo combination therapy efficiency and pharmacokinetic activities require specific studying with further work. Advanced co-delivering modified hydrophilic and hydrophobic drugs into a single drug delivery system is expected to play an important role in future cancer treatments.
Abbreviations
| GEM | Gemcitabine |
| PTX | Paclitaxel |
| VE | (+)-α-Tocopherol |
| FA | Folic acid |
| FA–PEG–VE | Folic acid–poly(ethylene glycol)–(+)-α-Tocopherol |
| MPEG–PLGA | Methoxy poly(ethylene glycol)–poly(lactide-co-glycolide) |
| GEM–VE | Gemcitabine–(+)-α-tocopherol |
| PTX–VE | Paclitaxel–(+)-α-tocopherol |
| SRB–VE | Sulforhodamine B–(+)-α-tocopherol (SRB–VE) |
| EDCI | 3-(Ethyliminomethylideneamino)-N,N-dimethylpropan-1-amine |
| CDI | Di(imidazol-1-yl)methanone |
| TEA | 2,2,2-Trifluoroacetic acid |
| DMAP | N,N-Dimethylpyridin-4-amine |
| DMF | N,N-Dimethylformamide |
| HBTU | O-Benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium hexafluorophosphate |
| DMSO | Dimethyl sulfoxide |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide |
| CMC | Critical micelle concentration |
| EE | Encapsulation efficiency |
| DL | Drug loading capacity |
| TEM | Transmission electron microscopy |
| CLSM | Confocal laser scanning microscopy |
Acknowledgements
This work was supported by the Natural Science Foundation of Liaoning Province (2015020729) and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry.
Notes and references
- S.-S. Feng, Cell. Mol. Bioeng., 2011, 4, 708–716 CrossRef CAS.
- H. Wang, Y. Zhao, Y. Wu, Y.-l. Hu, K. Nan, G. Nie and H. Chen, Biomaterials, 2011, 32, 8281–8290 CrossRef CAS PubMed.
- Y. Ma, X. Fan and L. Li, Carbohydr. Polym., 2016, 137, 19–29 CrossRef CAS PubMed.
- Y. Ge, Y. Ma and L. Li, Colloids Surf., B, 2016, 146, 482–489 CrossRef CAS PubMed.
- F. Greco and M. J. Vicent, Adv. Drug Delivery Rev., 2009, 61, 1203–1213 CrossRef CAS PubMed.
- X. Zhao, Q. Chen, Y. Li, H. Tang, W. Liu and X. Yang, Eur. J. Pharm. Biopharm., 2015, 93, 27–36 CrossRef CAS PubMed.
- Y. Ma, D. Liu, D. Wang, Y. Wang, Q. Fu, J. K. Fallon, X. Yang, Z. He and F. Liu, Mol. Pharm., 2014, 11, 2623–2630 CrossRef CAS PubMed.
- K. S. Albain, S. M. Nag, G. Calderillo-Ruiz, J. P. Jordaan, A. C. Llombart, A. Pluzanska, J. Rolski, A. S. Melemed, J. M. Reyes-Vidal, J. S. Sekhon, L. Simms and J. O'Shaughnessy, J. Clin. Oncol., 2008, 26, 3950–3957 CrossRef PubMed.
- J. W. Lee, S. C. Lee, G. Acharya, C. J. Chang and K. Park, Pharm. Res., 2003, 20, 1022–1030 CrossRef CAS.
- S. Vrignaud, J.-P. Benoit and P. Saulnier, Biomaterials, 2011, 32, 8593–8604 CrossRef CAS PubMed.
- M. H. Smith and L. A. Lyon, Acc. Chem. Res., 2012, 45, 985–993 CrossRef CAS PubMed.
- I. Noh, H.-O. Kim, J. Choi, Y. Choi, D. K. Lee, Y.-M. Huh and S. Haam, Biomaterials, 2015, 53, 763–774 CrossRef CAS PubMed.
- C. M. Hu and L. Zhang, Biochem. Pharmacol., 2012, 83, 1104–1110 CrossRef CAS PubMed.
- V. Bala, S. Rao, B. J. Boyd and C. A. Prestidge, J. Controlled Release, 2013, 172, 48–61 CrossRef CAS PubMed.
- C. Yanzuo, Z. Wei, H. Yukun, G. Feng, S. Xiany and F. Xiaoling, Int. J. Pharm., 2015, 488, 44–58 CrossRef PubMed.
- J. L. Tang, Q. Fu, Y. J. Wang, K. Racette, D. Wang and F. Liu, Cancer Lett., 2013, 336, 149–157 CrossRef CAS PubMed.
- D. Wang, J. L. Tang, Y. J. Wang, S. Ramishetti, Q. Fu, K. Racette and F. Liu, Mol. Pharm., 2013, 10, 1465–1469 CrossRef CAS PubMed.
- S. Kunjachan, B. Rychlik, G. Storm, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2013, 65, 1852–1865 CrossRef CAS PubMed.
- J. Savage, M. Meaney, G. P. Brennan, E. Hoey, A. Trudgett and I. Fairweather, Exp. Parasitol., 2013, 135, 642–653 CrossRef CAS PubMed.
- N. Wiradharma, Y. W. Tong and Y.-Y. Yang, Biomaterials, 2009, 30, 3100–3109 CrossRef CAS PubMed.
- W. Xiao, X. Chen, L. Yang, Y. Mao, Y. Wei and L. Chen, Int. J. Pharm., 2010, 393, 120–127 CrossRef PubMed.
- X. Duan, J. Xiao, Q. Yin, Z. Zhang, H. Yu, S. Mao and Y. Li, ACS Nano, 2013, 7, 5858–5869 CrossRef CAS PubMed.
- X. Yang, D. Wang, Y. Ma, Q. Zhao, J. K. Fallon, D. Liu, X. E. Xu, Y. Wang, Z. He and F. Liu, Nanomedicine, 2014, 9, 2773–2785 CrossRef CAS PubMed.
- B. Asadishad, M. Vosoughi, I. Alamzadeh and A. Tavakoli, J. Dispersion Sci. Technol., 2010, 31, 492–500 CrossRef CAS.
- T. Hao, D. Chen, K. Liu, Y. Qi, Y. Tian, P. Sun, Y. Liu and Z. Li, ACS Appl. Mater. Interfaces, 2015, 7, 18064–18075 CAS.
- S. Liu, N. Wiradharma, S. Gao, Y. Tong and Y. Yang, Biomaterials, 2007, 28, 1423–1433 CrossRef CAS PubMed.
- H. Aliabadi, S. Elhasi, A. Mahmud, R. Gulamhusein, P. Mahdipoor and A. Lavasanifar, Int. J. Pharm., 2007, 329, 158–165 CrossRef CAS PubMed.
- D. S. Booth, A.-S. Agustin and Y. Cheng, J. Visualized Exp., 2011, 58, e3227 Search PubMed.
- S. Mingshuang, G. Yunyun, Z. Zhihong, W. Huixin, H. Cuiyan, Y. Xinggang and W. Pan, Asian J. Pharm. Sci., 2016, 12, 51–58 Search PubMed.
- L. Qiu, M. Qiao, Q. Chen, C. Tian, M. Long, M. Wang, Z. Li, W. Hu, G. Li, L. Cheng, L. Cheng, H. Hu, X. Zhao and D. Chen, Biomaterials, 2014, 35, 9877–9887 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505–515 CrossRef CAS PubMed.
- F. M. Kievit, F. Y. Wang, C. Fang, H. Mok, K. Wang, J. R. Silber, R. G. Ellenbogen and M. Miqin Zhang, J. Controlled Release, 2011, 152, 76–83 CrossRef CAS PubMed.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3–27 CrossRef CAS.
- D. Wang, Q. Fu, J. Tang, M. Hackett, Y. Wang and F. Liu, Nanomedicine, 2015, 10, 3003–3013 CrossRef CAS PubMed.
- X. Han, Z. Li, J. Sun, C. Luo, L. Li, Y. Liu, Y. Du, S. Qiu, X. Ai, C. Wu, H. Lian and Z. He, J. Controlled Release, 2015, 197, 29–40 CrossRef CAS PubMed.
- U. S. Huth, R. Schubert and R. Peschka-Suss, J. Controlled Release, 2006, 110, 490–504 CrossRef CAS PubMed.
- I. Legen, S. Žakelj and A. Kristl, Int. J. Pharm., 2003, 256, 161–166 CrossRef CAS PubMed.
- J. S. Kim, T. J. Yoon, K. N. Yu, M. S. Noh, M. Woo, B. G. Kim, K. H. Lee, B. H. Sohn, S. B. Park, J. K. Lee and M. H. Cho, J. Vet. Sci., 2006, 7, 321–326 CrossRef PubMed.
- V. P. Torchilin, R. Rammohan, V. Weissig and T. S. Levchenko, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8786–8791 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02909h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yunyun Gaoa,
Xiumei Gaia,
Dun Wangb,
Yingying Wanga,
Xiaoguang Yangb,
Dan Zhangc,
Weisan Pan
a,
Yunyun Gaoa,
Xiumei Gaia,
Dun Wangb,
Yingying Wanga,
Xiaoguang Yangb,
Dan Zhangc,
Weisan Pan