DOI:
10.1039/C7RA02802D
(Paper)
RSC Adv., 2017,
7, 23073-23082
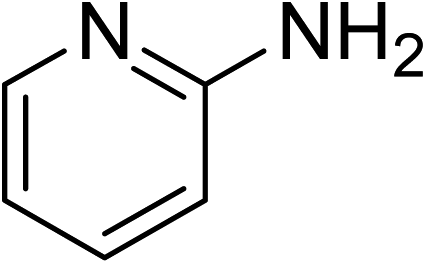
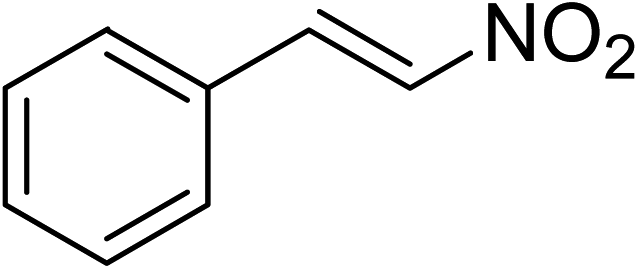
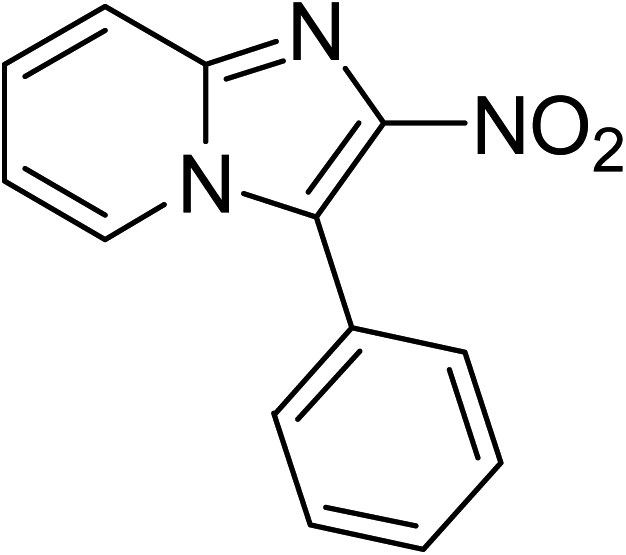
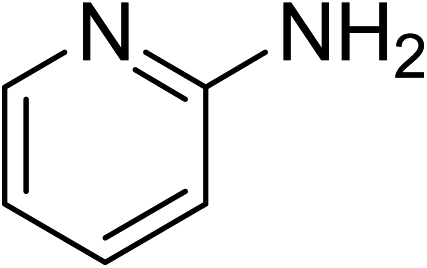
Indium-based metal–organic frameworks as catalysts: synthesis of 2-nitro-3-arylimidazo[1,2-a]pyridines via oxidative amination under air using MIL-68(In) as an effective heterogeneous catalyst†
Received
8th March 2017
, Accepted 10th April 2017
First published on 26th April 2017
Abstract
The metal–organic framework MIL-68(In) has emerged as a productive heterogeneous catalyst for the synthesis of 2-nitro-3-arylimidazo[1,2-a]pyridines via oxidative amination between 2-aminopyridines and nitroalkenes using air as an oxidation agent. The catalyst displayed impressively higher efficiency for the generation of 2-nitro-3-arylimidazo[1,2-a]pyridines than various other MOFs and several common homogeneous catalysts. Heterogeneous catalysis was verified for the reaction of 2-aminopyridines with nitroalkenes employing the framework-based catalyst. It was possible to collect and reuse the catalyst for the production of 2-nitro-3-arylimidazo[1,2-a]pyridines without a pronounced deterioration in catalytic efficiency. To the best of our knowledge, the preparation of 2-nitro-3-arylimidazo[1,2-a]pyridines has not been formerly conducted utilizing heterogeneous catalysts. Furthermore, indium-based catalysts were not previously employed for this transformation.
1. Introduction
Imidazo[1,2-a]pyridines are a prominent collection of biologically active nitrogen-containing heterocycles that exhibit universal applications in medicinal chemistry, agro chemistry, and materials science.1–6 A variety of synthetic strategies, including multicomponent condensation, oxidative coupling, and tandem reaction protocols, have been developed to produce imidazo[1,2-a]pyridine derivatives with numerous substituents at the 2 and 3-positions of this moiety.7–9 Gryko et al. reported a productive path to a wide range of imidazo[1,2-a]pyridines via the condensation of acetophenones with 2-aminopyridines.10 Huang et al. demonstrated an FeCl3-catalyzed tandem three-component coupling nitration transformation of nitroalkanes, aldehydes, and 2-aminopyridines providing imidazo[1,2-a]pyridine derivatives.11 Volkova et al. developed the preparation of functionalized imidazo[1,2-a]pyridines by the Cu(OAc)2-catalyzed one-pot three-component reactions of 2-aminopyridines, propiolate derivatives, and benzaldehydes.12 Liang et al. performed the first illustration of a CuBr-catalyzed production of 3-nitro-2-arylimidazo[1,2-a]pyridines from nitroolefins and aminopyridines using air as oxidation agent in a one-pot procedure.13 Interestingly, using similar starting materials, Hajra et al. recently demonstrated, for the first time, a unique Fe(NO3)3-catalyzed generation of 2-nitro-3-arylimidazo[1,2-a]pyridine derivatives with rare selectivity via C–H amination in ambient air.14 Although interesting results were achieved, this homogeneous catalyst could not be recycled and reutilized. To promote more environmentally beneficial methods for the generation of 2-nitro-3-arylimidazo[1,2-a]pyridines, recyclable heterogeneous catalysts must be investigated.
Metal–organic frameworks (MOFs) are established by joining metal-containing units with organic ligands, producing crystalline materials with permanent porosity.15–19 An abundant number of MOFs could be synthesized using multifarious organic linking ligands and innumerable metal cations, generating disparate unique properties.20–22 Although challenges in evolution still remain to be addressed, probable applications of MOFs have been substantially explored.23–25 Throughout the last few years, tremendous endeavors have been directed to investigate the critical aspects of MOFs in catalysis.26–29 Some indium-based MOFs have been synthesized and explored for applications in gas and vapor adsorption and separation,30 proton conductivity,31 and removal of tetracycline from waste water.32 However, reports on applications of these frameworks in catalysis have been very uncommon in the literature. Monge et al. performed the acetalization of carbonyl compounds,33 and the reduction transformations of nitroarenes34 using indium framework as a catalyst. Zheng et al. employed this material as a catalyst for the synthesis of numerous amino acids.35 Sun et al. recently employed anthracene-based In-MOFs for visible-light-induced atom transfer radical polymerization.36 MIL-68(In) possessed a three-dimensional network with infinite chains of the octahedral units of InO4(OH)2 connected via terephthalate linkers, generating triangular and hexagonal channels.37,38 In this manuscript, we would like to present the synthesis of 2-nitro-3-arylimidazo[1,2-a]pyridines via the oxidative amination between 2-aminopyridines and nitroalkenes utilizing the framework MIL-68(In) as an effective heterogeneous catalyst using air as an oxidation agent. To our best judgment, solid catalysts have not been used for the synthetic preparation of 2-nitro-3-arylimidazo[1,2-a]pyridines.
2. Experimental
2.1. Catalyst synthesis
The MIL-68(In) was prepared from terephthalic acid and indium nitrate by a solvothermal protocol, using a literature procedure formerly published by Latroche et al.37 In a representative experiment, In(NO3)3·6H2O (0.408 g, 1.35 mmol) and H2BDC (H2BDC = terephthalic acid; 0.200 g, 1.20 mmol) were dissolved in DMF (DMF = N,N′-dimethylformamide; 5 mL). The resulting solution was added to a 10 mL vial. The vial was then covered carefully and heated to 100 °C in an oven for 48 h, generating crystals during the course of the experiment. Following this step, the vial was cooled to room temperature. The frameworks were isolated by decantation and washed in DMF (3 × 10 mL). Solvent interchange was carried out with dichloromethane (3 × 10 mL) at room temperature. The framework was then heated on a Schlenk line under vacuum at 150 °C for 6 h, producing 0.33 g of MIL-68(In) in the form of white crystals (71% based on terephthalic acid). The MIL-68(In) was then characterized by various techniques (see ESI†).
2.2. Catalytic studies
In an exemplary experiment, 2-aminopyridine (0.0940 g, 1 mmol), trans-β-nitrostyrene (0.1788 g, 1.2 mmol), and 4-bromoanisole (0.0187 g, 1 mmol) were dissolved in dichloroethane (2.0 mL), and the solution was then added to a 10 mL pressurized vial enclosing the pre-determined quantity of catalyst. The catalyst portion was employed based on the indium/2-aminopyridine mole correlation. The reaction mixture was stirred magnetically in air at 100 °C for 6 h. Samples were withdrawn, diluted with ethylacetate (3 mL), dried with anhydrous Na2SO4, and analyzed by GC using 4-bromoanisole as a standard. The mixture remains were purified by column chromatography to achieve 2-nitro-3-phenylimidazo[1,2-a]pyridine. GC-MS, 1H NMR and 13C NMR analyses were performed to validate the structure of the product. To look into the reusability of catalyst in the generation of 2-nitro-3-phenylimidazo[1,2-a]pyridine, the catalyst was isolated, collected, and washed with large amounts of DMF and dichloromethane to remove any physisorped reagent, dried in a Schlenk line at 150 °C under vacuum in 2 h, and then reused for new reactions.
3. Results and discussion
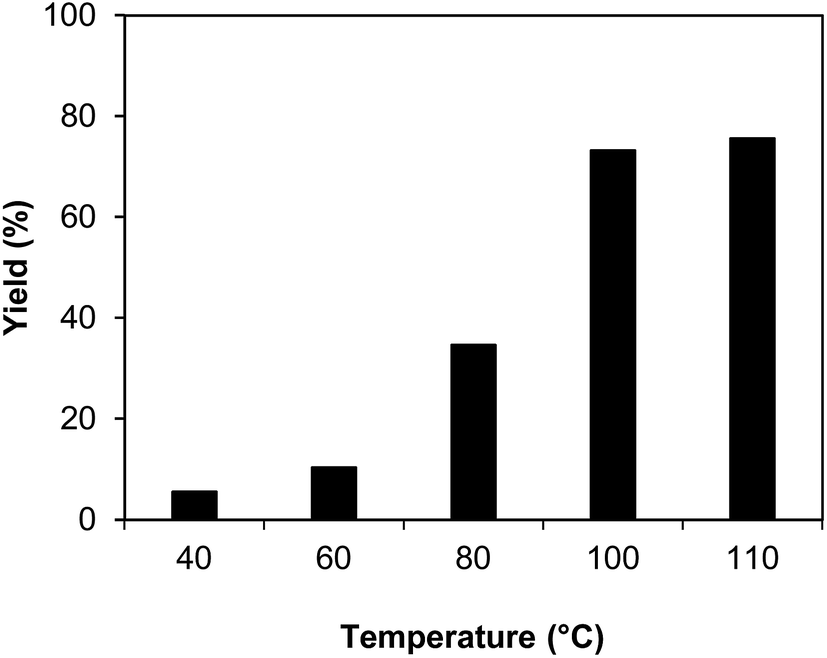
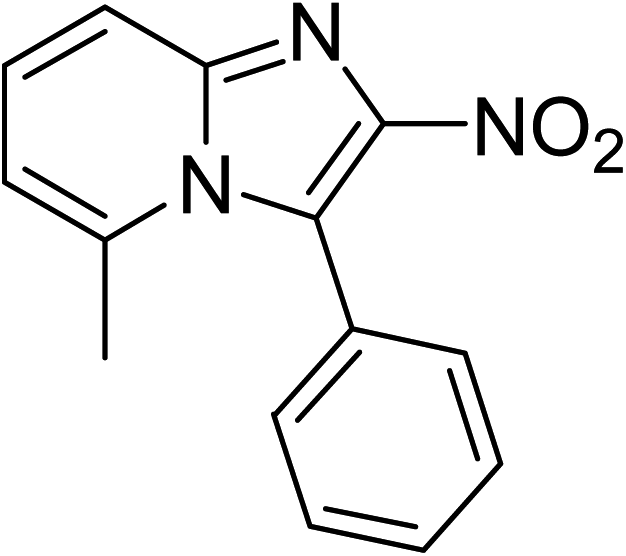
The indium framework was evaluated for its catalytic activity in the cyclization of trans-β-nitrostyrene with 2-aminopyridine under atmospheric air to provide 2-nitro-3-phenylimidazo[1,2-a]pyridine as the major product (Scheme 1). The initial studies investigated the impact of temperature on the yield of 2-nitro-3-phenylimidazo[1,2-a]pyridine. The reaction was implemented under air in dichloroethane for 6 h with 10 mol% catalyst and 10 mol% acetic acid as a co-catalyst using 2-aminopyridine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans-β-nitrostyrene mole proportion of 1:1.2, at a 2-aminopyridine concentration of 0.5 M at ambient temperature, 40 °C, 60 °C, 80 °C, 100 °C, and 110 °C. No reaction occurred at ambient temperature, with no trace quantity of the anticipated product after 6 h. The conversion progressed with difficulty at 40 °C, providing only 5% yield after 6 h. The generation of 2-nitro-3-phenylimidazo[1,2-a]pyridine was not increased when the temperature was boosted to 60 °C. The reaction conducted at 80 °C produced a 35% yield after 6 h. It was possible to improve the yield of the expected product to 74% by raising the reaction temperature to 100 °C. However, experimental data revealed that performing the cyclization reaction at 110 °C did not bring about an impressive augmentation in the production of 2-nitro-3-phenylimidazo[1,2-a]pyridine, though 75% yield was noticed after 6 h (Fig. 1).
:trans-β-nitrostyrene mole proportion of 1:1.2, at a 2-aminopyridine concentration of 0.5 M at ambient temperature, 40 °C, 60 °C, 80 °C, 100 °C, and 110 °C. No reaction occurred at ambient temperature, with no trace quantity of the anticipated product after 6 h. The conversion progressed with difficulty at 40 °C, providing only 5% yield after 6 h. The generation of 2-nitro-3-phenylimidazo[1,2-a]pyridine was not increased when the temperature was boosted to 60 °C. The reaction conducted at 80 °C produced a 35% yield after 6 h. It was possible to improve the yield of the expected product to 74% by raising the reaction temperature to 100 °C. However, experimental data revealed that performing the cyclization reaction at 110 °C did not bring about an impressive augmentation in the production of 2-nitro-3-phenylimidazo[1,2-a]pyridine, though 75% yield was noticed after 6 h (Fig. 1).
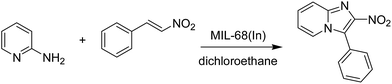 |
| | Scheme 1 Synthesis of 2-nitro-3-phenylimidazo[1,2-a]pyridine utilizing MIL-68(In) catalyst. | |
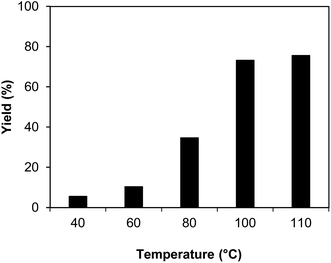 |
| | Fig. 1 Yields of 2-nitro-3-phenylimidazo[1,2-a]pyridine at varied temperatures. | |
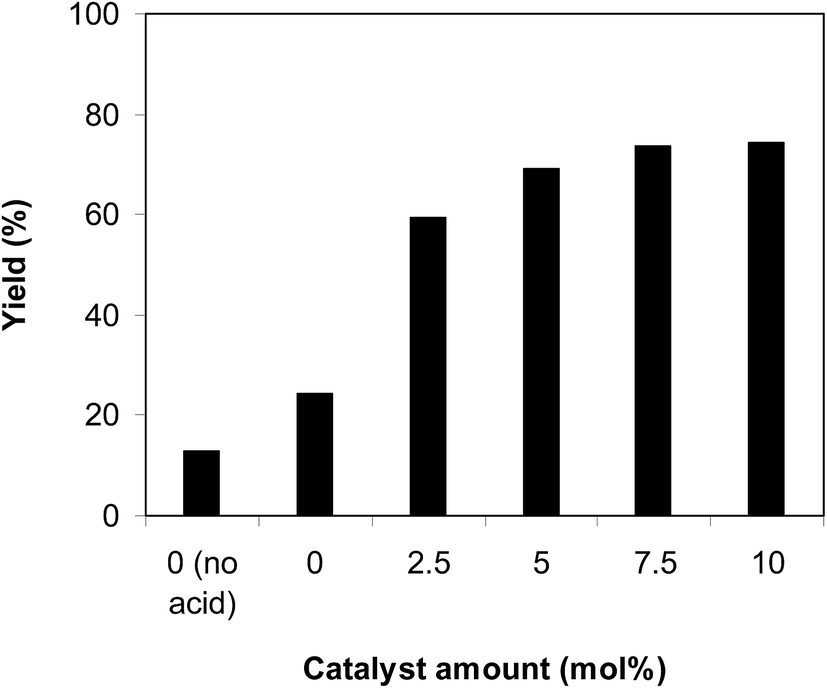
One more concern that must be studied for the cyclization of trans-β-nitrostyrene with 2-aminopyridine to generate 2-nitro-3-phenylimidazo[1,2-a]pyridine is the required catalyst quantity. In the first instance of the synthesis of 2-nitro-3-arylimidazo[1,2-a]pyridine derivatives from nitroolefins and aminopyridines via C–H amination in ambient air, Hajra et al. employed 20 mol% Fe(NO3)3 as a catalyst.14 Liang et al. performed the preparation of 3-nitro-2-arylimidazo[1,2-a]pyridines from the same starting materials employing 10 mol% CuBr catalyst.13 The reaction was implemented at 100 °C in air in dichloroethane for 6 h with 10 mol% acetic acid as a co-catalyst with 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1:1.2, at 2-aminopyridine concentration of 0.5 M, at 2.5 mol%, 5 mol%, 7.5 mol%, and 10 mol% catalyst, respectively. A 13% yield of 2-nitro-3-phenylimidazo[1,2-a]pyridine was recorded after 6 h in the absence of both the framework and acetic acid catalysts. In addition, 24% yield was obtained after 6 h in the presence of 10 mol% acetic acid without using the framework as a catalyst. These data highlight the need for the indium framework as a catalyst for the conversion. Positively, deploying the framework catalyst for the reaction caused an impressive advancement in the reaction yield. The reaction using 2.5 mol% catalyst could continue to 59% yield after 6 h, while 69% yield was obtained on employing 5 mol% catalyst. The yield could be improved to 73% after 6 h in the presence of 7.5 mol% catalyst. Using more than 7.5 mol% catalyst was found to be non-essential because the generation of 2-nitro-3-phenylimidazo[1,2-a]pyridine was not enhanced significantly (Fig. 2).
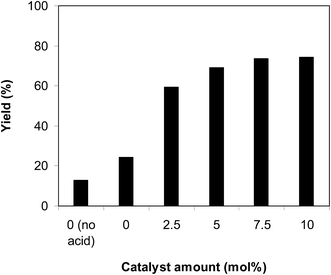 |
| | Fig. 2 Yields of 2-nitro-3-phenylimidazo[1,2-a]pyridine at various catalyst amounts. | |
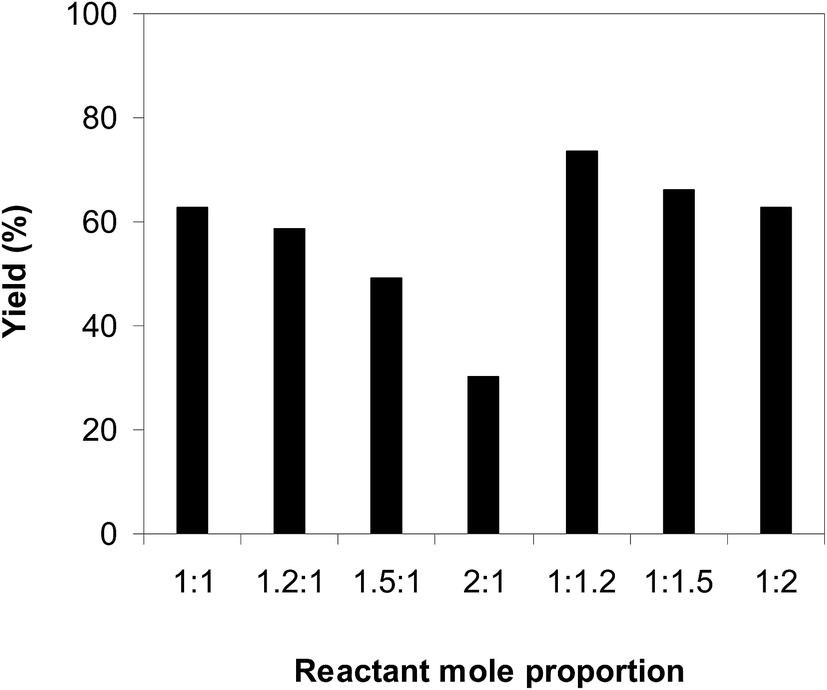
Afterwards, we investigated the consequence of the reactant mole proportion to the generation of 2-nitro-3-phenylimidazo[1,2-a]pyridine. The reaction was performed at 100 °C in air in dichloroethane for 6 h with 10 mol% catalyst and 10 mol% acetic acid as a co-catalyst at a 2-aminopyridine concentration of 0.5 M, employing a 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1:1, 1.2:1, 1.5:1, 2:1, 1:1.2, 1:1.5, and 1:2, respectively. The experimental data showed that the reactant mole proportion exhibited a noteworthy impact on the yield of the expected product. The reaction utilizing 1 equivalent of trans-β-nitrostyrene afforded 62% yield after 6 h. Using an excess of 2-aminopyridine resulted in a decrease in the yield of 2-nitro-3-phenylimidazo[1,2-a]pyridine. Indeed, the reaction using a 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1.2:1 progressed to 58% yield after 6 h, while 49% yield was noted on using 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1.5:1. The yield was decreased significantly to only 30% when 2 equivalents of 2-aminopyridine was employed. It was noticed that the reaction was favored using an excess of trans-β-nitrostyrene. The yield could be improved to 73% after 6 h with an aminopyridine:trans-β-nitrostyrene mole proportion of 1:1.2. However, utilizing more than 1.2 equivalents of trans-β-nitrostyrene provoked lower yields. Certainly, the 66% yield of the anticipated product was recorded for the reaction with a aminopyridine:trans-β-nitrostyrene mole proportion of 1:1.5, while a 62% yield was realized for that employing 2 equivalents of trans-β-nitrostyrene (Fig. 3).
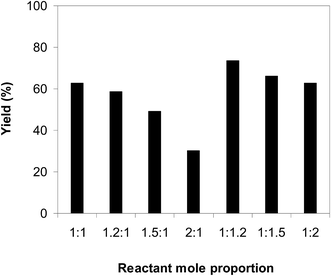 |
| | Fig. 3 Yields of 2-nitro-3-phenylimidazo[1,2-a]pyridine with diverse reactant mole proportions. | |
As the cyclization of trans-β-nitrostyrene with 2-aminopyridine to produce 2-nitro-3-phenylimidazo[1,2-a]pyridine was implemented in the liquid phase, the solvent might have showed a significant impact on the conversion. In the first illustration of the synthesis of 2-nitro-3-arylimidazo[1,2-a]pyridine derivatives from aminopyridines and nitroolefins, Hajra et al. performed the reaction in diverse solvents and demonstrated that dichloroethane should be used.14 For the CuBr-catalyzed synthesis of 3-nitro-2-arylimidazo[1,2-a]pyridines from the same starting materials, Liang et al. pointed out that DMF was the solvent of choice.13 It was accordingly decided to study the impact of the solvent on the generation of 2-nitro-3-phenylimidazo[1,2-a]pyridine. The reaction was conducted at 100 °C in air for 6 h with 7.5 mol% catalyst in the presence of 10 mol% acetic acid as a co-catalyst using a 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1:1.2 at a 2-aminopyridine concentration of 0.5 M, in DMF, DMSO, dichloroethane, dichlorobenzene, chlorobenzene, toluene, and diglyme, separately, as the solvents. Both DMF and DMSO were incompatible for the reaction using the framework catalyst with only 9% and 10% yields of 2-nitro-3-phenylimidazo[1,2-a]pyridine, respectively, after 6 h. The reaction continued slowly in dichlorobenzene and chlorobenzene, providing 42% and 46% yields, respectively, after 6 h. The reaction conducted in toluene produced 2-nitro-3-phenylimidazo[1,2-a]pyridine in 46% yield, whereas 50% yield was observed for diglyme. Among these solvents, dichloroethane produced the best results with 73% yield after 6 h (Fig. 4).
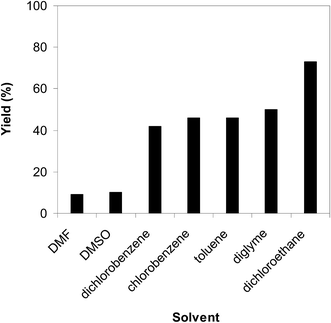 |
| | Fig. 4 Yields of 2-nitro-3-phenylimidazo[1,2-a]pyridine in the miscellaneous solvents. | |
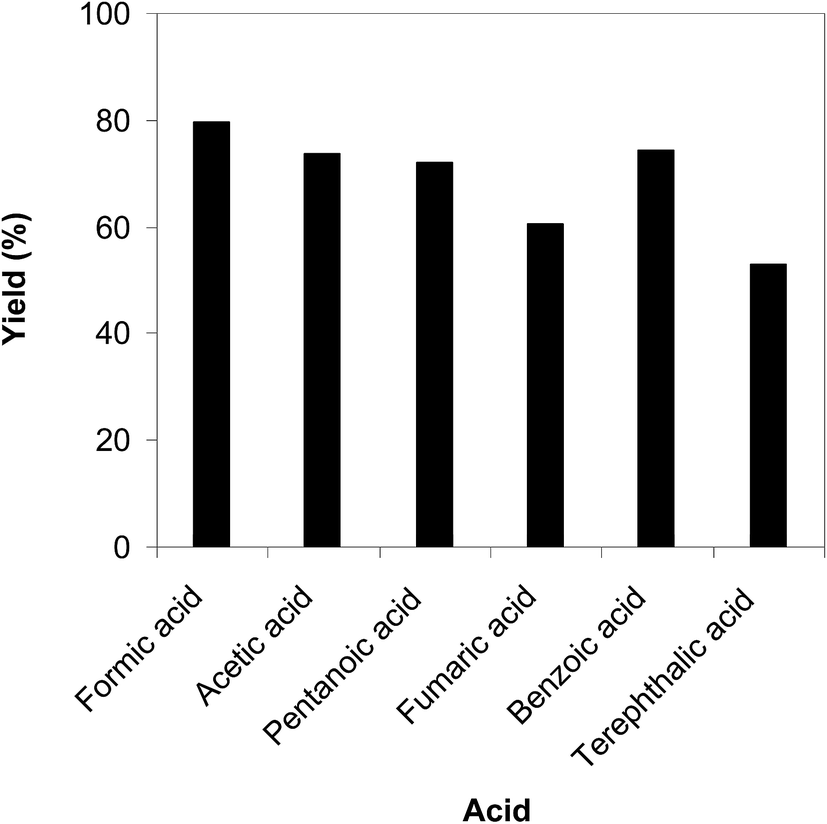
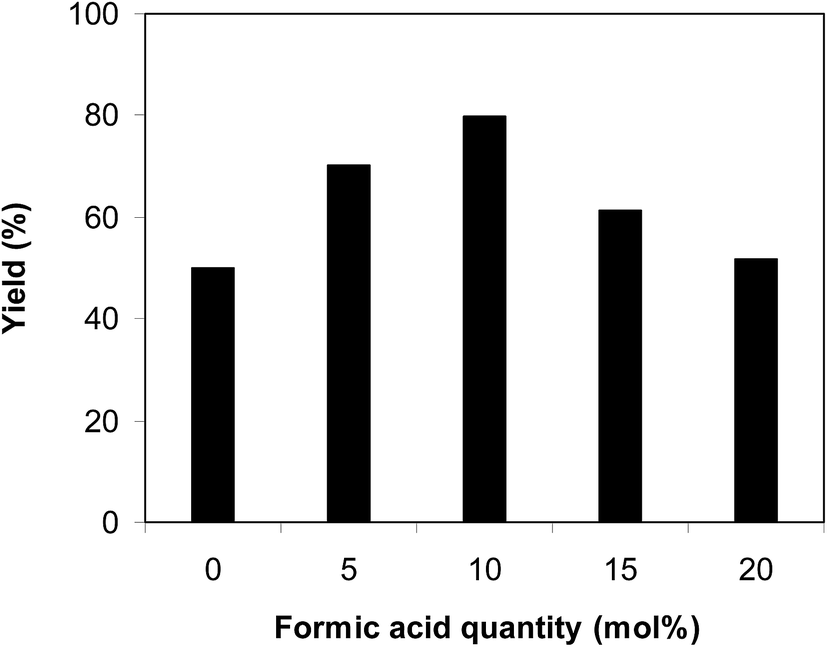
The cyclization of trans-β-nitrostyrene with 2-aminopyridine to produce 2-nitro-3-phenylimidazo[1,2-a]pyridine could provide only 51% yield after 6 h in the absence of acetic acid, hence denoting the usefulness of the Brønsted acid co-catalyst. Different Brønsted acid co-catalysts were then assessed for the synthesis of 2-nitro-3-phenylimidazo[1,2-a]pyridine. The reaction was conducted in air at 100 °C in dichloroethane for 6 h with 7.5 mol% catalyst, 10 mol% co-catalyst, and a 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1:1.2 at a 2-aminopyridine concentration of 0.5 M, using formic acid, acetic acid, pentanoic acid, fumaric acid, benzoic acid, and terephthalic acid as the co-catalysts, separately. A 73% yield of 2-nitro-3-phenylimidazo[1,2-a]pyridine was obtained after 6 h for the reaction using acetic acid as a co-catalyst. This yield was 74% yield for the reaction using benzoic acid as the co-catalyst. Employing pentanoic acid, the conversion was 72% after 6 h. Fumaric acid and terephthalic acid produced only 61% and 53% yields, respectively, after 6 h. Among these co-catalysts, formic acid showed the highest efficiency, 80% after 6 h (Fig. 5). In addition, the quantity of the co-catalyst also regulated the production of 2-nitro-3-phenylimidazo[1,2-a]pyridine. The best result was observed for the reaction using 10 mol% formic acid as a co-catalyst. Expanding or lowering the quantity of formic acid caused a decrease in the reaction yield (Fig. 6). In addition, the concentration of 2-aminopyridine also affected the reaction, and the reaction was conducted with a 2-aminopyridine concentration of 0.5 M (Fig. 7).
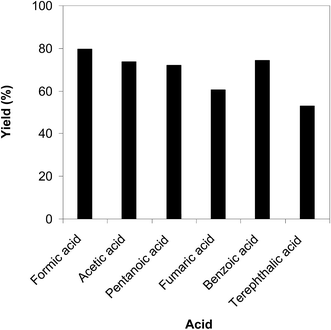 |
| | Fig. 5 Yields of 2-nitro-3-phenylimidazo[1,2-a]pyridine with different co-catalysts. | |
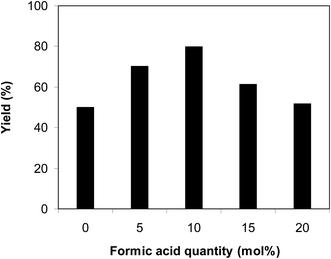 |
| | Fig. 6 Yields of 2-nitro-3-phenylimidazo[1,2-a]pyridine at varied co-catalyst quantities. | |
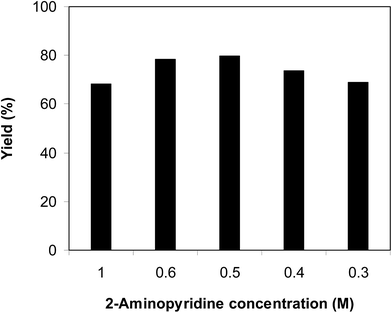 |
| | Fig. 7 Yields of 2-nitro-3-phenylimidazo[1,2-a]pyridine at diverse 2-aminopyridine concentrations. | |
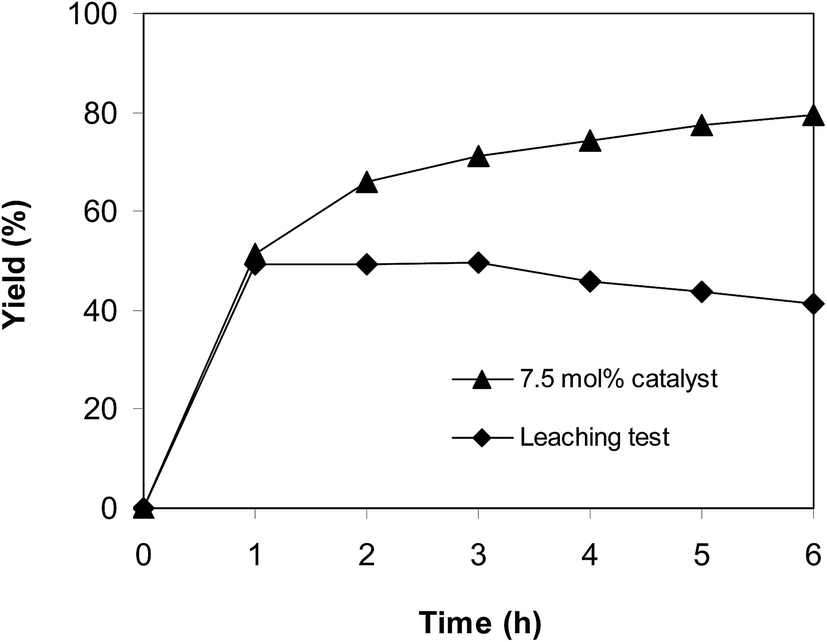
Because the reaction of trans-β-nitrostyrene with 2-aminopyridine to produce 2-nitro-3-phenylimidazo[1,2-a]pyridine was conducted in the liquid phase, a leaching phenomenon might have occurred throughout the course of the reaction, which must be considered. To determine if the active sites dissolved from the solid catalyst promoted the generation of 2-nitro-3-phenylimidazo[1,2-a]pyridine, a leaching assessment was accordingly performed. The reaction was implemented in air at 100 °C in dichloroethane for 6 h, 7.5 mol% catalyst, 10 mol% formic acid as co-catalyst, utilizing 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1:1.2, and a 2-aminopyridine concentration of 0.5 M. After the initial 1 h period with 49% yield being noted, the catalyst was collected by centrifugation. The solution phase was then added to a new and clean pressurized vial and stirred magnetically for an additional 5 h at 100 °C. No more 2-nitro-3-phenylimidazo[1,2-a]pyridine was generated after the catalyst was isolated (Fig. 8). These data suggest that cyclization of trans-β-nitrostyrene with 2-aminopyridine to produce 2-nitro-3-phenylimidazo[1,2-a]pyridine only continued in the existence of the framework-based catalyst, and the donation of homogeneous catalysis to the generation of 2-nitro-3-phenylimidazo[1,2-a]pyridine was insignificant.
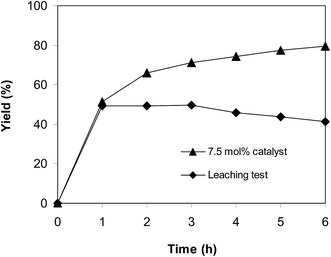 |
| | Fig. 8 Leaching assessment showed that 2-nitro-3-phenylimidazo[1,2-a]pyridine was not generated in the absence of a solid catalyst. | |
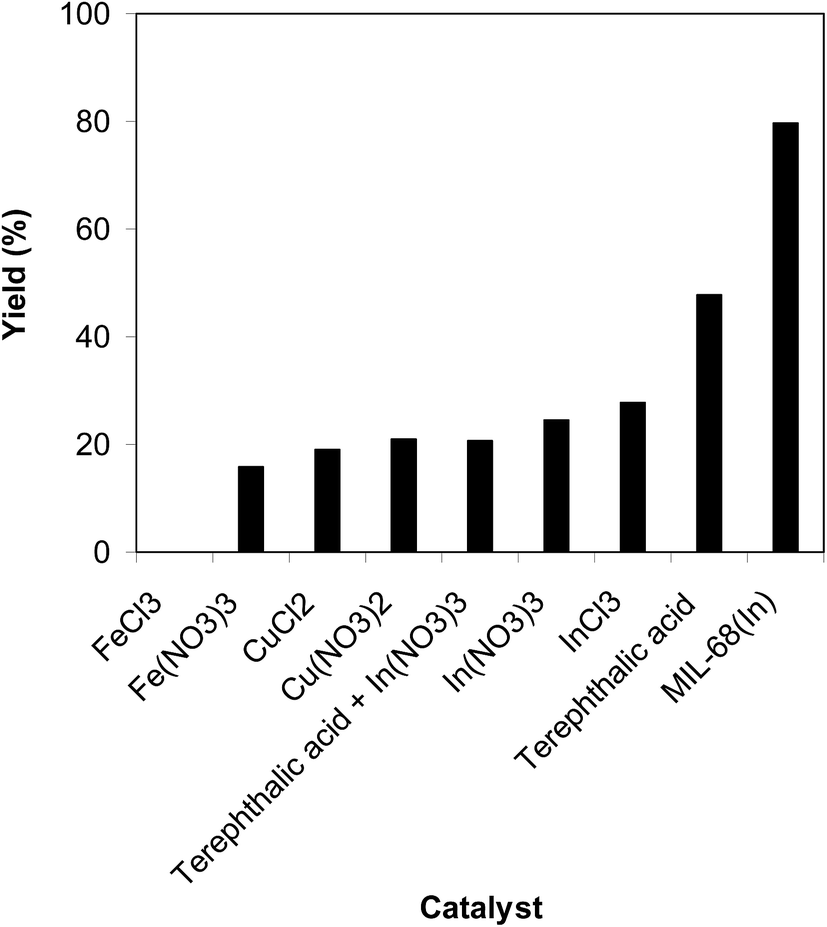
The efficiency of the indium framework in the cyclization of trans-β-nitrostyrene with 2-aminopyridine to produce 2-nitro-3-phenylimidazo[1,2-a]pyridine was correlated to a number of regular homogeneous catalysts. The reaction was conducted in air at 100 °C in dichloroethane for 6 h using 7.5 mol% catalyst, 10 mol% formic acid as a co-catalyst, 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1:1.2, and a 2-aminopyridine concentration of 0.5 M. FeCl3 was inactive for the conversion, while a 16% yield was noted after 6 h in the reaction using Fe(NO3)3 catalyst. Cu(NO3)2 and CuCl2 showed low activity for the reaction with 19% and 21% yields of the heterocyclic product, respectively, after 6 h. The reaction utilizing the indium framework could continue to 80% yield after 6 h. However, homogeneous indium salts displayed substantially lower catalytic activity than the solid indium framework catalyst. Certainly, the InCl3-catalyzed reaction of trans-β-nitrostyrene with 2-aminopyridine afforded only 28% yield after 6 h. To clarify the active sites for the reaction, several control experiments were conducted. The reaction using In(NO3)3, the precursor of the framework, expressed low activity with only 24% yield after 6 h, while the reaction under terephthalic acid, a framework linker, afforded only 47% yield. Remarkably, the conversion utilizing the mixture of terephthalic acid with In(NO3)3 as a catalyst proceeded to 21% yield of 2-nitro-3-phenylimidazo[1,2-a]pyridine after 6 h (Fig. 9). In addition, almost no progress was observed in the reaction using the MIL-68(In) soaked and poisoned by pyridine. The above observations support the active In sites and the cooperative effect between the metal cluster and organic linker within the MIL-68 framework.
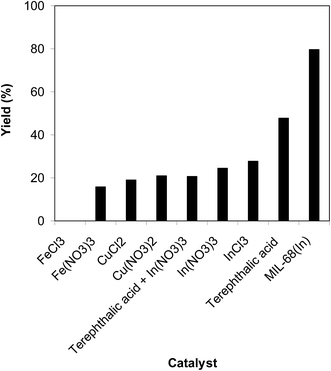 |
| | Fig. 9 Yields of 2-nitro-3-phenylimidazo[1,2-a]pyridine with miscellaneous homogeneous catalysts. | |
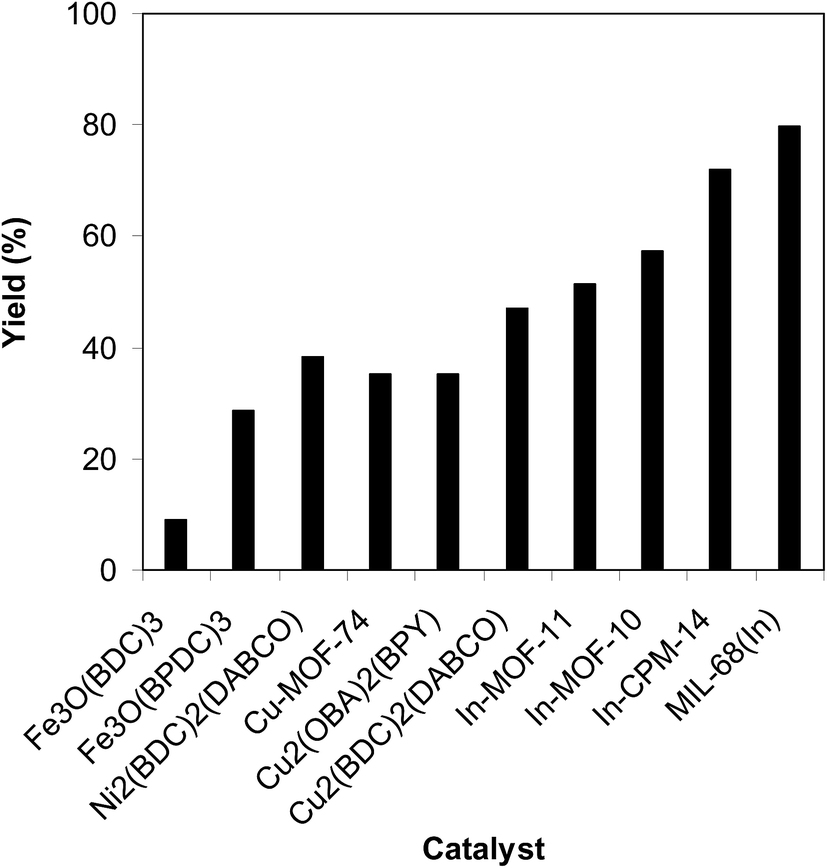
To determine the benefits of employing MIL-68(In) as a catalyst for the reaction between trans-β-nitrostyrene and 2-aminopyridine to produce 2-nitro-3-phenylimidazo[1,2-a]pyridine, its productivity was compared with that of other MOFs. The reaction was implemented in air in dichloroethane for 6 h using 7.5 mol% catalyst, 10 mol% formic acid as co-catalyst, 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1:1.2, and a 2-aminopyridine concentration of 0.5 M. Among these MOFs, Fe3O(BDC)3 revealed the lowest catalytic activity, reaching only 9% yield of 2-nitro-3-phenylimidazo[1,2-a]pyridine after 6 h. Fe3O(BPDC)3 was more active than Fe3O(BDC)3 with 29% yield achieved after 6 h. The Ni2(BDC)2(DABCO)-catalyzed reaction could continue to 38% yield after 6 h, while 35%, 36%, and 47% yields were noted for Cu-MOF-74, Cu2(OBA)2(BPY), and Cu2(BDC)2(DABCO), respectively. To confirm the effect of structure on the catalytic activity, reactions using various In-MOFs catalysts constructed with different linkers were performed. In particular, reaction using In-MOF-10 catalyst with a 4,4′-biphenyldicarboxylate linker provided 57% yield after 6 h. Similarly, only 51% yield of the heterocyclic product was achieved after 6 h for the reaction using In-MOF-11, which possesses 1,3,5,7-adamantanetetracarboxylate in the framework. In-CPM-14 synthesized using 4,4′-oxybis(benzoic acid) and In(NO3)3 was more active. However, only 72% yield of 2-nitro-3-phenylimidazo[1,2-a]pyridine was produced after 6 h (Fig. 10). These results confirm the hypothesis in the excellent structure-activity relationship in the MIL-68(In) for the cyclic reaction.
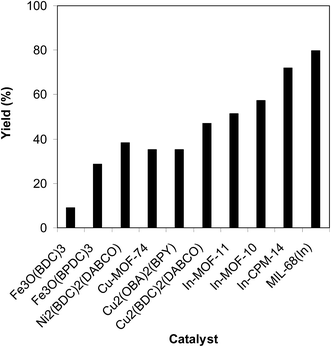 |
| | Fig. 10 Yields of 2-nitro-3-phenylimidazo[1,2-a]pyridine with miscellaneous MOF-based catalysts. | |
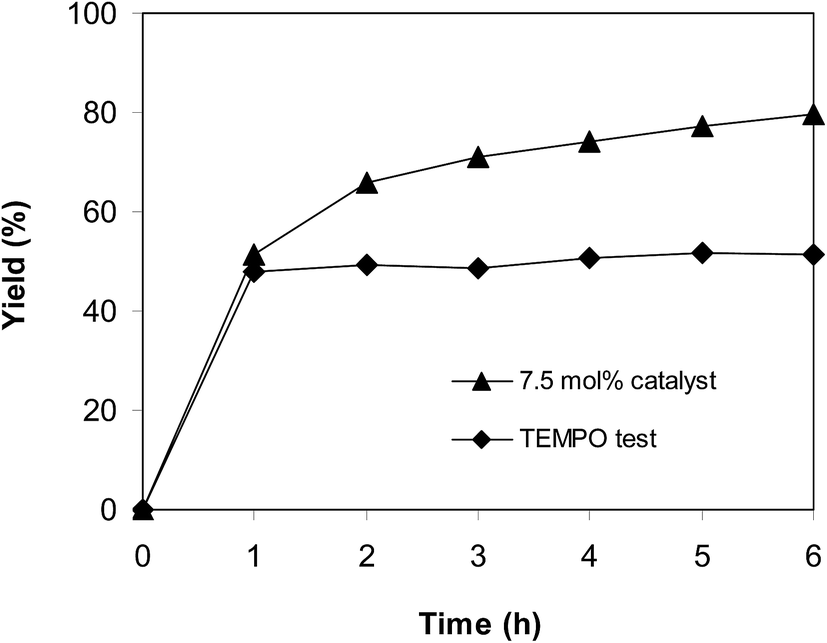
To obtain more information about the pathway of the cyclization reaction between trans-β-nitrostyrene and 2-aminopyridine to produce 2-nitro-3-phenylimidazo[1,2-a]pyridine, further mechanistic studies were conducted. The reaction was performed at 100 °C in air for 6 h in dichloroethane using 7.5 mol% catalyst, 10 mol% formic acid as co-catalyst, a 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1:1.2, and a 2-aminopyridine concentration of 0.5 M. After the first hour with 48% yield being recorded, 15 mol% of (2,2,6,6-tetramethylpiperidin-1-yl)oxy (TEMPO) as a radical trapping reagent was introduced, and the mixture was heated at 100 °C for an additional 5 h. Only 51% yield was obtained (Fig. 11). Up to 80% yield of 2-nitro-3-phenylimidazo[1,2-a]pyridine was achieved after 6 h for the reaction in the absence of TEMPO.
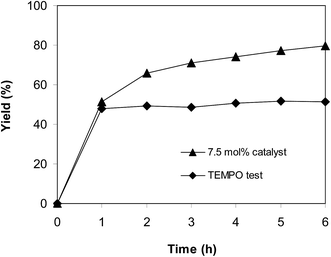 |
| | Fig. 11 TEMPO test showing that the reaction could not proceed in the presence of radical trapping reagent. | |
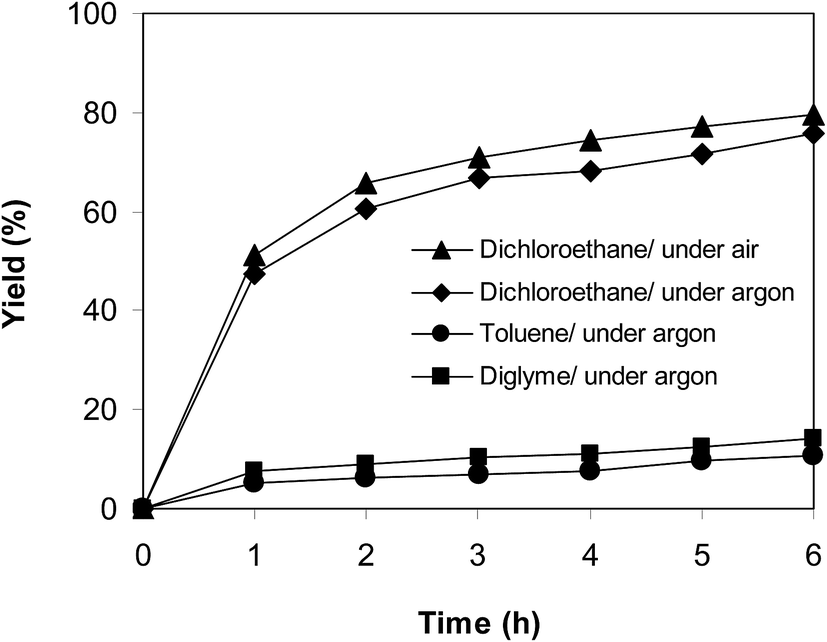
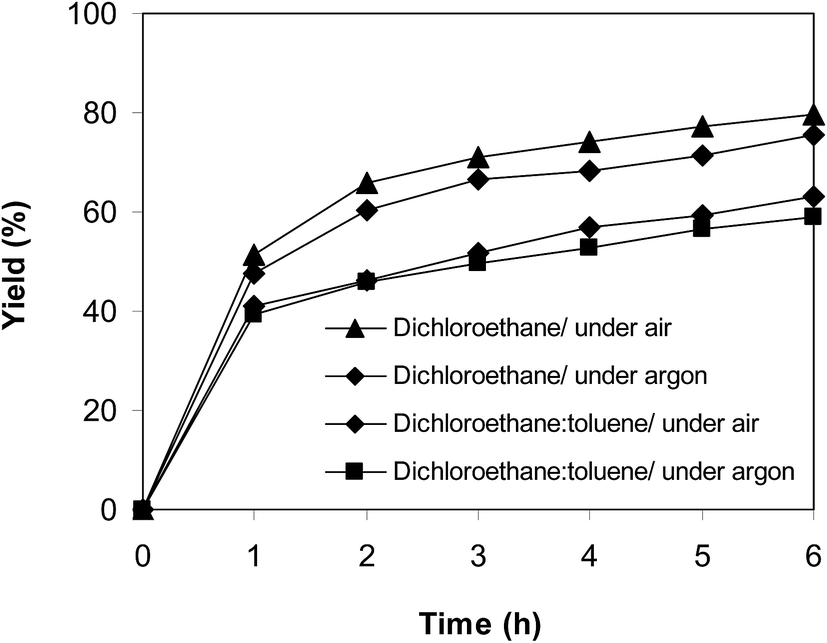
In another experiment, the reaction was performed under an argon atmosphere. The desired product 2-nitro-3-phenylimidazo[1,2-a]pyridine was achieved in 75% yield after 6 h (Fig. 12), implying that the cyclization reaction could still proceed in the absence of air. Indeed, Hajra et al. suggested that dichloroethane also displayed the function as an oxidant for the transformation.14 To clarify the role of dichloroethane in the formation of 2-nitro-3-phenylimidazo[1,2-a]pyridine, the reaction was conducted in toluene or diglyme under argon for 6 h, at 7.5 mol% catalyst with 10 mol% formic acid as a co-catalyst, employing 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1:1.2, at 2-aminopyridine concentration of 0.5 M. Toluene and diglyme possessed no oxidizing properties. The transformation proceeded slowly in these solvents under argon, generating the desired product in only 14% and 10% yield, respectively, after 6 h (Fig. 12). To further address this issue, the reaction was also performed in a mixture of dichloroethane and toluene (1:1, v/v) in air and argon. The results showed 63% yield of 2-nitro-3-phenylimidazo[1,2-a]pyridine for the reaction under air, whereas 59% yield was recorded for the reaction under argon (Fig. 13). These data confirmed that dichlorothane would work as both solvent and oxidant for the oxidative amination between trans-β-nitrostyrene and 2-aminopyridine, and that the contribution of dichloroethane to the yield of the expected product was significant. The fact that 75% yield was achieved for the reaction conducted in dichloroethane under argon, while 80% yield was obtained for the reaction under air suggested that the oxygen in air also contributed to the transformation, but the role of dichloroethane was more important.
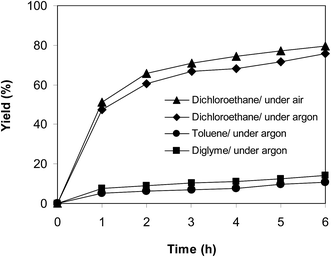 |
| | Fig. 12 Argon test indicating that the reaction could proceed in dichloroethane under argon. | |
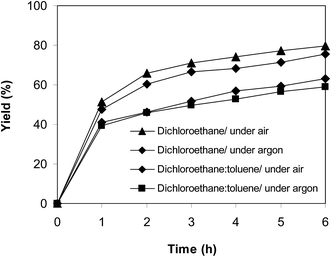 |
| | Fig. 13 Argon test indicating that yield was adjusted by the amount of dichloroethane. | |
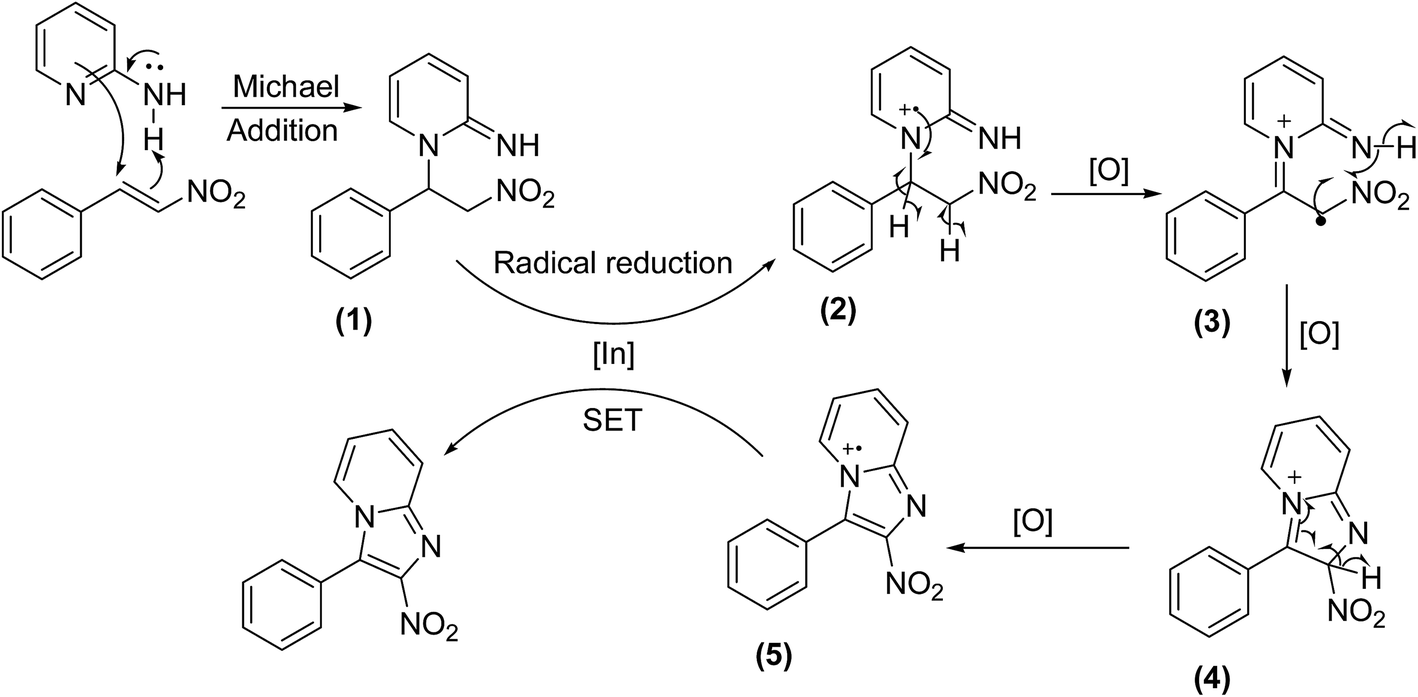
The reaction mechanism is depicted in Scheme 2. First, 2-aminopyridine underwent Michael addition in endocyclic manner to form a Michael adduct (1).14 Radical reduction through single electron transfer (SET) subsequently occurred in the presence of the indium catalyst to form intermediate (2).39 Next, radical dehydrogenation of (2) proceeded to generate (3), which was further oxidized to afford the radial cation (5) through the formation of cyclic (4).40 Another single electron transfer from (5) generated the indium catalyst to the catalytic cycle and provided the desired product.
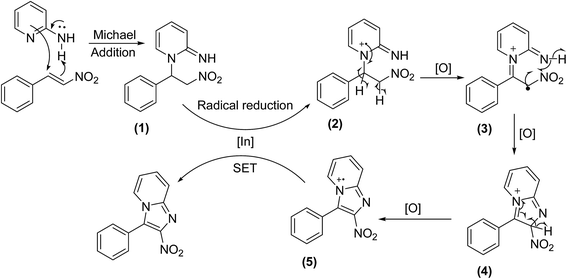 |
| | Scheme 2 Plausible reaction mechanism. | |
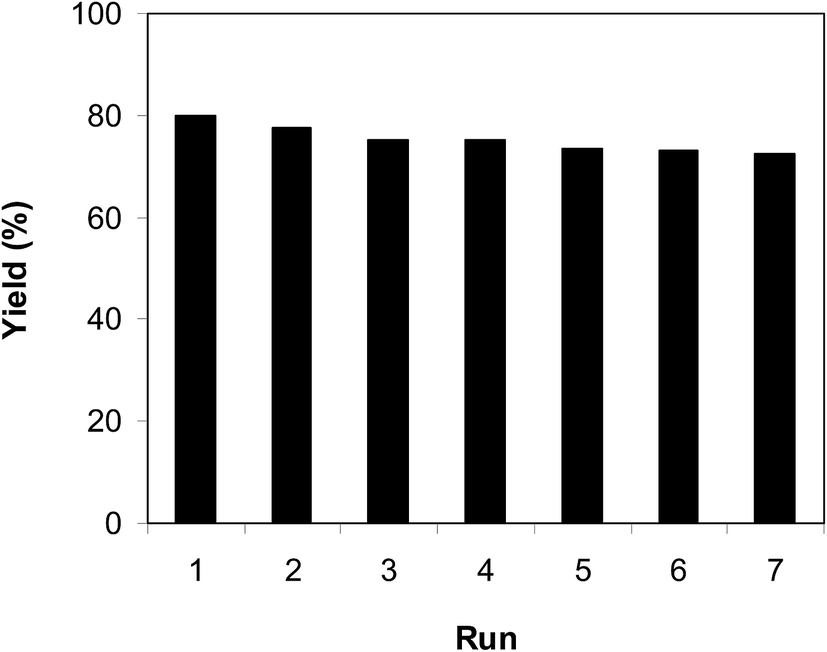
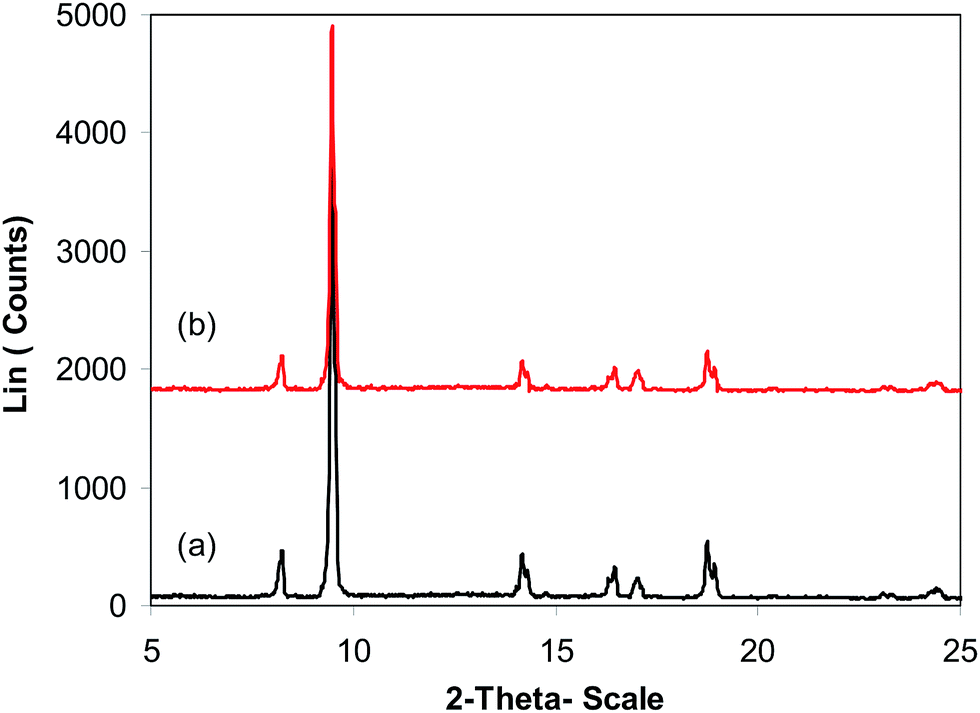
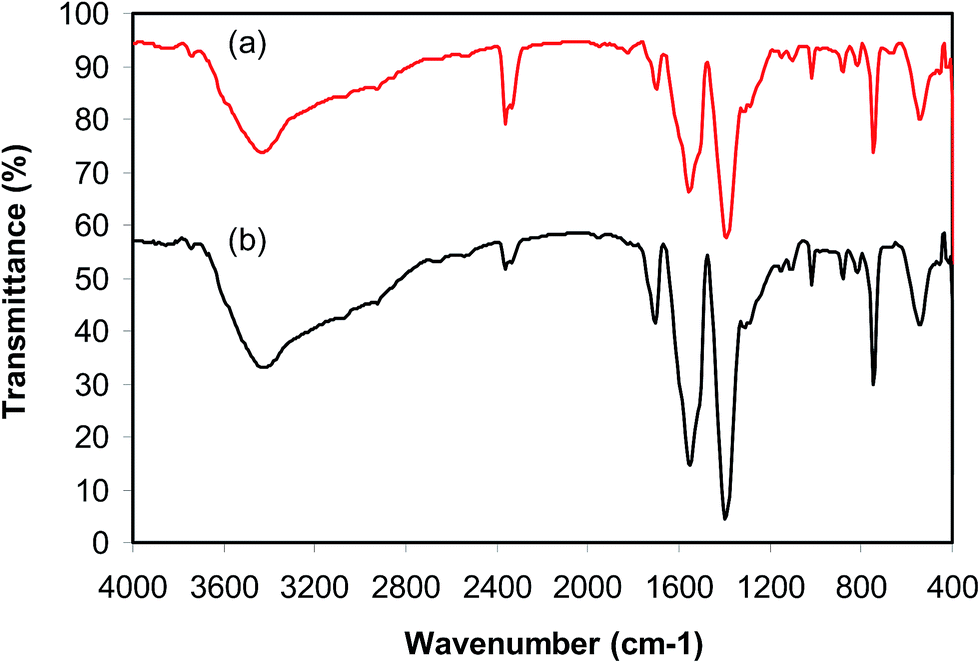
As previously pointed out, the indium framework displayed better efficiency in the cyclization reaction than various homogeneous and heterogeneous catalysts. To determine the benefits of this catalyst in the reaction between trans-β-nitrostyrene and 2-aminopyridine to generate 2-nitro-3-phenylimidazo[1,2-a]pyridine, one factor that should be considered is the possibility of the catalyst reusability. The catalyst was accordingly explored for reusability in the reaction over 7 consecutive runs. The reaction was conducted in air at 100 °C in dichloroethane for 6 h using 7.5 mol% catalyst, 10 mol% formic acid as co-catalyst, a 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1:1.2, and 2-aminopyridine concentration of 0.5 M. Upon completion of the first run, the catalyst was collected, washed carefully with DMF and dichloromethane, and desiccated at 150 °C under vacuum in a Schlenk line for 2 h. The recovered catalyst was subsequently reused in new experiments. The experimental data proved that the catalyst could be reused numerous times in the reaction of trans-β-nitrostyrene with 2-aminopyridine to generate 2-nitro-3-phenylimidazo[1,2-a]pyridine without considerable deterioration in efficiency. Undoubtedly, 72% yield of 2-nitro-3-phenylimidazo[1,2-a]pyridine was still recorded in the 7th run (Fig. 14). Furthermore, the integrity of the catalyst could be retained after the reaction, as indicated by XRD (Fig. 15) and FT-IR (Fig. 16) spectra of the recovered framework.
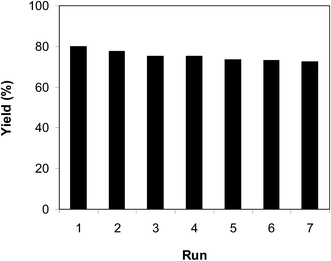 |
| | Fig. 14 Catalyst reusability. | |
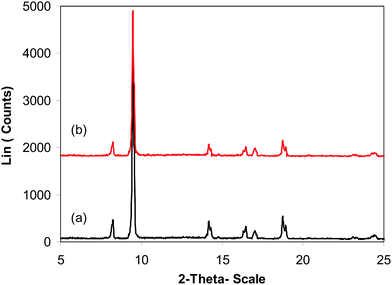 |
| | Fig. 15 X-ray powder diffraction patterns of the new (a) and recovered (b) catalyst. | |
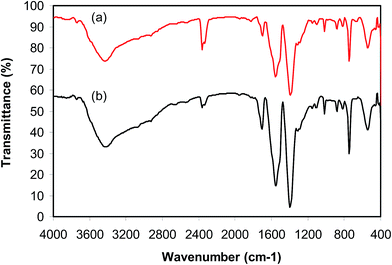 |
| | Fig. 16 FT-IR spectra of the new (a) and recovered (b) catalyst. | |
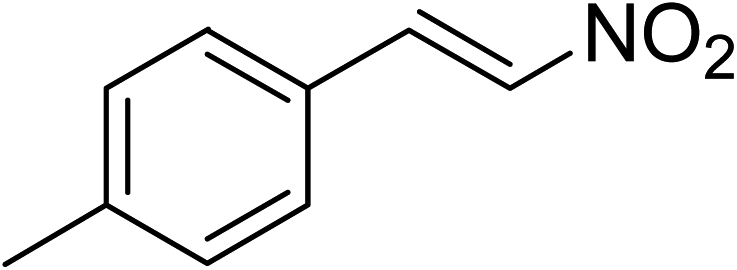
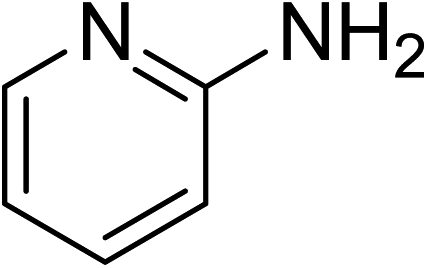
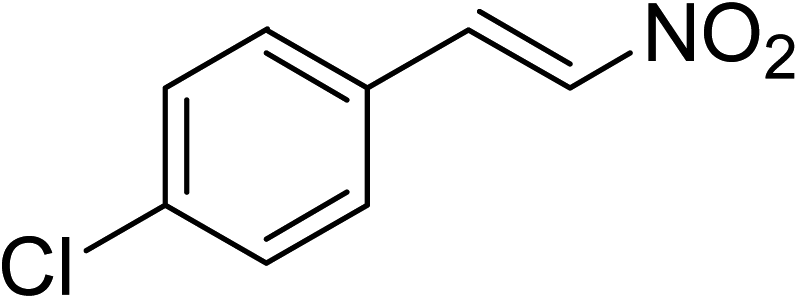
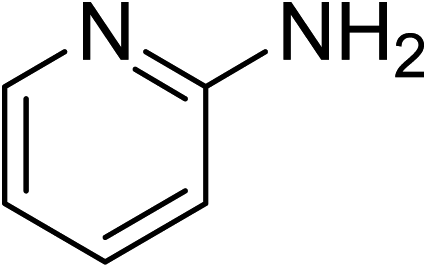
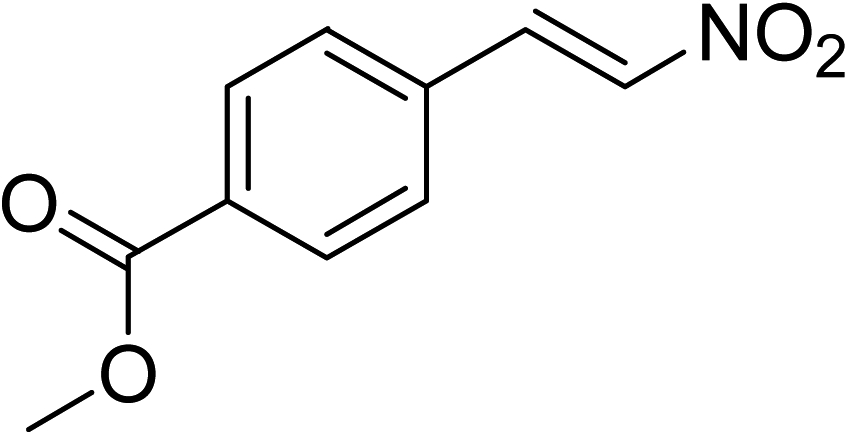
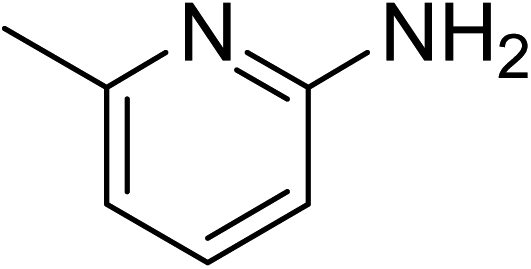
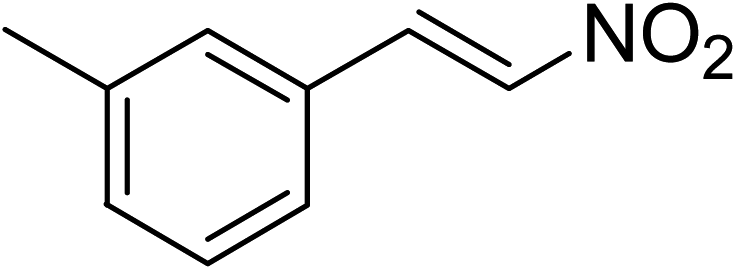
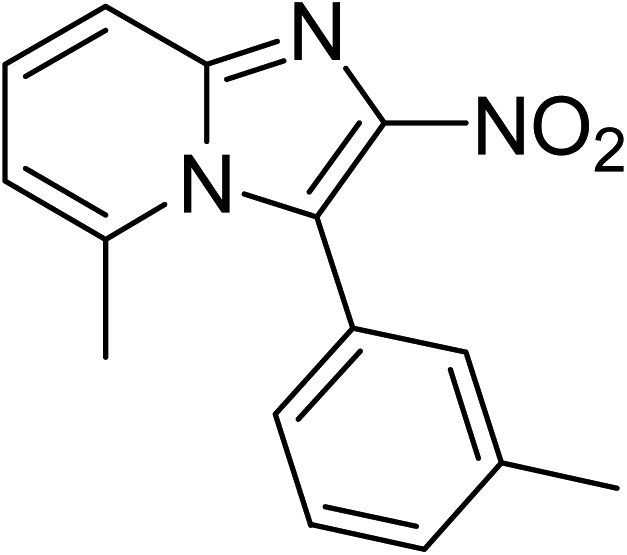
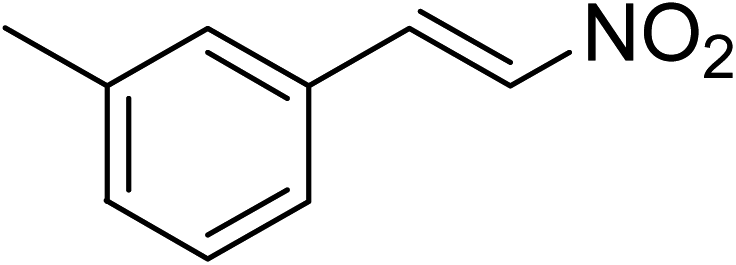
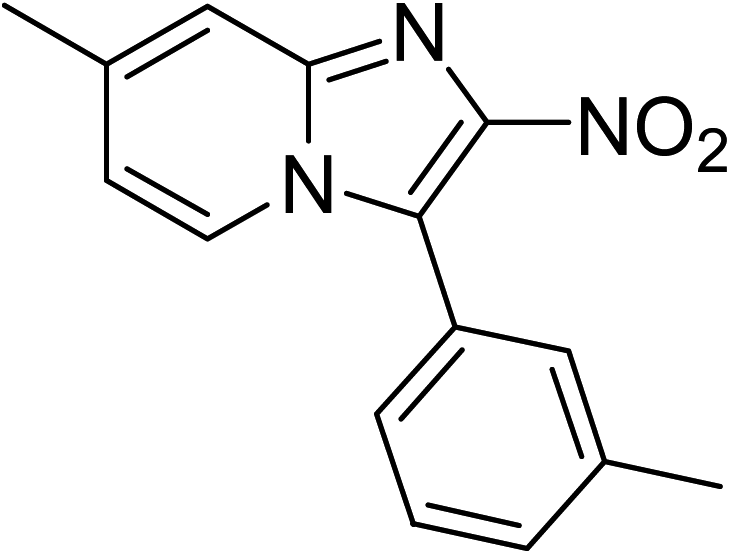
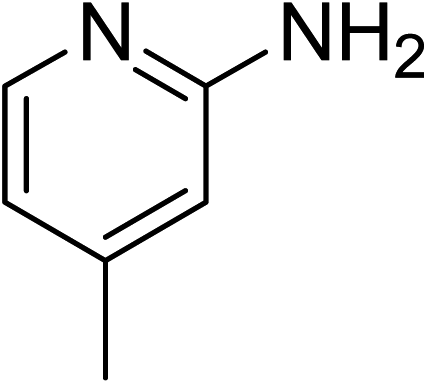
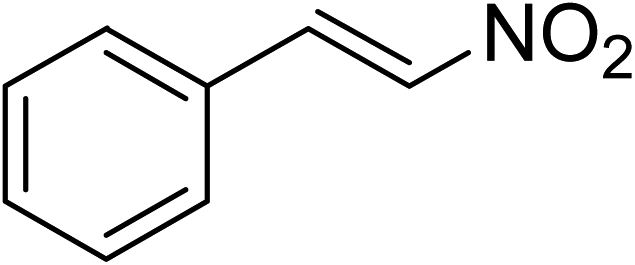
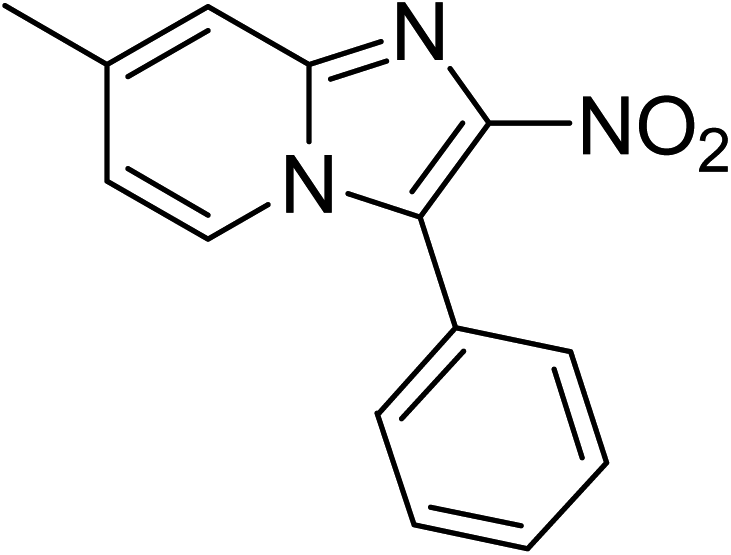
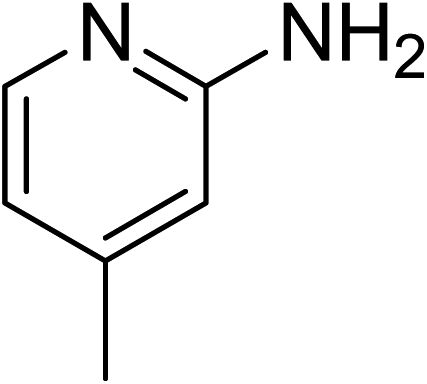
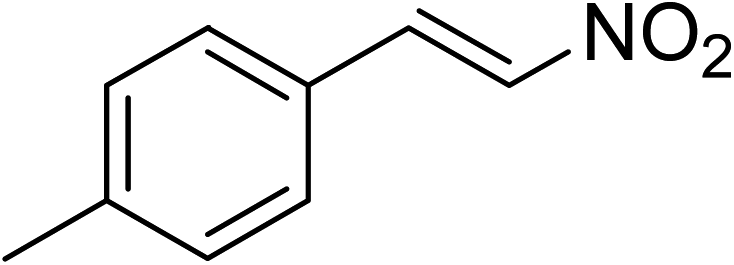
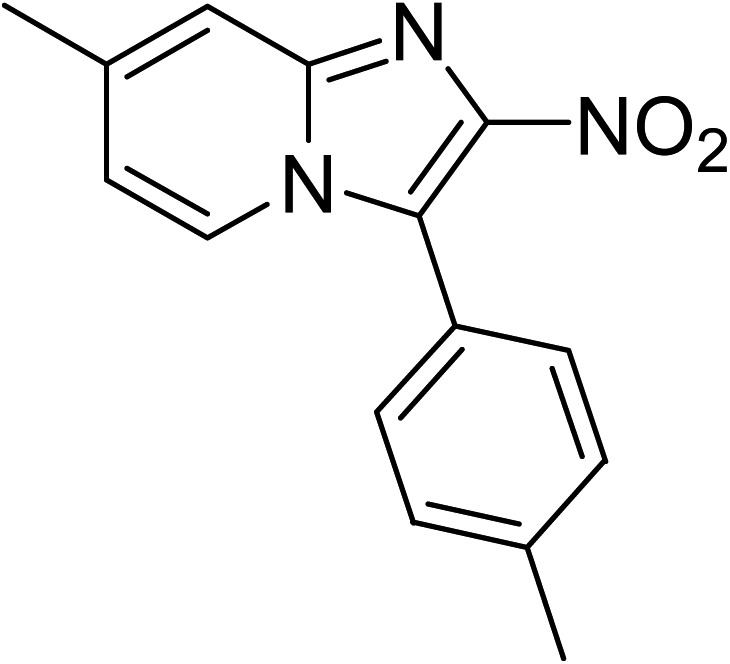
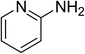
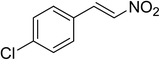
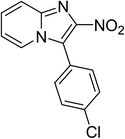
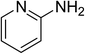
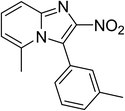
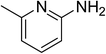
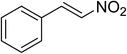
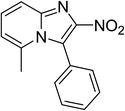
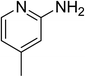
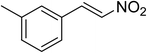
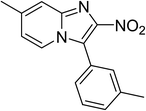
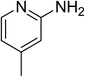
The research scope was subsequently expanded to the synthesis of different 2-nitro-3-arylimidazo[1,2-a]pyridines utilizing MIL-68(In) catalyst via the cyclization reaction. In the first experiment series, reactions between 2-aminopyridine and various trans-β-nitrostyrenes possessing substituent were performed. The reaction was conducted in air at 100 °C in dichloroethane for 6 h using 7.5 mol% catalyst, 10 mol% formic acid as co-catalyst, a 2-aminopyridine:trans-β-nitrostyrene mole proportion of 1:1.2, and 2-aminopyridine concentration of 0.5 M. The products were purified by column chromatography, and the isolated yields were then recorded. Under these conditions, 2-nitro-3-phenylimidazo[1,2-a]pyridine was generated in 77% yield (entry 1). The presence of a substituent on the benzene ring in trans-β-nitrostyrene decreased the yield slightly. The reaction between 2-aminopyridine and 4-methyl-trans-nitrostyrene produced 2-nitro-3-p-tolylimidazo[1,2-a]pyridine in 74% yield (entry 2), while 72% yield of 3-(4-chlorophenyl)-2-nitroimidazo[1,2-a]pyridine was achieved for the reaction between 2-aminopyridine and 4-chloro-trans-nitrostyrene (entry 3). The ester group was tolerated in this reaction, and 75% yield of methyl 4-(2-nitroimidazo[1,2-a]pyridin-3-yl)benzoate was obtained via the reaction of 2-aminopyridine with (E)-methyl 4-(2-nitrovinyl)benzoate (entry 4). Next, 2-amino-6-methylpyridine was used as a starting material for the cyclization reaction under similar conditions, producing 5-methyl-2-nitro-3-m-tolylimidazo[1,2-a]pyridine (entry 5) and 5-methyl-2-nitro-3-phenylimidazo[1,2-a]pyridine (entry 6) in 76% and 70% yield, respectively. In another experimental series, reactions between 2-amino-4-methylpyridine and different trans-β-nitrostyrenes were conducted. Similarly, 7-methyl-2-nitro-3-m-tolylimidazo[1,2-a]pyridine (entry 7), 7-methyl-2-nitro-3-phenylimidazo[1,2-a]pyridine (entry 8), and 7-methyl-2-nitro-3-p-tolylimidazo[1,2-a]pyridine (entry 9) were produced in 73%, 74%, and 69% yield, respectively (Table 1).
Table 1 Synthesis of 2-nitro-3-arylimidazo[1,2-a]pyridines utilizing MIL-68(In) catalyst
| Entry |
Reactant 1 |
Reactant 2 |
Product |
Isolated yields (%) |
| 1 |
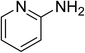 |
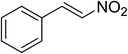 |
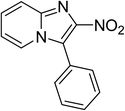 |
77 |
| 2 |
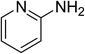 |
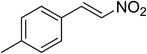 |
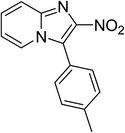 |
74 |
| 3 |
 |
 |
 |
72 |
| 4 |
 |
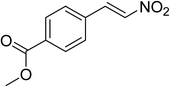 |
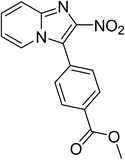 |
75 |
| 5 |
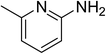 |
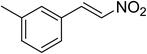 |
 |
76 |
| 6 |
 |
 |
 |
70 |
| 7 |
 |
 |
 |
73 |
| 8 |
 |
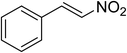 |
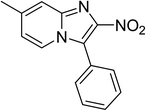 |
74 |
| 9 |
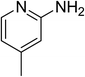 |
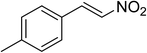 |
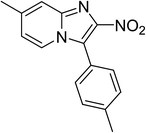 |
69 |
4. Conclusions
Metal–organic framework MIL-68(In) has emerged as a productive heterogeneous catalyst for the synthesis of 2-nitro-3-arylimidazo[1,2-a]pyridines via oxidative amination between 2-aminopyridines and nitroalkenes using air as an oxidation agent. The presence of formic acid as a co-catalyst accelerated the transformation remarkably. The indium framework showed higher efficiency for the synthesis of 2-nitro-3-arylimidazo[1,2-a]pyridines than miscellaneous MOF-based catalysts. The catalyst also displayed higher productivity than a number of homogeneous catalysts. Heterogeneous catalysis was verified for the cyclization reaction employing the framework-based catalyst. The catalyst could be collected and reused for the production of 2-nitro-3-arylimidazo[1,2-a]pyridines without a pronounced deterioration in catalytic efficiency. To the best of our knowledge, the preparation of 2-nitro-3-arylimidazo[1,2-a]pyridines was not formerly achieved using heterogeneous catalysts.
Acknowledgements
The Vietnam National University – Ho Chi Minh City (VNU-HCM) is acknowledged for financial support through project No. B2017-20-08.
References
- C. Ravi, D. C. Mohan and S. Adimurthy, Org. Biomol. Chem., 2016, 14, 2282–2290 CAS.
- S. Samanta, S. Jana, S. Mondal, K. Monir, S. K. Chandra and A. Hajra, Org. Biomol. Chem., 2016, 14, 5073–5078 CAS.
- D. Dheer, K. R. Reddy, S. K. Rath, P. L. Sangwan, P. Das and R. Shankar, RSC Adv., 2016, 6, 38033–38036 RSC.
- T. R. Mandlimath and K. I. Sathiyanarayanan, RSC Adv., 2016, 6, 3117–3125 RSC.
- X. Meng, J. Zhang, B. Chen, Z. Jing and P. Zhao, Catal. Sci. Technol., 2016, 6, 890–896 CAS.
- M. H. Shinde and U. A. Kshirsagar, Green Chem., 2016, 18, 1455–1458 RSC.
- A. K. Bagdi, S. Santra, K. Monir and A. Hajra, Chem. Commun., 2015, 51, 1555–1575 RSC.
- J. Koubachi, S. E. Kazzouli, M. Bousmina and G. Guillaumet, Eur. J. Org. Chem., 2014, 5119–5138 CrossRef CAS.
- P. Zhou, L. Jiang, F. Wang, K. Deng, K. Lv and Z. Zhang, Sci. Adv., 2017, e1601945 Search PubMed.
- A. J. Stasyuk, M. Banasiewicz, M. K. Cyranski and D. T. Gryko, J. Org. Chem., 2012, 77, 5552–5558 CrossRef CAS PubMed.
- H. Yan, Y. Wang, C. Pan, H. Zhang, S. Yang, X. Ren, J. Li and G. Huang, Eur. J. Org. Chem., 2014, 2754–2763 CrossRef CAS.
- I. V. Rassokhina, V. Z. Shirinian, I. V. Zavarzin, V. Gevorgyan and Y. A. Volkova, J. Org. Chem., 2015, 80, 11212–11218 CrossRef CAS PubMed.
- R.-L. Yan, H. Yan, C. Ma, Z.-Y. Ren, X.-A. Gao, G.-S. Huang and Y.-M. Liang, J. Org. Chem., 2012, 77, 2024–2028 CrossRef CAS PubMed.
- K. Monir, A. K. Bagdi, M. Ghosh and A. Hajra, Org. Lett., 2014, 16, 4630–4633 CrossRef CAS PubMed.
- A. R. Chowdhuri, D. Bhattacharya and S. K. Sahu, Dalton Trans., 2016, 45, 2963–2973 RSC.
- J.-Z. Gu, Y.-H. Cui, J. Wu and A. M. Kirillov, RSC Adv., 2015, 5, 78889–78901 RSC.
- C. L. Jones, A. J. Tansell and T. L. Easun, J. Mater. Chem. A, 2016, 4, 6714–6723 CAS.
- L. Zhang, Z. Kang, X. Xin and D. Sun, CrystEngComm, 2016, 18, 193–206 RSC.
- J. Du and G. Zou, Inorg. Chem. Commun., 2016, 69, 20–23 CrossRef CAS.
- L. J. Dunne and G. Manos, Dalton Trans., 2016, 45, 4213–4217 RSC.
- X.-D. Yang, C. Chen, Y.-J. Zhang, L.-X. Cai, B. Tan and J. Zhang, Dalton Trans., 2016, 45, 4522–4527 RSC.
- P. Kumar, K.-H. Kim, E. E. Kwon and J. E. Szulejko, J. Mater. Chem. A, 2016, 4, 345–361 CAS.
- C. H. Hendon and A. Walsh, Chem. Sci., 2015, 6, 3674–3683 RSC.
- A. K. Cheetham, T. D. Bennett, F.-X. Coudert and A. L. Goodwin, Dalton Trans., 2016, 45, 4113–4126 RSC.
- M. R. Mani, R. Chellaswamy, Y. N. Marathe and V. K. Pillai, RSC Adv., 2016, 6, 1907–1912 RSC.
- R. Babu, A. C. Kathalikkattil, R. Roshan, J. Tharun, D.-W. Kim and D.-W. Park, Green Chem., 2016, 18, 232–242 RSC.
- S. Zhang, L. Li, S. Zhao, Z. Sun, M. Hong and J. Luo, J. Mater. Chem. A, 2015, 3, 15764–15768 CAS.
- J. Zheng, M. Wu, F. Jiang, W. Su and M. Hong, Chem. Sci., 2015, 6, 3466–3470 RSC.
- W. Long, W. Qiu, C. Li, L. Song, G. Bai, G. Zhang and H. He, RSC Adv., 2016, 6, 40945–40952 RSC.
- Y. Huang, Z. Lin, H. Fu, F. Wang, M. Shen, X. Wang and R. Cao, ChemSusChem, 2014, 7, 2647–2653 CrossRef CAS PubMed.
- Q.-G. Zhai, C. Mao, X. Zhao, Q. Lin, F. Bu, X. Chen, X. Bu and P. Feng, Angew. Chem., Int. Ed., 2015, 54, 7886–7890 CrossRef CAS PubMed.
- H. Wang, X. Yuan, Y. Wu, G. Zeng, H. Dong, X. Chen, L. Leng, Z. Wu and L. Peng, Appl. Catal., B, 2015, 186, 19–29 CrossRef.
- F. Gándara, B. Gomez-Lor, E. Gutiérrez-Puebla, M. Iglesias, M. A. Monge, D. M. Proserpio and N. Snejko, Chem. Mater., 2008, 20, 70–76 CrossRef.
- B. Gomez-Lor, E. Gutiérrez-Puebla, M. Iglesias, M. A. Monge, C. Ruiz-Valero and N. Snejko, Inorg. Chem., 2012, 41, 2429–2432 CrossRef.
- J. Xia, J. Xu, Y. Fan, T. Song, L. Wang and J. Zheng, Inorg. Chem., 2014, 53, 10024–10026 CrossRef CAS PubMed.
- X. Li, D. Chen, Y. Liu, Z. Yu, Q. Xia, H. Xing and W. Sun, CrystEngComm, 2016, 18, 3696–3702 RSC.
- C. Volkringer, M. Meddouri, T. Loiseau, N. Guillou, J. Marrot, G. Férey, M. Haouas, F. Taulelle, N. Audebrand and M. Latroche, Inorg. Chem., 2008, 47, 11892–11901 CrossRef CAS PubMed.
- T. Lee, Y. H. Chang and H. L. Lee, CrystEngComm, 2017, 19, 426–441 RSC.
- K. Miura, M. Tomita, Y. Yamada and A. Hosomi, J. Org. Chem., 2007, 72, 787–792 CrossRef CAS PubMed.
- S. Yadav, M. Srivastava, P. Rai, B. P. Tripathi, A. Mishra, J. Singh and J. Singh, New J. Chem., 2016, 40, 9694–9701 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02802d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*