
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen backed silicon hydride in correlation with the photoluminescence of silicon nano-crystals
Cui-Li Chenab,
Jiang Zengb,
Ning Baob,
Hong Daic and
Hai-Ying Gu *ab
*ab
aCollege of Chemistry, Chemical Engineering and Material Science, Soochow University, 199 Ren'ai Road, Suzhou, Jiangsu 215123, China. E-mail: guhy99@21cn.com
bSchool of Public Health, Nantong University, 9 Se Yuan Road, Nantong, Jiangsu 226019, China
cCollege of Chemistry and Chemical Engineering, Nantong University, Nantong 226019, China
First published on 19th September 2017
Abstract
Converting silicon hydride (–SiH) to oxygen backed silicon hydride (–OSiH) on porous silicon leads to a shift in the wavelength of photoluminescence (PL) maximum from 670 to 605 nm, corresponding to an increase of 0.2 eV on emission energy. The results implied that silicon hydride, which links to the surfaces of silicon nano-crystals (SiNCs) via oxygen atoms, is directly responsible for the wavelength change of the PL peak.
Silicon nano-crystals (SiNCs) are one of the most attractive nano-functional materials due to their electronic and photonic properties, their compatibility in biological environments, as well as their popularity in the semiconductor microelectronics industry.1–4 Understanding the origin of the photoluminescence (PL) of SiNCs is fundamentally important, considering the possibility of their widespread applications. The surface of SiNCs containing silicon hydride groups could readily react with 1-ene compounds to form relatively stable Si–C bonds,5,6 introducing organic functional groups on the surface, facilitating the covalent attachment of organic and biological moieties that open the potential of such inorganic nano-materials to many biological applications.1,4,7,8
Since the discovery of the PL of SiNCs at room temperature in 1990,9 the mechanism of the PL generation has been the centre of many studies. Various theories have been proposed in order to reveal the physical and chemical grounds. To date, however, research data have seldom led to the complete understanding of such emission. Quantum confinement (QC) theory10 proposed in earlier years has been challenged by many experimental observations, which showed that PL was influenced by not only geometric dimensions of SiNCs, but also surface states of chemical groups, such as silicon oxide species.11–14 Crystal defects and dangling bonds have been regarded as the causes of the PL emission, but the effect of the oxidation of surface silicon hydride was not fully accounted for.15,16 It becomes well accepted that the PL of SiNCs is a complicated process and QC theory cannot be adequately applied for full explanation. Besides surface states of chemical groups, other factors, such as particle sizes, also play important roles in the mechanism of the PL.7,11,17–20
Time resolved PL spectra revealed that there are two types of PL emissions from SiNCs, namely, fast decaying “F band” blue emissions and slow decaying “S band” red emissions.21 Although short life-time F band emissions were believed to be originated from the core of SiNCs,20 both F and S bands were found to be influenced by oxygen on SiNCs.22,23 Recently a long lived blue band and a UV band were also found to be associated with oxidized silicon.24 Although the oxidation of silicon in ambient air has been known to cause wavelength shifts of PL maximum ever since the discovery of the PL of porous silicon for S band emission decades ago, the detailed mechanism was unclear. There are too many unknown factors for both S and F band emissions when oxygen is involved. Therefore, revealing the role of the oxidation on surface of SiNCs is an important step for fully understanding the nature of PL of SiNCs.
Based on the fact that oxidation of silicon hydride on porous silicon significantly influences the wavelength of PL maximum, the theory of Quantum Confinement–Luminescence Centre (QC–LC)25 was proposed to modify the QC theory, taking into account of oxidation effects. Although this model separated the excitation and emission centres, their chemical structures were not clearly defined.
Various chemical structures have been considered as models in attempts to shed light on the oxygen containing species that contribute to PL. For instance, Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species on the surface of SiNCs was proposed to be responsible for changes in both the intensity and the wavelength of PL. The study of PL spectra, oxygen isotopes, infrared characterization, and the quantum chemistry model indicated that silanone based SiO affected S band of PL and hydroxylated silicon was considered to be the precursor of SiO groups that were excited to provide PL.10,26,27 However, other investigations challenged the idea of the SiO structure being the centre of PL emission for the S band,28 and attributed this oxidation state to nonradiative relaxation processes.29 Thus, current reports on understanding of the effect of oxygen on the PL of SiNCs are far from conclusive.
O species on the surface of SiNCs was proposed to be responsible for changes in both the intensity and the wavelength of PL. The study of PL spectra, oxygen isotopes, infrared characterization, and the quantum chemistry model indicated that silanone based SiO affected S band of PL and hydroxylated silicon was considered to be the precursor of SiO groups that were excited to provide PL.10,26,27 However, other investigations challenged the idea of the SiO structure being the centre of PL emission for the S band,28 and attributed this oxidation state to nonradiative relaxation processes.29 Thus, current reports on understanding of the effect of oxygen on the PL of SiNCs are far from conclusive.
In this paper, four types of silicon species were prepared based on oxidation of silicon hydride in order to study the PL mechanism. Fourier transform infrared spectroscopy (FTIR) and steady state PL spectra were utilized to characterize the surface groups and the emission. Different silicon hydride species were studied in relation to the steady state emissions of porous silicon.
Porous silicon samples were prepared by immersing in or floating on HF solutions for chemical etching under ambient temperature. It needs to be emphasized that such treatment is critical for PL of SiNCs. Silicon wafers (boron-doped p-type 〈100〉 with resistivity of 20 Ω cm−1, from Wafer World, FL., USA) were diced to the dimension of 4 mm by 4 mm and cleaned, before etching, with RCA hydrogen peroxide solutions followed by acid chemical cleaning in a 5% HF solution (ACS grade from Aladdin, Shanghai, China) for 2 minutes, and finally rinsed with de-ionized water for 5 times. The cleaned wafer was placed in the solution of 48% HF containing 1% of nitric acid (analytical grade from Aladdin, Shanghai, China) and etched for 60 minutes. The atmosphere above the solution was either air or purged nitrogen with small amounts. The etched porous silicon was washed with de-ionized water for 5 times and anhydrous ethanol for 3 times before being dried in either ambient air or nitrogen for half an hour. The obtained porous silicon was further oxidized by ozone for 15 minutes to remove silicon hydride. FTIR measurements were performed in transmission mode under the background of air with a Nicolet Avatar 380 FTIR spectrometer. An integration of 32 scans was used to acquire the spectra. Steady state PL was obtained using a PG2000-PG fibre optic spectrometer (Ideaoptics Shanghai, China) equipped with a diode laser of 488 nm as the excitation source with the integration time of 4000 ms.
All the data were collected on the surface of porous silicon, which were used as models for examining the effect of oxygen on PL of SiNCs. Porous silicon samples prepared by chemical etching under different atmospheres were found to contain different oxidized species on the surface. Formation of the oxygen containing species could also take place during etching processes, or in post-etching storage stages. Four types of the oxygen containing species could be identified on the freshly prepared porous silicon surface: (I) silicon hydride –SiHx (x = 1 to 3) directly linking to the crystal substrate of silicon via Si–Si bond; (II) oxygen backed silicon hydride –OSiHx, in which silicon hydride connecting the crystal substrate via the bond of Si–O–Si; (III) co-existence of two groups, –SiHx and –OSiHx; (IV) when silicon hydride was completely oxidized by ozone treatment, all the silicon hydride groups were removed.
Fig. 1 illustrates transmission FTIR spectra of the above four types of porous silicon samples. Attention was paid to the wavelength range of 2110 to 2210 cm−1 where silicon hydrides were well characterized. SiHx exhibited typical silicon hydride bond stretches at around 2116 cm−1, while OSiHx groups showed OSi-Hx stretches at around 2251 cm−1. In case of porous silicon samples containing both SiHx and OSiHx, two bands at 2116 cm−1 and 2251 cm−1 region could be observed. For completely oxidized samples with mostly Si(OH)x groups, no peak was found in above regions, whereas an obvious abroad peak of Si(OH)x at around 3400 cm−1 could be seen. Such results are in agreement with those reported before.29,30
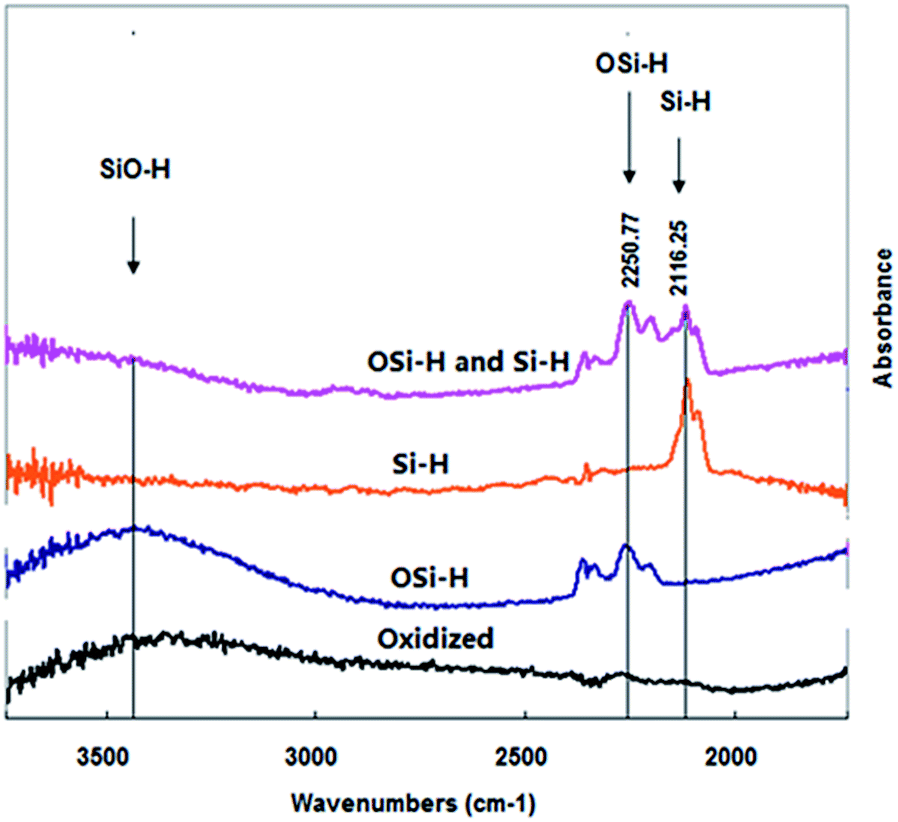 | ||
| Fig. 1 Transmission infrared spectra of porous silicon samples prepared by chemical etching: (I) wafer etched by floating on HF solution (purple); (II) silicon wafer etched in HF solution under nitrogen atmosphere (orange); (III) wafer etched by immersing in HF solution (blue); (IV) porous silicon sample oxidized by ozone (black). | ||
Freshly prepared, or etched, porous silicon, frequently referred as “hydrogen terminated”, contains silicon hydride groups on the surface. Depending on producing procedures of porous silicon, some “freshly prepared” samples may already contain certain amounts of OSiHx although they are still hydrogen terminated. Thus, hydrogen terminated groups can be SiHx, or OSiHx, or both. Differentiation of their PL would significantly help our understanding on the role of oxygen on the PL of SiNCs.
PL of porous silicon samples containing the above mentioned four oxidized species of silicon hydride were further studied. Fig. 2 shows a PL peak at 670 nm with a shoulder peak at ∼610 nm when there were both SiHx and OSiHx on the surface of the porous silicon, indicating emissions from a combination of processes. However, when samples containing either silicon hydride (SiHx) or oxygen backed silicon hydride (OSiHx), no shoulder peak could be found for PL emissions (Fig. 3).
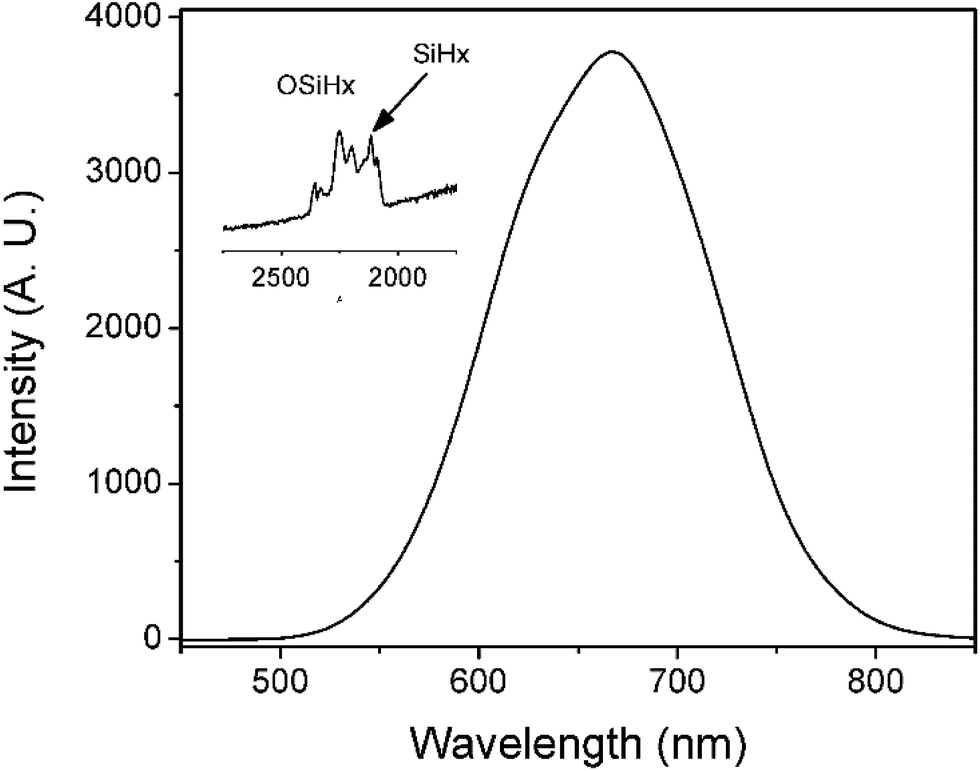 | ||
| Fig. 2 PL spectrum of porous silicon with a mixture of both SiHx and OSiHx groups on the surface. Inset: FTIR spectrum corresponding to this surface. | ||
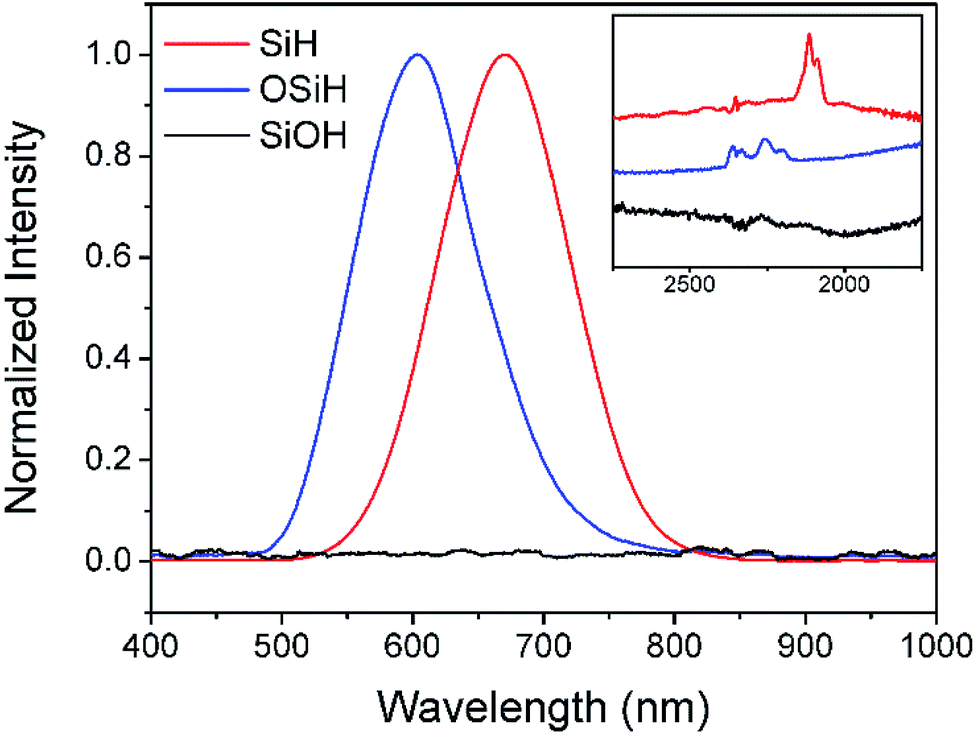 | ||
| Fig. 3 PL spectra of porous silicon samples containing different silicon hydride groups: type I sample with pure SiHx (red spectrum) emitted at 670 nm, type II sample with OSiHx (blue spectrum) emitted at 605 nm, while no emission could be found for type IV sample with pure SiOH (black spectrum). Inset: FTIR spectra corresponding to the three types of silicon hydride groups. | ||
It could be seen from Fig. 3 that pure silicon hydride SiHx was responsible for the emission peak at 670 nm while oxygen backed silicon hydride was responsible for the peak at 605 nm. By substituting OSiHx for SiHx, the wavelength of the emission shifted towards the higher energy end by 65 nm, or about 0.2 eV. Such results suggested that oxygen backed silicon hydride might be one of the causes for the blue shift of PL of porous silicon upon oxidation.
It could also be noticed in Fig. 1 that when hydrogen terminated groups were completely removed by ozone oxidation, silicon hydride was completely converted to be hydroxyls, Si(OH)x, as indicated by a broad FTIR peak at around at 3400 cm−1. No PL could be observed from these samples (Fig. 3). As listed in Table 1, various optical properties via different groups on the surface could be obtained by treating silicon wafers with different approaches based on HF solutions.
| Surface type | Group | IR region (cm−1) | The wavelength of PL max (nm) | Preparing methods |
|---|---|---|---|---|
| a Means shoulder peak and N/A means no peak was found. | ||||
| I | SiH | 2116 | 670 | Nitrogen above the HF solution |
| II | OSiH | 2251 | 605 | Immersing in the HF solution under ambient air |
| SiOH | 3400 | N/A | ||
| III | SiH | 2216 | 670 peak | Floating on the HF solution under ambient air |
| OSiH | 2251 | ∼610a peak | ||
| IV | SiOH | 3400 | N/A | Treated with ozone after etching |
The oxidation process might also remove other components such as dangling bonds that were associated with the emission. This is in agreement with previous reports that full oxidation would deplete PL of porous silicon. It is interesting to note that dipping completely oxidized samples to HF etching solution for just 1 s would recover the strong visible PL.31 The recovery is possibly caused by reappearance of silicon hydride after treatment.
The results of this study indicated that SiNCs with hydrogen terminated silicon hydride exhibited PL peaks with different bands depending on how SiH was linked onto the substrate of silicon crystals. When SiH was directly linked to silicon crystals, PL emission was at about 670 nm. When SiH was linked to crystal silicon via an oxygen bridge in the form of –OSiH, PL has an emission peak at about 605 nm. The influence of oxygen backed silicon hydride on PL emission suggested the importance of linking chemistry in silicon hydride when attached to the crystal substrates. This study might provide additional evidences for the study of PL emission of silicon materials and improve our understanding on related mechanisms.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key Research and Development Program of China (No. 2016YFF0203703-05) and National Natural Science Foundation of China (No. 21175075, 21475070).References
- C.-F. Wang, M. P. Sarparanta, E. M. Makila, M. L. K. Hyvonen, P. M. Laakkonen, J. J. Salonen, J. T. Hirvonen, A. J. Airaksinen and H. A. Santos, Biomaterials, 2015, 48, 108–118 CrossRef CAS PubMed.
- A. A. Ensafi, N. Ahmadi, B. Rezaei and M. M. Abarghoui, Talanta, 2015, 134, 745–753 CrossRef CAS PubMed.
- S. M. Haidary, A. B. Mohammed, E. P. Corcoles, N. K. Ali and M. R. Ahmad, Microelectron. Eng., 2016, 161, 1–6 CrossRef CAS.
- O. Syshchyk, V. A. Skryshevsky, O. O. Soldatkin and A. P. Soldatkin, Biosens. Bioelectron., 2015, 66, 89–94 CrossRef CAS PubMed.
- J. M. Buriak, M. P. Stewart, T. W. Geders, M. J. Allen, H. C. Choi, J. Smith, D. Raftery and L. T. Canham, J. Am. Chem. Soc., 1999, 121, 11491–11502 CrossRef CAS.
- B. R. Hart, S. E. Letant, S. R. Kane, M. Z. Hadi, S. J. Shields and J. G. Reynolds, Chem. Commun., 2003, 322–323 RSC.
- A. Angi, R. Sinelnikov, A. Meldrum, J. G. C. Veinot, I. Balberg, D. Azulay, O. Millo and B. Rieger, Nanoscale, 2016, 8, 7849–7853 RSC.
- J. Ahn, B. Cho, S. Kim and H. Sohn, J. Nanosci. Nanotechnol., 2015, 15, 4999–5003 CrossRef CAS PubMed.
- L. T. Canham, Appl. Phys. Lett., 1990, 57, 1046–1048 CrossRef CAS.
- J. L. Gole and D. A. Dixon, J. Phys. Chem. B, 1997, 101, 8098–8102 CrossRef.
- T. Nakamura, T. Ogawa, N. Hosoya and S. Adachi, J. Lumin., 2010, 130, 682–687 CrossRef CAS.
- V. S. Vendamani, S. V. S. Nageswara Rao and A. P. Pathak, Nucl. Instrum. Methods Phys. Res., Sect. B, 2013, 315, 188–191 CrossRef CAS.
- C. A. Caras, J. M. Reynard and F. V. Bright, Appl. Spectrosc., 2013, 67, 570–577 CrossRef CAS PubMed.
- C. A. Caras, J. M. Reynard, R. E. Deuro and F. V. Bright, Appl. Spectrosc., 2012, 66, 951–957 CrossRef CAS PubMed.
- B. E. B. Al-Jumaili, Z. A. Talib, L. Y. Josephine, S. B. Paiman, N. M. Ahmed, A. H. J. Al-Jumaily, A. Ramizy, S. A. Abdulateef, I. B. Muh'd and M. E. E. Mofdal, J. Nanomater., 2016, 9, 1–8 Search PubMed.
- K. Kulathuraan, K. Mohanraj and B. Natarajan, Spectrochim. Acta, Part A, 2016, 152, 51–57 CrossRef CAS PubMed.
- M. Aghajamali, M. Iqbal, T. K. Purkait, L. Hadidi, R. Sinelnikov and J. G. C. Veinot, Chem. Mater., 2016, 28, 3877–3886 CrossRef CAS.
- S. L. Brown, D. J. Vogel, J. B. Miller, T. M. Inerbaev, R. J. Anthony, U. R. Kortshagen, D. S. Kilin and E. K. Hobbie, J. Phys. Chem. C, 2016, 120, 18909–18916 CAS.
- S. Mitra, V. Svrcek, M. Macias-Montero, T. Velusamy and D. Mariotti, Sci. Rep., 2016, 6, 27727 CrossRef CAS PubMed.
- L. Ondic, K. Kusova, M. Ziegler, L. Fekete, V. Gaertnerova, V. Chab, V. Holy, O. Cibulka, K. Herynkova, M. Gallart, P. Gilliot, B. Hoenerlage and I. Pelant, Nanoscale, 2014, 6, 3837–3845 RSC.
- K. Kusova, I. Pelant and J. Valenta, Light: Sci. Appl., 2015, 4, e336 CrossRef CAS.
- M. Dasog, Z. Yang, S. Regli, T. M. Atkins, A. Faramus, M. P. Singh, E. Muthuswamy, S. M. Kauzlarich, R. D. Tilley and J. G. C. Veinot, ACS Nano, 2013, 7, 2676–2685 CrossRef CAS PubMed.
- K. Kusova, O. Cibulka, K. Dohnalova, I. Pelant, J. Valenta, A. Fucikova, K. Zidek, J. Lang, J. Englich, P. Matejka, P. Stepanek and S. Bakardjieva, ACS Nano, 2010, 4, 4495–4504 CrossRef CAS PubMed.
- B. Gelloz, R. Mentek and N. Koshida, ECS J. Solid State Sci. Technol., 2014, 3, R83–R88 CrossRef CAS.
- G. G. Qin, J. Infrared, Millimeter, Terahertz Waves, 2005, 24, 165–173 CAS.
- Y. A. Shu and B. G. Levine, J. Phys. Chem. C, 2014, 118, 7669–7677 CAS.
- M. V. Wolkin, J. Jorne, P. M. Fauchet, G. Allan and C. Delerue, Phys. Rev. Lett., 1999, 82, 197–200 CrossRef CAS.
- N. Arad-Vosk and A. Sa'ar, Nanoscale Res. Lett., 2014, 9, 47–53 CrossRef PubMed.
- D. B. Mawhinney, J. A. Glass and J. T. Yates, J. Phys. Chem. B, 1997, 101, 1202–1206 CrossRef CAS.
- D. S. Xu, L. Sun, H. L. Li, L. Zhang, G. L. Guo, X. S. Zhao and L. L. Gui, New J. Chem., 2003, 27, 300–306 RSC.
- S. M. Prokes, J. Appl. Phys., 1993, 73, 407–413 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |